

Abstract
Duchenne and Becker muscular dystrophies (DMD and BMD) are X-linked recessive neuromuscular disorders caused by mutations in the dystrophin gene affecting approximately 1 in 3,500 males. The human dystrophin gene spans > 2,200 kb, or roughly 0.1% of the genome, and is composed of 79 exons. The mutational spectrum of disease-causing alleles, including exonic copy number variations (CNVs), is complex. Deletions account for approximately 65% of DMD mutations and 85% of BMD mutations. Duplications occur in approximately 6–10% of males with either DMD or BMD. The remaining 30–35% of mutations consist of small deletions, insertions, point mutations, or splicing mutations, most of which introduce a premature stop codon. Laboratory analysis of dystrophin can be used to confirm a clinical diagnosis of DMD, characterize the type of dystrophin mutation, and perform prenatal testing and carrier testing for females. Current dystrophin diagnostic assays involve a variety of methodologies, including multiplex PCR, Southern blot analysis, MLPA, DOVAM-S, and SCAIP; however, these methods are time-consuming, laborious, and do not accurately detect duplication mutations in the dystrophin gene. Furthermore, carrier testing in females is often difficult when a related affected male is unavailable. Here we describe the development, design, validation, and implementation of a high-resolution CGH microarray-based approach capable of accurately detecting both deletions and duplications in the dystrophin gene. This assay can be readily adopted by clinical molecular testing laboratories and represents a rapid, cost-effective approach for screening a large gene, such as dystrophin.
Keywords: dystrophin, microarray, CGH array, Duchenne Muscular Dystrophy, DMD, Becker Muscular Dystrophy, BMD, exonic-copy number variation, CNV
Introduction
Duchenne and Becker muscular dystrophies (DMD; MIM# 310200 and BMD; MIM# 300376) are X-linked recessive neuromuscular disorders affecting approximately 1 in 3,500 and 1 in 30,000 live male births, respectively (Mehler, 2000). DMD and BMD are both characterized by progressive symmetrical muscular weakness, often with calf hypertrophy. DMD symptoms typically appear before age 5, with wheelchair dependency seen by age 12. Patients usually succumb to the disorder by their late teens or early 20s. For BMD, symptoms have a much later age of onset, and wheelchair dependency, if present, typically occurs after age 16. More than 90% of BMD patients are still alive in their 20s (Kunkel, et al., 1986; Mehler, 2000).
Both DMD and BMD are caused by mutations in the dystrophin gene, which is the largest human gene, spanning > 2,200 kb on the X chromosome and occupying roughly 0.1% of the genome. The gene is composed of 79 exons that together account for only 0.6% of its sequence (Koenig, et al., 1987). The extremely large size of the dystrophin gene leads to a complex mutational spectrum (Buzin, et al., 2005; White, et al., 2006; White and den Dunnen, 2006).
Previous reports suggest that large deletions account for approximately 65% of DMD mutations and 85% of BMD mutations. Duplications occur in roughly 6–10% of males with either DMD or BMD. The remaining mutations are small deletions, insertions, point mutations, or splicing mutations, most of which introduce premature stop codons (Mendell, et al., 2001; Prior and Bridgeman, 2005). Unlike the large deletions that cluster in just two regions of the dystrophin gene, small deletions and point mutations appear to be evenly distributed throughout. To date, 501 deletions, 84 duplications, and 989 point mutations have been documented in the dystrophin gene (Leiden muscular dystrophy database; http://www.dmd.nl).
The current methodologies used for detecting mutations in the dystrophin gene include multiplex PCR, Southern blotting (Stockley, et al., 2006), multiplex ligation-dependent probe amplification (MLPA) (Gatta, et al., 2005; Janssen, et al., 2005; Schwartz and Duno, 2004), detection of virtually all mutations-SSCP (DOVAM-S) (Buzin, et al., 2005; Buzin, et al., 2000; Liu, et al., 1999), denaturing high-performance liquid chromatography (DHPLC) (Bennett, et al., 2001), single condition amplification/internal primer sequencing (SCAIP) (Flanigan, et al., 2003), and Sanger sequencing (Hamed and Hoffman, 2006; Stockley, et al., 2006).
Multiplex PCR tests the most commonly deleted regions of the dystrophin gene; the original multiplex PCR only tested for about 20 out of the 79 total regions and could not test for duplications. Recent technical improvements now allow for the assay of all 79 exons using more than one multiplex reaction in males. Whereas deletion detection is fairly robust with this approach, small deletions and most duplications remain difficult to pick up. Moreover, the precise boundaries of a deletion cannot be identified to determine the reading frame. Female carriers are also difficult to identify by this method. Southern blotting can reveal large dystrophin gene deletions and duplications in males; however, Southern blotting is time-consuming, requires hazardous reagents, and is limited to only relatively large deletions/duplications. Although Southern blotting can sometimes detect female carriers, its sensitivity is generally low. MLPA is used to detect both deletions and duplications of coding regions of the dystrophin gene: however, finding duplications in males and some deletions in females is difficult. Single-exon deletions must be confirmed by a second method (such as multiplex PCR or sequencing), because single nucleotide polymorphisms in some regions tested will appear falsely as deletions. Most of these methods are suitable for detecting mutations in males. Testing for females is problematic with all these approaches, especially testing for deletions and duplications. Moreover, duplications are not easily detected by these methods for either sex.
Microarray-based genomic analysis has revolutionized cytogenetics (Gunn, et al., 2007; Shaffer and Bejjani, 2006). Recently, BAC arrays have been replaced in some applications by oligonucleotide arrays, which have proved to be robust and sensitive. Oligonucleotide arrays are known to be extremely effective in the detection of known and new microdeletion syndromes (Gunn, et al., 2007).
Here we describe the development and validation of a targeted, high-density oligonucleotide comparative genomic hybridization (CGH) microarray that permits a high-resolution analysis of the dystrophin gene. The CGH can identify not only deletions and duplications, but also previously unidentified deep intronic mutations. Furthermore, the sensitivity and specificity of the CGH array allow accurate testing for females. Thus our findings establish a CGH approach as the superior clinical laboratory test.
Material and Methods
Validation samples
A retrospective analysis of 29 patient samples was performed for validation (Table 1). These previously characterized samples were obtained from the Emory Genetics Laboratory, the OHSU DNA Diagnostic Laboratory (Portland, Oregon), and LabPLUS (Auckland, New Zealand). Samples were characterized using multiplex PCR of 32 exons and Southern blotting. These included 15 male patients, 11 with deletions and 4 with duplications in the dystrophin gene. Also included were 14 female samples, 12 with deletions and 2 with duplications. The technician performing the analysis was blinded to patient data for all samples.
Table 1.
List of deletions and duplications in the dystrophin gene
| Average Nimblescan/GLAD predicted breakpoint | Size of breakpoint | Array Results | Southern/Multiplex/MLPA Results | HGVS Nomenclature | ||
|---|---|---|---|---|---|---|
| Sample | Start | End | ||||
| 1Mv | 31,995,179 | 31,584,297 | 410,882 | del Ex45 - Ex54 | del Ex45 - Ex52 | c.6439−?_8027+?del |
| 2Mv | 32,469,353 | 32,073,457 | 395,897 | del Ex17 - Ex44 | del Ex17 - Ex44 | c.1993−?_6438+?del |
| 3Mv | 31,811,249 | 31,608,059 | 203,190 | del Ex48 - Ex53 | del Ex48 - Ex52 | c.6913−?_7872+?del |
| 4Mv | 32,842,800 | 32,662,600 | 180,200 | del Ex3 - Ex7 | del Ex3 - Ex7 | c.94−?_649+?del |
| 5Mv | 32,181,888 | 32,098,595 | 83,293 | del Ex44 | del Ex44 | c.6291−?_6438+?del |
| 6Mv | 32,003,438 | 31,855,778 | 147,661 | del Ex45 - Ex47 | del Ex45 - Ex47 | c.6439−?_6912+?del |
| 7Mv | 31,877,576 | 31,620,852 | 256,724 | del Ex46 - Ex52 | del Ex46 - Ex52 | c.6615−?_7660+?del |
| 8Mv | 31,824,256 | 31,637,403 | 186,853 | del Ex48 - Ex52 | del Ex48 - Ex52 | c.6913−?_7660+?del |
| 9Mv | 33,013,846 | 32,761,242 | 252,604 | dup Ex2 - Ex4 | dup Ex2 - Ex4 | c.32−?_264+?dup |
| 10Mv | 31,784,750 | 31,736,250 | 48,500 | del Ex49 - Ex50 | del Ex49 - Ex50 | c.7099−?_7309+?del |
| 11Mv | 32,474,250 | 32,094,750 | 379,500 | del Ex17 - Ex44 | del Ex17 - Ex44 | c.1993−?_6438+?del |
| 12Mv | 32,794,750 | 32,676,750 | 118,000 | dup Ex3 - Ex7 | dup Ex3 - Ex7 | c.94−?_649+?dup |
| 13Mv | 31,715,250 | 31,503,750 | 211,500 | dup Ex51 - Ex55 | dup Ex51 - 55 | c.7310−?_8217+?dup |
| 14Mv | 31,645,750 | 31,533,250 | 112,500 | del Ex53 - Ex55 | del Ex53 - Ex55 | c.7661−?_8217+?del |
| 15Mv | 33,026,750 | 32,761,250 | 265,500 | dup Ex2 - Ex4 | dup Ex2 - Ex4 | c.32−?_264+?dup |
| 16Fv | 31,864,405 | 31,555,638 | 308,767 | del Ex46 - Ex55 | del Ex46 - Ex55 | c.6615+?_8217+?del |
| 17Fv | 31,781,840 | 31,736,096 | 45,744 | del Ex49 - Ex50 | del Ex49 - Ex50 | c.7099−?_7309+?del |
| 18Fv | 32,779,413 | 32,706,289 | 73,125 | del Ex4 - Ex7 | del Ex3 - Ex7 | c.94−?_649+?del |
| 19Fv | 31,948,537 | 31,518,523 | 430,014 | del Ex45 - Ex55 | del Ex45 - Ex52 | c.6439−?_8217+?del |
| 20Fv | 31,799,723 | 31,723,560 | 76,164 | del Ex49 - Ex50 | del Ex48 - Ex50 | c.7099−?_7309+?del |
| 21Fv | 31,499,751 | 31,046,000 | 453,751 | del Ex56 - Ex79 | del Ex56 - Ex79 | c.8218−?_11058+?del |
| 22Fv | 31,495,434 | 31,046,000 | 449,434 | del Ex56 - Ex79 | del Ex56 - Ex79 | c.8218−?_11058+?del |
| 23Fv | 32,453,261 | 32,275,827 | 177,434 | dup Ex18 - Ex38 | dup Ex18 - Ex38 | c.2169−?_5448+?dup |
| 24Fv | 32,037,000 | 31,867,000 | 170,000 | del Ex45 | del Ex45 | c.6439+?_6614+?del |
| 25Fv | 32,191,750 | 32,030,750 | 161,000 | dup Ex44 | dup Ex44 | c.6291−?_6438+?dup |
| 26Fv | 32,633,250 | 32,521,250 | 112,000 | del Ex8 - Ex13 | del Ex8 - Ex13 | c.831−?1602+?del |
| 27Fv | 32,622,750 | 32,313,750 | 309,000 | del Ex10 - Ex33 | del Ex10 - Ex33 | c.961−?_4674+?del |
| 28Fv | 31,969,750 | 31,703,750 | 266,000 | del Ex45 - Ex50 | del Ex45 - Ex50 | c.6439−?_7309+?del |
| 29Fv | 32,488,250 | 32,425,250 | 63,000 | del Ex17 - Ex19 | del Ex17 - Ex19 | c.1993−?_2380+?del |
| 1Mc | 32,719,450 | 32,575,950 | 143,500 | del Ex8 - Ex11 | del Ex8 - Ex11 | c.650−?_1331+?del |
| 2Mc | 33,026,750 | 32,761,250 | 265,500 | dup Ex2 - Ex4 | dup Ex2 - Ex4 | c.32−?_264+?dup |
| 3Mc | 32,037,000 | 31,867,000 | 170,000 | del Ex45 | del Ex45 | c.6439−?_6614+?del |
| 4Mc | 31,981,750 | 31,563,750 | 418,000 | del Ex45 - Ex54 | del Ex45 - Ex54 | c.6439−?_8027+?del |
| 5Mc | 32,633,250 | 32,521,250 | 112,000 | del Ex8 - Ex13 | del Ex8 - Ex13 | c.650−?_1602+?del |
| 6Mc | 31,715,250 | 31,503,750 | 211,500 | dup Ex51 - Ex55 | dup Ex51 - Ex55 | c.7310−?_8217+?dup |
| 7Mc | 32,622,750 | 32,313,750 | 309,000 | del Ex10 - Ex33 | del Ex10 - Ex33 | c.961−?_4674+?del |
| 8Mc | 31,645,750 | 31,533,250 | 112,500 | del Ex53 - Ex55 | del Ex53 - Ex55 | c.7661−?_8217+?del |
| 9Mc | 31,969,750 | 31,703,750 | 266,000 | del Ex45 - Ex50 | del Ex45 - Ex50 | c.6439−?_7309+?del |
| 10Mc | 32,455,250 | 32,444,250 | 11,000 | del Ex18 | del Ex18 | c.2169−?_2292+?del |
| 11Mc | 32,488,250 | 32,425,250 | 63,000 | del Ex17 - Ex19 | del Ex17 - Ex19 | c.1993−?_2380+?del |
| 12Mc | 32,629,450 | 32,572,850 | 56,600 | dup Ex8 - Ex10 | dup Ex8 - Ex10 | c.650−?_1149+?del |
| 13Mc | 31,702,250 | 31,573,250 | 129,000 | del Ex54 | del Ex54 | c.7873−?_8027+?del |
| 14Mc | 31,989,750 | 31,578,750 | 411,000 | del Ex45 - Ex54 | del Ex45 - Ex54 | c.6439−?_8027+?del |
| 15Mc | 32,628,500 | 32,561,500 | 67,000 | dup Ex8 - Ex11 | dup Ex8 - Ex11 | c.650−?_1331+?dup |
| 16Mc | 31,661,000 | 31,641,000 | 20,000 | del Ex52 | del Ex52 | c.7543−?_7660+?del |
| 17Mc | 32,014,750 | 31,838,750 | 176,000 | del Ex45 - Ex47 | del Ex45 - Ex47 | c.6439−?_6912+?del |
| 18Mc | 32,212,750 | 31,720,250 | 492,500 | dup Ex44 - Ex50 | dup Ex44 - Ex50 | c.6291−?_7390+?dup |
| 19Mc | 31,615,750 | 31,607,250 | 8,500 | del Ex51 - Ex53 | del Ex51 - Ex53 | c.7310−?_7872+?del |
| 20Mc | 31,939,750 | 31,797,750 | 142,000 | del Ex45-Ex48 | del Ex45-Ex48 | c.6439−?_7098+?del |
| 21Mc | 32,000,750 | 31,874,250 | 126,500 | del Ex45 | del Ex45 | c.6439−?_6614+?del |
| 22Mc | 31,438,250 | 31,434,250 | 4,000 | del Ex56 | del Ex56 | c.8218−?_8390+?del |
| 23Mc | 32,092,250 | 31,979,750 | 112,500 | dup Ex35 - Ex44 | dup Ex35 - Ex44 | c.4846−?_6438+?dup |
| 24Mc | 32,518,250 | 32,484,250 | 34,000 | dup Ex3 - Ex16 | dup Ex3 - Ex16 | c.94−?_1992+?dup |
| 25Mc | 32,728,750 | 32,500,750 | 228,000 | dup Ex3 - Ex15 | dup Ex3 - Ex15 | c.94−?_1812+?dup |
| 26Mc | 31,921,250 | 31,886,250 | 35,000 | del Ex45 | del Ex45 | c.6439−?_6614+?del |
| 27Mc | 31,764,750 | 31,623,750 | 141,000 | del Ex49 - Ex52 | del Ex49 - Ex52 | c.7099−?_7660+?del |
| 28Mc | 32,590,500 | 32,452,500 | 138,000 | dup Ex10 -Ex17 | dup Ex10 -Ex17 | c.961−?_2168+?dup |
| 29Mc | 32,609,500 | 32,453,500 | 156,000 | dup Ex10 -Ex17 | dup Ex10 -Ex17 | c.961−?_2168+?dup |
| 30Mc | 31,870,250 | 31,667,750 | 202,500 | del Ex46 - Ex51 | del Ex46 - Ex51 | c.6615−?_7542+?del |
| 31Mc | 33,013,750 | 32,980,750 | 33,000 | del intron 1 | c.31+?_32−?del | |
| 32,899,250 | 32,888,250 | 11,000 | del intron 2 | c.93+?_94−?del | ||
| 32Fc | 32,794,750 | 32,676,750 | 118,000 | dup Ex3 - Ex7 | dup Ex3 - Ex7 | c.94−?_649+?dup |
| 33Fc | 32,191,750 | 32,030,750 | 161,000 | dup Ex44 | dup Ex44 | c.6291−?_6438+?dup |
| 34Fc | 32,628,750 | 32,557,750 | 71,000 | dup Ex8 - Ex11 | dup Ex8 - Ex11 | c.650−?_1331+?dup |
| 35Fc | 31,877,750 | 31,729,250 | 148,500 | del Ex46 - Ex50 | del Ex46 - Ex50 | c.6615−?_7309+?del |
| 36Fc | 32,834,250 | 32,485,250 | 349,000 | dup Ex3 - Ex16 | dup Ex3 - Ex16 | c.94−?_1992+?dup |
Samples listed included validation samples and mutations identified in clinical sample (GenBank file NM_004006.1). Key: Mv –Male validation, Fv – Female Validation, Mc – Male Clinical, Fc – Female Clinical. HGVS- Human Genome Variation Society
Clinical samples
Following this validation, prospective studies were performed on samples from patients referred for DMD or BMD evaluation based on clinical presentation and family history (Table 1).
Case study
A blood sample from a 4-year-old male was sent to our laboratory for microarray-based molecular testing. We identified a deletion mutation encompassing exons 8–13 (c.831−?_1602+?del) and subsequently sent the mother’s sample for carrier testing. All results were confirmed by MLPA and sequencing. The MLPA kit was obtained from MRC-Holland, and the assay was performed according to the manufacturer’s instructions. Exon 13 sequencing was performed using standard Sanger sequencing with the primers in Table 2.
Table 2.
Primers for Exon 13
| DMD Ex13F | TGT AAA ACG ACG GCC AGT GAGATGTAGCAGAAATAAATTTCACCAT |
| DMD Ex13R | CAG GAA ACA GCT ATG ACC TACTTTTCAAGTTATAGTTCTTTTAAAGGACATAT |
CGH high-density dystrophin array design
NimbleGen manufactures high-density DNA arrays based on its proprietary Maskless Array Synthesizer (MAS) technology. The arrays have long oligonucleotides (~50–75mer) to ensure greater sensitivity and specificity. A custom-designed 385K array was used for detecting deletions and duplications in the dystrophin gene. The array has 4,115 internal control probes and 385,474 probes spanning the 2,222,000 bases of the dystrophin gene on chromosome X: 31,046,000–33,268,000. Probe lengths range from 45–60 bases with isothermal Tm across the array. The average spacing between probes is 5 bases. The vast number of probes permits oversampling of the region (Figure 1).
Figure 1.
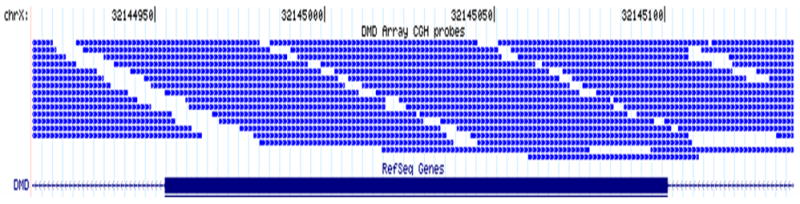
Representative example of dystrophin CGH array design. A sample 240-bp region, including the entire exon 44 and 92 bp of flanking intronic sequence, of the dystrophin gene shows array CGH probe distribution. Each thin blue line represents one probe. Exon 44 is represented by the thick blue line at the bottom of the figure.
CGH protocol and analysis
DNA was extracted from patient samples using the Puregene DNA Extraction Kit (Gentra Systems, Minneapolis, MN) according to the manufacturer’s instructions. Male and female wild-type control DNA was obtained from Promega Inc. Each patient and reference DNA sample was sonicated such that fragment size was between 500–2,000 bases, as verified on a 1% agarose gel. Patient and reference DNA samples were labeled using Klenow enzyme (NEB) and Cy3 or Cy5 9mer wobble primers (TriLink BioTechnologies, San Diego, CA), respectively. After labeling, each sample was purified by isopropanol precipitation and reconstituted in ultra-pure water. Next we combined 13 ug of labeled patient and reference DNA, and the products were desiccated in a vacufuge (Savant DNA 120), then resuspended in appropriate hybridization buffer, along with Cy3 and Cy5 control CPK6 50mer oligonucleotides. This mixture was hybridized to the array for 16–20 h at 42°C in a MAUI Hybridization System (BioMicro Systems, Salt Lake City, UT). Arrays were then washed according to the manufacturer’s recommendation and immediately scanned on a GenePix 4000 scanner (Molecular Devices).
After scanning, we extracted data from images and achieved within-array normalization using the manufacturer-provided software (NimbleScan). Normalized log2 ratio data were analyzed using two different analysis programs: (SegMNT or DNA copy) NimbleScan (NimbleGen Systems Inc) and GLAD (www.bioconductor.org; Hupe, et al., 2004). Both software programs report breakpoints for predicted deletions or duplications in the patient (or test) sample relative to the reference (GenBank File NM_004006.1) and also display results graphically in a bar graph, where the y-axis indicates gain or loss of material (1 – gain, 0 – normal, −1 – loss), while the x-axis indicates the position of each feature on the chromosome. For subsequent clinical testing, analysis was performed using only the NimbleScan software program.
Assessment of array quality
Array quality is assessed by control resequencing oligonucleotides on each array that correspond to synthetic sequence designed to have no cross-hybridization potential with any known sequence. This sequence was designed to have 3 distinct sequencing domains with different characteristics: the A, B, and C domains. We resequenced both the forward and the reverse strands, so the resequencing report has 6 distinct scores for the Cy3 channel and 6 distinct scores for the Cy5 channel: A-forward and A-reverse, B-forward and B-reverse, C-forward and C-reverse.
The ‘A’ domain contains long runs of G nucleotides that can be difficult to synthesize; the ‘B’ domain contains a large, perfect hairpin sequence, whereas the ‘C’ domain contains a straightforward domain that should always hybridize. Failure of domain ‘C’ would indicate a catastrophic failure. Control DNA is spiked into each experiment. A score of 0–100% is obtained, which represents the sequencing fidelity and is a good indicator of the quality of the microarray processes. This correlates well with the overall performance of a microarray experiment.
Results
Validation of CGH array
Males
We performed array CGH on 15 male samples with previously characterized structural mutations at the dystrophin locus. All 11 deletion mutations, including the single-exon mutation, were successfully detected (Table 1). Additionally, all 4 duplications were identified by our CGH array. Overall, our CGH array successfully identified mutations in all 15 samples. Representative profiles for deletion and duplication mutations using CGH are shown in Figure 2.
Figure 2.


Validation of targeted CGH dystrophin array for males and females. The dystrophin coordinates are represented at the top, with exon 1 to 79 from right to left. The representative array results shown here for males and females are displayed in the scatter plot. Each pair shows results of CGH analysis using NimbleGen DNACopy (A) and GLAD analysis (B). 1) 15 Mv - male with duplication of exons 2–4; 2) 5 Mv - male with deletion of exon 44; 3) 2 Mv - male with deletion of exons 17–44; 4) 8 Mv - male with deletion of exons 48–52; 5) 16 Fv - female with deletion of exons 46–55; 6) 20 Fv - female with deletion of exons 49–50; and 7) 23 Fv - female with duplication of exons 18–38.
Reassignment of breakpoints in male controls
Using our CGH microarray, we were able to reassign the deletion breakpoint for 2 samples more precisely (Table 1). All results were reconfirmed by multiplex PCR using exons included in the deletion and exons flanking the deleted exons. Sample 1Mv was previously found to have a deletion of exons 45 to 52 by multiplex PCR of 32 exons. The CGH array reclassified this 3′ deletion boundary to exon 54 (c.6439−?_8027+?del). Sample 3Mv was previously found to have a deletion of exons 48 to 52 by multiplex PCR of 32 exons. The CGH array showed that exon 53 was also deleted in this sample (c.6913−?_7872+?del).
Females
We performed array CGH on 14 female samples with previously characterized structural mutations at the dystrophin locus. All 12 deletion mutations, including one single-gene exon deletion, were detected (Table 1). Two samples containing duplications at the dystrophin locus were identified. Overall, our CGH array successfully identified mutations in all 14 samples. Representative profiles for deletion and duplication mutations using CGH are shown in Figure 2.
Reassignment of breakpoints in female controls
Using the CGH methodology we were able to reassign deletion breakpoints for 2 samples (Table 1). Sample 18Fv was previously found to have a deletion of exons 3 to 7 by Southern blot. The CGH array reclassified this 5′ deletion boundary to exon 4 and showed that exon 3 was not involved in the deletion (c.94−?_649+?del). Sample 19Fv was previously found to have a deletion of exons 45 to 52 by Southern blot. The CGH array showed that exons 53–55 were also deleted in this sample (c.6439−?_8217+?del). All results were confirmed by MLPA.
Clinical samples
A total of 35 deletions and 14 duplications were detected in the clinical samples received at the Emory Genetics Laboratory for clinical testing. There were 3 deletions and 4 duplications in-frame and 32 deletions and 10 duplications out-of-frame in the dystrophin transcript. Samples for which there was no deletion or duplication detected were subsequently sequenced for the entire coding region of the dystrophin gene. From a total of 102 samples received in the period from May 2006 to December 2007, 40% had deletions, 25% had duplications, and 33% had point mutations, taking the combined detection rate above 98% for patients with a clinical diagnosis of DMD/BMD. This 98% detection rate is based on some deletions and duplications being detected more than once in the patients analyzed. There were no mutations detected in an affected female with a possible but unconfirmed diagnosis of DMD, nor were any detected in 2 females with a family history of muscular dystrophy, type not specified. Duplications occurred more frequently than noted in the literature, but the sample size was insufficient to draw meaningful conclusions from this.
A novel double deletion was identified in an 18-year-old male (Sample 31Mc) with the BMD phenotype. These deletions were located in intron 1 (33013750–32980750: size 33 kb, c.31+?_32−?del) and intron 2 (32899250–32888250: size 11 kb, c.93+?_94−?del). A muscle biopsy sample from this patient was not available to study the effect of these 2 deletions on the dystrophin transcript. It is possible that these 2 deletions impair proper splicing of exons 1–3 in the dystrophin gene. Most samples were reported in an average turnaround time of 7–10 calendar days.
A summary of deletions and duplications identified in the clinical laboratory are listed in Table 1. Representative single-exon and multiple-exon deletions and duplications are shown in Figure 3. These samples served as a complete validation for the CGH-based approach for detecting deletions and duplications in the dystrophin gene. The deletions and duplications used for validation spanned the entire dystrophin gene sequence, establishing that the CGH array-based design is sensitive for detecting mutations across the entire dystrophin gene.
Figure 3.

Targeted CGH dystrophin array for clinical samples. The dystrophin gene coordinates are represented at the top, with exon 1 to 79 from right to left. The representative array results shown here for males and females are displayed in the scatter plot. Each pair shows results of CGH analysis using NimbleGen SegMNT. 1) 10 Mc - male with deletion of exon 18 (c.2169−?_2292+?del); 2) 4 Mc - male with deletion of exons 45–54 (c.6439−?_8027+?del); 3) 2 Mc -male with duplication of exon 2–4 (c.32−?_264+?dup); 4) 23 Mc - male with duplication of exons 35–44 (c.4846−?_6438+?dup); 5) 31 Mc - male with a 33-kb deletion in intron 1(c.31+?_32−?del) and an 11-kb deletion in intron 2 (c.93+?_94−?del); 6) 35 Fc - female with deletion of exons 49–50 (c.6615−?_7309+?del); and 7) 33 Fc - female with duplication of exon 44 (c.6291−?_6438+?dup); and 8) 34 Fc - female with duplication of exons 8–11 (c.650−?_1331+?dup).
Case study
A deletion mutation of exons 8–13 was identified in a male proband by CGH. Subsequent carrier testing on the mother’s sample by CGH did not detect the exon 8–13 (c.831−?_1602+?del) deletion. MLPA was used for confirmation of the mother’s result. MLPA showed no deletion of exons 8–12, but did indicate a deletion of exon 13. Sequencing of exon 13 on the mother’s sample detected a SNP (c.1554T> A: p.D518E) at the 3′ end of the forward probe (Figure 4).
Figure 4.


CGH and MLPA analysis of exon 8–13 (c.650−?_1602+?del) familial deletion mutation. Panel A - Exons 1 to 79 are represented on the CGH scatter plot from right to left. Scatter plots shown are for CGH analysis on the male proband with deletion of exons 8–13 (above) and carrier testing for the proband’s mother for exons 8–13 showing no deletion of exons 8–13 (below). Panel B -Confirmatory analysis of the negative findings for the mother using MLPA showed no deletion of exons 8–12 and a deletion of exon 13. Exon peaks are marked with black arrows, and exon 13 is marked with a red arrow. Other peaks on the panel are internal controls for the MLPA reaction. Panel C - Subsequent sequencing of exon 13 on the maternal sample detected a SNP (c.1554T> A: p.D518E) where the MLPA probe binds. Presence of the SNP interfered with hybridization of the probe, giving the appearance of an exon 13 deletion.
Discussion
Here we describe a novel CGH microarray-based testing strategy that enables detection of deletions and duplications in the dystrophin gene for both males and females. Completion of the first stage of diagnostic testing is achieved with a custom-designed, targeted CGH microarray capable of exhaustive detection of deletions and duplications in the dystrophin genomic region. This targeted array contains probes that cover the entire 2.2-MB genomic region of the dystrophin gene. All the unique sequences in this 2.2-MB genomic region are queried in our CGH assay, including all exons, introns, 5′ UTR, 3′ UTR, and promoters of the dystrophin gene, thereby enabling comprehensive detection of all disease-causing deletions and duplications. If the presence of a disease-causing deletion or duplication in a patient sample is ruled out by the CGH assay, there is a second diagnostic step: sequencing the dystrophin gene. In rare cases when a novel intronic change is identified and a muscle biopsy is available, RNA can be extracted from the muscle biopsy to search for evidence of a splicing error (Figure 5). In many cases a muscle biopsy may not be available, but in light of the novel findings, one might be warranted; another test that could be considered for this purpose is the less invasive needle biopsy, which should be adequate for isolating RNA.
Figure 5.

Dystrophin testing algorithm.
Our diagnostic protocol using targeted CGH permits an unmatched high-resolution analysis of the dystrophin gene. The CGH microarray will not only identify deletions and duplications, but also reveal previously unidentified deep intronic mutations. Furthermore, our CGH array allows for enhanced detection of duplications that may be missed by other technologies. Because of the density of oligonucleotide probes on our CGH array, all mutations are mapped very close to their exact nucleotide breakpoint. The superior sensitivity and specificity of microarrays allow accurate testing for female carriers, which is especially useful when no affected male is available for testing. The CGH microarray-based approach provides rapid results, is easy to perform, overcomes the limitations inherent to other diagnostic testing methodologies, and at the same time offers equivalent detection sensitivity for males and females.
Most importantly, the CGH array can be reported in 7–10 calendar days, in marked contrast to the current turnaround of 3–4 weeks common to most clinical laboratories, hence the rapidity of our testing procedure significantly improves the turnaround time compared with other available methodologies. Furthermore, the sensitivity of this array technology means improved access to carrier and prenatal testing.
CGH arrays can be obtained from several manufacturers, so their availability is not limited to a single source. The cost of performing the CGH array is comparable to other methods, such as Southern blot or MLPA, but the CGH array offers a significant improvement in the sensitivity and accuracy of mutation detection over the other techniques. The initial investment in this technology is the requirement for a scanner, the cost of which ranges from $50,000–$100,000; however, most cytogenetic laboratories and core facilities already have a scanner. The other necessary but smaller equipment is usually readily available in most clinical laboratories.
CGH array design
CGH array design is critical to the success of our assay. The CGH array allows oversampling of the region, so that instead of relying on a single probe to detect a deletion or duplication event, it is possible to have multiple probes covering an exon. The availability of thousands of oligos means the ability to cover the gene of interest with high density, thereby enabling higher sensitivity and accuracy for mutation detection. Thus, the ability to oversample a region gives the array-based assays a tremendous advantage over other methods for detecting deletions and duplications.
Advantages of CGH microarray
Use of a full genomic region CGH array confers a number of advantages over an exon array or other diagnostic testing technologies, like MLPA. The full genomic region array gives approximate intronic boundaries of the mutation event, which may prove useful in the future for targeted gene therapies, such as exon-skipping strategies (Table 1). Reassignment of the breakpoints for four previously characterized samples, two males and two females, clearly demonstrates the sensitivity of this methodology. These samples were characterized using multiplex PCR of 32 exons and Southern blotting. Multiplex PCR of 32 exons of the dystrophin gene does not allow precise determination of the exact exon breakpoint. Data generated from Southern blot is difficult to interpret for duplications in males and both deletions and duplications in females. Unlike MLPA, the full gene region CGH microarray does not require probes to bind to specific genome sequences, thereby reducing the chance of a false-positive finding (Figure 4). Since MLPA is available in a kit-based format from MRC-Holland, it is not feasible for clinical laboratories to easily replace troublesome probes in the kit. Most laboratories perform MLPA assays in duplicate or triplicate to obtain consistent results. Finally, the full genomic region microarray promises a more comprehensive assessment of the mutational spectrum at the dystrophin locus, including copy number variants (CNVs) within the dystrophin genomic sequence (White and den Dunnen, 2006).
In about 1–2% of DMD/BMD patients, there is no causative mutation identified in the dystrophin gene. In these cases the diagnosis can be confirmed by immunohistochemical analysis of a muscle biopsy. Over the past few years, several deep intronic mutations have been described (Adachi, et al., 2003; Buzin, et al., 2005; Chaturvedi, et al., 2001; Fajkusova, et al., 2001; Hofstra, et al., 2004; Tuffery-Giraud, et al., 1999; van Essen, et al., 2003). Our targeted CGH array will allow for detection of these previously missed deep intronic deletions and duplications. Futher functional studies, such as cDNA sequencing, will be required to predict the effect of novel changes in the dystrophin gene.
CGH arrays have been used extensively for detecting genomic rearrangements, and they are quickly becoming the preferred methodology in cytogenetics laboratories worldwide (Cheung, et al., 2007). Recently Dhami et al. (Dhami, et al., 2005) have described the utility of an exon array for identifying mutations. Using an exon array, 31 patient samples were screened across an array containing a total of 162 exons for five disease genes, including dystrophin; the array detected copy number changes ranging from whole-gene deletions and duplications to single-exon deletions and duplications in 100% of the cases.
Our custom-designed, high-density oligonucleotide microarray will allow a detailed characterization of the mutational spectrum of the dystrophin gene and will help us better understand its basic genomic structure. While evidence suggests that deletions and duplications occur nonrandomly, they may have a different distribution across the genomic region. Two deletion hot spots in the dystrophin gene are already known, but the mechanisms that determine chromosome breaks in these regions remain a mystery. Our inability to rapidly survey the large introns in the dystrophin gene has hampered the description of breakpoint sequences (Ferlini and Muntoni, 1998; Gualandi, et al., 2003; Gualandi, et al., 2006).
At the same time, the extent and significance of duplication mutations in DMD/BMD has long been a matter of debate. Duplication frequency is highest near the 5′ end of the gene, with a duplication of exon 2 being the single most common duplication identified. Recently, White and den Dunnen (White and den Dunnen, 2006) reviewed the duplication mutations in the dystrophin gene. In an unselected patient series, the duplication frequency was 7%. In patients already screened for deletions and point mutations, duplications were detected in 87% of cases. Four complex, noncontiguous rearrangements were found, two of which involved a partial triplication. In one of the few cases where RNA was analyzed, a seemingly contiguous duplication turned out to be a duplication/deletion case generating a transcript with an unexpected single-exon deletion and an initially undetected duplication. These findings indicate that, for clinical laboratory testing, an assay should allow for optimal detection of duplications and, where possible, cDNA sequencing should be performed.
Microarray-based DNA diagnostic testing
The great flexibility of microarrays and the recent major reductions in their cost, coupled with their need for minimal laboratory space and personnel effort, make them a highly attractive platform for diagnostic use. Microarray-based CGH and sequencing could be combined for comprehensive mutation detection in all known Mendelian single-gene disorders. We envision different testing strategies depending upon the mutation spectrum of the gene of interest. The dystrophin gene is a unique example wherein the majority of the mutations are large deletions and duplications; therefore, performing the CGH array is the first logical step. For genes where a majority of the mutations are point mutations, sequencing could be used first, followed by CGH array to detect large deletions and duplications. It is worth considering that prior technological limitations on the detection of deletions and duplications may have led to an underestimation of the role these mutations play in disease13. Furthermore, for large genes or in cases where collections of genes are known to harbor deletions or duplications that cause a disease phenotype, development of a CGH can assess the entire collection of genes in a rapid and cost-effective manner. Current CGH arrays can assay more than 2 MB of sequence per microarray, which translates to a substantial capacity for diagnostic testing.
Recent advances in therapeutic interventions make identifying mutations in the dystrophin gene more urgent. PTC124 was recently described as a drug that induces readthrough of a premature stop codon, without affecting the normal termination codon (Hamed, 2006; Welch, et al., 2007). In addition, several exon-skipping strategies are being pursued (Aartsma-Rus, et al., 2007; Wilton and Fletcher, 2006).
A microarray-based DMD and BMD test represents a huge leap forward in DMD and BMD diagnosis, offering major advantages over the other molecular diagnostic methodologies available today. Furthermore, this assay significantly enhances mutation detection in carrier females and in prenatal testing. Our findings lay the groundwork for future studies to explore other affected pathways by examining a large repertoire of dystrophin gene mutational events. Understanding the biology of the way dystrophin gene variants influence the expression of other genes in multiple pathways would prove invaluable to the design, development, and testing of customized therapeutic interventions for patients suffering with DMD or BMD.
Acknowledgments
We thank Dr. Sue Richards of the OHSU DNA Diagnostic Laboratory and Dr. Don Love of LabPLUS, Auckland Hospital, New Zealand for providing positive controls. This work was supported, in part, by NIH grants MH805832 to JGM and MH076439 to MEZ.
References
- Aartsma-Rus A, Janson AA, van Ommen GJ, van Deutekom JC. Antisense-induced exon skipping for duplications in Duchenne muscular dystrophy. BMC Med Genet. 2007;8:43. doi: 10.1186/1471-2350-8-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adachi K, Takeshima Y, Wada H, Yagi M, Nakamura H, Matsuo M. Heterogous dystrophin mRNA produced by a novel splice acceptor site mutation in intermediate dystrophinopathy. Pediatr Res. 2003;53(1):125–31. doi: 10.1203/00006450-200301000-00021. [DOI] [PubMed] [Google Scholar]
- Bennett RR, den Dunnen J, O’Brien KF, Darras BT, Kunkel LM. Detection of mutations in the dystrophin gene via automated DHPLC screening and direct sequencing. BMC Genet. 2001;2:17. doi: 10.1186/1471-2156-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzin CH, Feng J, Yan J, Scaringe W, Liu Q, den Dunnen J, Mendell JR, Sommer SS. Mutation rates in the dystrophin gene: a hotspot of mutation at a CpG dinucleotide. Hum Mutat. 2005;25(2):177–88. doi: 10.1002/humu.20132. [DOI] [PubMed] [Google Scholar]
- Buzin CH, Wen CY, Nguyen VQ, Nozari G, Mengos A, Li X, Chen JS, Liu Q, Gatti RA, Fujimura FK, et al. Scanning by DOVAM-S detects all unique sequence changes in blinded analyses: evidence that the scanning conditions are generic. Biotechniques. 2000;28(4):746–50. 752–3. doi: 10.2144/00284rr04. [DOI] [PubMed] [Google Scholar]
- Chaturvedi LS, Mukherjee M, Srivastava S, Mittal RD, Mittal B. Point mutation and polymorphism in Duchenne/Becker muscular dystrophy (D/BMD) patients. Exp Mol Med. 2001;33(4):251–6. doi: 10.1038/emm.2001.41. [DOI] [PubMed] [Google Scholar]
- Cheung SW, Shaw CA, Scott DA, Patel A, Sahoo T, Bacino CA, Pursley A, Li J, Erickson R, Gropman AL, et al. Microarray-based CGH detects chromosomal mosaicism not revealed by conventional cytogenetics. Am J Med Genet A. 2007;143(15):1679–86. doi: 10.1002/ajmg.a.31740. [DOI] [PubMed] [Google Scholar]
- Dhami P, Coffey AJ, Abbs S, Vermeesch JR, Dumanski JP, Woodward KJ, Andrews RM, Langford C, Vetrie D. Exon array CGH: detection of copy-number changes at the resolution of individual exons in the human genome. Am J Hum Genet. 2005;76(5):750–62. doi: 10.1086/429588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajkusova L, Lukas Z, Tvrdikova M, Kuhrova V, Hajek J, Fajkus J. Novel dystrophin mutations revealed by analysis of dystrophin mRNA: alternative splicing suppresses the phenotypic effect of a nonsense mutation. Neuromuscul Disord. 2001;11(2):133–8. doi: 10.1016/s0960-8966(00)00169-3. [DOI] [PubMed] [Google Scholar]
- Ferlini A, Muntoni F. The 5′ region of intron 11 of the dystrophin gene contains target sequences for mobile elements and three overlapping ORFs. Biochem Biophys Res Commun. 1998;242(2):401–6. doi: 10.1006/bbrc.1997.7976. [DOI] [PubMed] [Google Scholar]
- Flanigan KM, von Niederhausern A, Dunn DM, Alder J, Mendell JR, Weiss RB. Rapid direct sequence analysis of the dystrophin gene. Am J Hum Genet. 2003;72(4):931–9. doi: 10.1086/374176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatta V, Scarciolla O, Gaspari AR, Palka C, De Angelis MV, Di Muzio A, Guanciali-Franchi P, Calabrese G, Uncini A, Stuppia L. Identification of deletions and duplications of the DMD gene in affected males and carrier females by multiple ligation probe amplification (MLPA) Hum Genet. 2005;117(1):92–8. doi: 10.1007/s00439-005-1270-7. [DOI] [PubMed] [Google Scholar]
- Gualandi F, Rimessi P, Cardazzo B, Toffolatti L, Dunckley MG, Calzolari E, Patarnello T, Muntoni F, Ferlini A. Genomic definition of a pure intronic dystrophin deletion responsible for an XLDC splicing mutation: in vitro mimicking and antisense modulation of the splicing abnormality. Gene. 2003;311:25–33. doi: 10.1016/s0378-1119(03)00527-4. [DOI] [PubMed] [Google Scholar]
- Gualandi F, Rimessi P, Trabanelli C, Spitali P, Neri M, Patarnello T, Angelini C, Yau SC, Abbs S, Muntoni F, et al. Intronic breakpoint definition and transcription analysis in DMD/BMD patients with deletion/duplication at the 5′ mutation hot spot of the dystrophin gene. Gene. 2006;370:26–33. doi: 10.1016/j.gene.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Gunn SR, Robetorye RS, Mohammed MS. Comparative genomic hybridization arrays in clinical pathology: progress and challenges. Mol Diagn Ther. 2007;11(2):73–7. doi: 10.1007/BF03256225. [DOI] [PubMed] [Google Scholar]
- Hamed SA. Drug evaluation: PTC-124--a potential treatment of cystic fibrosis and Duchenne muscular dystrophy. IDrugs. 2006;9(11):783–9. [PubMed] [Google Scholar]
- Hamed SA, Hoffman EP. Automated sequence screening of the entire dystrophin cDNA in Duchenne dystrophy: point mutation detection. Am J Med Genet B Neuropsychiatr Genet. 2006;141(1):44–50. doi: 10.1002/ajmg.b.30234. [DOI] [PubMed] [Google Scholar]
- Hofstra RM, Mulder IM, Vossen R, de Koning-Gans PA, Kraak M, Ginjaar IB, van der Hout AH, Bakker E, Buys CH, van Ommen GJ, et al. DGGE-based whole-gene mutation scanning of the dystrophin gene in Duchenne and Becker muscular dystrophy patients. Hum Mutat. 2004;23(1):57–66. doi: 10.1002/humu.10283. [DOI] [PubMed] [Google Scholar]
- Hupe P, Stransky N, Thiery JP, Radvanyi F, Barillot E. Analysis of array CGH data: from signal ratio to gain and loss of DNA regions. Bioinformatics. 2004;20(18):3413–22. doi: 10.1093/bioinformatics/bth418. [DOI] [PubMed] [Google Scholar]
- Janssen B, Hartmann C, Scholz V, Jauch A, Zschocke J. MLPA analysis for the detection of deletions, duplications and complex rearrangements in the dystrophin gene: potential and pitfalls. Neurogenetics. 2005;6(1):29–35. doi: 10.1007/s10048-004-0204-1. [DOI] [PubMed] [Google Scholar]
- Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50(3):509–17. doi: 10.1016/0092-8674(87)90504-6. [DOI] [PubMed] [Google Scholar]
- Kunkel LM, Monaco AP, Bertelson CJ, Colletti CA. Molecular genetics of Duchenne muscular dystrophy. Cold Spring Harb Symp Quant Biol. 1986;51(Pt 1):349–51. doi: 10.1101/sqb.1986.051.01.041. [DOI] [PubMed] [Google Scholar]
- Liu Q, Feng J, Buzin C, Wen C, Nozari G, Mengos A, Nguyen V, Liu J, Crawford L, Fujimura FK, et al. Detection of virtually all mutations-SSCP (DOVAM-S): a rapid method for mutation scanning with virtually 100% sensitivity. Biotechniques. 1999;26(5)(8):932–936. 940–2. doi: 10.2144/99265rr03. [DOI] [PubMed] [Google Scholar]
- Mehler MF. Brain dystrophin, neurogenetics and mental retardation. Brain Res Brain Res Rev. 2000;32(1):277–307. doi: 10.1016/s0165-0173(99)00090-9. [DOI] [PubMed] [Google Scholar]
- Mendell JR, Buzin CH, Feng J, Yan J, Serrano C, Sangani DS, Wall C, Prior TW, Sommer SS. Diagnosis of Duchenne dystrophy by enhanced detection of small mutations. Neurology. 2001;57(4):645–50. doi: 10.1212/wnl.57.4.645. [DOI] [PubMed] [Google Scholar]
- Prior TW, Bridgeman SJ. Experience and strategy for the molecular testing of Duchenne muscular dystrophy. J Mol Diagn. 2005;7(3):317–26. doi: 10.1016/S1525-1578(10)60560-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz M, Duno M. Improved molecular diagnosis of dystrophin gene mutations using the multiplex ligation-dependent probe amplification method. Genet Test. 2004;8(4):361–7. doi: 10.1089/gte.2004.8.361. [DOI] [PubMed] [Google Scholar]
- Shaffer LG, Bejjani BA. Medical applications of array CGH and the transformation of clinical cytogenetics. Cytogenet Genome Res. 2006;115(3–4):303–9. doi: 10.1159/000095928. [DOI] [PubMed] [Google Scholar]
- Stockley TL, Akber S, Bulgin N, Ray PN. Strategy for comprehensive molecular testing for Duchenne and Becker muscular dystrophies. Genet Test. 2006;10(4):229–43. doi: 10.1089/gte.2006.10.229. [DOI] [PubMed] [Google Scholar]
- Tuffery-Giraud S, Chambert S, Demaille J, Claustres M. Point mutations in the dystrophin gene: evidence for frequent use of cryptic splice sites as a result of splicing defects. Hum Mutat. 1999;14(5):359–68. doi: 10.1002/(SICI)1098-1004(199911)14:5<359::AID-HUMU1>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- van Essen AJ, Mulder IM, van der Vlies P, van der Hout AH, Buys CH, Hofstra RM, den Dunnen JT. Detection of point mutation in dystrophin gene reveals somatic and germline mosaicism in the mother of a patient with Duchenne muscular dystrophy. Am J Med Genet A. 2003;118(3):296–8. doi: 10.1002/ajmg.a.10056. [DOI] [PubMed] [Google Scholar]
- Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, Paushkin S, Patel M, Trotta CR, Hwang S, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447(7140):87–91. doi: 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]
- White SJ, Aartsma-Rus A, Flanigan KM, Weiss RB, Kneppers AL, Lalic T, Janson AA, Ginjaar HB, Breuning MH, den Dunnen JT. Duplications in the DMD gene. Hum Mutat. 2006;27(9):938–45. doi: 10.1002/humu.20367. [DOI] [PubMed] [Google Scholar]
- White SJ, den Dunnen JT. Copy number variation in the genome; the human DMD gene as an example. Cytogenet Genome Res. 2006;115(3–4):240–6. doi: 10.1159/000095920. [DOI] [PubMed] [Google Scholar]
- Wilton SD, Fletcher S. Redirecting splicing to address dystrophin mutations: molecular by-pass surgery. Prog Mol Subcell Biol. 2006;44:161–97. doi: 10.1007/978-3-540-34449-0_8. [DOI] [PubMed] [Google Scholar]