

Abstract
We have previously shown that post-transcriptional mechanisms involving the 26S proteasome regulate the cyclin-dependent kinase inhibitors (CKIs), p21(Cip1) and p27(Kip1) during preadipocyte proliferation. Earlier studies further demonstrated that the anti-inflammatory, anti-carcinogenic phytochemical, helenalin is a potent inhibitor of periodic Skp2 accumulation, an F-box protein mediating SCF E3 ligase ubiquitylation and degradation of both CKIs during S phase progression. Data presented here demonstrate that helenalin dose-dependently induced G1 arrest of synchronously replicating 3T3-L1 preadipocytes. This effect occurred in the absence of discernable indices of cell toxicity or apoptosis under the conditions used in this study. Our results demonstrate that helenalin markedly increased p21 protein accumulation in both density-arrested and proliferating preadipocytes in a dose-dependent manner. This increase in p21 protein abundance occurred without change in mRNA transcript demonstrating that post-transcriptional mechanisms were involved. This notion was further supported by the modest accumulation of polyubiquitylated p21 following treatment with helenalin suggesting that suppression of targeted p21 proteolysis by the 26S proteasome contributed to helenalin-mediated p21 accumulation. The increase in p21 protein was compartmentalized to the nucleus where p21 is known to inhibit cell cycle progression. Finally, helenalin increased protein-protein interactions between p21 and cyclin-dependent kinase 2 (Cdk2) which may account in part for the anti-proliferative effect in 3T3-L1 preadipocytes.
Keywords: adipocyte, obesity, cell cycle, proliferation
INTRODUCTION
Helenalin is a naturally occurring phytochemical extracted from the aerial portion of the flowering plant, arnica montana L. Alcohol extracts containing this sesquiterpene lactone and its derivatives have been used in herbal medicine for many years to treat haematomas, sprains, rheumatic diseases, and superficial skin inflammation. Recently, helenalin has found principal use in cellular studies as a commercially available, potent inhibitor of IκB degradation and NF-κB transcriptional activity (Lyss et al., 1997). Apart from anti-inflammatory effects, helenalin is also known to suppress proliferation of cancer cells through multiple mechanisms including suppression of telomerase activity (Huang et al., 2005), elevation of intracellular free calcium (Powis et al., 1994), inhibition of protein and DNA synthesis (Hall et al., 1978;Hall et al., 1977), and promotion of apoptosis (Dirsch et al., 2001a). These and other studies suggest that helenalin has multiple modes of action within the cell aside from its ability to inhibit NF-κB DNA binding. Albeit shown to have anti-proliferative properties, mechanisms underlying helenalin’s ability to regulate cell cycle progression are largely unknown.
Cellular proliferation is driven by sequential expression of cyclin proteins that activate a series of Cdks that initiate timely events necessary for orderly cell cycle progression. Transition through the restriction point near the G1/S phase boundary represents a critical step of autonomous control where continued progression beyond this point becomes independent of mitogenic stimuli (Sherr and Roberts, 1999;Sherr and Roberts, 2004). Cell cycle progression through the restriction point is highly dependent on Cdk2 activity which is governed by cyclins E/A for activation and potently suppressed by the CKIs, p21 and p27. While the cyclins are sequentially expressed in response to mitogenic stimulation, p21 and p27 are highly regulated by various antimitotic stimuli such as nutrient deprivation, density arrest, differentiation, apoptosis, and tumor suppression. In addition to their well established role as inhibitors of Cdk2 activity, both CKIs can facilitate early cell cycle progression by serving as scaffolding proteins that facilitate the formation (Cheng et al., 1999;Zhang et al., 1994) and nuclear localization (LaBaer et al., 1997) of active Cyclin D1/Cdk4 complexes during mid-G1 progression. Mechanisms that balance and maintain optimal levels of p21 and p27 during preadipocyte replication are critical to understanding the etiology of obesity and various lipodystrophies as gene ablation has been shown to markedly increase fat mass due to adipocyte hyperplasia (Naaz et al., 2004).
We have previously reported that p27 is regulated during S phase progression of replicating 3T3-L1 preadipocytes by post-transcriptional mechanisms involving ubiquitylation and targeted protein degradation by the 26S proteasome (Auld et al., 2007). Furthermore, we have shown that Akt signaling and the periodic accumulation of Skp2, an F-box protein that links p27 to the SCF E3 ligase for ubiquitylation as well as p27 degradation during G1/S transition are suppressed in the presence of helenalin preventing the accumulation of Cyclin A and S phase progression (Auld et al., 2006). In the study reported here, we demonstrate that helenalin arrests proliferating preadipocytes in late-G1 phase and dramatically promotes p21 protein accumulation during density arrest and mid-G1 by post-transcriptional mechanisms. Furthermore, helenalin increases p21 accumulation in the nucleus where protein-protein interactions between p21 and Cdk2 may contribute to the anti-proliferative effect of helenalin on preadipocyte replication. These data represent the first study of the effect of helenalin on p21 accumulation in any cell type and provide a link between cell cycle progression, early signaling events, and adipocyte hyperplasia.
MATERIALS AND METHODS
Materials
Dulbecco’s Modified Eagle’s Medium (DMEM), calf bovine serum (CS), fetal bovine serum (FBS) and Trypsin-EDTA were purchased from Invitrogen. Antibodies used for immunoblotting were purchased as follows: p21 (Oncogene), Cyclin D1, α-tubulin, cleaved caspase-3, cleaved PARP, ubiquitin (Cell Signaling), GAPDH, Cyclin A (Santa Cruz), Nucleoporin p62, Cdk2, and Cdk4 (BD Biosciences). Polyclonal p21 antibody for immunoprecipitation was purchased from Santa Cruz Biotechnology. Helenalin was purchased from Biomol. Recombinant Protein-G beads for immunoprecipitation were from Pierce. Propidium Iodide and RNase A were purchased from Sigma. Polyvinylidene fluoride (PVDF) membrane was purchased from Millipore. Enhanced chemiluminescence (ECL) reagents, secondary antibodies and N-ethylmaleimide (NEM) were from Pierce.
Cell Culture and Induction of Differentiation
Murine 3T3-L1 preadipocytes were propagated in DMEM supplemented with 10% CS until reaching density arrest at 2 days post-confluence. Cells were subsequently induced to differentiate with DMEM containing 10% FBS supplemented with 0.5mM 3-isobutyl-1-methyxanthine, 1μM dexamethasone, and 1.7μM insulin (MDI). Throughout the study, the term “post-MDI” refers to the time elapsed since the addition of the differentiation cocktail to the culture medium. Additionally, the term “time 0” refers to density-arrested, 2 days post-confluent cells immediately before the addition of MDI to the culture medium. All experiments were repeated 3-5 times to validate results and ensure reliability.
Protein isolation
Cells were washed with phosphate-buffered saline (PBS) and harvested in ice cold lysis buffer containing 0.1M Tris (pH 7.4), 150mM NaCl, 10% sodium dodecyl sulfate (SDS), 1% Triton X, 0.5% NP40, 1mM EDTA, 1mM EGTA, and 10mM NEM. Phosphatase inhibitors (20mM β-glycerophosphate, 10mM sodium fluoride and 2μM sodium orthovanadate) and protease inhibitors (0.3μM aprotinin, 21μM leupeptin, 1μM pepstatin, 50μM phenanthroline, 0.5uM phenylmethylsulfonyl fluoride) were freshly added to the lysis buffer immediately before use. Lysates were sonicated and centrifuged at 13,000 × g for 10 min at 4°C. Protein content was determined by bicinchoninic acid (BCA) procedures according to manufacturer’s (Pierce) instructions.
Immunoblotting
Equal amounts of whole cell lysate protein were separated by SDS-PAGE electrophoresis. Protein samples were mixed with loading buffer containing 0.25M Tris, (pH 6.8), 4% SDS, 10% glycerol, 0.01% bromophenol blue, and 10% dithiothreitol, then heated at 80°C for 5 min prior to electrophoresis. Proteins were transferred to PVDF (Millipore), blocked in 4% milk and probed overnight at 4°C with specified primary antibodies. Membranes were subsequently probed for 1 hr at room temperature with secondary antibodies conjugated with horseradish peroxidase and results visualized with ECL using CL-XPosure film (Pierce).
RNA Isolation and Analysis
Total RNA was extracted from 3T3-L1 preadipocytes using the RNeasy Mini Kit according to manufacturer’s (QIAGEN) instructions. Briefly, cells were lysed and homogenized using the provided QIAshredders. Column-bound RNA was DNase-treated and eluted with RNase-free water. Total RNA (1ug) from the cells was subjected to Reverse-Transcriptase Polymerase Chain Reaction (RT-PCR) using One-Step RT-PCR kit (QIAGEN).Reactions were carried out in the presence of buffer containing dNTP’s (400μM), reverse transcriptase and DNA polymerase enzyme mix, and RNase inhibitor (10U). Gene specific primers designed for p21 (forward: 5’-GTCTGAGCGGCCTGAAGATT’3’ and reverse: 5’-TCCTGACCCACAGCAGAAGA-3’) were used at a final concentration of 0.6μM. Quantam RNA Classic II 18S primer/ competimer was used according to manufacturer’s (Ambion) instruction as an internal standard in the RT-PCR reactions at a concentration of 0.6μM. RT-PCR products were electrophoresed in 1.5% agarose in the presence of ethidium bromide and analyzed on a Kodak Digital Imager.
Flow Cytometry
Cell cycle progression was assessed by flow cytometry as described previously (Auld, Fernandes, and Morrison, 2007). Briefly, cells were washed with PBS, trypsinized, resuspended in ice cold PBS and washed twice by centrifugation at 300 × g for 5 min at 4°C. Pelleted cells were gently permeabilized and fixed with ice cold ethanol (70%), washed with PBS and incubated in the dark with propidium iodide stain (50μg/ml propidium iodide (PI) and 100μg/ml RNase A in PBS) for 30 min. DNA fluorescence was measured using flow cytometry (Becton Dickinson FACS Calibur). DNA histograms were extracted from width (FL2W) and area (FL2A) plots of PI fluorescence recorded for 10,000 events per data point.
Nuclear/Cytosolic Fractionation
Cells were washed with PBS and incubated with ice cold isotonic buffer containing 20mM Tris, pH7.4, 125mM NaCl, 1mM EGTA, 1mM EDTA and 1mM MgCl2 for 6 min on ice. The buffer was supplemented with fleshly prepared 0.1% NP40, and phosphatase/protease inhibitors as described above. Following detergent solubilization, cytosolic fractions were collected from the cell monolayers and clarified by centrifugation (13,000 × g for 5 min at 4°C). Intact nuclei were subsequently collected in PBS and gently pelleted by centrifugation at 300 × g for 3 min at 4°C. Nuclear proteins were extracted in ice cold buffer containing 20mM Tris (pH 7.4), 1% Triton X, 150mM NaCl, 1mM EDTA, 1mM EGTA supplemented with protease/phosphatase inhibitors and frozen at -80°C. Nuclear fraction were subsequently thawed on ice, sonicated, passed through a 21 gauge needle to shear DNA, and clarified by centrifugation at 13,000 × g for 10 min at 4°C.
Immunoflourescence
Cells, cultured on glass coverslips in 35mm plates, were washed with PBS, fixed with methanol free 3% Formaldehyde (Polysicences) for 20 min, permeabilized with 0.2% Triton X for 5 min, blocked for one hr with 3% BSA, and incubated overnight with primary antibody at 4°C. Coverslips were subsequently incubated with fluorescently-labeled secondary antibody, Alexa Fluor 488 anti-mouse (Molecular Probes) for 1 hr at room temperature, washed with PBS, mounted on glass slides using Antifade mounting solution (Molecular Probes) and visualized by confocal microscopy.
Immunoprecipitation/Co-immunoprecipitation
Cells were lysed with ice cold immunoprecipitation buffer containing 10mM Tris, 150mM NaCl, 1% TritonX, 0.5% NP-40, 1mM EDTA, 1mM EGTA and 10mM NEM as well as protease and phosphatase inhibitors as described above. Immunoprecipitations were performed using recombinant Protein G beads per manufacturer’s (Pierce) instructions. Briefly, lysate (1000μg) was incubated with the appropriate antibody for 2 hrs at 4°C, mixed with beads, and incubated for an additional 2 hrs at 4°C. Immune complexes were collected by centrifugation at 2500 × g for 3 min at 4°C, washed 6 times with PBS, resuspended in loading buffer, and heated at 70°C for 3 min. Samples were centrifuged at 2500 × g for 3 min and supernatants resolved with SDS-PAGE, transferred to PVDF membranes and immunoblotted. Recombinant beads were also incubated with whole cell lysates in the absence of antibody to rule out non-specific binding of proteins to the beads.
RESULTS
Helenalin dose-dependently arrest preadipocytes during G1-phase of cell cycle progression independent of apoptosis
Previous reports from our laboratory and others have demonstrated that density-arrested 3T3-L1 preadipocytes synchronously re-enter the cell cycle for a limited period of proliferation that precedes and is obligatory for adipocyte development (Morrison and Farmer, 1999;Auld, Fernandes, and Morrison, 2007;Tang et al., 2003b; Tang et al., 2003a). We have further shown that quiescent preadipocytes (G0 phase) transition G1/S and S/G2 phase boundaries at approximately 15 hrs and 19 hrs, respectively, following stimulation with a differentiation cocktail containing MDI (Auld, Fernandes, and Morrison, 2007). Using this model, we investigated the effects of helenalin on preadipocyte replication by stimulating density-arrested cells with MDI and increasing doses of helenalin. Cells were harvested 20 hr post-MDI and cell cycle progression was assessed by flow cytometry. As illustrated in Fig.1A, MDI-stimulated cells not treated with helenalin (0μM) displayed DNA histograms demonstrating decreased G1 phase content and increased S and G2/M phase content indicative of early-G2 phase progression as previously reported (Auld, Fernandes, and Morrison, 2007). Conversely, concomitant treatment with helenalin dramatically shifted the DNA profile toward an increase in G1 content in a dose-dependent manner clearly demonstrating cell cycle arrest prior to the G1/S phase transition. Moreover, DNA histograms presented no sub-2n cell populations at 20 hr post-treatment suggesting that helenalin impeded cell cycle progression independent of apoptosis. To confirm these observations, cells were stimulated with MDI in the absence and presence of increasing concentrations of helenalin and total cell lysates harvested at times indicated for immunoblotting (Fig.1B). Consistent with G1 arrest, helenalin suppressed the accumulation of Cyclin A which has been shown to occur during S phase in this cell model (Auld, Fernandes, and Morrison, 2007). To further confirm growth arrest in the absence of apoptosis, cell lysates were immunoblotted for cleavage of Caspase 3 and PARP; well established indices of apoptosis. As a positive control, lysates were collected from preadipocytes at 12 hr following transient exposure to UV radiation (100J/m2). As shown in Fig.1B, helenalin treatment did not lead to Caspase 3 or PARP cleavage under these conditions further supporting the premise of G1 growth arrest independent of apoptosis.
Figure 1. Helenalin mediates G1-arrest in a dose-dependent manner.

(A) Density-arrested 3T3-L1 preadipocytes were harvested 20 hr following exposure to MDI in presence of increasing concentration of helenalin as indicated and stained with propidium iodide for DNA histograms generated with flow cytometric analysis. (B) Total cell lysates were collected at 0, 12 and 20 hr from preadipocytes stimulated with MDI in the absence and presence of 3μM helenalin (HLN). Preadipocytes exposed to ultraviolet radiation (UV; 100 J/m2) were harvested as a positive control for apoptosis. Relative protein abundance of cleaved PARP, cleaved caspase 3 and Cyclin A were assessed by immunoblotting as shown. Abundance of GAPDH served as a loading control.
Helenalin dose-dependently enhances p21 protein accumulation
It is well established that cell cycle progression through the G1/S transition requires the timely decay of both p21 and p27. We have previously reported that helenalin inhibits p27 degradation through suppression of SCF E3 ligase activity responsible for polyubiquitylation targeting p27 for proteasomal degradation (Auld, Hopkins, Fernandes, and Morrison, 2006). To assess helenalin’s effect on p21, protein accumulation profiles were initially assessed during cell cycle progression in the absence of helenalin. Cells were harvested over time following MDI stimulation and analyzed by immunoblotting for p21 and Cyclin A as shown in Fig.2A. During density arrest (0 hr), p21 protein abundance was maintained at low levels. Upon stimulation with MDI, p21 transiently accumulated, peaked during mid-G1 (12 hrs), decreased during G1/S transition and S phase progression, and returned to basal levels by G2/M (24 hrs). The decrease of p21 during G1/S transition was confirmed by the onset of Cyclin A accumulation indicative of S and G2 phase progression. Since p21 is known to prohibit G1/S transition via potent inhibition of Cdk2 activity (Sherr and Roberts, 1999;Planas-Silva and Weinberg, 1997), the decrease in p21 protein levels beginning ~15 hrs post-MDI and preceding the accumulation of Cyclin A was consistent as an obligatory event for S phase progression. When cells were stimulated with MDI in the presence of helenalin, p21 accumulation was greatly induced over that observed during mid-G1 (12 hr) and early-G2 (20 hr) in a dose-dependent manner (Fig.2B). Increasing concentrations of helenalin did not affect total caspase-3 protein accumulation which served as a loading control in the experiment. As p21 is a well established, potent inhibitor of Cdk2 activity which is essential for G1/S transition, the excess accumulation of p21 during mid-G1 (12 hr) as well as during S/G2 phase transition (20 hr) supports the hypothesis that aberrant regulation of p21 mechanistically links helenalin with G1 growth arrest.
Figure 2. Helenalin enhances protein abundance of p21 at mid-G1 in a dose-dependent manner.

(A) Total cell lysates were harvested from 3T3-L1 preadipocytes over time following exposure to MDI and immunoblotted to assess relative protein accumulation of p21 and Cyclin A. (B) Total cell lysates, harvested from density-arrested preadipocytes at 0, 12 and 20 hr post-MDI supplemented without and with increasing doses of helenalin (HLN) as indicated, were separated by SDS-PAGE electrophoresis and immunoblotted for p21 as illustrated. Abundance of total caspase 3 served as a loading control.
Helenalin induces p21 protein accumulation post-transcriptionally during density arrest and mid-G1 phase progression
To determine the global mechanism linking helenalin and p21 regulation, total cell lysates and RNA were collected for comparative immunoblotting (Fig.3A) and RT-PCR (Fig.3B), respectively. Samples were harvested from untreated, density-arrested cells (lane 1) as well as cells treated for 12 hr with helenalin (lane 2), MDI (lane 3) or both (lane 4). As illustrated in Fig.3A, helenalin treatment dramatically increased p21 protein accumulation during density arrest (compare lanes 1 and 2) and at mid-G1 (compare lanes 3 and 4). Conversely, neither helenalin, MDI, nor the combination of the two increased p21 mRNA to any measurable extent over that observed in untreated, density-arrested cells (Fig.3B). 18S ribosomal RNA was determined simultaneously in the same PCR reaction using competimer technology (Ambion) to confirm equal loading. Primer specificity was also confirmed using p21 knockout mouse embryo fibroblasts (not shown). These data clearly demonstrate that the dramatic increase in p21 protein accumulation following MDI stimulation or helenalin treatment was independent of any detectable change in mRNA transcript. Collectively, these data strongly support a regulatory role for post-transcriptional mechanisms mediating p21 protein accumulation during mid-G1 of MDI-induced cell cycle progression as well as during growth arrest following helenalin treatment. As helenalin was shown above (Fig.2B) to have near maximal effects on p21 accumulation at 3 μM, the additive effect observed with MDI and helenalin (Fig.3A) presents the possibility that independent mechanisms regulate p21 protein abundance under each condition.
Figure 3. Helenalin promotes p21 protein accumulation by post-transcriptional mechanisms.
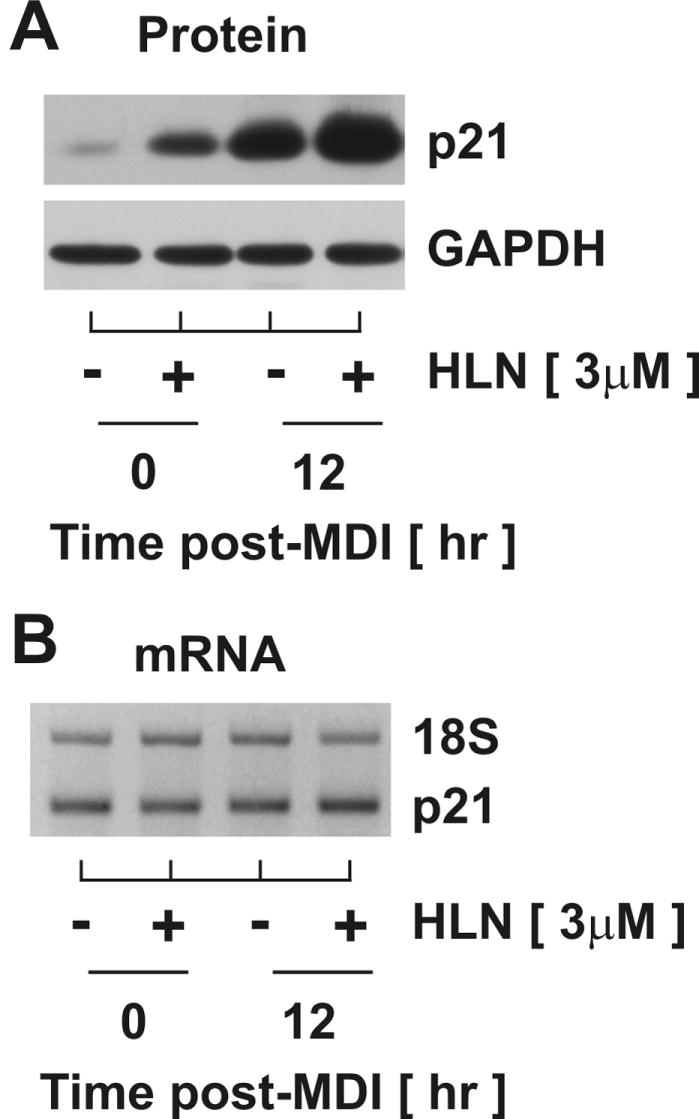
Preadipocytes were stimulated with MDI concomitantly with 3μM helenalin (HLN) for 12 hrs. Relative (A) protein and (B) mRNA accumulation was assessed through immunoblotting and RT-PCR, respectively. Abundance of GAPDH and 18S served as controls for equal loading.
Helenalin modestly promotes the accumulation of ubiquitylated p21
To determine if helenalin increased p21 protein accumulation through posttranscriptional mechanisms involving ubiquitylation and targeted proteasomal degradation, density-arrested were treated with and without helenalin in the presence and absence of MDI for 12 hr. Cell lysates were harvested under non-denaturing conditions, immunoprecipitated with a polyclonal p21 antibody, and immunoblotted with both ubiquitin and p21 (monoclonal) antibodies. For comparative purposes, lysates were also analyzed from density-arrested cells following 6 hr exposure to the potent, specific 26S proteasome inhibitor, epoxomicin. To enhance accumulation of ubiquitin-conjugated substrates, these studies were conducted in the presence of NEM to suppress deubiquitylating enzyme activity. As illustrated in Fig.4, helenalin treatment during density arrest (0hr) or during mid-G1 (12 hr) resulted in modest accumulation of polyubiquitylated p21 in comparison to the marked increase in ubiquitin-conjugated p21 observed in the presence of epoxomicin. Moreover, the apparent helenalin-mediated p21-ubiquitin conjugates were not elevated in the presence of MDI further suggesting that accumulation of p21 protein during mid-G1 and following helenalin treatment occurred through independent post-transcriptional mechanisms. While further experimentation will be required to determine if helenalin is a generic inhibitor of the 26S proteasome, the modest effects shown here are not likely to account in toto for the post-transcriptional mechanisms responsible for the dramatic increase in p21 protein accumulation.
Figure 4. Helenalin modestly promotes the accumulation of polyubiquitylated p21.

Total cells lysates were harvested at 0 hr and 12 hr post-MDI in the absence and presence of 1μM epoxomicin (Epox) or 3μM helenalin (HLN) for 12 hr, immunoprecipitated for p21 and immunoblotted for p21 and ubiquitin (Ub). 10% input protein was immunoblotted and designated by asterisks. Heavy chain (HC) and light chain (LC) immunoglobulins were noted.
Helenalin increases p21 protein accumulation in the nucleus
Subcellular protein localization is an important aspect governing p21 protein accumulation and function. To determine if helenalin leads to increased p21 compartmentalization, density-arrested cells were treated with or without helenalin either in presence or absence of MDI. Nuclear and cytosolic fractions were harvested 12 hr post-treatment and collected cell fractions were immunoblotted for p21. To confirm efficiency of fractionation, lysates were also immunoblotted for nucleoporin and α-tubulin as markers of nuclear and cytosolic fractions, respectively. As illustrated in Fig.5A, helenalin treatment promoted nuclear accumulation of p21 during density arrest and at mid-G1. The effect of helenalin on p21 compartmentalization was confirmed under identical test conditions by immunocytochemistry where cells were treated without and without MDI and helenalin for 12 hr, then fixed and stained for immunoflourescence visualization with confocal microscopy. As illustrated in Fig.5B, helenalin dramatically induced nuclear localization of p21 in density-arrested cells (0 hr) as well as during mid-G1 (12 hr). These data are consistent with the premise that helenalin promotes the nuclear accumulation of p21 by inhibiting proteasome-dependent degradation.
Figure 5. Helenalin increases p21 protein accumulation in the nucleus.

(A) Nuclear and cytosolic extracts were harvested from preadipocytes at indicated times after MDI stimulation in the absence and presence of 3μM helenalin (HLN) and immunoblotted as shown. (B) Subcellular p21 compartmentalization was confirmed by immunofluorescence where density-arrested 3T3-L1 preadipocytes were stimulated with and without MDI for 12 hrs in the absence and presence of 3μM helenalin and analyzed by confocal microscopy.
Helenalin enhances protein interactions between p21 and Cdk2
It is well established that nuclear accumulation of p21 inhibits cell cycle progression (Coqueret, 2003) through protein-protein interactions (Dotto, 2000). To elucidate the function of helenalin-induced nuclear p21 accumulation, density-arrested cells were treated with or without helenalin in presence or absence of MDI. Lysates were harvested 12 hr post-treatment under non-denaturing conditions, immunoprecipitated with p21, and immunoblotted for cell cycle proteins as indicated in Fig.6. Consistently, helenalin increased p21 protein accumulation to an extent greater than that observed under basal conditions of density arrest (0hr) as well as during MDI-induced G1 phase progression (12 hr). The increase in p21 protein accumulation in both cases was associated with increased protein-protein interaction between p21 and Cyclin D1 and Cdk2 and and between p21 and Cdk4 during density arrest. As p21 has been shown to facilitate Cyclin D1 and Cdk4 complex formation at specific stoichiometric ratios, further experimentation would be needed to determine if these interactions were facilitatory or inhibitory for cell cycle progression. The increased interaction with Cdk2, however, was less controversial as p21 is known only as a potent inhibitor of Cdk2 kinase activity. Increased interaction between p21 and Cdk2 was consistent with cell cycle arrest in G1 phase as Cdk2 activity is known to govern G1/S transition.
Figure 6. Increased interaction between p21 and Cyclin D1, Cdk2, and Cdk4 following helenalin exposure.

(A) Density-arrested cells stimulated with and without MDI in the absence and presence of 3μM helenalin (HLN) for 12 hr were harvested, immunoprecipitated for p21 and immunoblotted for Cyclin D1, Cdk4, Cdk2 and p21. 10% input protein was immunoblotted and designated by asterisks.
Helenalin does not inhibit mid-G1 accumulation of Cyclin D1
As shown above, helenalin increased protein-protein interaction between p21 and Cdk2, completely ablated the accumulation of Cyclin A known to occur during S phase progression, and increased the proportion of cells in G1 phase of the cell cycle following MDI stimulation. Each of these observations was consistent with the working model that p21 inhibition of Cdk2 activity suppressed G1/S phase transition. To further explore this premise, density-arrested cells were stimulated with and without MDI in the presence and absence of helenalin. Lysates were harvested at 12 hr post-treatment and cell cycle proteins immunoblotted as illustrated in Fig.7. Comparing protein profiles between cells collected at density arrest versus 12 hr post-MDI in the absence of helenalin demonstrated that mid-G1 phase progression was characterized by a dramatic accumulation of p21 and Cyclin D1 as well as a modest increase in Cdk4 and Cdk2 protein levels. Although concomitant treatment with helenalin suppressed the modest increase in both Cdks, it had no discernable effect of the marked increase in Cyclin D1 accumulation resulting from MDI stimulation. These data demonstrated that aspects of mid-G1 progression remained intact with helenalin treatment further supporting the notion of G1 phase growth arrest near the point of G1/S transition.
Figure 7. Helenalin does not inhibit accumulation of Cyclin D1 at mid-G1.
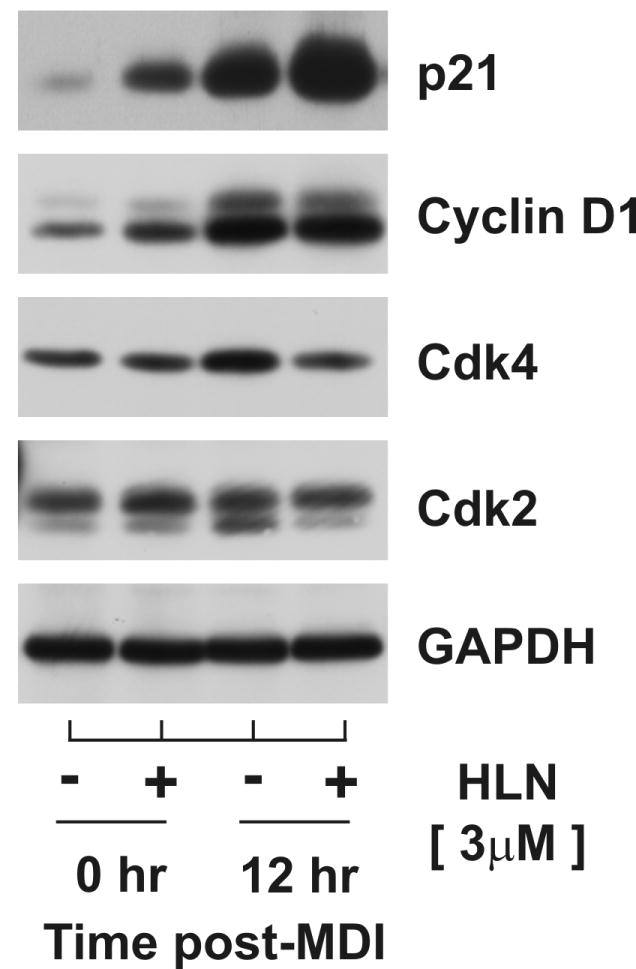
Total cell lysates were harvested from density-arrested 3T3-L1 preadipocytes at 0 hr and 12 hr following exposure to MDI in the absence and presence of 3μM helenalin (HLN) for 12 hr and immunoblotted as indicated.
DISCUSSION
Adipocyte hyperplasia, defined as the proliferation of preadipocytes and their subsequent differentiation into mature adipocytes, contributes to the development of obesity, a chronic disease that is reaching pandemic proportions in developed and developing nations (Hausman et al., 2001;Prins and O’Rahilly, 1997). The CKIs, p21 and 27 are critical in regulating adipocyte hyperplasia as gene ablation of p21 and/or p27 leads to a dramatic increase in adipocyte number and fat mass accumulation in mice (Naaz, Holsberger, Iwamoto, Nelson, Kiyokawa, and Cooke, 2004). Using 3T3-L1 as a cell model of adipocyte hyperplasia, we demonstrate that the phytochemical helenalin dose-dependently inhibits preadipocyte replication by mediating G1 phase arrest. While a cytotoxic effect of helenalin has been well documented in tumor cells (Dirsch et al., 2001b;Lee et al., 1977;Gertsch et al., 2003;Grippo et al., 1992), our data demonstrate that under the conditions used in this study, helenalin-mediated G1 arrest did not result in PARP and caspase-3 cleavage suggesting that growth arrest was not simply a result of apoptosis. The absence of sub-2n cell populations and morphological indices of cell stress further support the cytostatic, non-cytotoxic nature of helenalin at concentrations used in our study. Moreover, the doses of helenalin that caused apoptosis in cancer cells (10-100μM) (Huang, Yeh, and Wang, 2005;Powis, Gallegos, Abraham, Ashendel, Zalkow, Grindey, and Bonjouklian, 1994;Grippo, Hall, Kiyokawa, Muraoka, Shen, and Lee, 1992) and healthy PHA-stimulated PBMC cells (50μM) (Dirsch, Stuppner, and Vollmar, 2001b) were significantly higher as compared to the dose used in this study (3μM).
Although helenalin has been shown to have anti-tumorigenic effects in cancer cells, the mechanisms by which helenalin controls cell proliferation are yet to be determined. Our study is the first to demonstrate, in any cell type, that helenalin treatment can lead to a dramatic accumulation of p21 protein. Furthermore, data presented here support a novel mechanism whereby helenalin-induced accumulation of p21 occurs through post-transcriptional processes independent of changes in mRNA transcript categorically ruling out stress-induced p53 activation and consequential changes in p21 gene expression. Data presented here demonstrate that helenalin promotes nuclear accumulation of p21 as well as modest of polyubiquitylated p21 in a manner similar to that observed following blockade of the 26S proteasome. While the precise mechanisms remains to be determined, data presented here support the premise that helenalin-mediated suppression of proteasomal activity, either direct or indirect, contributes to the dramatic accumulation of p21 in preadipocytes.
We have previously reported that helenalin blocks mRNA and protein accumulation of Skp2, an F-box protein associated with the SCF E3 ligase that functions in p27 polyubiquitylation (Auld, Hopkins, Fernandes, and Morrison, 2006). As we have determined that Skp2 also contributes to proteasome-dependent degradation of p21 (unpublished data), inhibition of Skp2 expression following helenalin treatment may contribute to p21 protein accumulation. However, our observation that helenalin increased p21 ubiquitylation, in the face of helenalin-mediated Skp2 ablation, further suggests that helenalin blocks the degradation of ubiquitin conjugated p21 in addition to blocking processes that lead to p21 ubiquitylation. In a general sense, these data further demonstrate that Skp2-dependent E3 ligase activity is not obligatory for ubiquitylation of p21. Other E3 ligases, yet to be identified, are likely as p21 polyubiquitylation was shown in this report to occur during density arrest when Skp2 is not expressed. E3 ligases such as MDM2, shown to mediate p21 degradation of p21 independent of ubiquitylation (Zhang et al., 2004;Jin et al., 2003), are also potential candidates through which helenalin could increase p21 protein accumulation.
Helenalin is commercially available as a specific inhibitor of the NF-κB signaling pathway. NF-κB is a well studied transcription factor that regulates responses to inflammatory cytokines and its aberration leads to numerous chronic diseases. Helenalin inhibits the induction of NF-κB by multiple mechanisms, including alkylation of the p65 subunit which subsequently prevents DNA binding (Lyss et al., 1998) as well as modification of the NF-κB/IκB complex, thereby preventing the release of NF-κB form IκB and consequently inhibiting its nuclear localization (Lyss, Schmidt, Merfort, and Pahl, 1997). Studies investigating the effect of NF-κB on p21 expression in a variety of cell-types, including epithelial cells (Seitz et al., 2000), human Tleukemia cells (Chang and Miyamoto, 2006) and normal human keratinocytes (Basile et al.,2003), have found NF-κB activity, and not its inhibition as would be expected with helenalin treatment, to induce p21 mRNA expression. As the data presented in our study clearly demonstrate that helenalin dramatically modulates p21 accumulation independent of changes in mRNA transcript, it is probable that helenalin functions through NFκB independent mechanisms. However, transcriptional mechanisms cannot be ruled out as NFκB could regulate the expression of a protein (e.g., an E3 ligase) that in turn regulates p21 protein levels through controlled degradation.
Other potential mechanisms by which helenalin could induce post-transcriptional p21 protein accumulation might involve protein-protein interactions that either restricts p21 from the nucleus where it is degraded as well as interactions that restrict p21 from proteolytic machinery within the nucleus. Selective compartmentalization is unlikely as we also demonstrate that helenalin increased nuclear, as opposed to cytosolic, p21 accumulation. Within the nucleus, others have demonstrated that p21 interaction with proliferating cell nuclear antigen (PCNA) protects p21 from proteasome-dependent degradation and promotes its protein levels (Cayrol and Ducommun, 1998). Along similar lines, the association between Cyclin D1 and p21 within the nucleus has also been shown to increase the stability of p21 by preventing its 26S proteasome-dependent degradation (Coleman et al., 2003). The C8α subunit of the proteasome interacts with a degradation signal located at C-terminus of p21 and promotes its degradation (Touitou et al., 2001), however, the association of p21 with Cyclin D1 masks this degradation signal and prevents the proteasome-dependent turnover of p21 and increases its protein stability. As our results demonstrate increased association between p21 and Cyclin D1 in density-arrested and during mid-G1, this association may contribute to helenalin induced accumulation of p21 by suppressing its proteasome-dependent degradation.
Regulation of p21 protein accumulation represents a possible mechanism linking helenalin with G1 arrest. During mid-G1, p21 facilitates the assembly of Cyclin D1/Cdk4 complexes. However, under conditions of increased nuclear p21 accumulation, D-type cyclins become saturated (Coqueret, 2003) and p21 associates with Cyclin E/Cdk2 complexes, leading to conformational changes that mask the ATP binding site of Cdk2 rendering the complex inactive (El Deiry, 2001). As Cdk2 activity is essential for S phase progression, the decrease in p21 preceding G1/S phase transition is considered essential for cell cycle progression. Furthermore, numerous studies have determined elevated nuclear p21 binds to and inhibits PCNA activity and subsequently prevents DNA replication and suppresses cell cycle (Helt et al., 2004;Rousseau et al., 1999). As helenalin promotes nuclear p21 protein accumulation during S phase progression and increased p21-Cdk2 interactions, it is expected that elevated p21 functions to inhibit G1/S transition contributing to G1 arrest.
In summary, this study provides novel data that demonstrate how a naturally occurring phytochemical, helenalin, inhibits preadipocyte proliferation. Investigation of the mechanisms responsible for G1 arrest in 3T3-L1 preadipocytes revealed that helenalin post-transcriptionally induces p21 accumulation, at least in part, by suppressing its ubiquitin-dependent and proteasome-dependent degradation. Moreover, the enhanced nuclear abundance of p21 and increased associations between p21 and Cdk2 in response to helenalin are consistent with mechanisms that suppress preadipocyte proliferation.
Acknowledgments
We are grateful to Howard Green (Harvard Medical School) for the murine 3T3-L1 cell line. This work was supported by grants from the American Heart Association (0265418U) and National Institutes of Health (R21-DK072067) to R.F.M.
References
- Auld CA, Fernandes KM, Morrison RF. Skp2-mediated p27(Kip1) degradation during S/G(2) phase progression of adipocyte hyperplasia. J Cell Physiol. 2007;211:101–111. doi: 10.1002/jcp.20915. [DOI] [PubMed] [Google Scholar]
- Auld CA, Hopkins RG, Fernandes KM, Morrison RF. Novel effect of helenalin on Akt signaling and Skp2 expression in 3T3-L1 preadipocytes. Biochem Biophys Res Commun. 2006;346:314–320. doi: 10.1016/j.bbrc.2006.05.117. [DOI] [PubMed] [Google Scholar]
- Basile JR, Eichten A, Zacny V, Munger K. NF-kappaB-mediated induction of p21(Cip1/Waf1) by tumor necrosis factor alpha induces growth arrest and cytoprotection in normal human keratinocytes. Mol Cancer Res. 2003;1:262–270. [PubMed] [Google Scholar]
- Cayrol C, Ducommun B. Interaction with cyclin-dependent kinases and PCNA modulates proteasome-dependent degradation of p21. Oncogene. 1998;17:2437–2444. doi: 10.1038/sj.onc.1202189. [DOI] [PubMed] [Google Scholar]
- Chang PY, Miyamoto S. Nuclear factor-kappaB dimer exchange promotes a p21(waf1/cip1) superinduction response in human T leukemic cells. Mol Cancer Res. 2006;4:101–112. doi: 10.1158/1541-7786.MCR-05-0259. [DOI] [PubMed] [Google Scholar]
- Cheng M, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM, Sherr CJ. The p21(Cip1) and p27(Kip1) CDK ’inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 1999;18:1571–1583. doi: 10.1093/emboj/18.6.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman ML, Marshall CJ, Olson MF. Ras promotes p21(Waf1/Cip1) protein stability via a cyclin D1-imposed block in proteasome-mediated degradation. EMBO J. 2003;22:2036–2046. doi: 10.1093/emboj/cdg189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coqueret O. New roles for p21 and p27 cell-cycle inhibitors: a function for each cell compartment? Trends Cell Biol. 2003;13:65–70. doi: 10.1016/s0962-8924(02)00043-0. [DOI] [PubMed] [Google Scholar]
- Dirsch VM, Stuppner H, Vollmar AM. Cytotoxic sesquiterpene lactones mediate their death-inducing effect in leukemia T cells by triggering apoptosis. Planta Med. 2001a;67:557–559. doi: 10.1055/s-2001-16478. [DOI] [PubMed] [Google Scholar]
- Dirsch VM, Stuppner H, Vollmar AM. Helenalin triggers a CD95 death receptor-independent apoptosis that is not affected by overexpression of Bcl-x(L) or Bcl-2. Cancer Res. 2001b;61:5817–5823. [PubMed] [Google Scholar]
- Dotto GP. p21(WAF1/Cip1): more than a break to the cell cycle? Biochim Biophys Acta. 2000;1471:M43–M56. doi: 10.1016/s0304-419x(00)00019-6. [DOI] [PubMed] [Google Scholar]
- El Deiry WS. Akt takes centre stage in cell-cycle deregulation. Nat Cell Biol. 2001;3:E71–E73. doi: 10.1038/35060148. [DOI] [PubMed] [Google Scholar]
- Gertsch J, Sticher O, Schmidt T, Heilmann J. Influence of helenanolide-type sesquiterpene lactones on gene transcription profiles in Jurkat T cells and human peripheral blood cells: anti-inflammatory and cytotoxic effects. Biochem Pharmacol. 2003;66:2141–2153. doi: 10.1016/j.bcp.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Grippo AA, Hall IH, Kiyokawa H, Muraoka O, Shen YC, Lee KH. The cytotoxicity of helenalin, its mono and difunctional esters, and related sesquiterpene lactones in murine and human tumor cells. Drug Des Discov. 1992;8:191–206. [PubMed] [Google Scholar]
- Hall IH, Lee KH, Mar EC, Starnes CO, Waddell TG. Antitumor agents. 21. A proposed mechanism for inhibition of cancer growth by tenulin and helenalin and related cyclopentenones. J Med Chem. 1977;20:333–337. doi: 10.1021/jm00213a003. [DOI] [PubMed] [Google Scholar]
- Hall IH, Lee KH, Starnes CO, Eigebaly SA, Ibuka T, Wu YS, Kimura T, Haruna M. Antitumor agents XXX: Evaluation of alpha-methylene-gamma-lactone-containing agents for inhibition of tumor growth, respiration, and nucleic acid synthesis. J Pharm Sci. 1978;67:1235–1239. doi: 10.1002/jps.2600670912. [DOI] [PubMed] [Google Scholar]
- Hausman DB, DiGirolamo M, Bartness TJ, Hausman GJ, Martin RJ. The biology of white adipocyte proliferation. Obes Rev. 2001;2:239–254. doi: 10.1046/j.1467-789x.2001.00042.x. [DOI] [PubMed] [Google Scholar]
- Helt CE, Staversky RJ, Lee YJ, Bambara RA, Keng PC, O’Reilly MA. The Cdk and PCNA domains on p21Cip1 both function to inhibit G1/S progression during hyperoxia. Am J Physiol Lung Cell Mol Physiol. 2004;286:L506–L513. doi: 10.1152/ajplung.00243.2003. [DOI] [PubMed] [Google Scholar]
- Huang PR, Yeh YM, Wang TC. Potent inhibition of human telomerase by helenalin. Cancer Lett. 2005;227:169–174. doi: 10.1016/j.canlet.2004.11.045. [DOI] [PubMed] [Google Scholar]
- Jin Y, Lee H, Zeng SX, Dai MS, Lu H. MDM2 promotes p21waf1/cip1 proteasomal turnover independently of ubiquitylation. EMBO J. 2003;22:6365–6377. doi: 10.1093/emboj/cdg600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBaer J, Garrett MD, Stevenson LF, Slingerland JM, Sandhu C, Chou HS, Fattaey A, Harlow E. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997;11:847–862. doi: 10.1101/gad.11.7.847. [DOI] [PubMed] [Google Scholar]
- Lee KH, Hall IH, Mar EC, Starnes CO, ElGebaly SA, Waddell TG, HADGRAFT RI, Ruffner CG, Weidner I. Sesquiterpene antitumor agents: inhibitors of cellular metabolism. Science. 1977;196:533–536. doi: 10.1126/science.191909. [DOI] [PubMed] [Google Scholar]
- Lyss G, Knorre A, Schmidt TJ, Pahl HL, Merfort I. The anti-inflammatory sesquiterpene lactone helenalin inhibits the transcription factor NF-kappaB by directly targeting p65. J Biol Chem. 1998;273:33508–33516. doi: 10.1074/jbc.273.50.33508. [DOI] [PubMed] [Google Scholar]
- Lyss G, Schmidt TJ, Merfort I, Pahl HL. Helenalin, an anti-inflammatory sesquiterpene lactone from Arnica, selectively inhibits transcription factor NF-kappaB. Biol Chem. 1997;378:951–961. doi: 10.1515/bchm.1997.378.9.951. [DOI] [PubMed] [Google Scholar]
- Morrison RF, Farmer SR. Role of PPARgamma in regulating a cascade expression of cyclin-dependent kinase inhibitors, p18(INK4c) and p21(Waf1/Cip1), during adipogenesis. J Biol Chem. 1999;274:17088–17097. doi: 10.1074/jbc.274.24.17088. [DOI] [PubMed] [Google Scholar]
- Naaz A, Holsberger DR, Iwamoto GA, Nelson A, Kiyokawa H, Cooke PS. Loss of cyclin-dependent kinase inhibitors produces adipocyte hyperplasia and obesity. FASEB J. 2004;18:1925–1927. doi: 10.1096/fj.04-2631fje. [DOI] [PubMed] [Google Scholar]
- Planas-Silva MD, Weinberg RA. Estrogen-dependent cyclin E-cdk2 activation through p21 redistribution. Mol Cell Biol. 1997;17:4059–4069. doi: 10.1128/mcb.17.7.4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powis G, Gallegos A, Abraham RT, Ashendel CL, Zalkow LH, Grindey GB, Bonjouklian R. Increased intracellular Ca2+ signaling caused by the antitumor agent helenalin and its analogues. Cancer Chemother Pharmacol. 1994;34:344–350. doi: 10.1007/BF00686043. [DOI] [PubMed] [Google Scholar]
- Prins JB, O’Rahilly S. Regulation of adipose cell number in man. Clin Sci (Lond) 1997;92:3–11. doi: 10.1042/cs0920003. [DOI] [PubMed] [Google Scholar]
- Rousseau D, Cannella D, Boulaire J, Fitzgerald P, Fotedar A, Fotedar R. Growth inhibition by CDK-cyclin and PCNA binding domains of p21 occurs by distinct mechanisms and is regulated by ubiquitin-proteasome pathway. Oncogene. 1999;18:4313–4325. doi: 10.1038/sj.onc.1202686. [DOI] [PubMed] [Google Scholar]
- Seitz CS, Deng H, Hinata K, Lin Q, Khavari PA. Nuclear factor kappaB subunits induce epithelial cell growth arrest. Cancer Res. 2000;60:4085–4092. [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004;18:2699–2711. doi: 10.1101/gad.1256504. [DOI] [PubMed] [Google Scholar]
- Tang QQ, Otto TC, Lane MD. CCAAT/enhancer-binding protein beta is required for mitotic clonal expansion during adipogenesis. Proc Natl Acad Sci U S A. 2003a;100:850–855. doi: 10.1073/pnas.0337434100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang QQ, Otto TC, Lane MD. Mitotic clonal expansion: a synchronous process required for adipogenesis. Proc Natl Acad Sci U S A. 2003b;100:44–49. doi: 10.1073/pnas.0137044100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touitou R, Richardson J, Bose S, Nakanishi M, Rivett J, Allday MJ. A degradation signal located in the C-terminus of p21WAF1/CIP1 is a binding site for the C8 alpha-subunit of the 20S proteasome. EMBO J. 2001;20:2367–2375. doi: 10.1093/emboj/20.10.2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Hannon GJ, Casso D, Beach D. p21 is a component of active cell cycle kinases. Cold Spring Harb Symp Quant Biol. 1994;59:21–29. doi: 10.1101/sqb.1994.059.01.005. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Wang H, Li M, Agrawal S, Chen X, Zhang R. MDM2 is a negative regulator of p21WAF1/CIP1,independent of p53. J Biol Chem. 2004;279:16000–16006. doi: 10.1074/jbc.M312264200. [DOI] [PubMed] [Google Scholar]