

Abstract
The ability of human tumor cell lines to produce various cytokines, chemokines, angiogenic and growth factors was investigated using Luminex multiplex technology. Media conditioned by tumor cells protected tumor cells from drug-induced apoptosis and stimulated tumor cell proliferation. Antibodies neutralizing IL-6, CXCL8, CCL2 and CCL5 blocked this stimulation. Treatment of tumor cells with doxorubicin and cisplatin resulted in a substantial increase in the production of IL-6, CXCL8, CCL2, CCL5, BFGF, G-CSF, and VEGF. This stimulation was associated with drug-induced activation of NF-κB, AP-1, AP-2, CREB, HIF-1, STAT-1, STAT-3, STAT-5 and ATF-2 transcription factors and up-regulation of IL-6, CXCL8, FGF-2, CSF-3 and CCL5 gene expression. Treatment of tumor cells with doxorubicin and antibodies neutralizing G-CSF, CCL2 or CCL5 had higher inhibitory effects than each modality used alone. These results indicate that chemokines and growth factors produced by tumor by binding to the cognate receptors on tumor and stroma cells could provide proliferative and anti-apoptotic signals helping tumor to escape drug-mediated destruction. Clinical studies showed that antibodies neutralizing VEGF (Avastin/ Bevacizumab) or blocking HER2/neu signaling (Herceptin/ Trastuzumab) could increase the efficacy of chemotherapy although these beneficial effects have been limited. It is possible that drug-stimulated production of growth and pro-angiogenic factors could counterbalance the effects of antibody therapy. In addition, numerous growth factors and chemokines share angiogenic and growth-stimulating properties, and thus reduction of a single factor is insufficient to completely block tumor growth. Thus, a broad disruption of tumor cytokine network is needed to further increase the efficacy of cancer therapy.
Keywords: tumor cells, drugs, cytokines, chemokines, angiogenic factors
Introduction
The ability of tumor cells to produce various cytokines, chemokines, angiogenic and growth factors is crucial for tumor cell proliferation and the formation of stroma and blood vessel networks to provide oxygen and nutrients and support progressive tumor growth. Tumor-produced VEGF is the most potent angiogenic factor, which stimulates migration and proliferation of endothelial cells and formation of blood vessels 2, 3. Malignant transformation is often associated with overexpression of various growth factors including FGF, EGF, and HGF, all of which stimulate proliferation of tumor cells as well as stromal cells and manifest potent angiogenic effects 4-8.
Tumor cells also produce cytokines and chemokines such as IL-6, IL-8, IL-10, CCL2 (MCP-1), and CCL5 (RANTES) that have complex autocrine and paracrine effects in tumors. IL-6 is a pleiotropic pro-inflammatory cytokine that affects B- and T-cell differentiation, induces acute phase reactant production, and stimulates hematopoiesis. It has been shown that IL-6 could directly stimulate proliferation of tumor cells and manifests a potent angiogenic effect 9-11. Some reports indicate that IL-6 levels correlate with disease bulk and inversely correlate with response to treatment and survival 12. IL-8 belongs to the superfamily of CXC chemokines and has a wide range of pro-inflammatory effects. It stimulates migration of neutrophils, monocytes, and lymphocytes, and promotes tumor cell proliferation and metastasis 13-15. In addition, IL-8 exhibits strong angiogenic activity 13, 16, 17. Chemokines of the CC superfamily such as CCL5 (RANTES) and CCL2 (MCP-1) also are able to stimulate migration of normal and malignant cells, as well as promote tumor angiogenesis 18-22. Cytokines IL-10 and TGF-β are potent immunosuppressive factors that play an important role in protecting tumors from immune-mediated destruction 23, 24.
The production of cytokines, growth and angiogenic factors is not an exclusive property of tumor cells. Stroma cells such as endothelial cells, fibroblasts, lymphocytes, and macrophages also produce various cytokines, chemokines, growth and angiogenic factors that could affect survival and proliferation of stroma cells as well as tumor cells. Thus, there exists perpetual cross-talk between tumor and stromal cells. Tumor- and stroma-produced soluble factors represent a tumor cytokine network that plays an important role in tumor growth and tumor protection from endogenous (hypoxia, oxygen free radicals) and exogenous (drugs, x-irradiation) damage. Overproduction of these factors by growing tumors could lead to the observed increase in blood levels of cytokines, chemokines, angiogenic and growth factors often associated with resistance to therapy and overall poor prognosis 12, 13, 25-27.
Thus, different types of tumor and stroma producing factors (cytokines, chemokines, angiogenic and growth factors) have overlapping functions in promoting tumor growth and thus, blocking of a single factor may not be sufficient to inhibit tumor growth as other factors are able to compensate for lost function. However, the breadth and magnitude of the complete network of cytokine and tumor-produced factors remains largely uninvestigated, and a comprehensive analysis of the cytokine network signature of various human tumor cells is therefore well warranted.
The importance of targeting angiogenic and growth factor signaling is well established. Humanized monoclonal antibodies (Avastin/Bevacizumab) that neutralize the angiogenic factor VEGF used in combination with chemotherapy have demonstrated a significant clinical benefit. Similarly, blocking HER2/neu signaling with a monoclonal antibody (Herceptin/Trastuzumab) may improve the therapeutic effects of chemotherapy in patients with metastatic breast cancer.
It remains unclear what effects chemotherapy has on tumor-produced cytokines. It is conceivable that killing tumor cells and/or inhibiting their DNA, RNA, and protein synthesis by chemotherapeutic drugs could reduce intratumor production of cytokines, chemokines, angiogenic and growth factors, however direct confirmation of this possibility remains elusive. In fact, the few studies in which the effect of chemotherapeutic drugs on cytokine production by tumor cells was investigated suggested the opposite. Namely, dacarbazine treatment of melanoma cell lines stimulated IL-8 and VEGF production 30, 31. Augmentation of IL-8 and TNF-α production was observed when human epithelial carcinoma cell lines were treated with etoposide and mitomycin C 32 and doxorubicin increased production of IL-8 and MCP-1 in human small cell lung cancer cell lines 33.
It is possible that drug treatment induces a cellular stress response resulting in the production of cytokines, chemokines, angiogenic and growth factors that, by binding to their receptors on tumor and stroma cells, provide pro-survival signaling that protects these cells from drug-induced apoptosis and stimulates their proliferation.
The broad investigation of tumor cell cytokine networks and analysis of the effects of drug on cytokine production is largely limited by the low throughput of conventional ELISA, which typically allows testing of only one cytokine at a time. In order to overcome this problem, we applied Fluorokine® MultiAnalyte Profiling (xMAP™) technology developed by Luminex, Inc. (Austin, TX) to analyze simultaneously the production of 30 different cytokines, chemokines, angiogenic and growth factors by human H460 and A549 non-small cell lung cancer (NSCLC) lines, as well as MCF-7 and CAMA-1 breast tumor cell lines. The ability of chemotherapeutic drugs to affect production of these factors and mechanisms responsible for the drug-induced effect on cytokine production were investigated.
Materials and Methods
Cell Lines
Human H460 and A549 non-small cell lung cancer (NSCLC) cell lines, MCF-7 and CAMA-1 breast tumor, K562 erythroleukemia, LOX and FEMx melanoma and OVCAR-3 ovarian cancer cells tumor cell lines were obtained from the American Type Culture Collection (ATCC, Rockville, MD) and cultured in RPMI 1640 supplemented with 10% FBS and antibiotics (Millipore Inc., Billerica, MA).
Reagents
Doxorubicin, cisplatin, bleomycin, etoposide, and monensin were purchased from Sigma-Aldrich (Sigma-Aldrich, St. Louis, MO). Neutralizing monoclonal antibodies (mAbs) against IL-10, G-CSF and CCL5 (RANTES) were obtained from BioVision Inc. (BioVision, Mountain View, CA). Neutralizing mAbs against TGF-β1 and TGF-β2 were obtained from R&D Systems (R&D Systems Inc., Minneapolis, MN). MAbs against human MCP-1 (CCL2), VEGF, bFGF, IL-6 were purchased from Calbiochem (EMD Biosciences, Inc., San Diego, CA). Anti-IL-8 mAb was obtained from Abcam Inc. (Abcam, Cambridge, MA).
Drug treatment and proliferation assays
Tumor cells were seeded into 96-well or 24-well plates. The next day, drugs were added at indicated concentrations. Three days later, plates were centrifuged and supernatants from 4-6 wells were collected into 2-3 pools and frozen. Cell counts were estimated using the One Solution Cell Proliferation Kit (Promega, Madison, WI). In some experiments, cells harvested from T-25 flasks or 24-well plates were counted using a hemacytometer, whereas cells growing in 96-well plates were fixed with 2% paraformaldehyde (PFA), stained with Hoechst 33342 (2μg/mL), and counted using Cellomics Array Scan VTI (Cellomics/ ThermoFisher, Pittsburgh, PA), as described.
Multiplex analysis of cytokine production by tumor cells
Concentrations of various tumor cell-produced factors (TPFs) in media conditioned by tumor cells were analyzed using multiplex bead-based sandwich immunoassay technology (xMAP™, Luminex Corp., Austin, TX) using a 30-plex kit from Invitrogen/Biosource (Camarillo, CA) for testing the following cytokines: IL-1α, IL-1β, IL-4, IL-5, IL-6, IL-7, IL-10, IL-12p40, IL-13, IL-15, IL-17, GM-CSF, IFN-α, IFN-γ, TNFα; chemokines: CCL2 (MCP-1), CCL8 (MCP-2), CCL7 (MCP-3), CCL3 (MIP-1α), CCL4 (MIP-1β), CCL5 (RANTES), CCL11 (EOTAXIN), CXCL8 (IL-8), CXCL9 (MIG), CXCL10 (IP-10); growth factors: bFGF, EGF, G-CSF, HGF and VEGF. Analyses of tumor supernatants were performed in a 96-well microplate format according to the protocol by Invitrogen, as previously described 35, 36.
The analyte concentrations (pg/ml) were calculated as pg/1×106 tumor cells as follows:
Multiplex analysis of transcription factors
H460 cells were treated with doxorubicin for 0, 15, 30 or 60 min, nuclear extracts were prepared and levels of nine transcription factors (AP-1, AP-2, ATF-2, CREB, HIF-1, NFκB, STAT 1, STAT 3, and STAT-5) were analyzed using the multiplex Procarta Transcription Factor Assay Kit (Panomics, Fremont, CA; P/N PC5101). Briefly, nuclear extracts were normalized for protein content and incubated with a mixture of biotin-labeled double-stranded oligonucleotide probes to form protein/DNA complexes. Unbound probes were then removed by filtration followed by elution of the complexes from the filter matrix. Bound probes were denatured and hybridized with transcription factor-specific antisense oligonucleotide-conjugated microbeads. Probe-bound microbeads were detected with streptavidin-conjugated R-phycoerythrin and analyzed using the Bio-Plex Luminex 100 system (Bio-Rad, Hercules, CA).
Quantative real-time PCR of cytokines gene expression
H460 tumor cells were treated with doxorubicin (0.125 μg/ml) for 6h. RNA was extracted and cDNA was produced using the SuperArray ReactionReady First Strand cDNA Synthesis kit (SuperArray Bioscience). Real-time PCR was carried out using the SuperArray RT2 Real-Time ™ SYBR Green PCR Master Mix (SuperArray Bioscience) and was performed on the ABI Prism™ 7700 sequence detector real-time PCR system (AB Applied Biosystems). PCR conditions were as follows: denaturation at 95°C for 10 min, 40 cycles at 95°C for 15 s, and 60°C for 1 min. Primers for human IL-6, IL-8, VEGF, CCL5 (RANTES), FGF2, CSF3 (G-CSF) and GAPDH genes were from SuperArray RT2 PCR Primer Sets. Standard curves were generated from five 10-fold serial dilutions of tumor cells’ cDNA, and no product could be seen in the no-template control. Differences in gene expression in doxorubicin-treated and untreated H460 tumor cells were calculated using the 2(-ΔΔCt) method according to the manual from SuperArray Bioscience.
Analysis of the intracellular cytokines in tumor cells
H460 tumor cells (1×104/well) grown in 96-well plates were incubated overnight with doxorubicin (0.125 μg/ml) then monensin (2 μM) was added for 3h. Cells were fixed in 2% PFA, washed, permeabilazed with 0.1% Triton X-100, washed with FACS buffer and then incubated with antibodies against various cytokines for 1h and after washing with secondary anti-mouse antibodies conjugated with Alexa 488, 546, or 680 fluorochromes (Molecular Probes/Invitrogen) for 1 h. Cell nuclei were stained with Hoechst 33342 at 2μg/ml for 20 min to identify individual cells.
Cell images were acquired using the Cellomics ArrayScan HCS Reader (Cellomics/ThermoFisher, Pittsburgh, PA) and analyzed using the Target Activation BioApplication Software Module. The Cellomics ArrayScan HCS Reader collects information concerning distribution of fluorescently labeled components in stained cells. The scanner is equipped with emission and excitation filters (XF93, Omega Optical, Brattleboro, VT, USA) for selectively imaging fluorescent signals emitted by Hoechst 33342, Alexa 488, and Alexa 680. Data were captured, extracted and analyzed with ArrayScan II Data Acquisition and Data Viewer version 3.0 (Cellomics), Quattro Pro version 10.0.0 (Corel, Ottawa, Ontario, Canada), and MS Excel 2002 (Microsoft, Redmond, WA).
Effect of tumor-conditioned media (TCM) on tumor cell proliferation and drug-induced apoptosis
H460 tumor cells were cultured for 3 days in RPMI 1640 supplemented with 10% or 1% FBS until 80% confluence was achieved. TCM was collected and stored at -80° C. H460 cells were plated into 96 wells (1×104cells/well) in fresh medium (FM) or TCM. The next day, doxorubicin (0.125 μg/ml) was added. The numbers of cells was determined after 3 days in culture using the One Solution Cell Proliferation Kit (Promega) or the Cellomics ArrayScan HCS Reader.
To assess the ability of TCM to protect tumor cells from doxorubicin-induced apoptosis, H460 tumor cells were cultured in FM or TCM in the presence of doxorubicin (0.125 μg/ml) for 6 or 20 h. Cells were stained with Annexin V-FITC and propidium iodine (PI) and by flow cytometry.
Effect of neutralizing antibodies (Abs) on tumor cells proliferation and sensitivity to doxorubicin
H460 cells were plated in 96-well plates (1.0 × 104 cells/ml) in culture medium with 10% FBS. Next day culture medium was replaced by FM supplemented with 1% FBS or TCM with 1% FBS. Cells were pre-treated with mAbs neutralizing different cytokines (100 ng/ml) for 1 h, and then doxorubicin (0.125 μg/ml) was added. After 72h of cultivation, samples of conditioned media were collected and cells were fixed in PFA and stained with Hoechst 33342. Cells were counted using Cellomics Array Scan VTI.
Statistical analysis
Data presented as mean ± SD. Comparisons between the values were performed using a two-tailed Student’s t-test. For the comparison of multiple groups, a one- or two-way ANOVA test was applied. For all statistical analyses, the level of significance was set at a probability of P< 0.05. All experiments were repeated 2-3 times.
Results
Analysis of soluble factors produced by human tumor cell lines
Human H460 and A549 non-small cell lung cancer, MCF-7 breast tumor, K562 erythroleukemia, LOX and FEMx melanoma, and OVCAR-3 ovarian cancer line were cultured for 3 days in T-25 flasks, supernatants were collected, tumor cells were counted and concentrations of 30 different cytokines, chemokines, angiogenic and growth factors in the supernatants were analyzed using Multiplex xMAP™ technology. In comparison to other cell lines, H460 lung tumor and OVCAR-3 ovarian cancer cell lines produced high levels of proangiogenic IL-6 and CXCL8 (IL-8). Most of tumor cell lines produced high levels of angiogenic factor VEGF (Table1). The highest level of VEGF was found in the supernatant of OVCAR-3 cells, whereas LOX melanoma was the lowest producer of VEGF. All tested lines, except MCF-7 and OVCAR-3, produced high levels of G-CSF. FEMx melanoma line was a high producer of chemokine CCL5 (RANTES). Slow growing MCF-7 cells produced lower levels of growth and angiogenic factors compared with other cell lines (Table1).
Table 1.
Multiplex Analysis of Soluble Factors Produced by Human Tumor Cell Lines
| Tumor cells produced factors * | H460 | A549 | FEMX | LOX | MCF7 | OVCAR-3 | K562 |
|---|---|---|---|---|---|---|---|
| Cytokines (pg/1×106 cells) | |||||||
| IL-6 | 12,385 | 205 | 11 | 18 | 0.8 | 6,646 | 51 |
| IL-8 | 13,600 | 444 | 217 | 354 | 3 | 12,281 | 2 |
| IL-10 | 10 | 20 | 86 | 33 | 2 | 23 | 26 |
| G-CSF | 1,746 | 2,772 | 2,162 | 2,039 | 516 | 306 | 1,416 |
| HGF | 31 | 131 | 161 | 98 | 12 | 70 | 70 |
| EGF | 140 | 259 | 332 | 274 | 49 | 96 | 146 |
| VEGF | 2,049 | 1,450 | 1,504 | 81 | 615 | 13,331 | 1,074 |
| bFGF | 667 | 918 | 830 | 656 | 222 | 193 | 533 |
| MIP-1a | 13 | 41 | 51 | 54 | 3. | 0 | 24 |
| MIP-1b | 119 | 280 | 0 | 163 | 28 | 0 | 133 |
| CCL2 | 17 | 675 | 0 | 0 | 2 | 0 | 30 |
| CCL5 | 416 | 0 | 1,259 | 780 | 93 | 188 | 209 |
| IFN-a | 164 | 417 | 696 | 610 | 54 | 193 | 305 |
Human tumor cell lines H460 and A549 non-small cell lung cancer, LOX and FEMx melanoma, MCF7 breast, OVCAR-3 ovarian and K562 erythroleukemia cancer cells were cultured 3 days. Supernatants were collected and cells were counted. The concentrations of various tumor cell produced factors were analyzed using MAPx multiplex technology. Production of cytokines (pg/1×106tumor cells) was calculated. Inter-assay coefficients of variation for individual cytokines in the multiplex assay were in the range of 3.5-9.8% and intra-assay coefficients of variation were in the range of 3.6-12.6%.
Thus, tumor cells of different histological origin vary in their ability to produce various molecules essential for tumor cell proliferation, angiogenesis and migration of tumor and inflammatory cells. However, tumor cell lines of the same histological origin also showed different capacity in production of some factors. Some tested factors, such as IL-1β, IL-2, IL-4, IL-5, IL-12, IFN-γ, TNF-α, GM-CSF, Eotoxin, IL-13, IP-10, MIG were undetectable or produced in very low quantities by tested cell lines.
Effect of anticancer drugs on the production of TPFs
The level of tumor cells produced factors (TPFs) could be a function of tumor cell number. To test this, H460 cells were seeded at 10×104 to 1.25×104 cells/well onto 96-well plate and supernatants were collected after 24 h of culture. Production of VEGF, IL-6 and IL-8 by H460 tumor cells was found to be proportional to the number of tumor cells. A reduction in cell number by only 2.5 ×104 tumor cells resulted in a proportional reduction in VEGF, IL-6 and IL-8 levels (Fig 1). Based on the correlation between tumor cell number and levels of produced factors, it is reasonable to expect that drugs that cause a reduction in tumor cell number might likewise reduce concentrations of the secreted TPFs. To investigate this possibility, H460 cells were treated for 3 days with the following drugs: doxorubicin (1.0, 0.5 and 0.25 μg/ml), cisplatin (1.5, 0.75, 0.38 μg/ml), bleomycin (4, 2, and 1 μg/ml) and etoposide (1.0, 0.5, and 0.25 μg/ml). Bleomycin is not a drug of choice for NSCLC and was used in our experiments to test whether any chemotherapeutic drugs are able to affect cytokine production and whether the patterns of changes are similar or different for different drugs. Bleomycin in comparison to doxorubicin, cisplatin and etoposide was less efficient in reduction of H460 cell numbers (Fig. 2). All drugs were able to significantly reduce the overall concentration of secreted VEGF, although bleomycin and etoposide were less effective than doxorubicin and cisplatin (Fig 1). IL-6 production was more resistant to drug treatment and was inhibited only at the two highest concentrations of doxorubicin and cisplatin. No inhibition of IL-6 was observed in the groups treated with bleomycin and etoposide at any tested concentration (Fig. 2). Similar results were obtained when CXCL8 (IL-8) levels were analyzed (data not shown). Although treatment of H460 cells with doxorubicin and cisplatin substantially reduced total cell numbers, the surviving cells showed a significant (p<0.05) increase in production of bFGF (Fig. 2). In contrast, treatment of H460 cells with bleomycin and etoposide did not affect production of bFGF (Fig 2). All tested drugs increased production of G-CSF (Fig 2). High doses of doxorubicin (1 μg/ml) and cisplatin (1.5 μg/ml) decreased production of the chemokine CCL5 (RANTES), while lower doses (0.25 and 0.38 μg/ml, respectively) increased the production CCL5. Bleomycin and etoposide stimulated production of CCL5 at all tested concentrations (Fig 2).
Fig. 1. Production of VEGF, IL-6 and IL-8 as a function of tumor cell number.
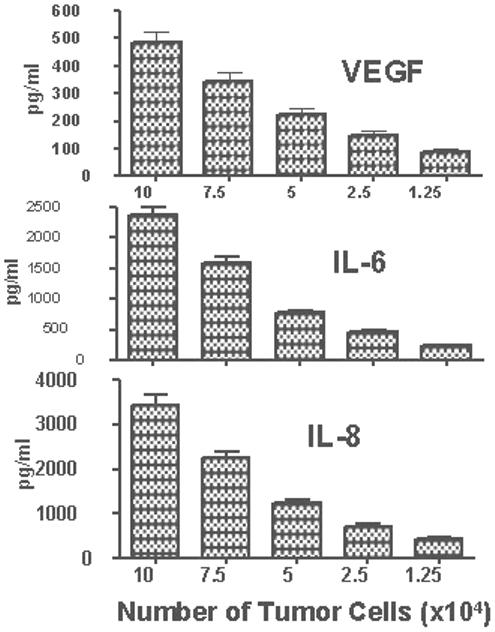
H460 tumor cells were plated onto 96-well plate at 10×104, 7.5×104, 5×104, 2.5×104 and 1.25×104cells/well. After 24 h the supernatants were collected and concentrations of VEGF, IL-6, and IL-8 were determined using multiplexed immunobeads technology. Data presented as mean concentrations (pg/ml) ± SD.
Fig 2. Effect of anticancer drugs on cytokine production by H460 cell line.
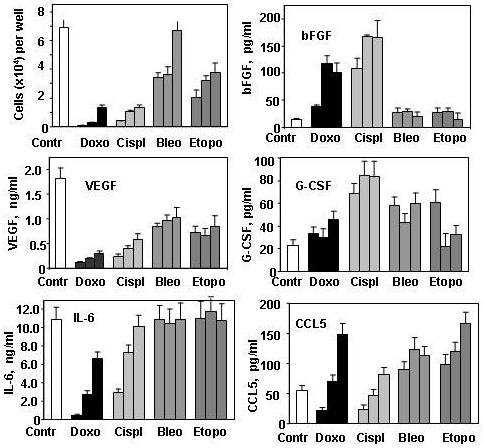
H460 cells were seeded into a 96-well plate (1×104 cells/ well) and treated with doxorubicin (1, 0.5 and 0.25 μg/ml), cisplatin (1.5, 0.75, 0.38 μg/ml), bleomycin (4, 2, and 1 μg/ml) and etoposide (1, 0.5, and 0.25 μg/ml). After three days, conditioned media were collected, cell numbers were determined using the One Solution Cell Proliferation Kit (Promega) and concentrations of cytokines and growth factors were measured using a multiplex cytokine assay. * Significantly (p<0.05) differed from the control group.
Post-treatment effects on TPF production
We next tested whether drug-induced stimulation of TPFs is sustained after drug withdrawal by surviving tumor cells. H460 cells were cultured in 24-well plates (2×105cells/well) with doxorubicin (0.125 μg/ml) or cisplatin (0.3 μg/ml) for 3 days. Both drugs exerted substantial cytotoxicity, resulting in about a 90% reduction in cell number, which was two-fold below the initial seeded level (Table 1S, Experiment A1, supplemented data). In spite of this, levels of IL-6, CXCL8 and bFGF were higher than those found in non-treated cells. Production of G-CSF and CCL5 in non-treated cells was below detectable levels, but was stimulated by drug treatment (Table 1S, supplemental data). To normalize cytokine production according to cell number, we calculated cytokine production per 1×106 cells. The data obtained revealed that doxorubicin and cisplatin induced dramatic per cell increases in IL-6, CXCL8, VEGF, BFGF, G-CSF and CCL5 (Table 2, Experiment A1).
Table 2.
Effect of drugs on cell proliferation and cytokine production by H460 cells
| Ex p# | Groups | No. cells (× 103) | Cytokines, (pg/1×106cells) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| IL-6 | IL-8 | VEGF | bFGF | G-CSF | CCL2 | CCL5 | |||
| A1 | Control | 1,000±212 | 5,118 | 7,594 | 596 | 24 | 0 | 0 | 0 |
| Doxo | 90±23* | 116,955* | 94,484* | 4,108* | 518* | 514* | 139* | 276* | |
| Cispl | 100±31* | 213,068* | 107,964* | 6,664 | 742* | 1,104* | 356* | 920* | |
| A2 | Control | 47±8 | 5,970 | 15,349 | 638 | 34 | 0 | 11 | 0 |
| PostDoxo | 9 ±1* | 137,689* | 103,911* | 3,177* | 267* | 623* | 59* | 111* | |
| PostCispl | 20±3* | 121,120* | 57,590* | 3,530* | 280* | 60* | 178* | 80* | |
Experiment A1: H460 tumor cells were seeded into a 24-well plate (2×105/well), with 8 wells per group, and cultured for 3 days in the presence of doxorubicin (0.125μg/ml) or cisplatin (0.3μg/ml). Supernatants were collected and cells were counted. The concentrations of TPFs in the supernatants were analyzed using a multiplex immunobeads kit and data were calculated as pg/1×106 cells.
Experiment A2: Cells collected from the control and drug-treated groups (experiment A1) were seeded into a 96-well plate (1×104 cells/well) in fresh media without any drug. After 3 days, supernatants were collected and the numbers of cells were estimated using the One Solution Cell Proliferation Kit (Promega).
Differences between control and experimental groups were significant (p<0.05).
After 3 days, tumor cells that survived drug treatment were harvested and viable cells were seeded into a 96-well plate (1×104cells/well) in fresh media without drugs. Growth of tumor cells previously treated with doxorubicin or cisplatin was still impaired even in the absence of drugs, but these cells continued to produce significantly high levels of IL-6, CXCL8, VEGF, bFGF, G-CSF and CCL5 on a per cell basis (Table 2, Experiment A2, for total levels of cytokine see Table 1S, supplemental data). However, the production of these factors 3 days following cisplatin withdrawal significantly reduced, whereas in doxorubicin pre-treated cells no reduction of IL-6 and CXCL8 was found (Table 2, compare A1 and A2). Thus, these data indicate that drug treatment stimulates cytokine production by H460 NSCLC cells.
Effect of doxorubicin on cytokine production by tumor cells of different origin
We next sought to determine whether drug treatment stimulated cytokine production in other tumor cell lines. Another non-small cell lung cancer line A549 and breast tumor cell lines MCF-7 and CAMA-1 were treated with doxorubicin (0.125 μg/ml) for 3 days that resulted in a substantial increase in the production of IL-6, CXCL8, VEGF, bFGF, CCL-2 and CCL-5 (Fig 3). Thus, drug treatment of tumor cells of different origin resulted in stimulation of cytokine, chemokine, growth and angiogenic factor production.
Fig 3. Cytokine production by human lung and breast tumor cells treated with doxorubicin.
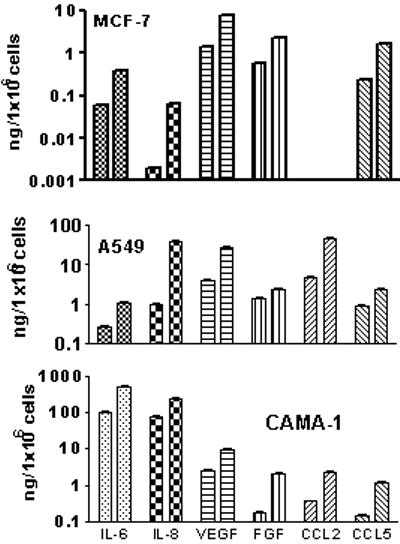
MCF7 and CAMA-1 breast tumor cells, and A549, non-small cell lung cancer cells were seeded into 96-well plates, six wells per group (5×103cells/well). After 24h, media was changed and doxorubicin (0.125 μg/ml) was added. After three days, conditioned media were collected; cells were stained with Hoechst 33342 and counted using Cellomics Array Scan VTI. Concentrations of various TPFs in the supernatants were determined using multiplex immunobeads technology. Data presented as mean ± SE pg/1×106 cells. Only cytokines with a significant (p<0.05) drug-induced increased production are presented. Left columns for each cytokine show untreated control cells; right columns for each cytokine - doxorubicin-treated tumor cells.
It is possible that increased levels of TPFs in the culture medium following drug treatment resulted from their massive release from dead cells. To test this, we analyzed media conditioned by H460 cells or by the lysates of these cells. The high levels of cytokines and chemokines were found in media conditioned by tumor cells (TCM) that were even higher in media conditioned by doxorubicin-treated tumor cells (TCMD) (Table 3). In fresh media (FM) conditioned by the lysates of tumor cells very low levels of cytokines were found (Table 3). It is important to note that a similarly low levels of tested cytokines and chemokines were found in lysates of doxorubicin-treated or untreated tumor cells probably due to the fact that these factors are not stored in cells but are quickly secreted. Thus, the increased levels of cytokines, chemokines and growth factors found in culture media were due to higher levels of production and secretion by surviving cells rather than the release from drug-killed tumor cells
Table 3.
Concentration of TPFs in medium conditioned by tumor cells (TCM), by doxorubicin-treated tumor cells (DTCM) and in fresh medium (FM) supplemented with tumor cell lysate
| Media | Cytokines (pg/1×106cells) | ||||
|---|---|---|---|---|---|
| IL-6 | CXCL8 | CCL2 | CXCL1 | VEGF | |
| TCM | 59,044 | 13,934 | 178 | 182 | 2098 |
| DTCM | 108,263* | 198,000* | 271* | 447* | 2968* |
| FM + tumor cells lysates | 122 | 68 | 12 | 78 | 4 |
| FM + drug-treated cell lysates | 150 | 67 | 10 | 60 | 4 |
H460 tumor cells were cultured in a 6-well plate (2×106/well/2 ml) in the presence or absence of doxorubicin (0.125 μg/ml) for 24h. Supernatants were collected, remaining cells were harvested and sonicated, and tumor lysates were centrifuged to remove cell debris. Tumor cell lysates (equivalent to 1×106tumor cells) were added to 1 ml of fresh culture medium. The concentrations of cytokines in media conditioned by untreated tumor cells (TCM), doxorubicin-treated tumor cells (DTCM) or fresh media (FM) conditioned by tumor lysates were determined using a Luminex multiplex assay.
Significantly (p<0.05) differ from other groups.
Effect of doxorubicin on the intracellular cytokine production
To further verify the effects of drug treatment on cytokine production, we analyzed the levels of various cytokines in cells pretreated with monensin to block cytokine secretion. Intracellular staining of multiple cytokines revealed that doxorubicin treatment of H460 cells increased production of CXCL8, CCL2, G-CSF, CCL5 and bFGF (Fig. 4A). Analysis of VEGF production showed that populations of H460 tumor cells consist of low and high producers (Fig 4B). Doxorubicin treatment increased VEGF production in both subpopulations that still remained distinct (Fig 4B).
Fig 4. Intracellular levels of cytokines in doxorubicin-treated and untreated H460 tumor cells.
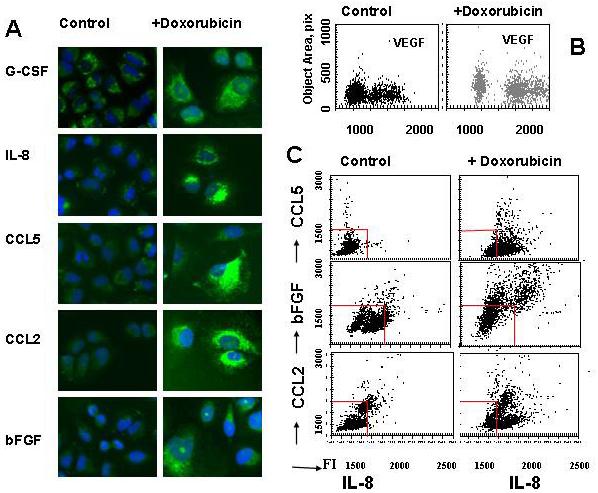
H460 cells growing in 96-well plates were treated with doxorubicin (0.125μg/ml) for 16 h and then were incubated with monensin (2 μM) for 3h. Cells were fixed, permeabilized and incubated with primary Abs against CXCL8, CCL2, CCL5, G-CSF, bFGF or VEGF and then with secondary Alexa Fluor 488 or Alexa 680-conjugated Abs. Cell nuclei were stained with Hoechst 33342 and cell images were acquired using the Cellomics ArrayScan HCS Reader (20X objective) and analyzed using the Target Activation BioApplication Software Module. A. Images of immunofluorescently stained H460 cells for CXCL8, CCL2, CCL5, G-CSF, or BFGF (Alexa488). B Fluorescence intensity of VEGF (Alexa488) is plotted against object area, pix. Each point represents a single cell. C. Doxorubicin-treated and untreated cells were incubated with monensin and double-stained for CCL5, bFGF or CCL2 (Alexa-488) and CXCL8 (Alexa-680). Fluorescence intensities of CCL5, bFGF or CCL2 were plotted against fluorescence intensity of CXCL8. Each point represents a single cell. The red lanes show the boundaries of the fluorescence intensity of untreated cells.
To test whether the same cells produce different cytokines we applied double fluorescent staining of doxorubicin-treated and untreated H460 cells (Fig 4C). Double staining with CXCL8 and CCL5 showed that a majority of tumor cells produced both CCL5 and CXCL8, but some cells preferentially produce CCL5 or CXCL8. After doxorubicin treatment, production of both CCL5 and CXCL8 substantially increased. Furthermore, cells that preferentially produced CXCL8 started to produce CCL5 (Fig 4C). Similarly, double staining showed that a vast majority of cells produce both CXCL8 and bFGF or CCL2 and doxorubicin treatment increased intracellular production of these factors (Fig 4B). These data brought additional confirmation that drug treatment stimulates cytokine production by tumor cells.
Effect of doxorubicin on transcription factor activation
Cytokine and chemokine production is regulated via activation of transcription factors 38-41. We analyzed the ability of doxorubicin to activate nine different transcription factors using the Procarta Transcription Factor Assay Kit (Panomics). H460 tumor cells were treated with doxorubicin (0.125 μg/ml). After 15 min of doxorubicin treatment, a significant (p<0.05) increase in the activity of NF-κB, AP1, AP-2, CREB, HIF-1, STAT-1, STAT-3, STAT5 and ATF-2 was observed (Fig 5A). Drug-induced activation of NF-κB, AP-1, AP-2, CREB, HIF-1 and STAT-3 was maintained even after 30 min of doxorubicin treatment (Fig 5A). These transcription factors play an important role in the expression of various genes that encode cytokines, chemokines, angiogenic and growth factors 38-42.
Fig 5.
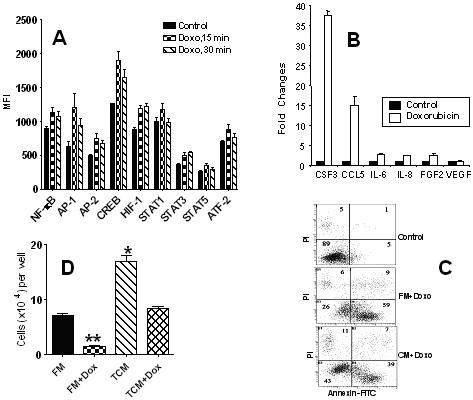
A. Doxorubicin activation of transcription factors in human H460 tumor cells. H460 tumor cells were treated with doxorubicin (0.125 μg/ml) for 0, 15 and 30 min. Nuclear extracts were prepared and an analysis of transcription factors was performed using the multiplex Procarta Transcription Factor Assay Kit. Only transcription factors with significant differences (p<0.05) after the doxorubicin treatment are presented.
B. Real-time PCR of cytokine gene expression in doxorubicin-treated and untreated H460 tumor cells. H460 tumor cells were treated with doxorubicin (0.125μg/ml) for 6h. RNA from drug-treated and untreated tumor cells was extracted, and cDNA was produced from 2 μg of RNA using the SuperArray ReactionReady First Strand cDNA Synthesis kit (SuperArray Bioscience). Real-time PCR was carried out using the SuperArray RT2 Real-Time ™ SYBR Green PCR Master Mix (SuperArray Bioscience) and was performed on the ABI Prism™ 7700 sequence detector real-time PCR system (AB Applied Biosystems). Fold changes in gene expression in doxorubicin-treated and untreated H460 tumor cells were calculated using the Δ Δ Ct method. Control level was set at 1.0.
C. Tumor cell-conditioned medium (TCM) protects tumor cells from doxorubicin-induced apoptosis. H460 cells were incubated with doxorubicin (0.125μg/ml) in fresh medium (FM) or TCM. After 6 h, cells were harvested, stained with Annexin V-FITC and PI and analyzed by flow cytometry.
D. Effect of TCM on tumor cell proliferation and drug survival. H460 cells (1×104/well) were cultured in FM or TCM for 3 days in a 96-well plate with and without doxorubicin (0.125μg/ml). Cell count was determined using the One Solution Cell Proliferation Kit (Promega, Madison, WI). * Differences between FM and TCM groups are significant (p<0.01); ** Differences between doxorubicin-treated groups are significant (p<0.05).
Effect of doxorubicin on cytokine gene expression
To assess whether drug-induced activation of transcription factors is associated with up-regulation of cytokine gene expression, a real-time PCR analysis was performed. H460 cells were treated with doxorubicin (0.125 μg/ml) for 6h. Doxorubicin treatment increased expression of IL-6, CXCL8 and FGF-2 genes by more than two-fold, whereas CSF-3 (G-CSF) and CCL5 gene expression increased by 37- and 14-fold, respectively (Fig. 5B). No significant change in expression of the VEGF gene in H460 cells was found, suggesting that a doxorubicin-induced increase in VEGF production in H460 cells might be due to posttranslational mechanisms.
Drug-protective effects of TPFs
Production of cytokines and growth factors by tumor cells could be beneficial for their proliferation and survival. To test this, H460 tumor cells grown in FM or TCM were treated with doxorubicin (0.125 μg/ml) for 6h and then incubated in drug-free FM or TCM for the next 20h. After 6h of treatment, a high proportion (59%) of tumor cells cultured in a FM entered into apoptosis, whereas only 39% of cells were found to be apoptotic when they were cultured in the TCM (Fig 5C). After 20h of treatment, about 65% of cells cultured in FM entered into the necrosis phase as evidenced by propidium iodine (PI) staining, and only 26 % of these cells remained viable. In contrast, only 19% of H460 cells cultured in TCM were necrotic and 71% of these cells were viable (data not shown).
Next we tested whether TPFs could stimulate tumor cell proliferation. Indeed, H460 tumor cells cultured in TCM, in comparison with those cultured in FM, showed a higher rate of proliferation (Fig 5D). Doxorubicin (0.125 μg/ml) significantly (p<0.01) reduced the number of surviving cells. However, the number of doxorubicin-surviving cells was higher when H460 cells were cultured in TCM than in FM (Fig 5D).
Effects of cytokine neutralizing mAbs on H460 tumor cell proliferation and sensitivity to doxorubicin
To determine which TPFs are responsible for stimulation of tumor cell proliferation and survival, we tested various neutralizing mAbs for their ability to abrogate proliferation of H460 tumor cells and increase their sensitivity to doxorubicin. H460 tumor cells were cultured in RPMI 1640 media supplemented with 1% of FBS to reduce the exogenous source of growth factors. Anti-bFGF mAb showed no effect on H460 cell proliferation, whereas neutralization of IL-6, CXCL8 or CCL2 inhibited tumor cell proliferation (Fig 6A). The highest inhibition of cell proliferation was found with anti-CCL5 mAb (Fig 6A).
Fig 6. Effects of mAbs neutralizing growth factors and chemokines on tumor cell proliferation and drug sensitivity.
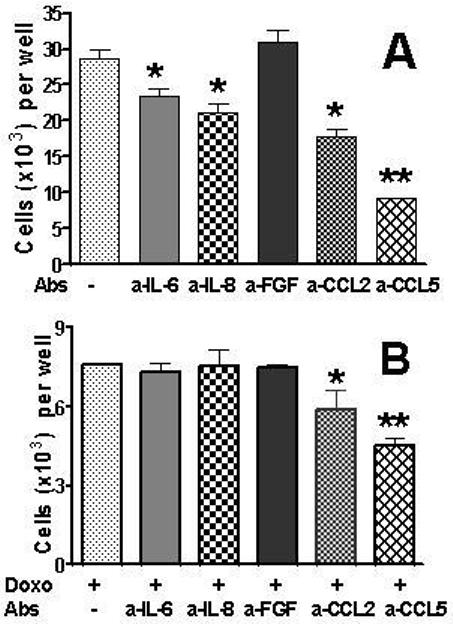
H460 cells were plated in 96-well plates at 1× 104 cells/well in FM with 10% FCS (six wells per group). The following day, cells were washed and FM with 1% FBS was added. Cells were preincubated with neutralizing mAbs (100 ng/ml) for 1 h before doxorubicin (0.125 μg/ml) was added. Cells were cultured for 48 h. After removal of the media, cells were fixed, stained with Hoechst 33342 (2 μg/mL), and counted using the Cellomics ArrayScan HCS Reader
A. Number of H460 tumor cells cultured in the presence of neutralizing mAbs. B. Number of H460 tumor cells cultured in the presence of neutralizing Abs and doxorubicin.
*Significantly differs (p<0.05) from control groups.
Treatment of H460 cells with doxorubicin (0.125 μg/ml) substantially reduced the number of tumor cells (see control in Fig 6A and doxorubicin-treated group in Fig 6B). Anti-IL-6, anti-CXCL8, and anti-bFGF mAbs had no effect on the number of surviving tumor cells treated with doxorubicin (Fig 6B). A slight but significant (p<0.05) reduction of the number of tumor cells was observed with anti-CCL2 mAb. Anti-G-CSF Ab had a similar inhibitory effect (data not shown). Anti-CCL5 Ab in combination with doxorubicn showed the most efficient reduction in number of surviving tumor cells (Fig 6). Anti-TGFβ1 and TGFβ2 Abs did not affect H460 cell proliferation or their doxorubicin survival (data not shown).
DISCUSSION
In this report we demonstrated that human tumor cell lines of different histological origin are able to produce numerous cytokines, chemokines, angiogenic and growth factors. These TPFs represent a cytokine network of tumor cells that play an important role in tumor cell proliferation and formation of supporting stroma in vivo. We found that treatment of H460 and A549 human NSCLC lines, MCF-7 and CAMA-1 breast carcinoma cell lines with anticancer drugs killed tumor cells or inhibited their proliferation. However, in surviving cells this treatment substantially stimulates production of IL-6, VEGF, bFGF, G-CSF, CXCL8, CCL2 and CCL5.
The observed drug-mediated stimulation of TPFs could be a result of an adaptive stress response through which tumor cells protect themselves from drug-mediated toxicity. Doxorubicin treatment induced multifactorial signaling with activation of various transcription factors, such as NF-κB, AP-1, AP-2, ATF-2, CREB, HIF-1, STAT-1, STAT-3, and STAT-5 that play an important role in the up-regulation of genes encoding various cytokine, chemokine and growth factors 38, 40-42. Indeed, activation of these transcription factors paralleled drug-induced up-regulation of IL-6, IL-8, bFGF, G-CSF, CCL2 and CCL5 gene expression. Thus, increased extracellular levels of cytokines and chemokines were not a result of their release from drug-killed tumor cells but due to drug-induced stimulation production of these factors.
Media conditioned by tumor cells stimulated tumor cell proliferation and protected them from drug-induced apoptosis. We found that blocking IL-6, CXCL8, CCL2 and CCL5 by specific antibodies reduced H460 tumor cell proliferation, indicating that these factors are involved in stimulation proliferation of these cells. However, blocking of IL-6 and CXCL8 did not affect survival of doxorubicin-treated tumor cells whereas neutralization of G-CSF, CCL2 and especially CCL5 increased the lethal effects of doxorubicin, suggesting that these factors are involved in protection of tumor cells from doxorubicin-mediated killing. Our recent experiments with knockdown of the CCL5 gene using specific siRNA transfection in H460 and CAMA-1 tumor cells also support the importance of CCL5 for tumor cell proliferation and sensitivity to doxorubicin (unpublished observations).
Initially, chemokine were identified for its ability to stimulate migration of proinflammatory cells. Recent studies demonstrated that they have broader biological effects both in normal and malignant cells. Chemokines, such as CXCL8, CCL2 and CCL5 are able to stimulate proliferation, migration and invasion of various tumor cells 18, 43, 44. They also manifest a potent angiogenic activity and could work in concert with other angiogenic factors to stimulate tumor angiogenesis 15, 18-21. Thus, different types of tumor producing factors (cytokines, chemokines, angiogenic and growth factors) have overlapping functions in promoting tumor growth. Blocking a single factor could be compensated by other factors with a similar functional activity and thus might be insufficient to block tumor growth.
Cellular stress response could be induced not only by drugs but also by various apoptosis-induced molecules that results in augmentation of cytokine production. FasL-Fas signaling stimulates production of IL-6 in tumor cells. We found that TRAIL by binding to cognate death receptors stimulates production of CXCL8, CCL2, CCL5 and bFGF in MCF-7, H460 and OVCAR-3 tumor cells. This stimulation was associated with activation of caspases 1 and 8 and various transcription factors. Media conditioned by tumor cells protects tumor cells from the apoptotic effect of TRAIL and this protected effect was blocked by antibodies neutralizing IL-8, CCL5 and bFGF. Our studies with doxorubicin and TRAIL showed that antibodies against TGFβ1 and TGFβ2 did not abrogate the protective effects of media conditioned by H460 lung cancer cells. However, TGFβ2 was a major factor in media conditioned by prostate cancer cells that protected fibroblasts from TNF-induced apoptosis. Media conditioned by thyroid cancer cells protected cancer cells from apoptosis induced by anticancer drugs (doxorubicin, cisplatin or taxol) or FasL and this protection was abrogated by antibodies against IL-4 and IL-10 48. These data suggest that in different histological types different factors could be involved in protection of tumor cells.
The mechanisms by which cytokines and growth factors block the apoptotic effects of drugs and other apoptotic molecules have been intensively investigated. Growth factors by binding to their respective receptors could activate PI-3 kinase/Akt, Ras/MAP kinase, and Jak/signal transducers and activators of transcription. This signaling could changes the balance between proapototic and antiapoptotic molecules, leading to increase in tumor cell surviving 49, 50.
Although all these studies are limited to in vitro cultured tumor cells we believe that drugs could induce similar effects in vivo. The tumor cytokine network of in vivo growing tumors is considerably more robust and contains factors produced by tumor cells as well as stroma cells (endothelial cells, pericytes, fibroblasts, lymphocytes, macrophages and other inflammatory cells). There is persistent cross-talk between tumor and stroma cells, producing factors that have autocrine and paracrine effects. These TPFs stimulate proliferation of tumor cells as well as endothelial cells, and drive the formation of new blood vessels to facilitate nutrient and oxygen supply and promote tumor survival. In addition, by binding to the receptors on tumor cells, cytokines, chemokines and growth factors provide proliferative signals that antagonize the harmful microenvironment (hypoxia, hypoglycemia, etc) or apoptotic effects of drugs. Thus, inhibition of such pro-survival responses might represent an important strategy for anticancer therapy.
Increased blood concentrations of cytokines, chemokines, angiogenic and growth factors were found in cancer patients, an observation often associated with resistance to therapy and a poor prognosis 12, 25, 26. It is possible that local, intra-tumor concentrations of these factors are higher than in circulation, and thus, TPFs could play an important role in tumor growth and resistance to drug therapy.
Chemotherapy is usually applied in several cycles, at three-week intervals in order to allow the body to restore hematopoietic and other normal cells damaged by drugs. However, some reports indicate that during this resting period tumor cells can aggressively repopulate the tumor and restore pre-treatment tumor size. Based on our in vitro findings, we believe that drug treatment may stimulate in vivo the production of various angiogenic and growth factors that stimulate tumor cell proliferation during resting periods, resulting in a rapid tumor repopulation. It remains undocumented whether chemotherapy results in the stimulation of cytokine production in vivo. It is unknown whether this stimulation is limited to the intra-tumor microenvironment or also results in a systemic increase of these factors in the circulation. However, some studies have shown that drug therapy stimulates the migration of endothelial precursors from the bone marrow into tumors, which is considered to be a result of drug stimulation of the production of pro-angiogenic factors. Therefore, targeting tumor-producing cytokines, chemokines, angiogenic and growth factors during drug-resting periods could reduce tumor repopulation by surviving tumor cells.
It was shown that conventional chemotherapy could be improved by combining it with a VEGF-neutralizing antibody (Avastin/ Bevacizumab) or blocking HER2/neu signaling using a mAb (Herceptin/ Trastuzumab). However, the beneficial effects of these strategies have been limited and manifested in slight increase in patient overall survival 28, 29. It is possible that drug-stimulated production of growth and pro-angiogenic factors could counterbalance the effects of antibody therapy. In addition, numerous growth factors and chemokines exhibit shared angiogenic and growth-stimulating properties, and thus reduction of a single factor is insufficient to completely block tumor growth. The efficacy of chemotherapy could be improved by modalities capable of broad inhibition or inactivation of multiple growth and proangiogenic factors involved in autocrine and paracrine regulation of tumor growth. Thus, it is necessary to develop a new approach for disrupting tumor cytokine networks in an effort to increase the efficacy of cancer therapy.
Supplementary Material
Acknowledgments
Grant support: DOD breast and Prostate Cancer Research Programs (DAMD17-03-1-0599 and DAMD-02-1-0127), Harry J. Lloyd Charitable Trust and Hillman Foundation (E.G.), NIH grants RO1 CA098642, R01 CA108990, and R21 CA102061 (L.A.). We are grateful to Mrs. Adele Marrangoni and Mrs. Liudmila Velikokhatnaya (Luminex Facility, University of Pittsburgh Cancer Institute) for their assistance with this work.
Abbreviations:
- TPFs
Tumor Produced Factors
- FM
Fresh Medium
- TCM
Tumor conditioned medium
References
- 1.Robinson SC, Coussens LM. Soluble mediators of inflammation during tumor development. Adv Cancer Res. 2005;93:59–87. doi: 10.1016/S0065-230X(05)93005-4. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15797447. [DOI] [PubMed] [Google Scholar]
- 2.Ferrara N, Alitalo K. Clinical applications of angiogenic growth factors and their inhibitors. Nat Med. 1999;5:1359–64. doi: 10.1038/70928. [DOI] [PubMed] [Google Scholar]
- 3.Kerbel R, Folkman J. Clinical translation of angiogenesis inhibitors. Nat Rev Cancer. 2002;2:727–39. doi: 10.1038/nrc905. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12360276. [DOI] [PubMed] [Google Scholar]
- 4.Basilico C, Moscatelli D. The FGF family of growth factors and oncogenes. Adv Cancer Res. 1992;59:115–65. doi: 10.1016/s0065-230x(08)60305-x. Available from http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&dopt=r&uid=1381547. [DOI] [PubMed] [Google Scholar]
- 5.Etscheid M, Beer N, Kress JA, Seitz R, Dodt J. Inhibition of bFGF/EGF-dependent endothelial cell proliferation by the hyaluronan-binding protease from human plasma. Eur J Cell Biol. 2004;82:597–604. doi: 10.1078/0171-9335-00349. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15035435. [DOI] [PubMed] [Google Scholar]
- 6.Seghezzi G, Patel S, Ren CJ, Gualandris A, Pintucci G, Robbins ES, Shapiro RL, Galloway AC, Rifkin DB, Mignatti P. Fibroblast growth factor-2 (FGF-2) induces vascular endothelial growth factor (VEGF) expression in the endothelial cells of forming capillaries: an autocrine mechanism contributing to angiogenesis. J Cell Biol. 1998;141:1659–73. doi: 10.1083/jcb.141.7.1659. Available from http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&dopt=r&uid=9647657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xin X, Yang S, Ingle G, Zlot C, Rangell L, Kowalski J, Schwall R, Ferrara N, Gerritsen ME. Hepatocyte growth factor enhances vascular endothelial growth factor-induced angiogenesis in vitro and in vivo. Am J Pathol. 2001;158:1111–20. doi: 10.1016/S0002-9440(10)64058-8. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11238059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zarnegar R, Michalopoulos GK. The many faces of hepatocyte growth factor: from hepatopoiesis to hematopoiesis. J Cell Biol. 1995;129:1177–80. doi: 10.1083/jcb.129.5.1177. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=7775566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nilsson MB, Langley RR, Fidler IJ. Interleukin-6, secreted by human ovarian carcinoma cells, is a potent proangiogenic cytokine. Cancer Res. 2005;65:10794–800. doi: 10.1158/0008-5472.CAN-05-0623. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16322225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ravoet C, DeGreve J, Vandewoude K, Kerger J, Sculier JP, Lacor P, Stryckmans P, Piccart M. Tumour stimulating effects of recombinant human interleukin-6. Lancet. 1994;344:1576–7. doi: 10.1016/s0140-6736(94)90387-5. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=7983975. [DOI] [PubMed] [Google Scholar]
- 11.Dankbar B, Padro T, Leo R, Feldmann B, Kropff M, Mesters RM, Serve H, Berdel WE, Kienast J. Vascular endothelial growth factor and interleukin-6 in paracrine tumor-stromal cell interactions in multiple myeloma. Blood. 2000;95:2630–6. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=10753844. [PubMed] [Google Scholar]
- 12.Salgado R, Junius S, Benoy I, Van Dam P, Vermeulen P, Van Marck E, Huget P, Dirix LY. Circulating interleukin-6 predicts survival in patients with metastatic breast cancer. Int J Cancer. 2003;103:642–6. doi: 10.1002/ijc.10833. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12494472. [DOI] [PubMed] [Google Scholar]
- 13.Bar-Eli M. Role of interleukin-8 in tumor growth and metastasis of human melanoma. Pathobiology. 1999;67:12–8. doi: 10.1159/000028045. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=9873223. [DOI] [PubMed] [Google Scholar]
- 14.Huang S, DeGuzman A, Bucana CD, Fidler IJ. Level of interleukin-8 expression by metastatic human melanoma cells directly correlates with constitutive NF-kappaB activity. Cytokines Cell Mol Ther. 2000;6:9–17. doi: 10.1080/13684730050515868. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=10976534. [DOI] [PubMed] [Google Scholar]
- 15.Xu L, Fidler IJ. Interleukin 8: an autocrine growth factor for human ovarian cancer. Oncol Res. 2000;12:97–106. doi: 10.3727/096504001108747567. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11132928. [DOI] [PubMed] [Google Scholar]
- 16.Koch AE, Polverini PJ, Kunkel SL, Harlow LA, DiPietro LA, Elner VM, Elner SG, Strieter RM. Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science. 1992;258:1798–801. doi: 10.1126/science.1281554. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=1281554. [DOI] [PubMed] [Google Scholar]
- 17.Strieter RM, Burdick MD, Mestas J, Gomperts B, Keane MP, Belperio JA. Cancer CXC chemokine networks and tumour angiogenesis. Eur J Cancer. 2006;42:768–78. doi: 10.1016/j.ejca.2006.01.006. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16510280. [DOI] [PubMed] [Google Scholar]
- 18.Azenshtein E, Luboshits G, Shina S, Neumark E, Shahbazian D, Weil M, Wigler N, Keydar I, Ben-Baruch A. The CC chemokine RANTES in breast carcinoma progression: regulation of expression and potential mechanisms of promalignant activity. Cancer Res. 2002;62:1093–102. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11861388. [PubMed] [Google Scholar]
- 19.Ben-Baruch A. The multifaceted roles of chemokines in malignancy. Cancer Metastasis Rev. 2006;25:357–71. doi: 10.1007/s10555-006-9003-5. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17016763. [DOI] [PubMed] [Google Scholar]
- 20.Salcedo R, Ponce ML, Young HA, Wasserman K, Ward JM, Kleinman HK, Oppenheim JJ, Murphy WJ. Human endothelial cells express CCR2 and respond to MCP-1: direct role of MCP-1 in angiogenesis and tumor progression. Blood. 2000;96:34–40. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=10891427. [PubMed] [Google Scholar]
- 21.Strieter RM, Belperio JA, Phillips RJ, Keane MP. CXC chemokines in angiogenesis of cancer. Semin Cancer Biol. 2004;14:195–200. doi: 10.1016/j.semcancer.2003.10.006. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15246055. [DOI] [PubMed] [Google Scholar]
- 22.Yaal-Hahoshen N, Shina S, Leider-Trejo L, Barnea I, Shabtai EL, Azenshtein E, Greenberg I, Keydar I, Ben-Baruch A. The chemokine CCL5 as a potential prognostic factor predicting disease progression in stage II breast cancer patients. Clin Cancer Res. 2006;12:4474–80. doi: 10.1158/1078-0432.CCR-06-0074. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16899591. [DOI] [PubMed] [Google Scholar]
- 23.Gorelik L, Flavell RA. Transforming growth factor-beta in T-cell biology. Nat Rev Immunol. 2002;2:46–53. doi: 10.1038/nri704. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11905837. [DOI] [PubMed] [Google Scholar]
- 24.Salazar-Onfray F. Interleukin-10: a cytokine used by tumors to escape immunosurveillance. Med Oncol. 1999;16:86–94. doi: 10.1007/BF02785841. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=10456656. [DOI] [PubMed] [Google Scholar]
- 25.Benoy IH, Salgado R, Van Dam P, Geboers K, Van Marck E, Scharpe S, Vermeulen PB, Dirix LY. Increased serum interleukin-8 in patients with early and metastatic breast cancer correlates with early dissemination and survival. Clin Cancer Res. 2004;10:7157–62. doi: 10.1158/1078-0432.CCR-04-0812. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15534087. [DOI] [PubMed] [Google Scholar]
- 26.Berek JS, Chung C, Kaldi K, Watson JM, Knox RM, Martinez-Maza O. Serum interleukin-6 levels correlate with disease status in patients with epithelial ovarian cancer. Am J Obstet Gynecol. 1991;164:1038–42. doi: 10.1016/0002-9378(91)90582-c. discussion 42-3. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=2014824. [DOI] [PubMed] [Google Scholar]
- 27.Schneider GP, Salcedo R, Welniak LA, Howard OM, Murphy WJ. The diverse role of chemokines in tumor progression: prospects for intervention (Review) Int J Mol Med. 2001;8:235–44. doi: 10.3892/ijmm.8.3.235. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11494048. [DOI] [PubMed] [Google Scholar]
- 28.Ferrara N, Hillan KJ, Novotny W. Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem Biophys Res Commun. 2005;333:328–35. doi: 10.1016/j.bbrc.2005.05.132. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15961063. [DOI] [PubMed] [Google Scholar]
- 29.Romond EH, Perez EA, Bryant J, Suman VJ, Geyer CE, Jr., Davidson NE, Tan-Chiu E, Martino S, Paik S, Kaufman PA, Swain SM, Pisansky TM, et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med. 2005;353:1673–84. doi: 10.1056/NEJMoa052122. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16236738. [DOI] [PubMed] [Google Scholar]
- 30.Lev DC, Onn A, Melinkova VO, Miller C, Stone V, Ruiz M, McGary EC, Ananthaswamy HN, Price JE, Bar-Eli M. Exposure of melanoma cells to dacarbazine results in enhanced tumor growth and metastasis in vivo. J Clin Oncol. 2004;22:2092–100. doi: 10.1200/JCO.2004.11.070. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=doptPubMed&=Citation&list_uids=15123733. [DOI] [PubMed] [Google Scholar]
- 31.Lev DC, Ruiz M, Mills L, McGary EC, Price JE, Bar-Eli M. Dacarbazine causes transcriptional up-regulation of interleukin 8 and vascular endothelial growth factor in melanoma cells: a possible escape mechanism from chemotherapy. Mol Cancer Ther. 2003;2:753–63. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12939465. [PubMed] [Google Scholar]
- 32.Darst M, Al-Hassani M, Li T, Yi Q, Travers JM, Lewis DA, Travers JB. Augmentation of chemotherapy-induced cytokine production by expression of the platelet-activating factor receptor in a human epithelial carcinoma cell line. J Immunol. 2004;172:6330–5. doi: 10.4049/jimmunol.172.10.6330. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15128823. [DOI] [PubMed] [Google Scholar]
- 33.Shibakura M, Niiya K, Kiguchi T, Kitajima I, Niiya M, Asaumi N, Huh NH, Nakata Y, Harada M, Tanimoto M. Induction of IL-8 and monoclyte chemoattractant protein-1 by doxorubicin in human small cell lung carcinoma cells. Int J Cancer. 2003;103:380–6. doi: 10.1002/ijc.10842. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12471621. [DOI] [PubMed] [Google Scholar]
- 34.Giuliano KA, Haskins JR, Taylor DL. Advances in high content screening for drug discovery. Assay Drug Dev Technol. 2003;1:565–77. doi: 10.1089/154065803322302826. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15090253. [DOI] [PubMed] [Google Scholar]
- 35.Gorelik E, Landsittel DP, Marrangoni AM, Modugno F, Velikokhatnaya L, Winans MT, Bigbee WL, Herberman RB, Lokshin AE. Multiplexed immunobead-based cytokine profiling for early detection of ovarian cancer. Cancer Epidemiol Biomarkers Prev. 2005;14:981–7. doi: 10.1158/1055-9965.EPI-04-0404. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15824174. [DOI] [PubMed] [Google Scholar]
- 36.Yurkovetsky Z, Kirkwood J, Edington H, Marrangoni A, Velikokhatnaya L, Winans M, Gorelik E, Lokshin AE. Multiplex analysis of serum cytokines in melanoma patients treated with interferon-alpha2b. Clin Cancer Res. 2007;13:2422–8. doi: 10.1158/1078-0432.CCR-06-1805. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17438101. [DOI] [PubMed] [Google Scholar]
- 37.Vogt A, Adachi T, Ducruet AP, Chesebrough J, Nemoto K, Carr BI, Lazo JS. Spatial analysis of key signaling proteins by high-content solid-phase cytometry in Hep3B cells treated with an inhibitor of Cdc25 dual-specificity phosphatases. J Biol Chem. 2001;276:20544–50. doi: 10.1074/jbc.M100078200. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11274178. [DOI] [PubMed] [Google Scholar]
- 38.Karin M. NF-kappaB and cancer: mechanisms and targets. Mol Carcinog. 2006;45:355–61. doi: 10.1002/mc.20217. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16673382. [DOI] [PubMed] [Google Scholar]
- 39.Richmond A. Nf-kappa B, chemokine gene transcription and tumour growth. Nat Rev Immunol. 2002;2:664–74. doi: 10.1038/nri887. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12209135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schindler C, Strehlow I. Cytokines and STAT signaling. Adv Pharmacol. 2000;47:113–74. doi: 10.1016/s1054-3589(08)60111-8. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=10582086. [DOI] [PubMed] [Google Scholar]
- 41.Shaulian E, Karin M. AP-1 in cell proliferation and survival. Oncogene. 2001;20:2390–400. doi: 10.1038/sj.onc.1204383. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11402335. [DOI] [PubMed] [Google Scholar]
- 42.Zarember KA, Malech HL. HIF-1alpha: a master regulator of innate host defenses? J Clin Invest. 2005;115:1702–4. doi: 10.1172/JCI25740. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16007247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vaday GG, Peehl DM, Kadam PA, Lawrence DM. Expression of CCL5 (RANTES) and CCR5 in prostate cancer. Prostate. 2006;66:124–34. doi: 10.1002/pros.20306. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16161154. [DOI] [PubMed] [Google Scholar]
- 44.Mrowietz U, Schwenk U, Maune S, Bartels J, Kupper M, Fichtner I, Schroder JM, Schadendorf D. The chemokine RANTES is secreted by human melanoma cells and is associated with enhanced tumour formation in nude mice. Br J Cancer. 1999;79:1025–31. doi: 10.1038/sj.bjc.6690164. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=10098731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choi C, Xu X, Oh JW, Lee SJ, Gillespie GY, Park H, Jo H, Benveniste EN. Fas-induced expression of chemokines in human glioma cells: involvement of extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase. Cancer Res. 2001;61:3084–91. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11306491. [PubMed] [Google Scholar]
- 46.Levina V, Marrangoni A, DeMarco R, Gorelik E, Lokshin A. Multiple effects of TRAIL in human carcinoma cells: induction of apoptosis, senescence, proliferation, and cytokine production. Experimental Cell Research. 2008;314:1605–16. doi: 10.1016/j.yexcr.2007.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu T, Burdelya LG, Swiatkowski SM, Boiko AD, Howe PH, Stark GR, Gudkov AV. Secreted transforming growth factor beta2 activates NF-kappaB, blocks apoptosis, and is essential for the survival of some tumor cells. Proc Natl Acad Sci U S A. 2004;101:7112–7. doi: 10.1073/pnas.0402048101. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15118089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Conticello C, Pedini F, Zeuner A, Patti M, Zerilli M, Stassi G, Messina A, Peschle C, De Maria R. IL-4 protects tumor cells from anti-CD95 and chemotherapeutic agents via up-regulation of antiapoptotic proteins. J Immunol. 2004;172:5467–77. doi: 10.4049/jimmunol.172.9.5467. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15100288. [DOI] [PubMed] [Google Scholar]
- 49.Lee TL, Yeh J, Friedman J, Yan B, Yang X, Yeh NT, Van Waes C, Chen Z. A signal network involving coactivated NF-kappaB and STAT3 and altered p53 modulates BAX/BCL-XL expression and promotes cell survival of head and neck squamous cell carcinomas. Int J Cancer. 2008;122:1987–98. doi: 10.1002/ijc.23324. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=18172861. [DOI] [PubMed] [Google Scholar]
- 50.Talapatra S, Thompson CB. Growth factor signaling in cell survival: implications for cancer treatment. J Pharmacol Exp Ther. 2001;298:873–8. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11504779. [PubMed] [Google Scholar]
- 51.Kim JJ, Tannock IF. Repopulation of cancer cells during therapy: an important cause of treatment failure. Nat Rev Cancer. 2005;5:516–25. doi: 10.1038/nrc1650. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15965493. [DOI] [PubMed] [Google Scholar]
- 52.Shaked Y, Kerbel RS. Antiangiogenic strategies on defense: on the possibility of blocking rebounds by the tumor vasculature after chemotherapy. Cancer Res. 2007;67:7055–8. doi: 10.1158/0008-5472.CAN-07-0905. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17671170. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.