

Abstract
Degenerating neurons of Parkinson’s disease (PD) patient brains exhibit granules of phosphorylated extracellular signal-regulated protein kinase 1/2 (ERK1/2) that localize to autophagocytosed mito-chondria. Here we show that 6-hydroxydopamine (6-OHDA) elicits activity-related localization of ERK1/2 in mitochondria of SH-SY5Y cells, and these events coincide with induction of autophagy and precede mitochondrial degradation. Transient transfection of wild-type (WT) ERK2 or constitutively active MAPK/ERK Kinase 2 (MEK2-CA) was sufficient to induce mitophagy to a degree comparable with that elicited by 6-OHDA, while constitutively active ERK2 (ERK2-CA) had a greater effect. We developed green fluorescent protein (GFP) fusion constructs of WT, CA, and kinase-deficient (KD) ERK2 to study the role of ERK2 localization in regulating mitophagy and cell death. Under basal conditions, cells transfected with GFP-ERK2-WT or GFP-ERK2-CA, but not GFP-ERK2-KD, displayed discrete cytoplasmic ERK2 granules of which a significant fraction colocalized with mitochondria and markers of autophago-lysosomal maturation. The colocalizing GFP-ERK2/mitochondria granules are further increased by 6-OHDA and undergo autophagic degradation, as bafilomycin-A, an inhibitor of autolysosomal degradation, robustly increased their detection. Interestingly, increasing ERK2-WT or ERK2-CA expression was sufficient to promote comparable levels of macroautophagy as assessed by analysis of the autophagy marker microtubule-associated protein 1 light chain 3 (LC3). In contrast, the level of mitophagy was more tightly correlated with ERK activity levels, potentially explained by the greater localization of ERK2-CA to mitochondria compared to ERK2-WT. These data indicate that mitochondrial localization of ERK2 activity is sufficient to recapitulate the effects of 6-OHDA on mitophagy and autophagic cell death.
Keywords: autophagy, oxidative stress, mitochondria, mitogen activated protein kinases, 6-hydroxydopamine, Parkinson’s disease
Introduction
Cellular degradation of long-lived proteins and organelles is mediated by two major processes: ubiquitin-proteasome-mediated degradation and autophagy. A proper balance in cellular degradation of long-lived proteins and organelles is critical for neuronal survival, and alterations in degradation lead to aberrant accumulation of ubiq-uitinated proteins as seen in Parkinson’s disease (PD).1,2
Autophagy plays important roles in cellular differentiation and survival.3,4 In mammalian cells, macroautophagy and chaperone-mediated autophagy (CMA) have been most intensively studied. Macroautophagy, commonly referred to as simply autophagy, is a regulated process that involves the engulfment of cytoplasmic constituents into autophagosomes or early autophagic vacuoles (AVs), which then mature into autolysosomes or late AVs for degradation. The human autophagic machinery is well conserved from yeast and includes conjugating enzymes, which mediate covalent attachment of Atg12 to Atg5 and of Atg8/microtubule-associated protein 1 light-chain 3 (LC3) to nascent autophagic membranes.5 The Atg12-Atg5 complex is lost upon completion of the autophago-some, but some LC3 remains associated with AVs until degraded in lysosomes.6 While upstream signals that regulate bulk degradation during nutrient deprivation are well established, signals that regulate degradation of unwanted or damaged organelles are less understood.7,8
A proper balance in catabolic and anabolic activities is critical for survival of neurons. In some instances, autophagy plays beneficial, homeostatic roles that include clearance of protein aggregates,9-12 and maintenance of mitochondria,13,14 and axonal-synaptic morphology.15 On the other hand, excessive or imbalanced autophagy creates a condition of “autophagic stress” that contributes to neurodegeneration and cell death.16 In particular, excessive mito-chondrial degradation may be detrimental to neurons due to their high dependence on oxidative phosphorylation.17,18 A dual role for autophagy and ERK signaling has also been described in C. elegans survival during starvation induced autophagy.19
The extracellular signal-regulated protein kinases 1/2 (ERK1/2) regulate several neuronal functions including synaptic/neuritic remodeling, differentiation and survival.20-23 Alterations in the kinetics and subcellular localization of ERK1/2 activity promote cell death during ischemic, traumatic, toxic, and neurodegenerative injuries.24 In human PD brain tissues, we observed increased cytoplasmic ERK1/2 activity.25 Degenerating substantia nigra neurons display phosphorylated-ERK1/2 granules, many of which colocalize with mitochondria and AVs.26 Cytoplasmic granules involving nuclear transcriptional regulators are also observed in PD neurons, suggesting that altered subcellular function of signaling proteins contributes to neurodegeneration.27,28 ERK1/2 activity is necessary for autophagy and mitochondrial degradation induced by MPP+,29 the active metabolite of a parkinsonian neurotoxin.30,31 However, it is unknown whether elevated ERK activity itself is sufficient to induce autophagy and mitochondrial turnover.
6-Hydroxydopamine (6-OHDA), a redox cycling dopamine analog, is an oxidative neurotoxin that recapitulates many features of PD in animal models.32,33 Treatment of dopaminergic cells with 6-OHDA elicits robust activation of ERK1/2, and pharmacological inhibition of ERK activation enhances neuronal survival.34,35 Interestingly, 6-OHDA elicits a delayed phase of mitochondrial ROS production35 that is associated with increased mitochondrial ERK activation.26,35 The potential function(s) of mitochondrially localized ERK activation during neuronal injury are still undefined.
In this report, we show that increased expression of ERK2 increases toxin-induced mitochondrial autophagy in the neuronal cell line SH-SY5Y cells. There is activity-dependent ERK2 association with mitochondria, autophagosomes and lysosomes that is stimulated by neurotoxin injury or by expression of a constitutively active ERK2 (ERK2-CA). Moreover, ERK2-CA expression recapitulates the level of mitophagy elicited by toxin treatment. While small increases in ERK2 activity induced by transfection with ERK2-WT is sufficient to induce autophagy and cause lysosomal expansion, the levels of mitochondrial ERK2 localization and of mitophagy are dependent upon the degree of increased ERK activity, with ERK2-CA showing much greater effects than ERK2-WT. Overall, our results suggest that ERK2 is a modulator of both macroautophagy and mitophagy, but higher levels of kinase activity and/or mitochondrial ERK2 localization are needed to induce mitophagy and autophagic cell stress.
Results
6-OHDA induces autophagy and ERK phosphorylation
Previous reports showed that MPP+, mutations in the PD gene leucine rich repeat kinase 2, or neurotoxic doses of dopamine elicit autophagy associated with neurodegeneration.23,29,42 Here, we found that 6-OHDA, another PD neurotoxin induces autophagy, demonstrated by transmission electron microscopy and alterations in the distribution and electrophoretic mobility of LC3, a marker for autophagy.43 The effects of 6-OHDA treatment of cells were analyzed at the ultrastructural level. Cells treated with vehicle exhibited normal mitochondrial morphology and near absence of AVs (Fig. 1A). However, cells exposed for 4 hrs. to 6-OHDA showed decreased mitochondrial matrix densities and an increase in the number of degradative AVs and lysosomes (Fig. 1B). Early AVs were also observed (inset, Fig. 1B).
Figure 1.

6-OHDA induces autophagy and ERK phosphorylation. Representative electron micrographs of cells treated with vehicle control (A) or 6-OHDA (B) for 4 hrs. Note that 6-OHDA increases the number of AVs and lysosomes (ly) and reduces mitochondria (m) content, while the integrity of the nucleus (n) is unaffected. Inset demonstrates an early AV containing distinct cytoplasmic material (scale bars: 1 μm). (C) Representative cells stably expressing GFP-LC3 in the presence or absence of 6-OHDA. The green channel was extracted to grayscale and inverted. (D) Effects of 6-OHDA treatment on GFP-LC3 puncta number (left bar graph) and size (right bar graph) were determined by using the NIH Image J macro described in Methods. For comparative purposes, the dotted line indicates the average number and size of GFP-LC3 puncta that accumulate due to bafilomycin-A treatment. Bar graphs show compiled means ± s.e.m. of at least 3 independent experiments (ø:p < 0.05 vs. untreated, ⊕:p < 0.001 vs. 6-OHDA). (E) SH-SY5Y cells were treated with 6-OHDA or with vehicle for the indicated time points. Following treatment with toxin, cells were immunoblotted for total and phospho-ERK1/2 and for LC3. (F) Quantification of the average number of GFP-LC3 puncta of transiently transfected cells co-treated with 6-OHDA for 4 h with or without the MEK inhibitor U0126. Bar graph shows means ± s.e.m. (30–40 cells analyzed per condition) and is representative of at least 3 independent experiments (ø: p < 0.0001 vs. basal, ⊗: p < 0.001 vs. 6-OHDA). Note that inhibition of MEK with U0126 suppresses 6-OHDA induction of GFP-LC3 puncta but has little effect on baseline autophagy.
At the single cell level, 6-OHDA increased both the number and size of GFP-LC3 puncta in SH-SY5Y cells at 4 h compared to untreated cells (Fig. 1C and D). Moreover, the size of GFP-LC3 puncta in 6-OHDA treated cells was greater than GFP-LC3 puncta in cells treated with bafilomycin-A (Fig. 1D, right graph), which does not induce autophagy but increases AV numbers by inhibiting degradation.39,40 The increase in AV size is consistent with AV induction,39,44 and return of AV numbers to baseline after 24 h of 6-OHDA treatment suggests that degradation is not impaired. Increased autophagic flux is further supported by observations that bafilomycin-A prevented the drop in puncta numbers at 24 h (Fig. 1D, left graph). The unconjugated form of LC3 (LC3-I) resides in the cytosol while the phosphatidylethanolamine-conjugated form (LC3-II) is localized to autophagosomes and exhibits greater mobility by SDS-PAGE.6 6-OHDA also promoted increased LC3-II by western blot, indicative of increased autophagosomes (Fig. 1E, LC3).
As previously observed in a different central nervous system cell line,34 we found that treatment of SH-SY5Y cells with 6-OHDA resulted in sustained activation of ERK, which coincided with elevated autophagy (Fig. 1E, P-ERK). Co-treating cells with U0126 suppressed the induction of GFP-LC3 puncta by 6-OHDA, suggesting the involvement of MEK dependent ERK phosphorylation (Fig. 1F).
6-OHDA promotes mitochondrial degradation
Autophagy is the primary mechanism by which organelles are targeted for lyso-somal degradation.45 Given that human PD brain tissues exhibit P-ERK1/2 in autophagocytosed mitochondria,26 we wished to investigate the effects of 6-OHDA on mitochondrial ERK phos-phorylation and mitophagy in SH-SY5Y cells. A 4 h exposure to 6-OHDA elicited increased phospho-ERK1/2 in mitochondrial fractions compared to untreated and vehicle treated controls (Fig. 2A). To determine whether 6-OHDA induces mitochondrial autophagy, we transfected cells with GFP-LC3 prior to exposure to 6-OHDA, and quantified the percent colocalization of GFP-LC3 with mito-chondria as an index of autophagic sequestration. We found that treating cells with 6-OHDA significantly increased colocalization of GFP-LC3 puncta with mitochondria compared to vehicle treated cells as analyzed by confocal microscopy (Fig. 2B and C). Moreover, 6-OHDA-mediated mitophagy requires activation of ERK1/2 since co-treating cells with the MEK inhibitor U0126 completely abolished the increase in mitochondrial-LC3 colocalization (Fig. 2C). Next, we studied effects of 6-OHDA on mitochondrial content in cells. We found that 6-OHDA treatment caused mitochondrial fragmentation (Fig. 2D). Quantitative image analysis confirms decreased cellular content of mitochondria (Fig. 2G) and immunoblotting for human mitochondrial antigen of 110 kDa (MITO-P110) demonstrated a time-dependent loss of mitochondrial protein relative to β-actin (Fig. 2E). Co-treating cells with the autolysosomal degradation inhibitor bafilomycin-A or RNAi mediated knock-down of Atg7 and LC3 all significantly reversed loss of mitochondria observed at 5 h of 6-OHDA (Fig. 2F and G), demonstrating a role of autophagy in 6-OHDA induced mitochondrial loss.
Figure 2.

6-OHDA promotes mitochondrial ERK1/2 phosphorylation and mitophagy. (A) Representative Western blot showing p-ERK1/2 in cytosolic (C) and in mitochondrial fractions (M) of SH-SY5Y cells treated with 6-OHDA for 4 hrs. compared to vehicle treated cells (V). (B) Representative confocal images of MitoTracker Red-stained (MTR) SH-SY5Y cells stably expressing GFP-LC3, treated with vehicle control or with 6-OHDA for 4 hrs. The insets, which have been enlarged by 50%, show the merged and separate channels for GFP-LC3 (1) and MTR (2) from a second 6-OHDA treated cell. (C) Quantification of the percent of GFP-LC3 puncta colocalizing with mitochondria. Bar graph shows means ± s.e.m. (25–30 cells analyzed per condition) and is representative of at least 3 independent experiments ø:p < 0.005 vs. vehicle ⊕:p < 0.005 vs. 6-OHDA). (D) Representative epifluorescence images of cells treated with vehicle control or with 4 hours of 6-OHDA, then immunolabeled using an antibody specific for human mitochondrial antigen of 60 kDa (MITO-P60). Note that 6-OHDA induces mitochondrial fragmentation. Scale bars: 20 μm. (E) SH-SY5Y cells were treated with 6-OHDA or with vehicle control for different time points. Cells were analyzed for total protein levels of mitochondria by immunoblotting for mitochondrial human antigen of 110 kDa (MITO-P110) and reprobed for β-actin as a loading control. (F) SH-SY5Y cells were treated with vehicle or 6-OHDA in the absence and presence of bafilomycin-A, and analyzed by Western blot for levels of the 60 kDa mitochondrial membrane protein (p60), and the matrix protein pyruvate dehydrogenase (PDH). (G, left) SH-SY5Y cells transiently transfected with GFP were treated with 6-OHDA for 5 h in the presence or absence of balilomycin-A. Cells were fixed, and immunostained for mitochondrial human antigen of 60 kDa. The effects of 6-OHDA on mitochondrial content was analyzed by measuring the percent cellular area occupied by mitochondria in GFP transfected cells using a custom made NIH Image J algorithm. Data was normalized to basal mitochondria content exhibited by non-treated cells. The representative bar graph shows means ± s.e.m. (22–50 cells analyzed per condition) (ø:p < 0.0001 vs. untreated, ⊕:p < 0.005 vs. 6-OHDA). (G, right) After 3 days, cells co-transfected with GFP and with siRNA targeting human Atg7 or LC3 were treated with 6-OHDA for 5 h and analyzed using Image J as described above. Data was normalized to basal mitochondria content exhibited by cells treated with each respective siRNA and not receiving toxin. The representative bar graph shows means ± s.e.m. (22–50 cells analyzed per condition) (ø:p < 0.0005 vs. untreated, ⊕:p < 0.0005 vs. scrambled siRNA with 6-OHDA, ⊗:p < 0.05 vs. scrambled siRNA with 6-OHDA).
These observations indicate that 6-OHDA mediated cell injury involves robust activation of cytosolic and mitochondrial ERK1/2, accompanied by toxin induced autophagy and degradation of mito-chondria.
Activation of ERK2 is sufficient to promote autophagy
Given that 6-OHDA induced autophagy and mitophagy requires MEK-ERK signaling, we wanted to assess whether ERK2 regulates autophagy and/or mitophagy in the absence of toxin-induced injury. Since ERK2 is the isoform that is phosphorylated in human PD brain tissues and ERK2 is phosphorylated to a greater extent than ERK1 during 6-OHDA injury in culture,25,34 we analyzed the impact of transiently transfecting constructs of ERK2 on autophagy.
We found that transient expression of ERK2-WT, which induces a 3–4 fold increase in basal intracellular ERK activity (Fig. 3D), increased the number of GFP-LC3 puncta per cell compared to an empty vector (Fig. 3A). Transient expression of an N-terminal GFP fusion of ERK2-WT (GFP-ERK2-WT) increased the LC3-II/β-actin ratio compared to GFP, indicating that the N-terminal GFP tag does not inhibit this function of ERK2 (Fig. 3B). Likewise, transient expression of GFP-ERK2 increased the average number of endogenous LC3 puncta compared to GFP transfected cells or untransfected cells, as determined by LC3 immunofluorescence (data not shown). We also transfected cells with ERK2-CA, which promoted an 82-fold increase in cellular ERK activity (Fig. 3D). Interestingly, transient expression of either ERK2-CA or GFP-ERK2-CA induced no further increases in GFP-LC3 puncta and LC3-II/β-actin ratios compared to cells transiently expressing the corresponding ERK2-WT plasmid, suggesting that a four-fold increase in cellular ERK2 activity is sufficient to promote autophagy (Fig. 3A and B).
Figure 3.

Activation of ERK2 is sufficient to promote macroautophagy and mitophagy. (A) Quantification of the average number of GFP-LC3 puncta per cell in SH-SY5Y cells transiently co-transfected with GFP-LC3 and either vector, wild-type ERK2, constitutively active ERK2 (ERK2-CA), or kinase deficient ERK2 (ERK2-KD). ⊘:p < 0.005 vs. Vector.+:p < 0.0001 vs. ERK2-WT). The bar graph shows means ± s.e.m. (25–30 cells analyzed per condition) and is representative of >3 independent experiments. Supplementary Figure S5C and D shows that pharmacologic inhibition of MEK inhibits ERK2-WT-induced GFP-LC3 puncta. (B) Representative LC3 Western blot of SH-SY5Y cells transfected with GFP control or the indicated N-terminal GFP fusion constructs of ERK2 and re-probed for β-actin. The bar graph demonstrates LC3-II/β-actin ratios for three replicates of each transfection condition analyzed on the same Western blot (ø:p < 0.014 vs GFP, ⊕:p < 0.05 vs.GFP-ERK2-WT). (C) Representative confocal microscopy images of cells transiently co-expressing GFP-LC3 and vector, wild-type or the indicated mutant plasmids of ERK2. To image mitochondria, cells were loaded with MitoTracker Red (MTR) dye for 30 minutes prior to imaging by confocal microscopy (scale bar: 20 μm). (D, left) Intracellular ERK activities measured using the Elk1 trans-Reporting System in SH-SY5Y cells expressing different forms of ERK2. The Elk-1 luciferase reporter plasmid was co-transfected with the indicated ERK2 plasmids and activity was measured 48 hrs. following transfection. Bar graph shows means ± s.e.m of the fold increase relative to vector control (4–8 wells per transfection condition) and is representative of 3 independent experiments. (⊗:p < 0.0001 vs Vector, ⊕:p < 0.0001 vs ERK2-WT, ø:p < 0.009 vs. ERK2-WT). (D, right) Representative bar graph showing the percent of GFP-LC3 puncta colocalizing with mitochondria in cells expressing the indicated plasmids in the presence or absence of 6-OHDA. Bar graph show means ± s.e.m. (25–30 cells analyzed per condition) and are representative of at least 3 independent experiments (⊗:p < 0.003 vs. vector, ⊕:p < 0.0005 vs ERK2-WT, ø:p < 0.01 vs. ERK2-WT). Supplementary Figure S3 shows that transient expression of active ERK or MEK elevates mitophagy to levels similar to 6-OHDA as measured by GFP-LC3/colocalization and loss of cellular mitochondrial content.
To determine the importance of ERK2 kinase activity, we used the K52R mutant of ERK2 (ERK2-KD), which shows reduced activity compared to wild type ERK2. Transient expression of untagged ERK2-KD or GFP-ERK2-KD failed to induce autophagy, suggesting that a basal level of ERK2 kinase activity is required for its effects on autophagy (Fig. 3A and B). Likewise, the MEK inhibitor U0126 completely abolished ERK2-WT mediated autophagy and mitophagy confirming that autophagy/mitophagy induced by ERK2 requires kinase activity (Fig. S5C and D).
Activation of ERK2 is sufficient to promote mitophagy in the absence of toxin injury
Transient expression of ERK2-WT increased the colocalization of GFP-LC3 to mitochondria when compared to cells expressing an empty vector (Fig. 3C and D, right graph), indicating that increased ERK2 activity is sufficient to promote mitophagy. The addition of 6-OHDA to ERK2-WT expressing cells showed only a trend towards a further increase, suggesting that 6-OHDA-induced mitophagy is predominantly mediated by ERK2 activation (Fig. S3A).
The effects of ERK2 on mitophagy is more tightly dependent upon the degree of elevated cellular ERK2 activity than those observed using LC3 measures of general autophagy, since transient expression of ERK2-CA significantly increased colocalization of GFP-LC3 puncta with mitochondria even further compared to ERK2-WT expression (Fig. 3D). Transient expression of constitutively active MEK2 S222D/S226D (MEK-CA), which elevates ERK-mediated Elk1 activation reporter activity by 23-fold, also increased mitophagy under basal conditions (Fig. S3A). On the other hand, we found that the ERK2-KD mutation abolished the effects of ERK2 on mitophagy (Fig. 3C and D) as did administration of the MEK inhibitor U0126 (Fig. S5D).
As the autophagic sequestration of mitochondria appears to be dependent upon the degree of ERK activity, we studied the effects of different ERK2 constructs on mitochondrial content in the presence or absence of 6-OHDA. Transient expression of GFP-ERK2-CA induced significantly decreased mitochondrial content under basal conditions (Fig. S3C). Treating cells with 6-OHDA caused a further decrease in cellular mitochondrial content in both GFP-ERK2-WT and GFP-ERK2-CA expressing cells compared to GFP transfected cells while GFP-ERK2-KD blocked the 6-OHDA induced mito-chondrial loss (Fig. S3C). In aggregate, these results demonstrate that activation of ERK2 is necessary and sufficient for inducing autophagic sequestration and degradation of mitochondria.
6-OHDA induces the formation of ERK2 granules that are degraded by lysosomal mechanisms
To begin addressing the potential role of mitochondrial ERK2 localization in regulating mitophagy, we constructed GFP-tagged derivatives of the different ERK2 plas-mids to study their subcellular distribution.
Confocal microscopy analysis of individual live cells demonstrated that GFP-ERK2-WT shows a mixed cytoplasmic and nuclear distribution with only 12% of cells showing an exclusively cytoplasmic distribution. However, treating cells with 6-OHDA for 4 h increased the proportion of cells containing exclusive cytoplasmic localization of GFP-ERK2-WT and decreased the number of cells displaying nuclear localization (Fig. 4A). Transient expression of GFP-ERK2-WT resulted in discrete GFP granules that were absent in cells transiently expressing GFP vector alone (data not shown), some of which colocalized to mitochondria as determined by confocal microscopy (Fig. 4B). Western blot analysis confirmed approximately equal expression of WT and mutant fusion constructs, and analysis using a pan-ERK antibody shows that the tagged constructs are expressed at low levels compared to endogenous ERK2, even after correcting for transfection efficiencies of 24% to 29% (Fig. 4C; Fig. S1A). There is no evidence of significant protein aggregation in the presence or absence of 6-OHDA, as the GFP-tagged proteins could be accounted for in Triton X-100-soluble fractions, as assessed by two independent methods (Fig. S1B and C).
Figure 4.

GFP-ERK2 exhibits activity dependent colocalization with mitochondria. (A) Quantification of the percentage of cells expressing GFP-ERK2-WT demonstrating the specified subcellular distribution of ERK2 for the indicated time points of 6-OHDA treatment. Note that 6-OHDA induces a time-dependent redistribution of GFP-ERK2-WT from the nucleus to the cytosol. The distribution bar graphs are representative of at least 3 independent experiments. (B) Representative confocal images of cells transiently expressing the indicated N-terminal GFP fusion constructs of ERK2 treated with vehicle control (left) or with 6-OHDA for 4 hrs. in the presence (right) or absence (middle) of bafilomycin-A. Mitochondria were imaged by loading cells with MitoTracker Red dye 30 min prior to imaging with the confocal microscope (scale bar: 20 μm). For some experiments mitochondria were visualized by co-transfecting cells with mito-RFP and GFP-ERK2 constructs, yielding similar colocalization results (not shown). White arrows point to ERK2-GFP puncta colocalizing with mitochondria. Note that 6-OHDA (top middle), but not vehicle control (top left), induces mitochondrial fragmentation and loss. Expression of ERK2-KD reduced mitochondrial loss, but not fragmentation (bottom middle). Supplementary Figure S1B and C shows that 6-OHDA does not induce any increase in triton-insoluble GFP or GFP-ERK2, and that comparable levels of GFP-ERK-WT and -CA are solubilized by triton X-100 versus SDS sample buffer, indicating that there is no aggregation occurring. (C) Representative Western blot of cells transfected with GFP or the indicated N-terminal GFP fusion constructs of ERK2 and immunoblotted for GFP and for total ERK1/2. Arrows heads point to immunoreactive bands corresponding to GFP-ERK2 constructs containing a predicted molecular weight of 73 kDa. Transfection efficiencies of all four GFP containing plasmids were ~25% across all experiments (Suppl. Fig. S1A). (D) Quantification of the average number of GFP-ERK2 granules per cell in untreated or 6-OHDA treated SH-SY5Y cells transiently transfected with GFP as a control or the indicated GFP fusion constructs of ERK2. Summary bar graph means ± s.e.m. of n = 3–7 experiments with 25–30 cells each (ø:p < 0.05 vs. GFP untreated, ⊕:p < 0.01 vs. untreated GFP-ERK2-WT, ⊗:p < 0.05 vs. GFP-ERK2-WT treated with 6-OHDA). (E) Summary quantification of the percent of GFP-ERK2 granules showing mitochondrial colocalization in the presence or absence of 6-OHDA (means ± s.e.m. of n = 3–7 experiments with 25–30 cells each; ø:p < 0.05 vs.GFP, ⊕:p < 0.005 vs untreated GFP-ERK2-WT, Φ:p < 0.05 vs. untreated GFP-ERK2-WT). Supplementary Figure S4 shows that the different plasmids show the same responses to MPP+, and Supplementary Figure S5A and B show that MEK activation of ERK2-WT is required for increase in number and mitochondrial colocalization of GFP-ERK2-WT granules. (F) Summary quantification of the percent of GFP-ERK2 granules colocalizing with mitochondria in the presence or absence of bafilomycin-A (means ± s.e.m. of n = 3–7 experiments with 25–30 cells each; ø:p < 0.02 vs. untreated GFP, ⊕:p < 0.01 vs. untreated GFP-ERK2-WT, ⊗:p < 0.05 vs. GFP-ERK2-WT with bafilomycin-A). Supplementary Figure S2 shows that bafilomycin-A also significantly increases numbers and mitochondrial colocalization of GFP-ERK2 granules in the presence of 6-OHDA.
Treating GFP-ERK2-WT transfected cells with 6-OHDA for 4 h significantly increased both number and mitochondrial localization of GFP-ERK2-WT granules per cell (Fig. 4B, D and E). Treatment with MPP+, another PD toxin that produces mitochondrial oxidative stress, gave similar results (Fig. S4). Furthermore, mitochondrial localization of GFP-ERK2 granules was dependent upon ERK activity levels, since transient expression of GFP-ERK2-CA promoted both a significant increase in the average number and mitochondrial localization of granules compared to GFP-ERK2-WT transfected cells, while GFP-ERK2-KD or pharmacological inhibition of MEK by U0126 blocked these effects (Fig. 4D and E; Fig. S5A and B) in either the presence and absence of 6-OHDA.
These results suggest that ERK2 forms activity-dependent cytoplasmic granules that are degraded by autophagy. Indeed, inhibition of autophagolysosomal fusion/degradation resulted in significantly increased levels of GFP-ERK2-WT granules in the presence or absence of 6-OHDA (Fig. S2A), indicating that ERK2 granules induced by either 6-OHDA treatment or the CA mutation are degraded by autophagolysosomal mechanisms.
Mitochondria colocalized with ERK2 granules undergo autophagic degradation
We had previously reported that endogenous ERK1/2 granules colocalized with mitochondria, AVs, intracellular filaments, and a small number of early endosomes in degenerating neurons of PD brain tissue.26 Moreover, ERK1/2 granules localized with aberrant mitochondria were engulfed by AVs raising the possibility that mitochondria associated with ERK2 granules undergo autophagic sequestration and degradation.26 To address this, we treated cells transiently expressing GFP fusion constructs of ERK2 with bafilomycin-A, and analyzed its effects on GFP-ERK2 puncta number and mitochondrial colocalization as a measure of flux.43 Inhibiting fusion/degradation with bafilomycin-A significantly increased mitochondria/GFP-ERK2 colocalizing granules in cells transiently expressing GFP-ERK2-WT compared to untreated cells, while bafilomycin-A treatment had no effect on cells transiently expressing GFP (Fig. 4F). The increase in colocalizing mitochondria/ GFP-ERK2 is also observed using a second independent inhibitor of lysosomal degradation E64-D (data not shown). We also found that co-treating cells with 6-OHDA and bafilomycin-A for 4 hrs. was sufficient to further elevate number and mitochondrial colocaliza-tion of GFP-ERK2 granules in cells expressing GFP-ERK2-WT or GFP-ERK2-CA compared to cells treated with 6-OHDA alone (Fig. S2A and B). Thus, mitochondria/GFP-ERK2 colocalizing granules are cleared by similar mechanisms in 6-OHDA treated cells.
Interestingly, while 6-OHDA treatment of GFP-ERK2-WT expressing cells increased even further the numbers of ERK2-mitochondria colocalizing granules, the numbers of granules elicited by GFP-ERK2-CA alone were equivalent to those elicited by a combination of ERK2-WT and toxin (Fig. 4D and E), suggesting that 6-OHDA mediated activation of ERK is the major mechanism of 6-OHDA-induced mitophagy. Paradoxically, the addition of 6-OHDA to GFP-ERK2-CA-expressing cells resulted in a non-significant trend of decreased percent of the GFP-ERK-CA granules showing mitochondrial colocalization (Fig. 4E), but had no effect on the average number of GFP-ERK2-CA granules per cell (Fig. 4D). One possibility is that the combination of constitutively active ERK2 and 6-OHDA resulted in more effective degradation of ERK2-mitochondria colocalizing particles. Indeed, co-treatment of ERK2-CA expressers with 6-OHDA and bafilomycin-A restored levels of mitochondrial colocalization (data not shown). Similarly, transfection with MEK-CA in the presence of 6-OHDA also shows reduced colocalization of GFP-LC3 with mitochondria compared to MEK-CA in the absence of toxin, and the addition of bafilomycin-A more than reverses this drop (Fig. S3B), suggesting that the combination of constitutively active kinase and 6-OHDA enhances mitophagic clearance.
GFP-ERK2 granules colocalized with mitochondria, lysosomes and autophagosomes
As ERK2 granules associated with mito-chondria undergo autophagic degradation, we wanted to determine whether GFP-ERK2 also co-localize with AVs and lysosomes, two components of the autophagolysosomal axis. Cells transfected with different constructs of GFP-ERK for two days were analyzed by confocal microscopy for colocalization of ERK2 with mitochondria and lysosomes. A fraction of ERK2 granules were associated with AVs, as observed by colocalization of GFP-ERK2 puncta with cherry-LC3 puncta under basal conditions (Fig. 5A). This was not due to nonspecific GFP aggregation, as cells expressing GFP showed mostly diffuse distribution of cherry-LC3. Conversely, transient transfec-tion of a N-terminal RFP fusion of wild-type ERK2 (RFP-ERK2) demonstrated partial colocalization of RFP-ERK2 granules with either GFP-LC3 (Fig. 5B) or endogenous LC3 puncta (Fig. 5C).
Figure 5.

GFP-ERK2 granules colocalize with autophagosomes and lysosomes. (A) Representative confocal sections of 6-OHDA treated cells transiently co-expressing GFP vector or GFP-ERK2 and an N-terminal RFP fusion of LC3 (Cherry-LC3), or (B) co-expressing an N-terminal RFP fusion of ERK2 (RFP-ERK2) and GFP-LC3, or (C) expressing RFP-ERK2 and stained for endogenous LC3 by immunofluorescence with blue nuclear counterstain (scale bar: 20 μm). Arrows point to LC3 puncta colocalizing with ERK2 granules. (D) Representative confocal sections of LysoTracker Red (LTR) stained cells expressing with GFP vector control or the indicated N-terminal GFP fusion constructs of ERK2 (scale bar: 20 μm). Arrows point to GFP-ERK2 granules colocalizing with lysosomes (E) Summary quantification of the percent of GFP-ERK2 granules colocalizing with lysosomes per cell (means ± s.e.m. of n = 3–5 experiments with 25–30 cells each; ⊕:p < 0.05 vs GFP, ø: p < 0.05 vs GFP-ERK2-WT. (F) Summary quantification of the average number of lysosomes per cell (means ± s.e.m. of n = 3–5 experiments with 25–30 cells each; ø:p < 0.05 vs. GFP, ⊗:p < 0.05 vs. GFP-ERK2-WT).
Confocal microscopy analyses also revealed that a fraction of ERK2 granules colocalized with lysosomes in cells transiently expressing GFP-ERK2-WT. Low levels of increased ERK2 activity were sufficient to induce colocalization of ERK2 granules with lysosomes since GFP-ERK2-CA did not elicit further increases in colocalization (Fig. 5D and F). However, some basal level of ERK2 activity is necessary for colocalization of GFP-ERK2 granules with lysosomes as transient expression of GFP-ERK-KD gave similar colocalization levels as GFP transfection alone (Fig. 5D and E). Lysosomal expansion is another indicator of macroautophagy upregulation in eukaryotes.44 Transient expression of either GFP-ERK2-WT or GFP-ERK2-CA increased the average number of lysosomes per cell compared to cells transiently expressing GFP or GFP-ERK2-KD (Fig. 5F).
Persistent ERK2 activation promotes autophagic stress
Given that transient expression of active forms of ERK2 promotes not only mitophagy, but also macroautophagy, both of which are central features of 6-OHDA toxicity (Fig. 3), we hypothesized that persistent autophagic cell stress mediated by sustained ERK2 activation is detrimental to cells. The percentages of GFP transfected cells that were labeled with propidium iodide (PI) as a marker of cell death were determined following an overnight exposure to vehicle or 6-OHDA (85 μM). Transient expression of GFP-ERK2-WT significantly increased both basal and 6-OHDA induced cell death compared to cells transfected with the GFP control plasmid (Fig 6). Interestingly, transient expression of GFP-ERK2-CA potentiated basal cell death to levels comparable with that of combined GFP-ERK2-WT and 6-OHDA exposure, suggesting that sustained ERK2 activation induced by either toxin treatment or mutation is toxic to cells (Fig. 6). Not surprisingly, GFP-ERK2-KD, which shows dominant inhibitory effects on endogenous ERK activity,38 also increased cell death compared to cells expressing GFP alone, indicating that a basal level of ERK2 activity is required for survival. On the other hand, ERK2 activity contributes to 6-OHDA induced cell death since expression of GFP-ERK2-KD had a partial, but significant, protective effect against 6-OHDA compared to GFP-ERK-WT transfected cells (Fig. 6), in accord with previous studies using MEK inhibitors.34 The dual effects of basal versus injury-induced ERK activation have also been reported with MPP+ toxicity.47
Figure 6.
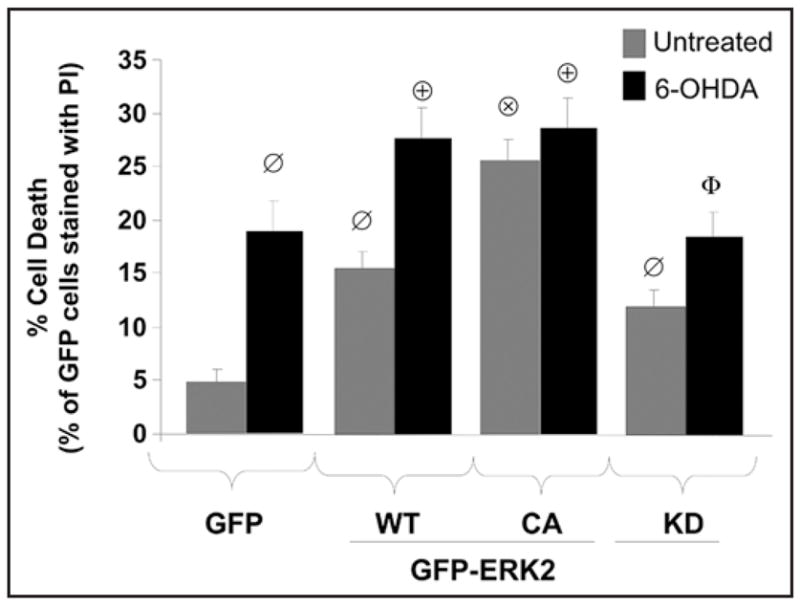
Autophagic cell stress induced by ERK2 expression promotes basal and 6-OHDA induced cell death. Cells transiently expressing GFP as a control or the indicated N-terminal GFP fusion constructs of ERK2 were analyzed for cell death by quantifying the percentage of GFP positive cells permeable to propidium iodide (PI) per epifluorescence micrograph field. The representative bar graph shows means ± s.e.m. (150–200 cells analyzed per condition collected from 7–10 random microscopic fields) and is representative of at least 3 independent experiments (ø:p < 0.005 vs. GFP, ⊗:p < 0.001 vs untreated GFP-ERK2-WT, ⊕:p < 0.05 vs. GFP with 6-OHDA, Φ:p < 0.05 vs. GFP-ERK2-WT with 6-OHDA).
Discussion
ERK1/2 has been implicated in critical neuronal functions including differentiation, plasticity and survival. On the other hand, sustained ERK1/2 activation plays a detrimental role in several central nervous system diseases.24 ERK phosphorylation is observed in PD and Lewy Body dementia 25, epilepsy 48, Alzheimer’s disease 49,50, and the penumbra region of ischemic strokes in patients and in rat models.51-53 ERK activation is also observed in 6-OHDA and MPP+ models of parkinsonian injury, and pharmacological MEK inhibitors confer neuroprotection in several neuronal cell lines and in primary midbrain dopaminergic neurons 29,34,47 (Fig. S6).
The disparate effects of ERK1/2 signaling on neuronal survival are likely explained by alterations in the subcellular compartmentalization, and therefore downstream targets, of ERK1/2 activity.24 Early transient ERK1/2 activation accompanied by nuclear translocation is associated with enhanced neuronal survival,21,22,54,55 while cytoplasmic ERK1/2 without evidence of nuclear localization is typical of degenerating neurons of Alzheimer’s disease,50 and in PD and related models.25,35 While it is not possible to tell from human autopsy studies whether ERK1/2 regulates mitochondrial turnover or is passively localized to mitochondria and AVs, the current study indicates that activity level-dependent localization of ERK2 to mitochondria is sufficient to induce mitophagy. Moreover, dominant interfering ERK2-KD expression and the MEK inhibitor U0126 prevented both generation and colocalization of ERK2 granules with mitochondria, suppressing 6-OHDA induced mitophagy (Fig. 4B, D and E; Fig. S5). Subsets of ERK2 granules also colo-calized with autophagosome and lysosome markers (Fig. 5), and bafilomycin experiments suggest that ERK2 granules are degraded with colocalizing mitochondria in these compartments (Fig. 4F and Fig. S2). These data support a model whereby mitophagy is induced by elevated mitochondrial ERK2 activity, and suggest that persistently dysregulated ERK2 signaling leads to autophagic/mitophagic stress (Fig. 7).
Figure 7.
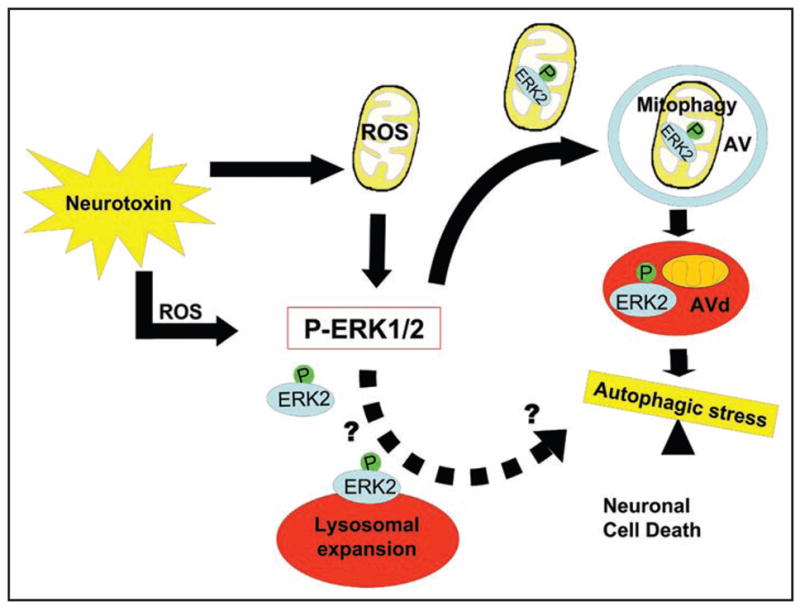
Schematic depicting a proposed role for ERK2 in mediating autophagic cell stress. The PD toxin 6-OHDA induces cytosolic and mitochondrial ROS, inducing robust phosphorylation of ERK2 and the formation of granules of ERK2 that colocalize with mitochondria, AVs and lysosomes (red ovals). Persistent activation of ERK2 induced by oxidative stress or by activating mutations promotes autophagy and mitophagy leading to autophagic cell stress and neurodegeneration. Although persistent ERK2 activation by toxin or experimental mutations induces lysosomal colocalization of ERK2 granules and lysosomal expansion, more evidence is needed to determine whether ERK2 might also be directly targeted to lysosomes and what role this signaling may play (hatched arrow).
The mechanism by which activated ERK2 is enriched in mito-chondria is still unknown. While the involvement of mitochondrial reactive oxygen species in redox activation of ERK1/2 suggests a role for in situ phosphorylation,35,56 the GFP-ERK2 data supports a mechanism involving mitochondrial translocation. Given that phospho-activated ERK represents only a few percent of total cellular ERK,57 trafficking of this component to a subset of mitochondria may not be detected by standard biochemical fractionation methods. Thus, it is possible that both mechanisms contribute to altered distribution of activated ERK1/2, in analogy with nuclear translocation in which individual proteins shuttle in and out with enhanced nuclear retention of the activated subset.58
In addition to being carried to lysosomes in association with autophagocytosed mitochondrial cargo, ERK1/2 has been reported to bind lysosomal scaffold proteins in non-neuronal cells.59 A C-terminal truncation in MEK1 is proposed to unmask a cryptic lysosomal targeting signal, resulting in lysosomal MEK/ERK signaling and decreased viability in Hela cells.60 Thus, ERK2 could conceivably contribute to autophagic stress through multiple mechanisms involving either induction or delayed lysosomal maturation, as suggested for the carcinogen lindane.61 In other systems, however, several lines of evidence suggest that ERK promotes rather than inhibits lysosomal degradation. In colon carcinoma cells, ERK activity is necessary for starvation-induced increases in both sequestration and degradation of [14C] valine-labeled long-lived proteins.62 In MPP+ treated neuronal cells, inhibition of MEK/ERK signaling blocks toxin-induced mitochondrial degradation assessed by EM, immunofluorescence, and western blot analysis.29 In 6-OHDA-treated cells, inhibition of either autophagy induction or lysosomal fusion/degradation significantly restored cellular mitochondrial content, as detected by antibodies to several mitochondrial proteins (Fig. 2F and G). While we cannot completely exclude the possibility that 6-OHDA may also reduce immunoreactivity to mitochondrial proteins through other mechanisms such as oxidative damage, the GFP-ERK2 bafilomycin studies indicate effective lysosomal degradation of ERK2-mitochondria colocalizing particles (Fig. 4F and Fig. S2B). Whether or not direct targeting of ERK to lysosomes occurs during parkinsonian neurodegeneration, and potential effects of ERK activity on lysosomal function, remain to be discovered.
What are the mechanisms by which ERK2 regulates mitophagy and macroautophagy? It is interesting to note that both ERK2 localization to mitochondria and its ability to stimulate mito-chondria-LC3 colocalization is highly dependent on the degree of increased ERK2 activity, with ERK-CA constructs showing much greater effects than ERK-WT (Figs. 3D, 4E and F). In contrast, the modest increase in total ERK activity afforded by transfection with ERK2-WT is sufficient to promote macroautophagy as evidenced by LC3-II shift, LC3 puncta numbers and lysosomal expansion, with no further increases induced by the CA mutation (Fig. 3A and B; Fig. 5E and F). ERK activity is required for both general macroau-tophagy and mitophagy, as transient transfection of GFP-ERK2-KD fails to increase basal autophagy while inhibiting 6-OHDA-induced autophagy, and the MEK inhibitor U0126 reduces both toxin- and ERK2 overexpression-induced macroautophagy and mitophagy (Figs. 1F, 2C, 3; Fig. S5C and D). The exact mechanism by which ERK activation promotes autophagy or mitophagy remain to be discovered, and may include phosphorylation of protein targets or activation-related alterations in conformation or binding partners. In either case, activation of ERK1/2 is necessary and sufficient to induce macroautophagy, mitochondrial localization and mitophagy.
Future studies are required to determine if mitochondrially localized ERK signaling functions upstream or downstream of mitochondrial injury and dysfunction. Neither pharmacological inhibition of MEK nor transient expression of GFP-ERK2-KD expression protects against the mitochondrial fragmentation and swelling induced by 6-OHDA (Fig. 4B; GFP-ERK2-KD/6-OHDA vs. GFP-ERK2-WT/6-OHDA). MEK inhibitors also do not reduce 6-OHDA-induced mitochondrial superoxide production35 or mito-chondrial swelling in the acute MPP+ model.29 Thus, we hypothesize that increased mitochondrial ERK activity may serve as a sensor of mitochondrial injury to initiate mitophagy. Given that mitochon-drial ERK signaling can promote mitochondrial dysfunction and decreased respiration in renal cells,63 however, it is also possible that ERK signaling promotes mitochondrial damage in other contexts.
Interestingly, our data show that localization of ERK2 granules to mitochondria plays a more essential role in increasing mitochondrial-LC3 colocalization than in inducing generalized measures of LC3 lipidation. As mitochondrial ROS production is temporally correlated with mitochondrial ERK activation,35 and antioxidants can prevent 6-OHDA-mediated ERK activation,25,35,56 candidate signals to trigger damage-induced mitophagy include oxidation of mitochondrial proteins or lipids,64 decreased mitochon-drial membrane potential65,66 and/or altered phosphorylation targets on mitochondria.18
Neurons are particularly vulnerable to autophagic stress,4,16 and factors that promote autophagic stress lead to the aberrant accumulations of AVs and lysosomes.18,67 As mitochondrial dysfunction plays a prominent role in PD pathogenesis,68,69 ROS mediated mitophagy may initially represent an adaptive response to “sequester” damaged mitochondria. However, excessive mitochondrial autophagy may commit neurons to cell death.70 Autophagy-induced neuritic remodeling would also be expected to play both physiologic71 and pathologic roles, as observed in a mutant leucine rich repeat kinase 2 model of PD.23 In both toxin and genetic models of PD, ERK1/2 serves a pivotal role in regulating autophagic stress (Fig. 7). Understanding the mechanisms by which activated ERK1/2 regulates autophagy and mitochondrial homeostasis will offer important insights for PD and other neurological diseases. In particular, identification of mito-chondrial signals that trigger ERK1/2-dependent mitophagy may be beneficial for developing future therapies to combat harmful levels of mitophagic stress in PD.16
Materials and Methods
DNA constructs
Wild-type ERK2, the matched constitutively active (CA) plasmid that has the dual activating mutations L73P and S151D36 and kinase deficient (KD) ERK2 (K52R) mutation in pCMV5.0 were obtained from Natalie Ahn (University of Colorado at Boulder). The construct for ERK2-WT in pEGFP-C1,37 was provided by Rony Seger (Weizmann Institute of Science). To generate constitutively active GFP tagged ERK2 (GFP-ERK2-CA), a BamHI-ApaI fragment of pCMV5-ERK2 (L73P, S151D) and a novel SspI site, was subcloned into the corresponding sites in GFP-ERK2-WT. Colonies were screened for the novel SspI site and SspI positive colonies were sequenced to confirm the introduction of mutations. To generate kinase-deficient GFP tagged ERK2 (GFP-ERK2-KD), pCEP4-ERK2 (K52R), a gift of Melanie Cobb (University of Texas Southwestern, Dallas, Texas),38 was digested with BamHI-ApaI and subcloned into the corresponding sites in GFP-ERK2-CA. Colonies were screened for the loss of the SspI site and sequenced to confirm the introduction of the kinase dead (KD) mutation. To generate pCMV5.0 empty vector, the DNA insert coding for ERK2-CA in pCMV5.0 was excised by digesting with EcoRI and XbaI. The empty vector was gel purified and religated following treatment with Klenow fragment of DNA polymerase I (Promega, Madison, Wisconsin).
Other DNA plasmids used in these studies include FLAG-tagged wild-type ERK2 in pCDNA3.1 from Scott T. Eblen (Medical University of South Carolina, Charleston, South Carolina), untar-geted RFP provided by Dr. Yongjian Liu (University of Pittsburgh, Pennsylvania), the mitochondrial targeting sequence of human cytochrome oxidase subunit VIII fused to the N-terminus of RFP (Mito-RFP) from Ian Reynolds (Merck Research Laboratories, West Point, Pennsylvania), RFP-ERK2 from Robert Lefkowitz (Duke University, Durham, North Carolina), cherry-LC3 from Dr. Jayanta Debnath (University of California, San Francisco) and LC3 fused to red (RFP-LC3) or green (GFP-LC3) fluorescent proteins from Tamotsu Yoshimori, (National Institute of Genetics, Japan). Constitutively active MEK2 (S222D/S226D) was purchased from Upstate Biotechnology, Lake Placid, New York).
Tissue culture
The SH-SY5Y cell line (American Type Culture Collection, Rockville, Maryland), a human neuroblastoma cell line that expresses tyrosine hydroxylase (TH) and dopamine transporter, was maintained in antibiotic-free Dulbecco’s modified Eagle’s medium (BioWhittaker, Walkerville, Maryland) supplemented with 10% fetal bovine serum (Gibco/Invitrogen, Carlsbad, California), 15 mmol/L HEPES, and 2 mmol/L glutamine (BioWhittaker), in at 37°C. SH-SY5Y cells were a humidified incubator with 5% CO2 treated with dH2O (vehicle) or freshly dissolved 6-hydroxydopamine concentration (85 μM) that (Sigma, St. Louis, Missouri) at the LD50 we determined for this cell line using the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium inner salt (MTS) assay (Promega, Madison, Wisconsin) at 18 h, or with a higher dose (120 μM). Some cultures also received media control, 1-methyl-4-phenylpyridinium (MPP+) at 2.5 mM (Sigma, St. Louis, Missouri), the MAPK/ERK kinase (MEK) inhibitor U0126 (10 μM; Cell Signaling, Beverly, Massachusetts), or bafi-lomycin-A (10 nM, Calbiochem, San Diego, California), which inhibits autolysosomal degradation through effects on fusion and/or lysosomal acidification.39,40
Generation of GFP-LC3 stable cell line
SH-SY5Y cells at a density of 20% to 30% confluency were transduced with moloney mouse leukemia virus (MoMLV) particles pseudotyped with vesicular stomatits virus G (VSV-G) protein and packaged with a puro-BABE vector containing the cDNA encoding a N-terminal GFP fusion of LC3 (GFP-LC3). At the time of transduction, conditioned media containing 8 ug/ml polybrene (Hexadimethrine bromide, Sigma) was used to dilute MoMLV particles to the desired MOI. Twenty-four hours following transduction, GFP-LC3 expressing cells were selected in media containing 2 μg/ml puromycin for three days followed by a recovery period in regular media. The GFP-LC3 line was amplified and seeded in larger tissue culture plates and tested for proper expression of GFP-LC3 and autophagic responsiveness.
Transmission electron microscopy
Cells grown in 6-well tissue culture dishes were rinsed with PBS, fixed in 2.5% gluteraldehyde overnight at 4°C, and processed as previously described29 for imaging on a JEOL JEM 1210 transmission electron microscope.
Liposome-mediated transfection
SH-SY5Y cells were trans-fected with the above DNA plasmids using LipofectAMINE 2000 (Invitrogen). For cells grown in 2-well chambered coverslips or twelve well plates, 1 μg of DNA diluted in OPTIMEM was mixed with liposomes at a final concentration of 0.10%. These transfection conditions do not induce LC3 gel shift or GFP-LC3 puncta.
Confocal microscopy
Two days following transfection, cells were stained with 100 nM of MitoTracker Red dye 580 or LysoTracker Red DND-99 (Molecular Probes, Eugene, Oregon) to analyze for colocalization of GFP-ERK2 with mitochondria and lysosomes, respectively, or for colocalization of GFP-LC3 with mitochondria as a measure of mitophagy. For some experiments, cells were treated with vehicle control or with 6-hydroxydopamine to induce injury, and live cells imaged using a FluoView 1000 laser scanning confocal microscope.
Immunofluorescence microscopy
For counting endogenous AVs in cells, SH-SY5Y cells co-transfected with GFP vector and untagged ERK2 constructs in four well-chambered coverglasses (Nunc) were washed once in PBS, fixed with 3.7% paraformaldehyde with 0.1% Triton X-100/PBS and then blocked in 5% normal donkey serum for at least 35 minutes, then stained with rabbit anti-LC3 (1:2000)41 overnight at 4°C. To analyze the effects of 6-OHDA on mitochon-dria, cells were incubated with mouse anti-mitochondrial antigen 60KD (clone 113–1; 1:100; BioGenex, San Ramon, California) overnight at 4°C. Cells were then washed in PBS 3 to 5 times for 5 minutes per washed followed by incubation with Alexa 568 conjugated donkey anti-rabbit antibodies (Molecular Probes, Eugene, Oregon) or with Cy3-conjugated donkey anti-mouse (1:400; Jackson Immuno-Research Laboratories, West Grove, Pennsylvania). Cells were counterstained with 1.25 μg/ml DAPI to visualize nuclei, and imaged for LC3 puncta and mitochondrial morphology using an IDX71 Olympus fluorescence microscope (Olympus America Inc., Melville, New York) equipped with FITC and rhodamine specific filters, excitation wavelength/emission filter (490 nm/520 nm; 541 nm/572 nm, respectively).
Mitochondrial isolation assay
Mitochondria were isolated from SH-SY5Y cells using an isolation kit for cultured cells (Pierce) as described previously.35
Western Blot analysis and densitometry
Following treatments, cells were washed with PBS, lysed in 0.1% Triton X-100 and a protease/phosphatase inhibitor cocktail, and 30 μg of protein loaded, as determined by Coomassie Plus Protein Assay (Pierce, Rockford, Illinois), for electrophoresis through 5% to 15% gradient polyacryl-amide gels. For sequential detergent extraction, the Triton X-100 pellet was further extracted by incubation in SDS loading buffer (4.0% SDS, 40% glycerol, 0.33 M Ammediol buffer, 0.1% bromo-phenol blue) followed by sonication for 10 min using a 60 Sonic Dismembrenator (Fisher Scientific, Waltham, Massachusetts). In other experiments, the cells were lysed directly in SDS loading buffer. Protein bands were transferred to Immobilon-PDVF membranes (Millipore, Bedford, Massachusetts) as previously described.29 The membranes were blocked for 1 hour in 5% non-fat dry milk mixed in PBST (20 mmol/L potassium phosphate, and 150 mmol/L potassium chloride, pH 7.4 containing 0.3% (w/v) Tween 20) and probed for endogenous LC3, transfected GFP or ERK1/2 by incubating membranes overnight at 4°C in rabbit anti-MAP-LC3 (1:4000),41 rabbit anti-GFP polyclonal antibody (1:1000) (Invitrogen, Carlsbad, California), mouse anti-phospho-ERK1/2 (1:1000 in phosphate-buffered saline/Tween 20; Cell Signaling) or rabbit anti-total ERK1/2 (1:30,000, Upstate Biotechnology, Lake Placid, New York) followed by incubation with horseradish peroxidase-conjugated sheep-anti-mouse or rabbit IgG (Amersham, Piscataway, NJ) at a (1:5000) dilution. For mitochondrial preparations, immunoblots were also probed with antibodies for lactate dehydrogenase (Santa Cruz Biotechnology, Santa Cruz, California), human mitochondrial antigen of 110 kDa (MITO-P110) (1:500, Oncogene, Boston, Massachusetts) and superoxide dismutase 2 (1:100, SOD2) (Upstate Biotechnology, Lake Placid, NY) to confirm the purity of the preparations as described previously.35 Immunoreactivity was detected using an ECL detection kit (Amersham, Piscataway, New Jersey). The membranes were stripped using 10% SDS, glycine, pH 2.0 for 30 minutes prior to PBST washes, and followed by incubation with mouse anti-β-actin monoclonal antibody (Sigma; 1:10,000). Densitometry was performed using NIH Image J software (NIH, USA).
ERK signaling reporter assay
The effects of different constructs of ERK2 on intracellular ERK activity levels were measured using the PathDetect Elk1 trans-Reporting System (Stratagene, La Jolla, California). In brief, cells cultured on 24 well plates were transfected with 0.25 μg/well of empty vector, wild-type, and mutant ERK2 cDNAs in pCMV5.0 vector, and constitutively active MEK1 (MEK-CA) as a control, and co-transfected with 0.25 μg/well of a cocktail mix (20:1 ratio) of an Elk-1-specific fusion trans-activator plasmid, which consists of the activation domain of Elk-1 transcription factor fused to the DNA binding domain of the yeast transcriptional activator GAL4, and a pFR-Luc reporter plasmid that expresses the Photinus pyralis (firefly) luciferase gene regulated by a promoter containing yeast GAL4 binding sites. These constructs measure ERK-dependent phosphorylation and transactivation of Elk-1 in living cells, which drives expression of luciferase. Two days following transfection, cells were prepared and measured for luciferase activity as described with some modifications.27 In brief, cells were washed with phosphate-buffered saline (PBS) with high glucose, followed by gentle agitation for 20 min in mammalian passive lysis buffer. Cell lysates were microcentrifuged at 20,000 x g for 15 minutes and light emitted from 20 μl of cell lysate was monitored for 20 s in a Lumat LB9507 luminometer.
Cell death assay
Transiently transfected SH-SY5Y cells were seeded on 24 well plates for 48 h prior to exposure with LD50 doses of 6-hydroxydopamine (85 μM) to induce cell injury. Cell death was quantified by staining cells with propidium iodide (PI) (Molecular Probes, Eugene, Oregon) at a final concentration of (1 μg/ml) and incubated for 5–10 minutes at 37°C. Cells were washed two times in PBS and immediately imaged for GFP and PI specific fluorescence using an inverted Olympus IDX71 microscope fitted with FITC and Rhodamine filters. Green and red channels were assembled and merged into RGB images using a built-in macro function of the MicroSuite software. Merged images were randomized and scored blindly to the experimental condition for the percentage of GFP transfected cells that colocalized with PI stain.
RNA interference (RNAi)
Small interfering RNA (siRNA) for human Atg proteins using published sequences (5 -GCCAGUGGGUUUGGAUCAA- 3 for Atg7, and 5-GAAGGCGCUUACAGCUCAA-3 for LC3)23,29 were synthesized by Invitrogen (Austin, Texas). Efficacy of transfection, Atg protein knockdown and suppressive effects on induced autophagy have been previously validated in our laboratory.23,29 In brief, cells were trans-fected at 30% confluence with 40 nmol/L LC3, 20 nmol/L Atg7 siRNA, or with siControl Nontargeting (scrambled) siRNA Pool (Dharmacon, Lafayette, Colorado) 72 hours before treating cells with 6-OHDA for 5 hours.
Quantification of number and size of GFP-LC3 puncta and of mitochondrial area
RGB images captured at a magnification of at least 40X were processed for algorithmic quantification of GFP-LC3 puncta per cell using a custom-written ImageJ macro containing plug-ins. (Chu et al., Autophagy in neurite and neuro-degeneration: in vitro and in vivo models. Methods in Enzymology: Autophagy, in press). In brief, the green channel of RGB images was extracted and converted to binary (grayscale) images by auto-thresholding, and GFP-LC3 granules were analyzed for number and for size (radius = sqrt (Area/π)) using the “Analyze Particles” function of the software. The macro-assisted algorithm was set to measure all particle sizes larger than background pixelation, but smaller than the average nuclear size. Cells containing apoptotic nuclei or necrotic cell bodies were excluded from the analyses.
To measure cellular mitochondrial area, RGB images were processed using another custom Image J macro that calculates total area occupied by mitochondrial particles in the cell of interest using the outlines algorithm of the “Analyze particles” function. The macro then computes the percentage of cellular area occupied by mitochondria using the formula: ((total mitochondrial area/cellular area) x 100).
Statistics
Unless otherwise indicated, results are expressed as mean ± SEM from at least three independent experiments. Two group comparisons were performed using a Student’s t-test (www.graphpad.com). Multiple group comparisons were performed using one-way analysis of variance and Fisher’s LSD. Values of p < 0.05 were defined as statistically significant.
Supplementary Material
Supplementary materials can be found at: www.landesbioscience.com/supplement/DagdaAUTO4-6-Sup.pdf
Acknowledgments
We thank Simon Watkins and the Center for Biological Imaging (CBI) at the University of Pittsburgh for technical expertise with live confocal and EM imaging, Christopher Fung for constructing stable GFP-LC3 cell lines, Charlotte Diges for technical assistance, and all of the investigators listed in the methods section for providing molecular reagents. This research was supported by funding from the National Institutes of Health (AG026389 and DC009120, CTC) and a Veterans Administration Advanced Career Development Award (SMK). RKD was supported in part by T32 NS07495 and F32 AG030821.
References
- 1.Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson’s disease. Science. 2003;302:819–22. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- 2.Ventruti A, Cuervo AM. Autophagy and neurodegeneration. Curr Neurol Neurosci Rep. 2007;7:443–51. doi: 10.1007/s11910-007-0068-5. [DOI] [PubMed] [Google Scholar]
- 3.Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol. 2004;36:2503–18. doi: 10.1016/j.biocel.2004.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chu CT. Autophagic stress in neuronal injury and disease. J Neuropathol Exp Neurol. 2006;65:423–32. doi: 10.1097/01.jnen.0000229233.75253.be. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mizushima N, Ohsumi Y, Yoshimori T. Autophagosome formation in mammalian cells. Cell Struct Funct. 2002;27:421–9. doi: 10.1247/csf.27.421. [DOI] [PubMed] [Google Scholar]
- 6.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nair U, Klionsky DJ. Molecular mechanisms and regulation of specific and nonspecific autophagy pathways in yeast. J Biol Chem. 2005;280:41785–8. doi: 10.1074/jbc.R500016200. [DOI] [PubMed] [Google Scholar]
- 8.Kissova I, Deffieu M, Manon S, Camougrand N. Uth1p is involved in the autophagic degradation of mitochondria. J Biol Chem. 2004;279:39068–74. doi: 10.1074/jbc.M406960200. [DOI] [PubMed] [Google Scholar]
- 9.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–9. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 10.Rideout HJ, Lang-Rollin I, Stefanis L. Involvement of macroautophagy in the dissolution of neuronal inclusions. Int J Biochem Cell Biol. 2004;36:2551–62. doi: 10.1016/j.biocel.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 11.Qin ZH, Wang Y, Kegel KB, Kazantsev A, Apostol BL, Thompson LM, Yoder J, Aronin N, DiFiglia M. Autophagy regulates the processing of amino terminal huntingtin fragments. Hum Mol Genet. 2003;12:3231–44. doi: 10.1093/hmg/ddg346. [DOI] [PubMed] [Google Scholar]
- 12.Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O’Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–95. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y, Qi H, Taylor R, Xu W, Liu LF, Jin S. The role of autophagy in mitochondria maintenance: characterization of mitochondrial functions in autophagy-deficient S. cerevisiae strains. Autophagy. 2007;3:337–46. doi: 10.4161/auto.4127. [DOI] [PubMed] [Google Scholar]
- 14.Jennings JJ, Jr, Zhu JH, Rbaibi Y, Luo X, Chu CT, Kiselyov K. Mitochondrial aberrations in mucolipidosis type IV. J Biol Chem. 2006 doi: 10.1074/jbc.M607982200. [DOI] [PubMed] [Google Scholar]
- 15.Komatsu M, Wang QJ, Holstein GR, Friedrich VL, Jr, Iwata J, Kominami E, Chait BT, Tanaka K, Yue Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci USA. 2007;104:14489–94. doi: 10.1073/pnas.0701311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cherra S, III, Chu CT. Autophagy in neuroprotection and neurodegeneration: A question of balance. Future Neurol. 2008;3:309–23. doi: 10.2217/14796708.3.3.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tolkovsky AM, Xue L, Fletcher GC, Borutaite V. Mitochondrial disappearance from cells: a clue to the role of autophagy in programmed cell death and disease? Biochimie. 2002;84:233–40. doi: 10.1016/s0300-9084(02)01371-8. [DOI] [PubMed] [Google Scholar]
- 18.Chu CT, Zhu J, Dagda R. Beclin 1-independent pathway of damage-induced mitophagy and autophagic stress: implications for neurodegeneration and cell death. Autophagy. 2007;3:663–6. doi: 10.4161/auto.4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang C, You YJ, Avery L. Dual roles of autophagy in the survival of Caenorhabditis elegans during starvation. Genes Dev. 2007;21:2161–71. doi: 10.1101/gad.1573107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trifilieff P, Calandreau L, Herry C, Mons N, Micheau J. Biphasic ERK1/2 activation in both the hippocampus and amygdala may reveal a system consolidation of contextual fear memory. Neurobiol Learn Mem. 2007;88:424–34. doi: 10.1016/j.nlm.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 21.Lu Z, Xu S. ERK1/2 MAP kinases in cell survival and apoptosis. IUBMB Life. 2006;58:621–31. doi: 10.1080/15216540600957438. [DOI] [PubMed] [Google Scholar]
- 22.Hetman M, Gozdz A. Role of extracellular signal regulated kinases 1 and 2 in neuronal survival. Eur J Biochem. 2004;271:2050–5. doi: 10.1111/j.1432-1033.2004.04133.x. [DOI] [PubMed] [Google Scholar]
- 23.Plowey ED, Cherra SJ, 3rd, Liu YJ, Chu CT. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem. 2008 doi: 10.1111/j.1471-4159.2008.05217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chu CT, Levinthal DJ, Kulich SM, Chalovich EM, DeFranco DB. Oxidative neuronal injury. The dark side of ERK1/2. Eur J Biochem. 2004;271:2060–6. doi: 10.1111/j.1432-1033.2004.04132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu JH, Kulich SM, Oury TD, Chu CT. Cytoplasmic aggregates of phosphorylated extra-cellular signal-regulated kinase in Lewy body diseases. Am J Pathol. 2002;161:2087–98. doi: 10.1016/S0002-9440(10)64487-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu JH, Guo F, Shelburne J, Watkins S, Chu CT. Localization of phosphorylated ERK/ MAP kinases to mitochondria and autophagosomes in Lewy body diseases. Brain Pathol. 2003;13:473–81. doi: 10.1111/j.1750-3639.2003.tb00478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chalovich EM, Zhu JH, Caltagarone J, Bowser R, Chu CT. Functional repression of cAMP response element in 6-hydroxydopamine-treated neuronal cells. J Biol Chem. 2006;281:17870–81. doi: 10.1074/jbc.M602632200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chu CT, Plowey ED, Wang Y, Patel V, Jordan-Sciutto KL. Location, location, location: altered transcription factor trafficking in neurodegeneration. J Neuropathol Exp Neurol. 2007;66:873–83. doi: 10.1097/nen.0b013e318156a3d7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu JH, Horbinski C, Guo F, Watkins S, Uchiyama Y, Chu CT. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am J Pathol. 2007;170:75–86. doi: 10.2353/ajpath.2007.060524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Przedborski S, Jackson-Lewis V. Mechanisms of MPTP toxicity. Movement Disorders. 1998;13:35–8. [PubMed] [Google Scholar]
- 31.Beal MF. Experimental models of Parkinson’s disease. Nature Reviews Neuroscience. 2001;2:325–34. doi: 10.1038/35072550. [DOI] [PubMed] [Google Scholar]
- 32.Segura Aguilar J, Kostrzewa RM. Neurotoxins and neurotoxic species implicated in neuro-degeneration. Neurotox Res. 2004;6:615–30. doi: 10.1007/BF03033456. [DOI] [PubMed] [Google Scholar]
- 33.Callio J, Oury TD, Chu CT. Manganese superoxide dismutase protects against 6-hydroxy-dopamine injury in mouse brains. J Biol Chem. 2005;280:18536–42. doi: 10.1074/jbc.M413224200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kulich SM, Chu CT. Sustained extracellular signal-regulated kinase activation by 6-hydroxy-dopamine: Implications for Parkinson’s disease. J Neurochem. 2001;77:1058–66. doi: 10.1046/j.1471-4159.2001.00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kulich SM, Horbinski C, Patel M, Chu CT. 6-Hydroxydopamine induces mitochondrial ERK activation. Free Radic Biol Med. 2007;43:372–83. doi: 10.1016/j.freeradbiomed.2007.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emrick MA, Hoofnagle AN, Miller AS, Ten Eyck LF, Ahn NG. Constitutive activation of extracellular signal-regulated kinase 2 by synergistic point mutations. J Biol Chem. 2001;276:46469–79. doi: 10.1074/jbc.M107708200. [DOI] [PubMed] [Google Scholar]
- 37.Wolf I, Rubinfeld H, Yoon S, Marmor G, Hanoch T, Seger R. Involvement of the activation loop of ERK in the detachment from cytosolic anchoring. J Biol Chem. 2001;276:24490–7. doi: 10.1074/jbc.M103352200. [DOI] [PubMed] [Google Scholar]
- 38.Frost JA, Geppert TD, Cobb MH, Feramisco JR. A requirement for extracellular signal-regulated kinase (ERK) function in the activation of AP-1 by Ha-Ras, phorbol 12-myristate 13-acetate, and serum. Proc Natl Acad Sci USA. 1994;91:3844–8. doi: 10.1073/pnas.91.9.3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998;23:33–42. doi: 10.1247/csf.23.33. [DOI] [PubMed] [Google Scholar]
- 40.Fass E, Shvets E, Degani I, Hirschberg K, Elazar Z. Microtubules support production of starvation-induced autophagosomes but not their targeting and fusion with lysosomes. J Biol Chem. 2006;281:36303–16. doi: 10.1074/jbc.M607031200. [DOI] [PubMed] [Google Scholar]
- 41.Koike M, Shibata M, Waguri S, Yoshimura K, Tanida I, Kominami E, Gotow T, Peters C, Figura KV, Mizushima N, Saftig P, Uchiyama Y. Participation of autophagy in storage of lysosomes in neurons from mouse models of neuronal ceroid-lipofuscinoses (Batten disease) Am J Pathol. 2005;167:1713–28. doi: 10.1016/S0002-9440(10)61253-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gomez-Santos C, Ferrer I, Santidrian AF, Barrachina M, Gil J, Ambrosio S. Dopamine induces autophagic cell death and alpha-synuclein increase in human neuroblastoma SH-SY5Y cells. J Neurosci Res. 2003;73:341–50. doi: 10.1002/jnr.10663. [DOI] [PubMed] [Google Scholar]
- 43.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, Bamber BA, Bassham DC, Bergamini E, Bi X, Biard-Piechaczyk M, Blum JS, Bredesen DE, Brodsky JL, Brumell JH, Brunk UT, Bursch W, Camougrand N, Cebollero E, Cecconi F, Chen Y, Chin LS, Choi A, Chu CT, Chung J, Clarke PG, Clark RS, Clarke SG, Clave C, Cleveland JL, Codogno P, Colombo MI, Coto-Montes A, Cregg JM, Cuervo AM, Debnath J, Demarchi F, Dennis PB, Dennis PA, Deretic V, Devenish RJ, Di Sano F, Dice JF, Difiglia M, Dinesh-Kumar S, Distelhorst CW, Djavaheri-Mergny M, Dorsey FC, Droge W, Dron M, Dunn WA, Jr, Duszenko M, Eissa NT, Elazar Z, Esclatine A, Eskelinen EL, Fesus L, Finley KD, Fuentes JM, Fueyo J, Fujisaki K, Galliot B, Gao FB, Gewirtz DA, Gibson SB, Gohla A, Goldberg AL, Gonzalez R, Gonzalez-Estevez C, Gorski S, Gottlieb RA, Haussinger D, He YW, Heidenreich K, Hill JA, Hoyer-Hansen M, Hu X, Huang WP, Iwasaki A, Jaattela M, Jackson WT, Jiang X, Jin S, Johansen T, Jung JU, Kadowaki M, Kang C, Kelekar A, Kessel DH, Kiel JA, Kim HP, Kimchi A, Kinsella TJ, Kiselyov K, Kitamoto K, Knecht E, Komatsu M, Kominami E, Kondo S, Kovacs AL, Kroemer G, Kuan CY, Kumar R, Kundu M, Landry J, Laporte M, Le W, Lei HY, Lenardo MJ, Levine B, Lieberman A, Lim KL, Lin FC, Liou W, Liu LF, Lopez-Berestein G, Lopez-Otin C, Lu B, Macleod KF, Malorni W, Martinet W, Matsuoka K, Mautner J, Meijer AJ, Melendez A, Michels P, Miotto G, Mistiaen WP, Mizushima N, Mograbi B, Monastyrska I, Moore MN, Moreira PI, Moriyasu Y, Motyl T, Munz C, Murphy LO, Naqvi NI, Neufeld TP, Nishino I, Nixon RA, Noda T, Nurnberg B, Ogawa M, Oleinick NL, Olsen LJ, Ozpolat B, Paglin S, Palmer GE, Papassideri I, Parkes M, Perlmutter DH, Perry G, Piacentini M, Pinkas-Kramarski R, Prescott M, Proikas-Cezanne T, Raben N, Rami A, Reggiori F, Rohrer B, Rubinsztein DC, Ryan KM, Sadoshima J, Sakagami H, Sakai Y, Sandri M, Sasakawa C, Sass M, Schneider C, Seglen PO, Seleverstov O, Settleman J, Shacka JJ, Shapiro IM, Sibirny A, Silva-Zacarin EC, Simon HU, Simone C, Simonsen A, Smith MA, Spanel-Borowski K, Srinivas V, Steeves M, Stenmark H, Stromhaug PE, Subauste CS, Sugimoto S, Sulzer D, Suzuki T, Swanson MS, Tabas I, Takeshita F, Talbot NJ, Talloczy Z, Tanaka K, Tanaka K, Tanida I, Taylor GS, Taylor JP, Terman A, Tettamanti G, Thompson CB, Thumm M, Tolkovsky AM, Tooze SA, Truant R, Tumanovska LV, Uchiyama Y, Ueno T, Uzcategui NL, van der Klei I, Vaquero EC, Vellai T, Vogel MW, Wang HG, Webster P, Wiley JW, Xi Z, Xiao G, Yahalom J, Yang JM, Yap G, Yin XM, Yoshimori T, Yu L, Yue Z, Yuzaki M, Zabirnyk O, Zheng X, Zhu X, Deter RL. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–75. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Munafo DB, Colombo MI. A novel assay to study autophagy: regulation of autophagosome vacuole size by amino acid deprivation. J Cell Sci. 2001;114:3619–29. doi: 10.1242/jcs.114.20.3619. [DOI] [PubMed] [Google Scholar]
- 45.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–21. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Larsen KE, Fon EA, Hastings TG, Edwards RH, Sulzer D. Methamphetamine-induced degeneration of dopaminergic neurons involves autophagy and upregulation of dopamine synthesis. J Neurosci. 2002;22:8951–60. doi: 10.1523/JNEUROSCI.22-20-08951.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gomez-Santos C, Ferrer I, Reiriz J, Vinals F, Barrachina M, Ambrosio S. MPP+ increases alpha-synuclein expression and ERK/MAP-kinase phosphorylation in human neuroblas-toma SH-SY5Y cells. Brain Res. 2002;935:32–9. doi: 10.1016/s0006-8993(02)02422-8. [DOI] [PubMed] [Google Scholar]
- 48.Xi ZQ, Wang XF, He RQ, Li MW, Liu XZ, Wang LY, Zhu X, Xiao F, Sun JJ, Li JM, Gong Y, Guan LF. Extracellular signal-regulated protein kinase in human intractable epilepsy. Eur J Neurol. 2007;14:865–72. doi: 10.1111/j.1468-1331.2007.01777.x. [DOI] [PubMed] [Google Scholar]
- 49.Zhu X, Lee HG, Raina AK, Perry G, Smith MA. The role of mitogen-activated protein kinase pathways in Alzheimer’s disease. Neurosignals. 2002;11:270–81. doi: 10.1159/000067426. [DOI] [PubMed] [Google Scholar]
- 50.Pei JJ, Braak H, An WL, Winblad B, Cowburn RF, Iqbal K, Grundke-Iqbal I. Upregulation of mitogen-activated protein kinases ERK1/2 and MEK1/2 is associated with the progression of neurofibrillary degeneration in Alzheimer’s disease. Brain Res Mol Brain Res. 2002;109:45–55. doi: 10.1016/s0169-328x(02)00488-6. [DOI] [PubMed] [Google Scholar]
- 51.Alessandrini A, Namura S, Moskowitz MA, Bonventre JV. MEK1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. Proc Natl Acad Sci USA. 1999;96:12866–9. doi: 10.1073/pnas.96.22.12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li PA, He QP, Yi-Bing O, Hu BR, Siesjo BK. Phosphorylation of extracellular signal-regulated kinase after transient cerebral ischemia in hyperglycemic rats. Neurobiol Dis. 2001;8:127–35. doi: 10.1006/nbdi.2000.0363. [DOI] [PubMed] [Google Scholar]
- 53.Wang X, Zhu C, Qiu L, Hagberg H, Sandberg M, Blomgren K. Activation of ERK1/2 after neonatal rat cerebral hypoxia-ischaemia. J Neurochem. 2003;86:351–62. doi: 10.1046/j.1471-4159.2003.01838.x. [DOI] [PubMed] [Google Scholar]
- 54.Lin E, Cavanaugh JE, Leak RK, Perez RG, Zigmond MJ. Rapid activation of ERK by 6-hydroxydopamine promotes survival of dopaminergic cells. J Neurosci Res. 2008;86:108–17. doi: 10.1002/jnr.21478. [DOI] [PubMed] [Google Scholar]
- 55.Luo Y, DeFranco DB. Opposing roles for ERK1/2 in neuronal oxidative toxicity: distinct mechanisms of ERK1/2 action at early versus late phases of oxidative stress. J Biol Chem. 2006;281:16436–42. doi: 10.1074/jbc.M512430200. [DOI] [PubMed] [Google Scholar]
- 56.Kulich SM, Chu CT. Role of reactive oxygen species in ERK phosphorylation and 6-hydroxydopamine cytotoxicity. J Biosci. 2003;28:83–9. doi: 10.1007/BF02970136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Philipova R, Whitaker M. Active ERK1 is dimerized in vivo: bisphosphodimers generate peak kinase activity and monophosphodimers maintain basal ERK1 activity. J Cell Sci. 2005;118:5767–76. doi: 10.1242/jcs.02683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu L, Kang Y, Col S, Massague J. Smad2 nucleocytoplasmic shuttling by nucleoporins CAN/Nup214 and Nup153 feeds TGFbeta signaling complexes in the cytoplasm and nucleus. Mol Cell. 2002;10:271–82. doi: 10.1016/s1097-2765(02)00586-5. [DOI] [PubMed] [Google Scholar]
- 59.Wunderlich W, Fialka I, Teis D, Alpi A, Pfeifer A, Parton RG, Lottspeich F, Huber LA. A novel 14-kilodalton protein interacts with the mitogen-activated protein kinase scaffold mp1 on a late endosomal/lysosomal compartment. J Cell Biol. 2001;152:765–76. doi: 10.1083/jcb.152.4.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cha H, Lee EK, Shapiro P. Identification of a C-terminal region that regulates mitogen-activated protein kinase kinase-1 cytoplasmic localization and ERK activation. J Biol Chem. 2001;276:48494–501. doi: 10.1074/jbc.M107601200. [DOI] [PubMed] [Google Scholar]
- 61.Corcelle E, Nebout M, Bekri S, Gauthier N, Hofman P, Poujeol P, Fenichel P, Mograbi B. Disruption of autophagy at the maturation step by the carcinogen lindane is associated with the sustained mitogen-activated protein kinase/extracellular signal-regulated kinase activity. Cancer Res. 2006;66:6861–70. doi: 10.1158/0008-5472.CAN-05-3557. [DOI] [PubMed] [Google Scholar]
- 62.Ogier-Denis E, Pattingre S, El Benna J, Codogno P. Erk1/2-dependent phosphorylation of Galpha-interacting protein stimulates its GTPase accelerating activity and autophagy in human colon cancer cells. J Biol Chem. 2000;275:39090–5. doi: 10.1074/jbc.M006198200. [DOI] [PubMed] [Google Scholar]
- 63.Nowak G, Clifton GL, Godwin ML, Bakajsova D. Activation of ERK1/2 pathway mediates oxidant-induced decreases in mitochondrial function in renal cells. Am J Physiol Renal Physiol. 2006;291:840–55. doi: 10.1152/ajprenal.00219.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kissova I, Deffieu M, Samokhvalov V, Velours G, Bessoule JJ, Manon S, Camougrand N. Lipid oxidation and autophagy in yeast. Free Radic Biol Med. 2006;41:1655–61. doi: 10.1016/j.freeradbiomed.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 65.Kim EH, Sohn S, Kwon HJ, Kim SU, Kim MJ, Lee SJ, Choi KS. Sodium selenite induces superoxide-mediated mitochondrial damage and subsequent autophagic cell death in malignant glioma cells. Cancer Res. 2007;67:6314–24. doi: 10.1158/0008-5472.CAN-06-4217. [DOI] [PubMed] [Google Scholar]
- 66.Rodriguez-Enriquez S, He L, Lemasters JJ. Role of mitochondrial permeability transition pores in mitochondrial autophagy. Int J Biochem Cell Biol. 2004;36:2463–72. doi: 10.1016/j.biocel.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 67.Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–22. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- 68.Mandemakers W, Morais VA, De Strooper B. A cell biological perspective on mitochondrial dysfunction in Parkinson disease and other neurodegenerative diseases. J Cell Sci. 2007;120:1707–16. doi: 10.1242/jcs.03443. [DOI] [PubMed] [Google Scholar]
- 69.Betarbet R, Sherer TB, Di Monte DA, Greenamyre JT. Mechanistic approaches to Parkinson’s disease pathogenesis. Brain Pathol. 2002;12:499–510. doi: 10.1111/j.1750-3639.2002.tb00468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xue L, Fletcher GC, Tolkovsky AM. Mitochondria are selectively eliminated from eukary-otic cells after blockade of caspases during apoptosis. Curr Biol. 2001;11:361–5. doi: 10.1016/s0960-9822(01)00100-2. [DOI] [PubMed] [Google Scholar]
- 71.Koike T, Yang Y, Suzuki K, Zheng X. Axon & dendrite degeneration: Its mechanisms and protective experimental paradigms. Neurochem Int. 2008;52:751–60. doi: 10.1016/j.neuint.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 72.Chu CT, Plowey ED, Dagda RK, Hickey RWSJC, III, Clark RS. Autophagy in neurite injury and neurodegeneration: in vitro and in vivo models. Methods Enzymol. 2008 doi: 10.1016/S0076-6879(08)04011-1. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials can be found at: www.landesbioscience.com/supplement/DagdaAUTO4-6-Sup.pdf