

Abstract
We have previously reported that the expression of Angiotensin II (Ang II) type 1 receptors (AT1R) was increased in the rostral ventrolateral medulla (RVLM) of rabbits with chronic heart failure (CHF) and in the RVLM of normal rabbits infused with intracerebroventricular (ICV) Ang II. The present study investigated if oxidant stress plays a role in Ang II induced AT1R up-regulation and its relationship to the transcription factor activator protein 1 (AP1) in CHF rabbits and in the CATHa neuronal cell line. In CATHa cells, Ang II significantly increased AT1R mRNA by 123 ± 11%, P<0.01; c-Jun mRNA by 90 ± 20%, P<0.01; c-fos mRNA by 148 ± 49%, P<0.01; NADPH oxidase activity by 126 ± 43%, P<0.01 versus untreated cells. Tempol and Apocynin reversed the increased expression of AT1R mRNA, c-Jun mRNA, c-fos mRNA, and superoxide production induced by Ang II. We also examined the effect of ICV Tempol on the RVLM of CHF rabbits. Compared to vehicle treated CHF rabbits, Tempol significantly decreased AT1R protein expression (1.6±0.29 vs 0.88±0.16, P<0.05), phosphorylated Jnk protein (0.4 ± 0.05 vs 0.2 ± 0.04, P<0.05), cytosolic phosphorylated c-Jun (0.56 ± 0.1 vs 0.36 ± 0.05, P<0.05), and nuclear phosphorylated c-Jun (0.67±0.1 vs 0.3±0.08, P<0.01). Tempol also significantly decreased the AP-1-DNA binding activity in the RVLM of CHF rabbits compared to the vehicle group (9.14 × 103 vs 41.95 × 103 grey level P<0.01). These data suggest that Ang II induces AT1R up-regulation at the transcriptional level by induction of oxidant stress and activation of AP1 in both cultured neuronal cells and in intact brain of rabbits. Antioxidant agents may be beneficial in CHF and other states where brain Ang II is elevated by decreasing AT1R expression through the Jnk and AP1 pathway.
Introduction
Angiotensin II (Ang II), the principle mediator of the renin-angiotensin system, plays a pivotal role in cardiovascular and renal homeostasis. Most of the physiological and pathophysiological effects of Ang II are medicated by the Angiotensin II type 1 receptor (AT1R). The activation of the renin-angiotensin system and the sympathetic nervous system in chronic heart failure (CHF) are critical factors that contribute to the development and progression of CHF in patients[1,2]. Evidence also demonstrates that AT1R blockers, when added to conventional treatment for patients with CHF, are associated with a reduction in morbidity and mortality as well as an improvement in quality of life[3]. Currently, blockade of the renin-angiotensin system has become the primary treatment for patients with CHF. Therefore, a comprehensive understanding of the regulation of the AT1R is necessary for developing new therapies to target this receptor in the setting of CHF.
The role of reactive oxygen species (ROS) in the cardiovascular system has been investigated extensively over the past decade[4]. It has been reported that, in the rostral ventrolateral medulla (RVLM), the levels of ROS are elevated in stroke-prone spontaneously hypertensive rats. Scavenging superoxide anion (O •−2) in the RVLM reduces both sympathetic nerve activity and blood pressure[5]. Similar evidence has been provided for rabbits with CHF[6] . Ang II induces a rapid and transient increase in ROS in a wide variety of cell types[7]. Ang II-induced ROS-formation is attenuated by the NADPH oxidase inhibitor diphenyleneiodonium chloride (DPI), suggesting that it is mediated through NADPH Oxidase[7]. In addition, Ang II-induced ROS-formation can also be blocked by losartan[8], suggesting that AT1R and its down-stream pathway are involved.
However, the downstream targets of ROS have remained largely unexplored, at least for the regulation of the AT1R. Extracellular administration of non-lethal concentrations of H2O2 has been demonstrated to activate mitogen-activated protein kinase (MAPK) as well as the c-Jun amino-terminal kinase (JNK) [9]. Endogenous ROS induced by Ang II has also been shown to activate stress activated protein kinase (SAPK), which can be inhibited by treating cells with chemical or enzymatic antioxidants[10-13]. This has also been demonstrated for JNK activation, where it has been noted in several cell types that activation of JNK is inhibited by pretreatment with the antioxidant N-acetylcysteine[14].
The transcription factor AP1 is stimulated by a host of ligands leading eventually to serine phosphorylation and subsequent AP1 translocation to the nucleus. ROS are important in AP1 activation since most of the activation can be blocked by antioxidant treatment[15].
We and other investigators have reported that Ang II infusion into the RVLM can alter sympathetic outflow. The expression of AT1R is up-regulated in the RVLM of rabbits with CHF or in normal rabbits subjected to chronic introcerebroventricular (ICV) infusion of Ang II. The up-regulation of central AT1R occurs with activation of the renin-angiotensin system and results in increased sympathetic nerve activity in CHF animals[16,17]. We observed that losartan attenuated the AT1R up regulation in rabbits with CHF or following ICV Ang II infusion in normal rabbits[18]. Central blockade of the AT1R in the CHF state reduced sympathetic nerve activity and increased baroreflex function[19,20]. In rat hypertensive models, losartan suppresses the expression of both basal and enhanced cardiac AT1R[21].
We hypothesized that Ang II up-regulates AT1R through ROS modulation of a downstream signaling pathway that includes AP1. The effects of losartan in CHF on the amelioration of AT1R upregulation may be mediated by blocking the formation of endogenous ROS. To test our hypothesis and provide additional molecular targets for central intervention in the CHF state, we investigated the relationship between oxidative stress, antioxidant treatment, AT1R regulation and the transcription factor AP1 in a neuronal cell line and in the RVLM of intact CHF rabbits.
Materials and Methods
These experiments were reviewed and approved by the University of Nebraska Medical Center Institutional Animal Care and Use Committee and conformed to the Guidelines for the Care and Use of Experimental Animals of the American Physiological Society and the National Institutes of Health.
Cell Culture
A neuronal cell line (CATH.a) was purchased from ATCC™ and the cells were grown in RPMI 1640 containing 8% horse serum, 4% FBS, 100 IU/l penicillin, at 37°C in 95% air and 5% CO2 in a humidified atmosphere. For experiments, cells were plated on polystyrene tissue culture dishes at a density of 1 × 107 cells/100-mm plate, or 1.5 × 106 cells/well in 6-well culture plates. After subculture, cells were allowed to grow for two days and then pretreated with either an AT1 receptor blocker (losartan, 10 μM, Merck), an NADPH oxidase inhibitor (Apocynin, 20μM, Calbiochem), an SOD mimetic (Tempol, 0.2μM, Calbiochem), or a SAPK inhibitor (Jnk inhibitor I, 1μM, Calbiochem) for 30 minutes, then treated with Ang II (100 nM, Sigma) for 3 hours. DMSO or water was used a vehicle control.
Induction of CHF and ICV Infusion in Rabbits
Ten male New Zealand White rabbits weighing between 3.5 and 4.3 Kg were induced to develop CHF by chronic ventricular tachycardia, as previously described[20]. The ten CHF rabbits were divided into two groups (n=5 per group): one treated with ICV vehicle infusion and one with ICV Tempol infusion (10μg/μl/h). For chronic ICV infusions, a 19-gauge cannula was implanted into a lateral cerebral ventricle as previously described[22]. An osmotic minipump (Model 2001, Durect Corp.) filled with Tempol or artificial cerebrospinal fluid (1μL/h) was implanted subcutaneously in the back of the neck and connected to the ICV cannula. The infusion was continued for 7 days.
Cardiac Function, Arterial Pressure, Heart Rate, LV Pressure, Ejection Fraction
Cardiac function was measured by echocardiography (Acuson Sequoia 512C), with the rabbits hand-held in the conscious state. Arterial pressure was measured with a radiotelemetry unit (Data Sciences International) the catheter of which was inserted into the right femoral artery. LV pressure was measured with a Millar transducer (Model SPR-524, Millar Instruments, Inc.) under anesthesia at the conclusion of the experiment.
Preparation of RVLM Tissue
At the end of the experiment, the rabbits were killed with pentobarbital sodium. The brain was removed and immediately frozen on dry ice, blocked in the coronal plane, and sectioned at 300-μm thickness in a cryostat. The RVLM was punched according to the method of Palkovits and Brownstein[23] for analysis of mRNA and protein of the AT1R receptor and other molecular studies.
RNA Extraction, cDNA Synthesis, and real-time PCR
Total RNA from 3 mm punches of RVLM or CATH.a cells was isolated using the RNeasy kit (Qiagen). cDNA was generated using the iScript™ cDNA Sysnthesis Kit (Bio-Rad). Gene Specific primers and probes were synthesized in the UNMC Eppley Cancer Center DNA synthesis Core Facility (please see Table 1 in the on line supplement for the sequences of the gene specific primers and probes). Gene specific probes were labeled with FAM at the 5’ site with Blackhole at the 3’ site in order to add specificity and sensitivity. β-actin was used as an internal control for calculation of relative expression levels of target genes in the rabbit studies. GAPDH was used as an internal control for calculation of relative expression levels of target genes in CATHa cells. Both β-actin and GAPDH are commonly used as internal controls in Ang II studies by others[22,24]. Real-time PCR was performed by using HotStarTaq® DNA polymerase (Qiagen) on CHROMO4TM Continuous Fluorescence Detector (Bio-Rad). Analysis of relative gene expression was based on the method described by Livak, et al.[25].
Table 1.
Baseline Hemodynamic Parameters in CHF Rabbits Treated with Vehicle or Tempol
| CHF + Vehicle (icv) | CHF + Tempol (icv) | |||
|---|---|---|---|---|
| n | 5 | 5 | ||
| Normal (pre pacing) | CHF (post pacing) | Normal (pre pacing) | CHF (post pacing) | |
| BW, kg | 3.7±0.2 | 3.6 ± 0.1 | 3.6±0.1 | 3.5± 0.3 |
| HW/BW, g/kg | - | 3.2 ± 0.2 | - | 3.6 ± 0.2 |
| WLW/BW, g/kg | - | 3.8 ± 0.3 | - | 3.7 ± 0.2 |
| DLW/BW, g/kg | - | 2.2 ± 0.3 | - | 2.5 ± 0.2 |
| MAP, mm Hg | 79.2±4.3 | 73.8 ± 5.5 | 76.2±2.2 | 70.2 ± 6.2 |
| HR, bpm | 235.9±4.5 | 266.7±8.4 * | 225±11.6 | 263.7 ± 8.2 * |
| LVSD, mm | 9.5±0.6 | 13.4± 0.6* | 9.2±0.9 | 13.7 ± 1.1* |
| LVEDD, mm | 13.1±0.8 | 16.4 ± 0.8 * | 13.9±0.7 | 17.0 ± 0.7* |
| EF, % | 60.5 ± 1.9 | 32.55 ± 1.2* | 62.1±1.4 | 33.8 ± 2.5* |
Values are mean±SEM. BW indicates body weight; HW, heart weight; MAP, mean arterial pressure; LVSD, left ventricle systolic diameter; LVEDD, left ventricle end-diastolic diameter, EF, ejection fraction.
Western Blot Analysis
Homogenates were prepared from RVLM. Protein concentration was measured using a bicinchoninic acid protein assay kit (Pierce; Rockford, IL). Equal amounts of homogenate were loaded and then separated on 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis. The proteins of these samples were electrophoretically transferred at 100 V for 1.5 h onto nitrocellulose membranes. The membrane were blocked with 5% non-fat milk in tris-buffered saline-tween 20, and probed at room temperature with primary antibodies against the AT1R, GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA), p-Jnk, Jnk, c-Jun, Phospho-c-Jun (Cell Signaling Technology®), and β-tubulin (Sigma). After washing, the membranes were incubated for 30 min with peroxidase-conjugated secondary antibody. The membranes were then washed and incubated for 5 min with Supersignal West Pico Chemiluminescent detection reagents (Pierce; Rockford, IL). The bands on the membrane were digitized and analyzed using UVP BioImaging Systems (UVP, Upland, CA).
Activity of NADPH Oxidase by Lucigenin-enhanced Chemiluminescence
Superoxide anion production by CATHa cells was measured by the lucigenin enhanced chemiluminescence (ECL) method (TD-20/20 luminometer; Turner Designs; Sunnyvale, CA). After subculture, CATHa cells were allowed to grow for two days and then treated with either Ang II (100nM) alone or with AngII for three hours following pretreatment with losartan (10 uM) for 30 minutes. Cells were collected and total protein concentration was determined using a bicinchoninic acid protein-assay kit (Pierce; Rockford, IL).NADPH (100 μM, Sigma) and dark-adapted lucigenin (5μM, Sigma) were added into 0.5-ml microcentrifugal tubes just before reading. Light emission was recorded over 5 min, and values were expressed as mean light units per minute per milligram of protein. Using this method, the superoxide anion production also represents NADPH oxidase activity. The NADPH oxidase activity was expressed in relative ratios by comparing the treatment groups to the control groups.
Detection of Superoxide Production by Dihydroethidium Fluorescence
To measure the production of superoxide anion, dihydroethidium (DHE; Molecular Probes, Eugene, OR) stock (10 mM) was made in DMSO, and dilutions of the stock were used only on the experimental day. Dilutions of DHE were made in reoxygenation solution that resulted in a final concentration of 1 μM DHE in each experimental well. CATH.a cells then were incubated with DHE at 37 °C in a light protected humidified chamber for 30 minutes. After washing with phosphate-buffered saline, DHE fluorescence was visualized by a Leica fluorescent microscope with appropriate excitation/emission filters. Pictures were captured by a digital camera system.
Electrophoretic Mobility Shift Assay (EMSA)
The AP1-DNA binding activity of rabbit RVLM was measured by EMSA, the protein specificity was tested by adding antibodies of c-fos and c-Jun (Santa Cruz Biotechnology). Detailed methods were described by this laboratory in a previous study[18].
Statistics
Data are expressed as mean ± SEM. The statistical analyses were performed by non-paired Student t test when comparing data between the two groups of CHF rabbits. A one-way analysis of variance (ANOVA) for the multiple groups of CATHa cell experiments was performed. The statistical software SigmaStat (SPSS, Chicago, IL) was used for analysis. If significance was found between groups post hoc analyses were carried out using the Bonferroni correction. A P value < 0.05 was considered significantly different
Results
AT1R mRNA expression in CATHa cells and effects of antioxidants
Ang II (100 nM) significantly increased AT1R mRNA expression by 123 ± 11% compared with the control group (figure 1). Ang II induced AT1R mRNA expression was inhibited by the AT1R blocker (Losartan, 10 μM), the NADPH oxidase inhibitor (Apocynin, 20 μM), the SOD mimetic (Tempol, 0.2 μM), and Jnk Inhibitor I (1 μM). These data suggest there is a positive feedback pathway for Ang II to stimulate AT1R expression and that this pathway involves oxidant stress and Jnk.
Figure 1. AT1R mRNA in CATHa Cells.

Real-time RT-PCR of AT1R mRNA expression. Fluorescent signals versus PCR cycles of AT1R (GAPDH as an internal control). Ang II induced AT1R mRNA expression was inhibited by the AT1R blocker Losartan, the NADPH oxidase inhibitor Apocynin, the SOD mimetic Tempol, and Jnk inhibitor I. * p<0.01 compared control group, # p<0.01 compared to Ang II treated group, n=5.
Superoxide production and NADPH Oxidase Activity in CATHa cells
Ang II induced superoxide production. Both the NADPH oxidase inhibitor Apocynin (20μM) and the SOD mimetic Tempol (0.2 μM) reversed Ang II induced superoxide production (Figure 2). This suggests Ang II induced superoxide is mediated by activation of NAPDH oxidase. The activity of NADPH oxidase in CATHa cells was measured by using lucigenin-enhanced chemiluminescence (Figure 3). Ang II increased NADPH oxidase activity by 126 ± 43%, * p<0.01 compared to control group. The increased activity was normalized by losartan (p<0.01) compared to the Ang II treated group. These further suggest that Ang II can induce superoxide by activation of NADPH oxidase through the AT1R.
Figure 2. Superoxide Production in CATHa cells.
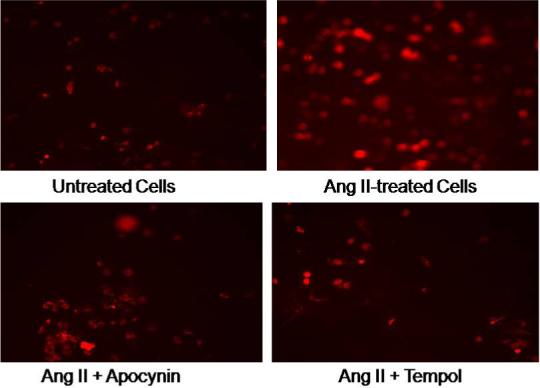
DHE staining of Ang II induced superoxide production. The NADPH oxidase inhibitor Apocynin, and the SOD mimetic Tempo reversed Ang II induced superoxide production.
Figure 3. NADPH Oxidase Activity in CATHa cells.

The activity of NADPH oxidase in CATHa cells was measured by using lucigenin-enhanced chemiluminescence. Ang II increased NADPH oxidase activity. The increased activity was normalized by losartan. * p<0.01 compared to control group, # p<0.01 compared to Ang II treated group, n=5.
c-Jun and c-fos mRNA Expression in CATHa Cells and the Effects of Antioxidants
Ang II (100nM) significantly increased c-Jun and c-fos mRNA expressions compared with the control group (figure 4). Ang II induced c-Jun and c-fos mRNA expressions were inhibited by the AT1R blocker Losartan (10 μM), the NADPH oxidase inhibitor Apocynin (20 μM) and the SOD mimetic Tempol (0.2 μM). These data suggest that Ang II can induce AP1 expression through ROS by activation of NADPH oxidase.
Figure 4. c-Jun and c-Fos mRNA in CATHa Cells.
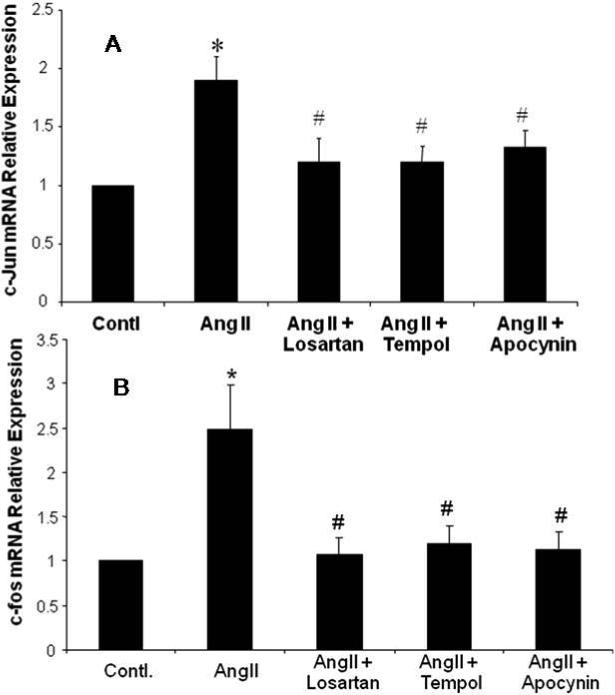
Real-time RT-PCR of c-Jun gene (A) and cfos gene (B) expressions. Fluorescent signals versus PCR cycles of c-Jun (GAPDH as an internal control). Ang II induced c-Jun or c-fos mRNA expression was inhibited by Losartan, NADPH oxidase inhibitor Apocynin, and SOD mimetic Tempol. * p<0.01 compared to control group, # p<0.01 compared to AngII treated group, n=5.
Body Weight, Heart Weight, Hemodynamics, and Echocardiographic Data in CHF Rabbits
Table 1 shows the values for body weight, ratio of organ weight to body weight, hemodynamics, and echocardiographic data of CHF rabbits treated with vehicle or Tempol. After three weeks of pacing, rabbits with CHF showed significant decreases in ejection fraction (EF), increases in heart rate (HR), left ventricle systolic diameter (LVSD), and left ventricle end-diastolic diameter (LVEDD). There were no differences between Vehicle and Tempol treatment groups in the pre paced or post paced states. These data also suggest that seven days of ICV Tempol treatment does not alter hemodynamic parameters of CHF rabbits.
The Effect of Tempol on AT1R mRNA, c-Jun mRNA and c-fos mRNA in the RVLM of Rabbits with CHF
After 7 days of ICV infusion of Tempol the RVLM was punched out and analyzed for AT1R mRNA, c-Jun mRNA and c-fos mRNA expressions using Real-time RT-PCR. Compared with the Vehicle treated group, AT1R mRNA expression was significantly decreased in the Tempol group, * p<0.01(Figure 5A). Compared with the ICV vehicle group, c-Jun mRNA and c-fos expressions in the Tempol treated group were significantly decreased, p<0.05 (Figure 5B and C). These data indicate that the change in c-Jun and c-fos occur in a similar fashion as the up regulation of the AT1R, and they are all dependent on superoxide production.
Figure 5. The Effect of Tempol on AT1R mRNA, c-Jun and c-fos in the RVLM of Rabbits with CHF.
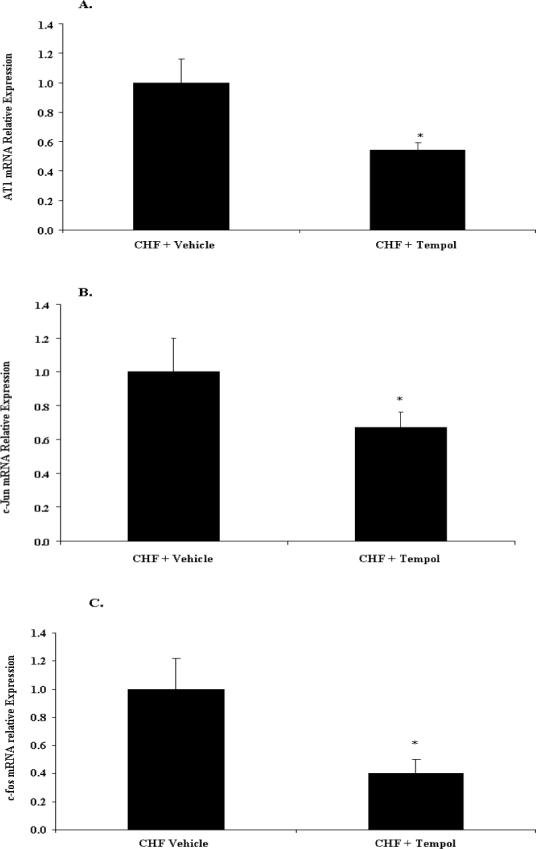
Real-time RT-PCR of mRNA expression. Fluorescent signals versus PCR cycles of AT1R, c-Jun or c-fos (β-actin as an internal control). The SOD mimetic, Tempol 10 μg/ul/h was given for seven days. A. AT1R mRNA expression, * p<0.01, n=10; B. c-jun mRNA expression. * p<0.01, n=10. C. c-fos mRNA expression. * p<0.01, n=10.
Effects of Tempol on Protein Expression of AT1R, p-JNK and p-c-Jun
Rabbit RVLM protein was extracted. The cytosolic protein and nuclear protein were separated by using the kit of NE-PER® (PIERCE). AT1R, phosphorylated c-Jun, phosphorylated Jnk were detected by Western blotting. Tempol decreased AT1R (1.6±0.29 vs 0.88±0.16, P<0.05) (figure 6A); decreased the ratio of p-c-Jun to total c-Jun in cytosolic compartment (0.56 ± 0.10 vs 0.36 ± 0.05, P<0.05) and in the nuclear compartment (0.67 ± 0.1 vs 0.30± 0.08, P<0.01) (figure 6C and D); decreased the ratio of p-Jnk protein to total Jnk (0.40 ± 0.05 vs 0.20 ± 0.04, P<0.05) compared to the Vehicle treated group (figure 6B). These data suggest that oxidant stress can up regulate AT1R through the Jnk-c-Jun-AP1 pathway in CHF.
Figure 6. Effects of Tempol on Protein Expression of AT1R and c-Jun in the RVLM of Rabbitswith CHF.
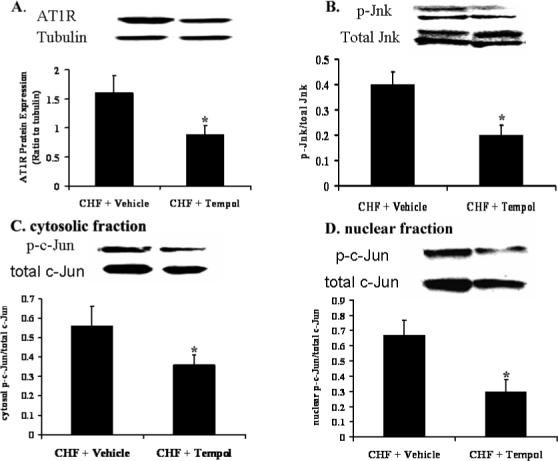
Protein expression in rabbits with CHF. A. AT1R, Tubulin is used as an internal control; B. p-Jnk to total Jnk; C. cytosolic p-c-Jun to total c-Jun. D. Nuclear p-c-Jun to total c-Jun. *p<0.01, n=10.
Effects of Tempol on AP1-DNA binding activity
In our previous study, we showed the binding activity of the transcription faction AP-1 was increased significantly in CHF compared to sham rabbits [18]. Here after giving ICV tempol to the CHF rabbits AP-1 DNA binding activity was significantly decreased compared to ICV vehicle CHF rabbits (Figure 7A and B). The DNA/protein complex could be suppressed by adding 200-fold molar excess of unlabeled competitor AP-1 DNA sequence. The DNA/protein complex could be supershifted by adding antibodies of c-fos and c-Jun (Figure 7A). This indicates the DNA/protein components have c-fos and c-Jun proteins that can form a dimer of AP1 transcription factor.
Figure 7. AP-1-DNA binding activity and super gell shift.
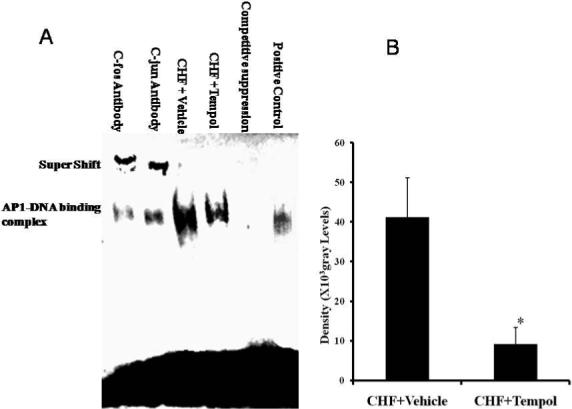
A. Electrophoretic mobility-shift assay (EMSA) and supershift analysis with c-fos or c-Jun antibody for rabbit RVLM. B. Mean data of the EMSA density. * p<0.01, n=10.
Discussion
We have previously reported that the expression of AT1R was up-regulated in the RVLM of rabbits with CHF. We also observed that the up-regulation depends on the increase in the transcription factor AP1 which is activated by the SAPK/Jnk pathway but not by p38 or ERK1/2 MAP Kinase pathway[18]. AP1 is redox-sensitive through the conserved cysteine residues located in its DNA-binding domain[26]. The present study provides further evidence for the role of ROS in up regulation of AT1R expression in CHF.
The NADPH oxidase enzymes are a particularly important source of reactive oxygen species that are implicated in redox signalling[27,28]. It is now well accepted that Ang II can stimulate NADPH oxidase dependent superoxide (O−2) generation [29]. Substantial clinical and experimental evidence support the important role of oxidant stress in cardiac remodeling and heart failure[30]. Markers of oxidant stress are elevated in patients with heart failure and correlate with the severity of contractile dysfunction [31,32]. Enhanced ROS generation has been directly documented in the failing heart using highly specific methods such as electron spin resonance spectroscopy[33]. However, there are few studies concerning the role of ROS and antioxidants in the central control of sympathetic nerve activity in CHF.
The up regulation of AT1R in CHF contributes to a positive feedback system that evokes a further deterioration of the CHF state. We have also reported that NADPH oxidase activity is increased in the cardiovascular control areas of the rabbit brain with CHF[34]. Accordingly, in the present study, we used a neuronal cell line to further explore the relationship between AT1R expression, NADPH Oxidase and ROS. We observed superoxide release following Ang II treatment in CATH.a cells. We also measured NADPH oxidase activity which was increased by Ang II treatment in CATH.a cells. The increased activity was attenuated by losartan. The expression of AT1R was up-regulated by treating CATHa cells with Ang II and was inhibited by losartan. Interestingly, the up regulation of AT1R mRNA expression was also blocked by treating the cells with Tempol and Apocynin. These data lead to the speculation that Ang II may induce the up-regulation of AT1R expression via a pathway dependent on ROS production. An important aspect of this work is that a similar conclusion can be derived from our experiments in rabbits with CHF following ICV administration of Tempol. These in vivo and in vitro studies suggest that ROS plays a crucial role in the central up-regulation of AT1R expression in CHF.
The influence of NADPH Oxidase-derived ROS on downstream cellular effects has been extensively studied in the failing heart [35,36]. These include alterations in the activity of redox-sensitive protein kinases, such as SAPK and other MAPKs, which may occur indirectly following inactivation of tyrosine phosphatases or through direct activation; These also include alterations in the activity of transcription factors, including NFkB, AP-1, hypoxia-inducible factor-1, and STATs. We have reported that the phosphorylation of Jnk was increased in the RVLM of CHF rabbits. Exogenous (ICV) Ang II also increased activity of phosphorylated Jnk[18]. In the present study, we investigated the effect of Tempol on the SAPK/Jnk pathway in the central sympathetic nervous system of CHF rabbits. We observed that ICV Tempol decreased Jnk phosphorylation in the RVLM of CHF rabbits compared to vehicle treated rabbits, providing evidence that ROS can regulate Jnk phosphorylation in CHF.
The exposure of mammalian cells to extracellular stress induces the expression of immediate early genes such as c-fos and c-jun and activates various transcription factors, among which is AP-1[37]. AP-1 is ubiquitously expressed in the brain and has been shown to be associated with a variety of physiological and pathophysiological states[38]. For instance, Wang et al. reported that Fos protein is required for the re-expression of AT1R in the nucleus tractus solitarii (NTS) after baroreceptor activation in the rat[39]. Chan et al. showed an augmented up-regulation by c-fos of AT1R in the NTS of spontaneously hypertensive rats[40]. Fleegal et al. demonstrated that Ang II induction of AP-1 in neurons requires stimulation of Jnk[41]. We previously showed that phosphorylated c-Jun and c-Jun mRNA increased in the RVLM of CHF rabbits. We also showed that ICV administration of Ang II to normal rabbits could induce c-Jun phosphorylation and its mRNA expression[18]. In the present study ICV Tempol attenuated the expression of phosphorylated c-Jun protein, c-Jun mRNA and c-fos mRNA compared to vehicle treated animals.
Therefore, the findings that Ang II treated CATHa cells exhibited an increase in c-Jun and cfos mRNA which was attenuated by Tempol or Apocynin strongly suggests that oxidant stress plays a pivotal role in this pathway. In CHF rabbits, Tempol decreased AP1 DNA binding activity. Given that c-Jun and c-fos are the major components of AP-1, these data support the idea that ROS is involved in AP-1 activation. Furthermore, Tempol also suppressed the p-Jnk level of CHF suggesting that that ROS activation of AP-1 may occur through phosphorylation of Jnk.
In order to put these data into clinical perspective, a variety of human diseases have been linked to an overproduction of ROS. The growing realization that oxidants may function in signaling pathways may, in turn, causes a re-evaluation of the pathways and sources of oxidant production linked to human disease. In addition, a variety of clinical and epidemiological evidence supports a protective role for antioxidants in cardiovascular disease[42-45]. Gao et al.[34] showed central Ang II activates sympathetic outflow by stimulation of NADPH oxidase and ROS in the CHF state. Chan et al. showed NADPH oxidase-derived superoxide anion in the RVLM mediates Ang II-induced pressor responses[46]. Likewise, Campese et al. demonstrated that oxidant stress mediates Ang II-dependent stimulation of sympathetic nerve activity[47]. In previous studies from this laboratory Gao et al. also showed that ICV Tempol could lower renal sympathetic nerve activity (RSNA)[22,34]. Xu et al. showed systemic high dose Tempol could decrease blood pressure and RSNA, which was mediated largely by NO-independent sympatho-inhibition in normotensive and hypertensive rats[48,49]. The dose of Tempol used in the present study did not lower blood pressure, most likely because arterial pressure was already lower in the CHF state. The fact that ICV Tempol inhibited the upregulation of the AT1R in the RVLM suggests that this intervention would also suppress sympathetic nerve activity as indicated in our previous work[22,34] . Infanger et al.[50] showed CuZnSOD transfection to the paraventricular nucleus of myocardiac infarcted mice improved the decreased ejection fraction and increased left ventricle end-volume. Ritchie et al.[51] showed Tempol treatment reduced morphological and molecular evidence of cardiac hypertrophy in the GLUT4-knockout insulin-resistant mouse in vivo. Curiously, there was no effect of ICV Tempol on cardiac function in our CHF rabbits. It is possible that other factors that modulate sympathetic outflow are dominant in this setting or that a reduction in sympathetic outflow following Tempol is targeted primarily to the periphery. Indeed our previous study clearly showed a reduction in RSNA following ICV Tempol[22,34]. In addition, in this model the continuation of cardiac pacing during the experiment may have overrode the beneficial effects of ICV Tempol on the cardiac hemodynamics.
The present study extends these findings and shows that the antioxidant Tempol decreased the expression of AT1R, phosphorylated Jnk, c-Jun and c-fos in the RVLM. The Jnk pathway may provide an explanation for how AP-1 is activated by oxidant stress. Taken together, these data strongly suggest that antioxidant agents may be beneficial in CHF by decreasing AT1R expression through inhibition of the Jnk and AP1 signaling pathway at the molecular level.
In the future, additional studies are needed to elucidate the effect of hydrogen peroxide, hydroxyl radical, and the specific molecular sites at which ROS act to exert their effects. Except for superoxide dismutase (SOD)-like activity, other possible mechanisms of Tempol action should be kept in mind, including scavenging oxy- and carbon based free radicals, and oxidation of reduced transition metals.
Supplementary Material
Acknowledgements
The authors would like to thank Ms Phyllis Anding, Ms Pamela Curry and Ms Johnnie Hackley for their excellent technical assistance.
Funding Support
This study was supported by NIH grant PO-1 HL62222. L. Gao was supported by a Scientist Development Grant 0635007N from the American Heart Association.
Footnotes
Publisher's Disclaimer: This is an un-copyedited author manuscript accepted for publication in Circulation Research, copyright The American Heart Association. This may not be duplicated or reproduced, other than for personal use or within the “Fair Use of Copyrighted Materials” (section 107, title 17, U.S. Code) without prior permission of the copyright owner, The American Heart Association. The final copyedited article, which is the version of record, can be found at http://circres.ahajournals.org/. The American Heart Association disclaims any responsibility or liability for errors or omissions in this version of the manuscript or in any version derived from it by the National Institutes of Health or other parties.
Disclosures None
References
- 1.Goldsmith SR, Garr M, McLaurin M. Regulation of regional norepinephrine spillover in heart failure: the effect of angiotensin II and beta-adrenergic agonists in the forearm circulation. J Card Fail. 1998;4:305–310. doi: 10.1016/s1071-9164(98)90236-6. [DOI] [PubMed] [Google Scholar]
- 2.Goldsmith SR. Angiotensin II and sympathoactivation in heart failure. J Card Fail. 1999;5:139–145. doi: 10.1016/s1071-9164(99)90036-2. [DOI] [PubMed] [Google Scholar]
- 3.Pitt B, Poole-Wilson PA, Segal R, Martinez FA, Dickstein K, Camm AJ, Konstam MA, Riegger G, Klinger GH, Neaton J, Sharma D, Thiyagarajan B. Effect of losartan compared with captopril on mortality in patients with symptomatic heart failure: randomised trial--the Losartan Heart Failure Survival Study ELITE II. Lancet. 2000;355:1582–1587. doi: 10.1016/s0140-6736(00)02213-3. [DOI] [PubMed] [Google Scholar]
- 4.Paravicini TM, Touyz RM. Redox signaling in hypertension. Cardiovasc Res. 2006;71:247–258. doi: 10.1016/j.cardiores.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 5.Kishi T, Hirooka Y, Kimura Y, Ito K, Shimokawa H, Takeshita A. Increased reactive oxygen species in rostral ventrolateral medulla contribute to neural mechanisms of hypertension in stroke-prone spontaneously hypertensive rats. Circulation. 2004;109:2357–2362. doi: 10.1161/01.CIR.0000128695.49900.12. [DOI] [PubMed] [Google Scholar]
- 6.Gao L, Wang W, Liu D, Zucker IH. Exercise training normalizes sympathetic outflow by central antioxidant mechanisms in rabbits with pacing-induced chronic heart failure. Circulation. 2007;115:3095–3102. doi: 10.1161/CIRCULATIONAHA.106.677989. [DOI] [PubMed] [Google Scholar]
- 7.Russell ST, Eley H, Tisdale MJ. Role of reactive oxygen species in protein degradation in murine myotubes induced by proteolysis-inducing factor and angiotensin II. Cell Signal. 2007;19:1797–1806. doi: 10.1016/j.cellsig.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 8.Kazama K, Anrather J, Zhou P, Girouard H, Frys K, Milner TA, Iadecola C. Angiotensin II impairs neurovascular coupling in neocortex through NADPH oxidase-derived radicals. Circ Res. 2004;95:1019–1026. doi: 10.1161/01.RES.0000148637.85595.c5. [DOI] [PubMed] [Google Scholar]
- 9.Kwon SH, Pimentel DR, Remondino A, Sawyer DB, Colucci WS. H(2)O(2) regulates cardiac myocyte phenotype via concentration-dependent activation of distinct kinase pathways. J Mol Cell Cardiol. 2003;35:615–621. doi: 10.1016/s0022-2828(03)00084-1. [DOI] [PubMed] [Google Scholar]
- 10.Taniyama Y, Ushio-Fukai M, Hitomi H, Rocic P, Kingsley MJ, Pfahnl C, Weber DS, Alexander RW, Griendling KK. Role of p38 MAPK and MAPKAPK-2 in angiotensin II-induced Akt activation in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2004;287:C494–499. doi: 10.1152/ajpcell.00439.2003. [DOI] [PubMed] [Google Scholar]
- 11.Griendling KK, Ushio-Fukai M. Reactive oxygen species as mediators of angiotensin II signaling. Regul Pept. 2000;91:21–27. doi: 10.1016/s0167-0115(00)00136-1. [DOI] [PubMed] [Google Scholar]
- 12.Griendling KK, Sorescu D, Lassegue B, Ushio-Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol. 2000;20:2175–2183. doi: 10.1161/01.atv.20.10.2175. [DOI] [PubMed] [Google Scholar]
- 13.Ushio-Fukai M, Alexander RW, Akers M, Griendling KK. p38 Mitogen-activated protein kinase is a critical component of the redox-sensitive signaling pathways activated by angiotensin II. Role in vascular smooth muscle cell hypertrophy. J Biol Chem. 1998;273:15022–15029. doi: 10.1074/jbc.273.24.15022. [DOI] [PubMed] [Google Scholar]
- 14.Zafarullah M, Li WQ, Sylvester J, Ahmad M. Molecular mechanisms of N-acetylcysteine actions. Cell Mol Life Sci. 2003;60:6–20. doi: 10.1007/s000180300001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Puga A, Barnes SJ, Chang C, Zhu H, Nephew KP, Khan SA, Shertzer HG. Activation of transcription factors activator protein-1 and nuclear factor-kappaB by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochem Pharmacol. 2000;59:997–1005. doi: 10.1016/s0006-2952(99)00406-2. [DOI] [PubMed] [Google Scholar]
- 16.Francis J, Wei SG, Weiss RM, Felder RB. Brain angiotensin-converting enzyme activity and autonomic regulation in heart failure. Am J Physiol Heart Circ Physiol. 2004;287:H2138–2146. doi: 10.1152/ajpheart.00112.2004. [DOI] [PubMed] [Google Scholar]
- 17.Gao L, Schultz HD, Patel KP, Zucker IH, Wang W. Augmented input from cardiac sympathetic afferents inhibits baroreflex in rats with heart failure. Hypertension. 2005;45:1173–1181. doi: 10.1161/01.HYP.0000168056.66981.c2. [DOI] [PubMed] [Google Scholar]
- 18.Liu D, Gao L, Roy SK, Cornish KG, Zucker IH. Neuronal angiotensin II type 1 receptor upregulation in heart failure. activation of activator protein 1 and Jun N-terminal kinase. Circ Res. 2006;99:1004–1011. doi: 10.1161/01.RES.0000247066.19878.93. [DOI] [PubMed] [Google Scholar]
- 19.Liu JL, Zucker IH. Regulation of sympathetic nerve activity in heart failure. a role for nitric oxide and angiotensin II. Circ Res. 1999;84:417–423. doi: 10.1161/01.res.84.4.417. [DOI] [PubMed] [Google Scholar]
- 20.Murakami H, Liu JL, Zucker IH. Angiotensin II blockade [corrected] enhances baroreflex control of sympathetic outflow in heart failure. Hypertension. 1997;29:564–569. doi: 10.1161/01.hyp.29.2.564. [DOI] [PubMed] [Google Scholar]
- 21.Sim MK, Chen WS. Effects of losartan on angiotensin receptors in the hypertrophic rat heart. Regul Pept. 2006;137:140–146. doi: 10.1016/j.regpep.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 22.Gao L, Wang W, Li YL, Schultz HD, Liu D, Cornish KG, Zucker IH. Sympathoexcitation by central ANG II: roles for AT1 receptor upregulation and NAD(P)H oxidase in RVLM. Am J Physiol Heart Circ Physiol. 2005;288:H2271–2279. doi: 10.1152/ajpheart.00949.2004. [DOI] [PubMed] [Google Scholar]
- 23.Palkovits M, Brownstein M. In: Brain microdissection techniques. Cuello AE, editor. Wiley, Chichester; UK: 1983. [Google Scholar]
- 24.Nakamura S, Moriguchi A, Morishita R, Yamada K, Nishii T, Tomita N, Ohishi M, Kaneda Y, Higaki J, Ogihara T. Activation of the brain angiotensin system by in vivo human angiotensin-converting enzyme gene transfer in rats. Hypertension. 1999;34:302–308. doi: 10.1161/01.hyp.34.2.302. [DOI] [PubMed] [Google Scholar]
- 25.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods Enzymol. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 26.Toone WM, Morgan BA, Jones N. Redox control of AP-1-like factors in yeast and beyond. Oncogene. 2001;20:2336–2346. doi: 10.1038/sj.onc.1204384. [DOI] [PubMed] [Google Scholar]
- 27.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 28.Lara-Ortiz T, Riveros-Rosas H, Aguirre J. Reactive oxygen species generated by microbial NADPH oxidase NoxA regulate sexual development in Aspergillus nidulans. Mol Microbiol. 2003;50:1241–1255. doi: 10.1046/j.1365-2958.2003.03800.x. [DOI] [PubMed] [Google Scholar]
- 29.Li JM, Wheatcroft S, Fan LM, Kearney MT, Shah AM. Opposing roles of p47phox in basal versus angiotensin II-stimulated alterations in vascular O2- production, vascular tone, and mitogen-activated protein kinase activation. Circulation. 2004;109:1307–1313. doi: 10.1161/01.CIR.0000118463.23388.B9. [DOI] [PubMed] [Google Scholar]
- 30.Byrne JA, Grieve DJ, Cave AC, Shah AM. Oxidative stress and heart failure. Arch Mal Coeur Vaiss. 2003;96:214–221. [PubMed] [Google Scholar]
- 31.Keith M, Geranmayegan A, Sole MJ, Kurian R, Robinson A, Omran AS, Jeejeebhoy KN. Increased oxidative stress in patients with congestive heart failure. J Am Coll Cardiol. 1998;31:1352–1356. doi: 10.1016/s0735-1097(98)00101-6. [DOI] [PubMed] [Google Scholar]
- 32.Heymes C, Bendall JK, Ratajczak P, Cave AC, Samuel JL, Hasenfuss G, Shah AM. Increased myocardial NADPH oxidase activity in human heart failure. J Am Coll Cardiol. 2003;41:2164–2171. doi: 10.1016/s0735-1097(03)00471-6. [DOI] [PubMed] [Google Scholar]
- 33.Mak S, Newton GE. The oxidative stress hypothesis of congestive heart failure: radical thoughts. Chest. 2001;120:2035–2046. doi: 10.1378/chest.120.6.2035. [DOI] [PubMed] [Google Scholar]
- 34.Gao L, Wang W, Li YL, Schultz HD, Liu D, Cornish KG, Zucker IH. Superoxide mediates sympathoexcitation in heart failure: roles of angiotensin II and NAD(P)H oxidase. Circ Res. 2004;95:937–944. doi: 10.1161/01.RES.0000146676.04359.64. [DOI] [PubMed] [Google Scholar]
- 35.Nediani C, Borchi E, Giordano C, Baruzzo S, Ponziani V, Sebastiani M, Nassi P, Mugelli A, d'Amati G, Cerbai E. NADPH oxidase-dependent redox signaling in human heart failure: relationship between the left and right ventricle. J Mol Cell Cardiol. 2007;42:826–834. doi: 10.1016/j.yjmcc.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 36.Murdoch CE, Zhang M, Cave AC, Shah AM. NADPH oxidase-dependent redox signalling in cardiac hypertrophy, remodelling and failure. Cardiovasc Res. 2006;71:208–215. doi: 10.1016/j.cardiores.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 37.Kunsch C, Medford RM. Oxidative stress as a regulator of gene expression in the vasculature. Circ Res. 1999;85:753–766. doi: 10.1161/01.res.85.8.753. [DOI] [PubMed] [Google Scholar]
- 38.Rylski M, Kaczmarek L. Ap-1 targets in the brain. Front Biosci. 2004;9:8–23. doi: 10.2741/1207. [DOI] [PubMed] [Google Scholar]
- 39.Wang LL, Chan SH, Chan JY. Fos protein is required for the re-expression of angiotensin II type 1 receptors in the nucleus tractus solitarii after baroreceptor activation in the rat. Neuroscience. 2001;103:143–151. doi: 10.1016/s0306-4522(00)00543-1. [DOI] [PubMed] [Google Scholar]
- 40.Chan JY, Wang LL, Lee HY, Chan SH. Augmented upregulation by c-fos of angiotensin subtype 1 receptor in nucleus tractus solitarii of spontaneously hypertensive rats. Hypertension. 2002;40:335–341. doi: 10.1161/01.hyp.0000029241.33421.eb. [DOI] [PubMed] [Google Scholar]
- 41.Fleegal MA, Sumners C. Angiotensin II induction of AP-1 in neurons requires stimulation of PI3-K and JNK. Biochem Biophys Res Commun. 2003;310:470–477. doi: 10.1016/j.bbrc.2003.09.047. [DOI] [PubMed] [Google Scholar]
- 42.Penckofer S, Schwertz D, Florczak K. Oxidative stress and cardiovascular disease in type 2 diabetes: the role of antioxidants and pro-oxidants. J Cardiovasc Nurs. 2002;16:68–85. doi: 10.1097/00005082-200201000-00007. [DOI] [PubMed] [Google Scholar]
- 43.Rimm EB, Stampfer MJ. Antioxidants for vascular disease. Med Clin North Am. 2000;84:239–249. doi: 10.1016/s0025-7125(05)70216-9. [DOI] [PubMed] [Google Scholar]
- 44.Hennekens CH, Gaziano JM, Manson JE, Buring JE. Antioxidant vitamin-cardiovascular disease hypothesis is still promising, but still unproven: the need for randomized trials. Am J Clin Nutr. 1995;62:1377S–1380S. doi: 10.1093/ajcn/62.6.1377S. [DOI] [PubMed] [Google Scholar]
- 45.Gaziano JM. Antioxidant vitamins and coronary artery disease risk. Am J Med. 1994;97:18S–28S. doi: 10.1016/0002-9343(94)90294-1. [DOI] [PubMed] [Google Scholar]
- 46.Chan SH, Hsu KS, Huang CC, Wang LL, Ou CC, Chan JY. NADPH oxidase-derived superoxide anion mediates angiotensin II-induced pressor effect via activation of p38 mitogen-activated protein kinase in the rostral ventrolateral medulla. Circ Res. 2005;97:772–780. doi: 10.1161/01.RES.0000185804.79157.C0. [DOI] [PubMed] [Google Scholar]
- 47.Campese VM, Shaohua Y, Huiquin Z. Oxidative stress mediates angiotensin II-dependent stimulation of sympathetic nerve activity. Hypertension. 2005;46:533–539. doi: 10.1161/01.HYP.0000179088.57586.26. [DOI] [PubMed] [Google Scholar]
- 48.Xu H, Fink GD, Galligan JJ. Nitric oxide-independent effects of tempol on sympathetic nerve activity and blood pressure in DOCA-salt rats. Am J Physiol Heart Circ Physiol. 2002;283:H885–H892. doi: 10.1152/ajpheart.00134.2002. [DOI] [PubMed] [Google Scholar]
- 49.Xu H, Fink GD, Chen A, Watts S, Galligan JJ. Nitric oxide-independent effects of tempol on sympathetic nerve activity and blood pressure in normotensive rats. Am J Physiol Heart Circ Physiol. 2001;281:H975–H980. doi: 10.1152/ajpheart.2001.281.2.H975. [DOI] [PubMed] [Google Scholar]
- 50.Infanger DW, Braga VA, Stupinski JA, Sharma RV, Davisson RL. Superoxide scavenging in the paraventricular nucleus (PVN) reduces sympathoexcitation and improves cardiac function following myocardial infarction. The FASEB Journal. 2008;22:951. (Abstract) [Google Scholar]
- 51.Ritchie RH, Quinn JM, Cao AH, Drummond GR, Kaye DM, Favaloro JM, Proietto J, Delbridge LM. The antioxidant tempol inhibits cardiac hypertrophy in the insulin-resistant GLUT4-deficient mouse in vivo. J Mol Cell Cardiol. 2007;42:1119–1128. doi: 10.1016/j.yjmcc.2007.03.900. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.