

Abstract
The risk of developing severe ventricular arrhythmias and/or organ toxicity by currently available drugs used to treat atrial fibrillation (AF) has prompted the development of atrial-selective antiarrhythmic agents. Until recently the principal focus has been on development of agents that selectively inhibit the ultra-rapid delayed rectifier outward potassium channels (IKur), taking advantage of the presence of these channels in atria but not ventricles. Recent experimental studies have demonstrated important atrioventricular differences in biophysical properties of the sodium channel and have identified sodium channel blockers such as ranolazine and chronic amiodarone that appear to take advantage of these electrophysiologic distinctions and act to specifically or predominantly depress sodium channel-mediated parameters in “healthy” canine atria versus ventricles. Atrial-selective/predominant sodium channel blockers such as ranolazine effectively suppress AF in experimental models of AF involving canine isolated right atrial preparations at concentrations that produce little to no effect on ventricular electrophysiologic parameters. These findings point to atrial-selective sodium channel block as a new strategy for the management of AF. The present review examines our current understanding of atrioventricular distinctions between atrial and ventricular sodium channels and our understanding of the basis for atrial selectively of the sodium channel blockers. A major focus will be on the ability of the atrial-selective sodium channel blocking properties of these agents, possibly in conjunction with IKur and/or IKr blocking properties, to suppress and prevent the reinduction of AF.
Keywords: atrial fibrillation, pharmacology, cardiac arrhythmias
INTRODUCTION
Antiarrhythmic agents remain the first-choice treatment of atrial fibrillation (AF), the most common clinical arrhythmia.1 Currently available agents used in the management of AF act largely via inhibition of the rapidly activating delayed rectified potassium current (IKr; eg, d-sotalol or dofetilide) and/or early sodium current (INa; eg, flecainide or propafenone) or via inhibition of multiple ion channels (potassium, sodium, and calcium channels; eg, amiodarone). An important limitation of currently available anti-AF agents is the risk of induction of severe ventricular arrhythmias and/or organ toxicity. The use of sodium channel blockers is contraindicated in patients with structural heart diseases (such as congestive heart failure, myocardial infarction, hypotrophy, etc), which accounts for more than 50% of patients with AF. IKr blockers may induce polymorphic ventricular tachycardia, known as Torsade de Pointes (TdP).1 Whereas amiodarone is a good choice for the maintenance of sinus rhythm following AF cardioversion (and safe for use in patients with structurally compromised ventricles), this agent can induce multiorgan toxicity. These limitations of currently available anti-AF agents have prompted the development of safer atrial-selective pharmacologic agents. Among atrial-specific targets under investigation are the ultra-rapid delayed rectified outward potassium current (IKur), acetylcholine-regulated potassium current (IKACh), connexin 40, and angiotensin II receptors.2 Although block of IKur is considered to be the most promising of these approaches, at concentrations that terminate AF, IKur blockers relatively potently inhibit transient outward current (Ito) and/or IKACh (eg, AVE0118)3 and/or INa (eg, AZD7009 and vernakalant).4,5
Since the Cardiac Arrhythmias Suppression Trial (CAST) demonstrated in 1989 an increased risk of mortality with the use of sodium channel blockers in patients with ventricular ectopy and nonsustained ventricular tachycardia after myocardial infarction,6 the development of new INa blockers had been largely abandoned and the focus shifted to development of potassium channel blockers. The present review describes recent experimental data indicating that atrial and ventricular sodium channels differ with respect to their biophysical properties and action potential characteristics and that sodium channel blockers that take advantage of these distinctions can exert atrial-selective effects to inhibit INa and to suppress AF.7-9
CHARACTERISTICS OF SODIUM CHANNEL BLOCKERS
Most antiarrhythmic agents are not ion channel selective in their actions. Antiarrhythmic agents that block INa as their primary action are generally classified as Class IA, IB, or IC based on their unbinding kinetics from the sodium channel and their effect on action potential duration (APD) in ventricular myocardium.10 Class IB agents such as lidocaine and mexelitine abbreviate APD and have rapidly unbinding kinetics from the sodium channel (τ < 1 sec). Class IA agents, such as procainamide, quinidine, and disopyramide, prolong APD (largely as a result of block of IKr) and have intermediate unbinding kinetics (τ > 1 but < 12 sec). Class IC agents, such as propafenone or flecainide, generally produce little to no effect on ventricular APD and manifest slow unbinding kinetics from the sodium channel (τ > 12 sec). Although amiodarone is classified as a Class III antiarrhythmic agent (prolonging APD), this agent potently blocks INa as well (with rapid kinetics),11,12 contributing to anti-AF properties of this drug. A critical and unique feature of INa blockers related to their antiarrhythmic actions is their ability to produce postrepolarization refractoriness (PRR; ie, ie, to prolong effective refractory period [ERP] without APD prolongation or to a greater extent than APD prolongation).
The effectiveness of most INa blockers to inhibit INa is typically enhanced following acceleration of activation rate, a phenomenon termed “use dependence.”12,13 It is related to a generally higher affinity of INa blockers for the open and/or inactivated state of the sodium channels (ie, during the action potential) than to the rested channels (ie, during the diastolic interval, when net unbinding occurs). Acceleration of heart/pacing rate increases the proportion of time during which the sodium channels are in open and/or inactivated states versus in a rested state. The efficacy of sodium channel blockade is normally augmented by depolarization of resting membrane potential (RMP) resulting from increases in the fraction of inactivated versus rested sodium channels. APD shortening tends to reduce the efficacy of sodium channel blockade as a result of relative decrease of the time during which the sodium channels remain in the inactivated state versus rested state.11-13
There are no “pure” open- or inactivated-state sodium channel blockers. As a general rule, predominantly inactivated-state blockers are Class IB and predominantly open-state blockers are Class IA and IC agents.12,13 Whereas recovery from sodium channel block occurs largely during the resting state, Class IA and IC agents when compared to Class IB agents may more readily unbind during the open and/or inactivated state.11-13
SODIUM CHANNEL BLOCK SUPPRESSES AF
In the clinic, Class IC and Class IA (which also reduce IKr), but not Class 1B (relatively selective INa blockers), agents can effectively suppress AF. Class IA agents, however, are rarely used because of the risk of induction of TdP due to delayed ventricular repolarization.1 Although there are experimental and theoretical studies suggesting that “pure” INa block can successfully suppress AF,14 clinical data supporting this notion are limited to the apparently selective INa blocker pilsicainide (a Class IC agent used in Japan).15
AF is thought to be initiated mainly by a focal mechanism (triggered activity or automaticity) and to be maintained by either reentrant or focal mechanism(s).16 INa blockers can effectively suppress both the reentry-mediated and the focally mediated AF, apparently simply not allowing closely coupled extrasystole(s) and/or rapid repetitive activation to occur. The antiarrhythmic mechanisms underlying anti-AF efficacy of INa blockers are not well understood and likely to be multifactorial, involving depression of excitability, impaired impulse propagation, prolongation of ERP (largely as a result of PRR), and non-INa-block influence (ie, the prolongation of APD) from inhibition of IKr.
There is an apparent contradiction between the classical thinking regarding the mechanism of AF (largely reentrant by nature) and the high anti-AF efficiency of INa blockers. Inhibition of INa slows down conduction velocity (CV), which as a single factor, reduces wavelength (defined as the product of CV and ERP) and should promote reentrant arrhythmias. INa blockers may also prolong ERP,11,13 which, in turn, could offset the effects of CV slowing on wavelength. The importance of wavelength for pharmacologic conversion of AF was, however, challenged by several studies. Wijffels et al in 200017 demonstrated that anti-AF efficacy of Class IC and III agents is associated with an increase of the excitable gap but not of the wavelength in remodeled goat atria in vivo. In this study, ERP and CV were measured at rapid rates equivalent to the AF cycle length (CL) or during AF. An experimental and mathematic model study by Kneller et al14 also observed that “pure” sodium channel block effectively suppresses AF despite abbreviation of wavelength, as a result of enlargement of the core of reentrant circuit, decrease of anchoring to functional obstacles, and reduction of a number of daughters wavelets.
ATRIAL-SELECTIVE SODIUM CHANNEL BLOCKADE
In recent studies, we examined atrioventricular differences of the effects of ranolazine, chronic amiodarone, lidocaine, and propafenone on sodium channel–dependent parameters, such as the maximum rate of rise of the action potential upstroke (Vmax), diastolic threshold of excitation (DTE), CV, and PRR.7-9 Using canine-isolated coronary-perfused atrial and ventricular preparations, we evaluated therapeutically relevant concentrations of these agents and tested physiologically relevant pacing rates. Ranolazine, a recently marketed antianginal agent, was found to depress Vmax, DTE, and CV and induce PRR exclusively or predominantly in atrial preparations (Figs. 1 and 2).7 Thus, when studied in beating multicellular preparations, ranolazine proved to be an atrial-selective sodium channel blocker (“an atrial selective Class I agent”). Within its therapeutic concentration range (2–10 μM), ranolazine significantly inhibited late INa (IC50 = 6 μM) and IKr (IC50 = 12 μM), and, to a lesser extent, late ICa (IC50 = 50 μM), in canine ventricular myocytes.18,19
Figure 1.

Ranolazine specifically induces prolongation of the effective refractory period (ERP) and development of postrepolarization refractoriness in atria (PRR, the difference between ERP and APD75 in atria and between ERP and APD90 in ventricles; ERP corresponds to APD75 in atria and APD90 in ventricles). CL = 500 ms. C, control. The arrows in panel A illustrate the position on the action potential corresponding to the end of the ERP in atria and ventricles and the effect of ranolazine to shift the end of the ERP in atria but not ventricles. *P < 0.05 versus control. †P < 0.05 versus APD75 values in atria and APD90 in ventricles; (n = 5–18). From Burashnikov et al7 with permission.
Figure 2.

Ranolazine produces a much greater rate-dependent inhibition of the maximal action potential upstroke velocity (Vmax) in atria than in ventricles. A, Normalized changes in Vmax of atrial and ventricular cardiac preparations paced at a cycle length (CL) of 500 ms. B, Ranolazine prolongs late repolarization in atria but not ventricles, and acceleration of rate leads to elimination of the diastolic interval, resulting in a more positive takeoff potential in atrium and contributing to atrial selectivity of ranolazine. The diastolic interval remains relatively long in ventricles. *P < 0.05 versus control. †P < 0.05 versus respective values of M cell and Purkinje (n = 7–21). From Burashnikov et al7 with permission.
Chronic amiodarone was found to depress sodium channel–dependent parameters in both atrial and ventricular preparations but much more effectively in atria.9 Lidocaine turned out to be also an atrial-predominant sodium channel blocker but much less atrial selective than ranolazine or amiodarone.7 Propafenone showed no chamber selectivity for INa block at a normal pacing rate (CL = 500 ms), but it did show some atrial predominance at rapid pacing rates, likely because of atrial-specific APD prolongation.8
It is interesting that both ranolazine and chronic amiodarone also produced specific or a more prominent APD prolongation in atria versus ventricles. At rapid rates, the greater slowing of Phase 3 repolarization in atria led to a depolarization of the takeoff potential, thus reducing the availability of sodium channels and accentuating the atrial-selective INa inhibitory effect of these sodium channel blockers (Fig. 2). The atrial-specific APD prolongation also eliminates the diastolic interval (during which recovery from block primarily occurs) specifically in atria, which further promotes atrial-selective sodium channel blockade, particularly at rapid activation rates (Fig. 2). In contrast, lidocaine abbreviates AP duration measured at 90% repolarization (APD90) in both atria and ventricles.
The atrial-specific/predominant APD90 prolongation induced by ranolazine, propafenone, and amiodarone is likely a result of the ability of these agents to block IKr.12,18 Indeed, selective IKr blockers are known to preferentially prolong atrial versus ventricular refractory period (at normal pacing CLs).20 At slow rates or pauses, however, IKr block can dramatically prolong APD and induce early afterdepolarizations (EAD) and TdP in ventricles but not in atria.21,22 The mean peak IKr density is larger in canine atrial versus ventricular myocytes (0.62 versus 0.44 pA/pF, respectively),23 which may contribute to atrial-predominant APD/ERP prolongation by IKr blockers.
Atrial-selective APD prolongation promotes, but does not solely mediate, atrial-selective suppression of INa at normal pacing rates. Ranolazine and amiodarone induce a much greater prolongation of PRR (a feature of INa, not IKr, blockers) than of APD90 in atria. Lidocaine abbreviates both atrial and ventricular APD90, but it produces an atrial-predominant suppression INa. Moreover, propafenone prolongs APD90 specifically in atria but is not atrial-selective in its suppression of INa at normal pacing rates.
Atrioventricular differences in response to sodium channel blockers are poorly studied. Lidocaine, quinidine, and prajmaline are unlike ranolazine in causing a frequency-dependent differential reduction in Vmax in rabbit superfused atrial and ventricular tissue slices (Fig. 3).24 Depression of Vmax by lidocaine was atrial selective at moderate rates of stimulation but not at fast rates. Prajmaline caused a similar depression of Vmax in atria and ventricles.24 Quinidine also produced a relatively larger decrease of Vmax in atria compared to ventricles.24 These 3 agents, particularly lidocaine, caused a larger resting state (tonic) Vmax reduction in atrial than ventricular preparations, which was attributed to the more positive RMP in atria.24 However, the lidocaine analog, Ro 22-9194, produced tonic block selectively in guinea pig atrial myocytes at the same holding potential.25 Under voltage-clamp conditions, lidocaine blocks INa similarly in human atrial and ventricular myocytes26 and moricizine blocks ventricular INa more effectively than atrial INa in guinea pig myocytes.27 GE 68, a propafenone analog lacking β-adrenoreceptor blocking activity, does not show any atrial selectivity in Vmax reduction in the guinea pig.28 Tedisamil reduces Vmax predominantly in human superfused ventricular versus atrial tissue slices.29 Mexelitine decreases Vmax primarily in ventricular and disopyramide causes similar Vmax reduction in ventricular and atrial superfused guinea pig tissue slice preparations.30 AZD7009, which blocks IKur, INa, and IKr, prolongs ERP and reduces DTE and CV predominantly in canine atria versus ventricles in vivo, demonstrating an atrial-predominant suppression of INa-mediated parameters in vivo, although a similar Vmax reduction was observed in isolated superfused atrial (pectinate muscle) and ventricular tissue preparations.4,31 A semiquantitative assessment of chamber selectivity of INa blockers studied in atrial and ventricular tissues under comparable conditions is shown in Fig. 4.
Figure 3.
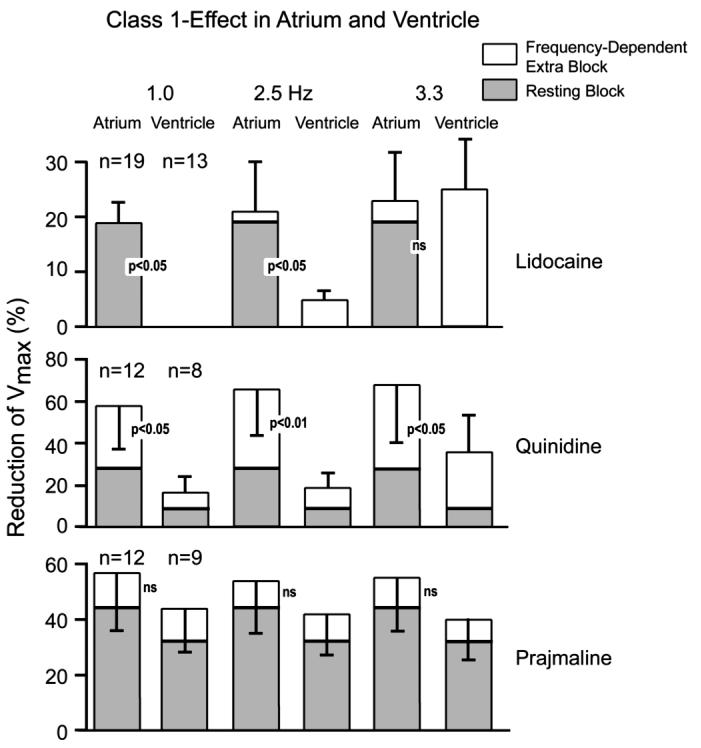
Frequency-dependent extra block (ie, phasic) and resting (ie, tonic) sodium channel block induced by lidocaine, quinidine, and prajmaline in rabbit superfused atrial and ventricular slice preparations. From Langenfeld et al24 with permission.
Figure 4.
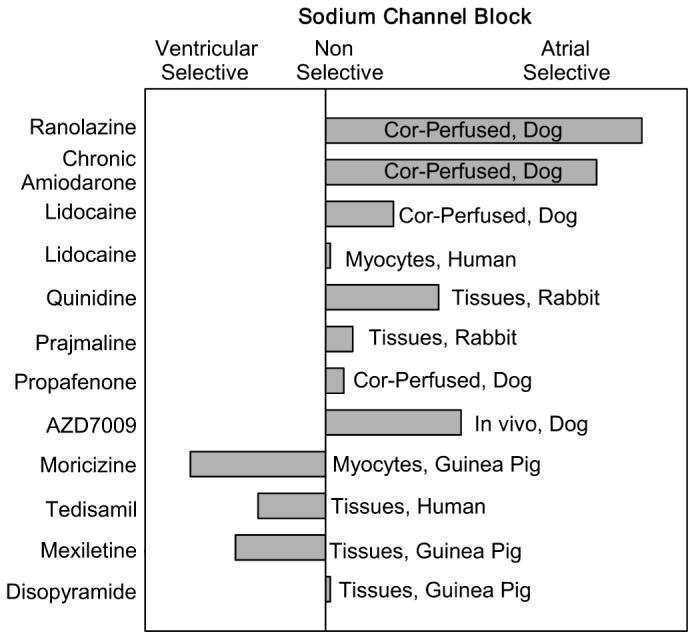
A semiquantitative assessment of atrial selectivity of INa blockers based on studies conducted in atrial and ventricular coronary-perfused (Cor-perfused) and superfused (Tissues) preparations, isolated myocytes, and in vivo (see text for details).
Thus, the available data point to the existence of atrial selective, ventricular selective, and nonchamber-selective sodium channel blockers. An important caveat to consider is that many of these studies were conducted using superfused preparations. In contrast to ventricular superfused slices, atrial ones (at least canine) are generally not viable, showing abnormal action potential parameters and pharmacologic responses,32 making the comparison of superfused atrial and ventricular preparations uncertain. Our experience with the atrial-selective INa blocker ranolazine suggests that evaluation of sodium channel activity is best done under physiologically relevant conditions (ie, coronary-perfused atrial preparations).7,18
MECHANISMS UNDERLYING ATRIAL SELECTIVITY OF SODIUM CHANNEL BLOCKERS
Differences in action potential morphology of atrial and ventricular cells are thought to contribute prominently to the manifestation of an atrial-selective response to sodium channel blockers. In addition to a more depolarized RMP, the atrial action potential displays a more gradual Phase 3 repolarization (Fig. 2).7,33 At progressively faster activation rates, diastolic interval is abolished and takeoff potential is progressively depolarized as a result of failure of Phase 3 to reach maximum diastolic potential. As a consequence, the availability of sodium channels is further compromised in atria because of the presence of a larger fraction of channels in the inactivated state. The elimination of the diastolic interval and the slow repolarization of Phase 3 (keeping membrane potential more positive in atria versus ventricles) also results in slower unbinding of drugs from the sodium channels, leading to significant accumulation of block at fast, but not slow, rates. This would be particularly true in the case of agents that dissociate rapidly from the resting state of the sodium channel, such as ranolazine (τ = 1.56 ± 0.56 sec), and less so for agents that dissociate slowly, such as propafenone.
Recent studies have uncovered major differences in the biophysical properties of atrial and ventricular sodium channels (Fig. 5).7,25,34 The half inactivation voltage (V0.5) in atrial myocytes is 9–14 mV more negative than that of ventricular myocytes,7,25,34 indicating that there is a larger fraction of inactivated sodium channels in atrial versus ventricular cells. Because atrial cells have an intrinsically more depolarized RMP, it is estimated that a sizable fraction of atrial sodium channels are inactivated in atria, but not in ventricles, at the normal RMP. A larger fraction of inactivated state sodium channels in atrial versus ventricular cells (which translates into a smaller fraction of resting sodium channels) could promote atrial-selective/predominant suppression of sodium channels via (1) greater binding of the inactivated atrial sodium channels with inactivated-state sodium channel blockers, and/or (2) by slowed dissociation of sodium channel blockers that normally unbind from the resting state.11-13
Figure 5.

Activation and steady-state inactivation in atrial versus ventricular myocytes. A, Current-voltage relation in ventricular and atrial myocytes. Voltage of peak INa is more positive and current density is larger in atrial versus ventricular myocytes. B, Summarized steady-state inactivation curves. C, Steady-state inactivation curves before and after addition of 15 μM ranolazine. From Burashnikov et al7 with permission.
Recovery from inactivation of the sodium channel is slower in atrial versus ventricular myocytes,34 which should delay sodium channel unblocking primarily in atria, thus promoting atrial-selective inhibition of INa. This would be particularly relevant in the case of rapid activation rates and/or premature impulses, contributing to rate-dependent atrial-selective ERP prolongation.
Sodium channel blockers are known to produce an apparent leftward shift in the steady state inactivation curve (ie, h-curve) or a negative shift in V0.5, increasing the fraction of inactivated channels and reducing the fraction of rested channels.12,13 Ranolazine produces a greater leftward shift in h-curve of atrial versus ventricular myocytes (Fig. 5),7 which further exaggerates the difference in voltage-dependence of inactivation between atrial and ventricular sodium channels, thus contributing to ranolazine's atrial selectivity. Similar shifts in the h-curve are produced by Ro 22-9194, the INa blocker reported to produce atrial-selective tonic block.25 In contrast, the ventricular-selective INa blocker moricizine produces a larger leftward shift in h-curve in ventricular versus atrial myocytes.27
The time constants for INa activation and inactivation are twice as rapid in atrial as in ventricular myocytes and INa density is much greater in atrial than in ventricular myocytes.7,34 A higher density of INa in atrial versus ventricular cells7 points to a larger “sodium channel reserve” in the former, which offsets the lower availability of sodium channels in atrial versus ventricular cells. Vmax values are comparable in per- fused atrial and ventricular muscles.32 It is interesting that DTE is lower in atria than in ventricles7,33 possibly also because of a lower density of inward rectifier current (IK1) in atria, as reported by Golod et al,33 and the voltage threshold for activation of the action potential in atrial cells is more negative that that of ventricular cells (−59 ± 1 and −46 ± 2 mV, respectively).33
The data described earlier point to marked differences in the sodium channels of atrial and ventricular cells both in terms of current density and biophysical properties, suggesting the possibility of tissue-specific cardiac sodium channel isoforms or differences in the stoichiometry of auxiliary subunits. This subject, however, is poorly investigated. The α-subunit of cardiac sodium channel (SCN5A) is likely to be the same in atrial and ventricular cells. Fahmi et al showed that SCN3B, a β-subunit of the sodium channel, is present in the ventricles but not in the atria of sheep hearts.35 Similar data were reported for the rat as well.36 SCN1B (Navβ1) is found both in atria and ventricles of guinea pigs, rat, and humans.35-37 Navβ1 was found to be more strongly expressed in atria versus ventricles in humans.37 It is interesting that the coexpression of SCN3B with SCN5A in Xenopus oocytes shifts the h-curve to the right, compared to SCN5A alone or SCN5A + SCN1B coexpression,35 which may underlie atrioventricular difference in the steady-state inactivation curves (Fig. 5). However, the coexpression of SCN5A with SCN3B in TSA201 cells was reported to shift the h-curve to the left, compared to SCN5A alone or SCN5A + SCN1B coexpression.38 A leftward shift of the h-curve was also observed when SCN5A was coexpressed with SCN3B in Chinese hamster ovary (CHO) cells.36
At present, it is not clear whether binding/unbinding rates or affinities to open or inactivated sodium channel state determine atrial selectivity of INa blockers. From the atrioventricular differences in RMP, h-curve, and recovery from inactivation, it is conceivable that the inactivated-state sodium channel blockers might be more atrial selective than open state blockers. Indeed, the effectiveness of inactivated-state sodium channel blockers is known to be enhanced by depolarization of RMP to a greater extent than that of open- state blockers.11-13 Data on atrial predominant effects of lidocaine, chronic amiodarone (predominantly inactivated-state blockers), and nonchamber-selective actions of propafenone (predominantly open-state blocker) are consistent with that line of thinking. It is not obvious with ranolazine, which has been reported to have a higher affinity for inac- tivated versus rested sodium channels,19 but it seems to be a predominantly open-state sodium channel blocker, staying trapped in the pore of the channel during inactivation and unbinding during the preopen/resting state (Nesterenko et al, unpublished). If recovery from block occurs rapidly during the resting state, INa block would be expected to be atrial-selective whether or not the agent binds to open or inactivated sodium channels as a result of a smaller fraction of rested sodium channels at RMP in atria versus ventricles.
ATRIAL-SELECTIVE SODIUM CHANNEL BLOCK AS A NOVEL STRATEGY FOR THE MANAGEMENT OF AF
In recent studies, we compared the effectiveness of therapeutically relevant concentrations of ranolazine, prop- afenone, and lidocaine in suppressing and preventing the reinduction of AF in isolated canine coronary-perfused right atrial preparations.7-9 The effectiveness of chronic amiodarone in preventing induction of AF was examined as well. Ranolazine effectively prevented the initiation acetylcholine-mediated AF, terminated persistent AF, and prevented its reinduction in coronary-perfused atrial preparations (Fig. 6).7 This anti-AF efficacy of ranolazine (10 μM) was greater than that of lidocaine (21 μM) and somewhat similar to that of propafenone (1.5 μM). In atria isolated from chronic amiodarone-treated dogs (40 mg/kg for 6 weeks), persistent ACh-mediated AF could be induced only in 1 out of 6 atria (versus 10/10 atria in controls).9 These antiarrhythmic effects of ranolazine, amiodarone, and propafenone were associated with both APD prolongation (in the presence of ACh) and the development of a significant PRR, with the duration of the latter being much longer than the extent of APD prolongation, suggesting that sodium channel block plays a more prominent role in the anti-AF actions of these agents.9 The concentrations of ranolazine that suppress AF produce little to no effect on electrophysiologic parameters in normally beating ventricular preparations. These findings suggest that atrial-selective sodium channel block may be a promising novel approach in the management of AF, deserving of further investigation.7
Figure 6.
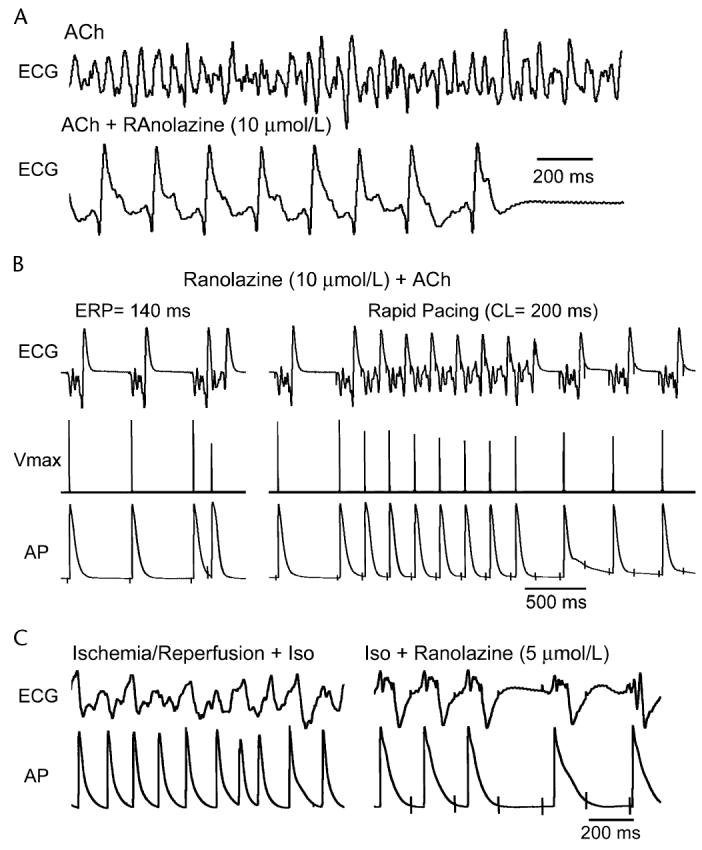
Ranolazine suppresses AF and/or prevents its induction in 2 experimental models involving isolated arterially perfused right atria. A, Ranolazine (10 μM) prevents rapid-pacing induction of AF following pretreatment with acetylcholine (ACh; 0.5 μM). Effective refractory period (ERP) is 140 ms at a cycle length (CL) of 500 ms (left panel). Acceleration of pacing rate from a CL of 500 to 200 ms permits a 1:1 response only during the first 7 beats (right panel). B, Persistent AF induced following pretreatment with ACh (0.5 μM) is suppressed by ranolazine (10 μM). AF is initially converted to flutter (within 17 min) and then to sinus rhythm (17 sec later). C, Rapid-pacing induced nonsustained AF (48-sec duration) induced following ischemia/reperfusion and isoproterenol (ISO, 0.2 μM) (left panel) and the effect of ranolazine to prevent the electrical induction of AF (right panel). In both models, ranolazine causes prominent use-dependent depression of excitability and induction of post-repolarization refractoriness. ECG, pseudoelectrocardiogram; AP, action potential. From Burashnikov et al7 with permission.
Are atrial-selective INa blockers such as ranolazine or amiodarone not effective in suppressing ventricular arrhythmias? On the contrary, these drugs are quite effective in the management of some ventricular arrhythmias.12,39 In the case of ranolazine, this has been shown to result from the potent action of the drug to inhibit late INa in ventricular myocardium, whereas in the case of amiodarone, this is believed to result from the effectiveness of the drug to inhibit late INa and potassium and calcium channels and adrenergic receptors in ventricles of the heart.
The recent MERLIN-TIMI 36 study evaluated the efficacy and safety of ranolazine during long-term treatment of patients with non-ST-segment elevation acute coronary syndrome (ACS).39 The study reported that ranolazine significantly reduced the incidence of both ventricular and supraventricular tachycardias and caused a 31% reduction of new onset of AF.39 The efficacy of ranolazine against ventricular arrhythmias was principally attributed to its action to block late INa.39,40 The study concluded that ranolazine is safe even in patients with severe ACS and appears to have antiarrhythmic effects.39
Thus, both preclinical and clinical data provide compelling evidence in support of an antiarrhythmic action of ranolazine and suggest that studies specifically designed to evaluate the potential role of ranolazine and similar agents as antiarrhythmics are warranted. Particularly in the management of AF, ranolazine may provide a safe alternative to currently available antiarrhythmic drugs, which have a potential for significant adverse effects and are contraindicated in specific populations.1,40,41 In theory, ranolazine might be expected to produce potent INa block in depolarized ventricular muscle; however, available data from several controlled clinical trails (MARISA, CARISA, ERICA, and MERLIN-TIMI-36) have failed to demonstrate proarrhythmic actions of ranolazine39 even in patients with severe ACS.
ATRIAL-SELECTIVE INa + IKur BLOCK FOR AF?
It stands to reason that a combination of both atrial-selective INa block and atrial-specific IKur block may yield a more potent agent than either approach alone in the management of AF. Support for this hypothesis derives from recent studies showing that AZD7009, which blocks IKur, INa, and IKr and depresses CV and DTE predominantly in canine atria in vivo,4 is effective in suppressing clinical AF.42
UNANSWERED QUESTIONS
It is important to recognize that the atrial sodium channel selectivity of ranolazine and chronic amiodarone were based on recordings made in “healthy” right atria and left ventricles.7,9 Clinical atrial and ventricular arrhythmias commonly occur in conjunction with a number of conditions (congestive heart failure, infarction, hypotrophy, dilatation, hypertension, etc) associated with electrical and/or structural remodeling in atria and ventricles. These pathophysiologic changes, and differences in rate of activation of atria versus ventricles during arrhythmia, may modify chamber selectivity of INa blockade. Intrachamber and interchamber differences in the development of electrical and structural remodeling may contribute as well. The selective effect of INa blockers on pulmonary veins in normal, and remodeled hearts also are of great interest. The density of INa is similar in healthy pulmonary vein muscle and left atrial muscles,43 but alterations in INa density have been reported in remodeled canine (left atria)44 but not goat (Bachmann Bundle)45 or human (right atrial appendage)46 atria. Of note, V0.5 of INa inactivation is shifted by +10 mV in cells isolated from AF versus sinus rhythm patients,46 which may reduce the sensitivity to INa blockers. The potency of Class IC agents appears not to be altered by atrial remodeling in goats.47 Thus, there are many possible permutations that could develop with disease states that could affect the atrial selectivity of sodium channel blockers. These and many other issues await future investigation.
CONCLUSIONS
Important differences exist in the action potential characteristics and biophysical properties of sodium channels of atrial and ventricular cells, and drugs that take advantage of these distinctions, such as ranolazine and chronic amiodarone, possess the ability to produce atrial-selective/predominant inhibition of sodium channels, useful in the management of atrial fibrillation in experimental models. Available data suggest that the addition of an IKr and possibly IKur inhibitory effect further potentiates the atrial selectivity and possibly the clinical effectiveness of such agents.
Acknowledgments
Supported by grant HL47678 from NHLBI and NYS and Florida Grand Lodges F. & A. M.
Footnotes
Conflict of Interest Disclosure: Dr. Antzelevitch received research support from and is a consultant to CV Therapeutics.
REFERENCES
- 1.Fuster V, Ryden LE, Cannom DS, et al. ACC/AHA/ESC 2006 guidelines for the management of patients with atrial fibrillation—Executive summary: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Revise the 2001 Guidelines for the Management of Patients With Atrial Fibrillation) J Am Coll Cardiol. 2006;48:854–906. doi: 10.1016/j.jacc.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 2.Nattel S, Carlsson L. Innovative approaches to anti-arrhythmic drug therapy. Nat Rev Drug Discov. 2006;5:1034–1049. doi: 10.1038/nrd2112. [DOI] [PubMed] [Google Scholar]
- 3.Blaauw Y, Gogelein H, Tieleman RG, et al. “Early” class III drugs for the treatment of atrial fibrillation: Efficacy and atrial selectivity of AVE0118 in remodeled atria of the goat. Circulation. 2004;110:1717–1724. doi: 10.1161/01.CIR.0000143050.22291.2E. [DOI] [PubMed] [Google Scholar]
- 4.Goldstein RN, Khrestian C, Carlsson L, et al. Azd7009: A new anti-arrhythmic drug with predominant effects on the atria effectively terminates and prevents reinduction of atrial fibrillation and flutter in the sterile pericarditis model. J Cardiovasc Electrophysiol. 2004;15:1444–1450. doi: 10.1046/j.1540-8167.2004.04354.x. [DOI] [PubMed] [Google Scholar]
- 5.Fedida D, Orth PM, Chen JY, et al. The mechanism of atrial antiarrhythmic action of RSD1235. J Cardiovasc Electrophysiol. 2005;16:1227–1238. doi: 10.1111/j.1540-8167.2005.50028.x. [DOI] [PubMed] [Google Scholar]
- 6.CAST Investigators Preliminary report: Effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N Engl J Med. 1989;321:406–412. doi: 10.1056/NEJM198908103210629. [DOI] [PubMed] [Google Scholar]
- 7.Burashnikov A, Di Diego JM, Zygmunt AC, et al. Atrium-selective sodium channel block as a strategy for suppression of atrial fibrillation: Differences in sodium channel inactivation between atria and ventricles and the role of ranolazine. Circulation. 2007;116:1449–1457. doi: 10.1161/CIRCULATIONAHA.107.704890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burashnikov A, Belardinelli L, Antzelevitch C. Ranolazine and propafenone both suppress atrial fibrillation but ranolazine unlike propafenone does it without prominent effects on ventricular myocardium. Heart Rhythm. 2007;4:S163. [Google Scholar]
- 9.Burashnikov A, Di Diego JM, Zygmunt AC, et al. Atrial-selective sodium channel block as a strategy for suppression of atrial fibrillation. Ann N Y Acad Sci. 2008;1123:105–112. doi: 10.1196/annals.1420.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vaughan Williams EM. A classification of antiarrhythmic actions reassessed after a decade of new drugs. J Clin Pharmacol. 1984;24:129–147. doi: 10.1002/j.1552-4604.1984.tb01822.x. [DOI] [PubMed] [Google Scholar]
- 11.Whalley DW, Wendt DJ, Grant AO. Basic concepts in cellular cardiac electrophysiology: Part II: Block of ion channels by antiarrhythmic drugs. PACE. 1995;18:1686–1704. doi: 10.1111/j.1540-8159.1995.tb06990.x. [DOI] [PubMed] [Google Scholar]
- 12.Carmeliet E, Mubagwa K. Antiarrhythmic drugs and cardiac ion channels: Mechanisms of action. Prog Biophys Mol Biol. 1998;70:1–72. doi: 10.1016/s0079-6107(98)00002-9. [DOI] [PubMed] [Google Scholar]
- 13.Hondeghem LM, Katzung BG. Mechanism of action of antiarrhythmic drugs. In: Sperelakis N, editor. Physiology and pathophysiology of the heart. 3rd ed. Kluwer Academic Publishers; Norwell, MA: 1995. pp. 589–603. [Google Scholar]
- 14.Kneller J, Kalifa J, Zou R, et al. Mechanisms of atrial fibrillation termination by pure sodium channel blockade in an ionically-realistic mathematical model. Circ Res. 2005;96:e35–e47. doi: 10.1161/01.RES.0000160709.49633.2b. [DOI] [PubMed] [Google Scholar]
- 15.Kumagai K, Nakashima H, Tojo H, et al. Pilsicainide for atrial fibrillation. Drugs. 2006;66:2067–2073. doi: 10.2165/00003495-200666160-00003. [DOI] [PubMed] [Google Scholar]
- 16.Nattel S. New ideas about atrial fibrillation 50 years on. Nature. 2002;415:219–226. doi: 10.1038/415219a. [DOI] [PubMed] [Google Scholar]
- 17.Wijffels MC, Dorland R, Mast F, et al. Widening of the excitable gap during pharmacological cardioversion of atrial fibrillation in the goat: Effects of cibenzoline, hydroquinidine, flecainide, and d-sotalol. Circulation. 2000;102:260–267. doi: 10.1161/01.cir.102.2.260. [DOI] [PubMed] [Google Scholar]
- 18.Antzelevitch C, Belardinelli L, Zygmunt AC, et al. Electrophysiologic effects of ranolazine: A novel anti-anginal agent with antiarrhythmic properties. Circulation. 2004;110:904–910. doi: 10.1161/01.CIR.0000139333.83620.5D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Undrovinas AI, Belardinelli L, Undrovinas NA, et al. Ranolazine improves abnormal repolarization and contraction in left ventricular myocytes of dogs with heart failure by inhibiting late sodium current. J Cardiovasc Electrophysiol. 2006;17:S161–S177. doi: 10.1111/j.1540-8167.2006.00401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spinelli W, Parsons RW, Colatsky TJ. Effects of WAY-123,398, a new Class-III antiarrhythmic agent, on cardiac refractoriness and ventricular fibrillation threshold in anesthetized dogs—A comparison with UK-68798, e-4031, and DL- Sotalol. J Cardiovasc Pharmacol. 1992;20:913–922. doi: 10.1097/00005344-199212000-00011. [DOI] [PubMed] [Google Scholar]
- 21.Antzelevitch C, Shimizu W, Yan GX, et al. The M cell: Its contribution to the ECG and to normal and abnormal electrical function of the heart. J Cardiovasc Electrophysiol. 1999;10:1124–1152. doi: 10.1111/j.1540-8167.1999.tb00287.x. [DOI] [PubMed] [Google Scholar]
- 22.Burashnikov A, Antzelevitch C. Late-Phase 3 EAD. A unique mechanism contributing to initiation of atrial fibrillation. PACE. 2006;29:290–295. doi: 10.1111/j.1540-8159.2006.00336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gintant GA. Characterization and functional consequences of delayed rectifier current transient in ventricular repolarization. Am J Physiol. 2000;278:H806–H817. doi: 10.1152/ajpheart.2000.278.3.H806. [DOI] [PubMed] [Google Scholar]
- 24.Langenfeld H, Weirich J, Kohler C, et al. Comparative analysis of the action of class I antiarrhythmic drugs (lidocaine, quinidine, and prajmaline) in rabbit atrial and ventricular myocardium. J Cardiovasc Pharmacol. 1990;15:338–345. doi: 10.1097/00005344-199002000-00023. [DOI] [PubMed] [Google Scholar]
- 25.Hiroe K, Hisatome I, Tanaka Y, et al. Tonic block of the Na+ current in single atrial and ventricular guinea-pig myocytes, by a new antiarrhythmic drug, Ro 22-9194. Fundam Clin Pharmacol. 1997;11:402–407. doi: 10.1111/j.1472-8206.1997.tb00202.x. [DOI] [PubMed] [Google Scholar]
- 26.Furukawa T, Koumi S, Sakakibara Y, et al. An analysis of lidocaine block of sodium current in isolated human atrial and ventricular myocytes. J Mol Cell Cardiol. 1995;27:831–846. doi: 10.1016/0022-2828(95)90090-x. [DOI] [PubMed] [Google Scholar]
- 27.Ahmmed GU, Hisatome I, Kurata Y, et al. Analysis of moricizine block of sodium current in isolated guinea-pig atrial myocytes. Atrioventricular difference of moricizine block. Vascul Pharmacol. 2002;38:131–141. doi: 10.1016/s1537-1891(02)00213-6. [DOI] [PubMed] [Google Scholar]
- 28.Lemmens-Gruber R, Marei H, Heistracher P. Electrophysiological properties of the propafenone-analogue GE 68 (1-[3-(phenylethyl)-2-benzofuryl]-2-(propylamino)-ethanol) in isolated preparations and ventricular myocytes of guinea-pig hearts. Naunyn Schmiedebergs Arch Pharmacol. 1997;355:230–238. doi: 10.1007/pl00004937. [DOI] [PubMed] [Google Scholar]
- 29.Nemeth M, Virag L, Hala O, et al. The cellular electrophysiological effects of tedisamil in human atrial and ventricular fibers. Cardiovasc Res. 1996;31:246–248. [PubMed] [Google Scholar]
- 30.Kodama I, Toyama J, Takanaka C, et al. Block of activated and inactivated sodium channels by class I antiarrhythmic drugs studied by using the maximum upstroke velocity (Vmax) of action potential in guinea-pig cardiac muscles. J Mol Cell Cardiol. 1987;19:367–377. doi: 10.1016/s0022-2828(87)80582-5. [DOI] [PubMed] [Google Scholar]
- 31.Carlsson L, Chartier D, Nattel S. Characterization of the in vivo and in vitro electrophysiological effects of the novel antiarrhythmic agent AZD7009 in atrial and ventricular tissue of the dog. J Cardiovasc Pharmacol. 2006;47:123–132. doi: 10.1097/01.fjc.0000196242.04384.c3. [DOI] [PubMed] [Google Scholar]
- 32.Burashnikov A, Mannava S, Antzelevitch C. Transmembrane action potential heterogeneity in the canine isolated arterially-perfused atrium: effect of IKr and Ito/IKur block. Am J Physiol. 2004;286:H2393–H2400. doi: 10.1152/ajpheart.01242.2003. [DOI] [PubMed] [Google Scholar]
- 33.Golod DA, Kumar R, Joyner RW. Determinants of action potential initiation in isolated rabbit atrial and ventricular myocytes. Am J Physiol. 1998;274:H1902–H1913. doi: 10.1152/ajpheart.1998.274.6.H1902. [DOI] [PubMed] [Google Scholar]
- 34.Li GR, Lau CP, Shrier A. Heterogeneity of sodium current in atrial vs epicardial ventricular myocytes of adult guinea pig hearts. J Mol Cell Cardiol. 2002;34:1185–1194. doi: 10.1006/jmcc.2002.2053. [DOI] [PubMed] [Google Scholar]
- 35.Fahmi AI, Patel M, Stevens EB, et al. The sodium channel beta-subunit SCN3b modulates the kinetics of SCN5a and is expressed heterogeneously in sheep heart. J Physiol. 2001;537:693–700. doi: 10.1111/j.1469-7793.2001.00693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ko SH, Lenkowski PW, Lee HC, et al. Modulation of Na(v)1.5 by beta1- and beta3-subunit co-expression in mammalian cells. Pflugers Arch. 2005;449:403–412. doi: 10.1007/s00424-004-1348-4. [DOI] [PubMed] [Google Scholar]
- 37.Gaborit N, Le BS, Szuts V, et al. Regional and tissue specific transcript signatures of ion channel genes in the non-diseased human heart. J Physiol. 2007;582:675–693. doi: 10.1113/jphysiol.2006.126714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu D, Zygmunt AC, Burashnikov A, et al. Sodium channel of canine atrial and ventricular cells differ with respect to voltage dependence of inactivation. Heart Rhythm. 2007;4:S148. [Google Scholar]
- 39.Scirica BM, Morrow DA, Hod H, et al. Effect of ranolazine, an antianginal agent with novel electrophysiological properties, on the incidence of arrhythmias in patients with non ST-segment elevation acute coronary syndrome: Results from the Metabolic Efficiency with Ranolazine for Less Ischemia in Non ST-Elevation Acute Coronary Syndrome Thrombolysis in Myocardial Infarction 36 (MERLIN-TIMI 36) randomized controlled trial. Circulation. 2007;116:1647–1652. doi: 10.1161/CIRCULATIONAHA.107.724880. [DOI] [PubMed] [Google Scholar]
- 40.Antzelevitch C. Ranolazine: a new antiarrhythmic agent for patients with non-ST-segment elevation acute coronary syndromes? Nat Clin Pract Cardiovasc Med. 2008;5:248–249. doi: 10.1038/ncpcardio1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zipes DP, Camm AJ, Borggrefe M, et al. ACC/AHA/ESC 2006 Guidelines for Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death—Executive Summary. J Am Coll Cardiol. 2006;48:1064–1108. doi: 10.1016/j.jacc.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 42.Crijns HJ, Van GI, Walfridsson H, et al. Safe and effective conversion of persistent atrial fibrillation to sinus rhythm by intravenous AZD7009. Heart Rhythm. 2006;3:1321–1331. doi: 10.1016/j.hrthm.2006.06.035. [DOI] [PubMed] [Google Scholar]
- 43.Ehrlich JR, Cha TJ, Zhang L, et al. Cellular electrophysiology of canine pulmonary vein cardiomyocytes: action potential and ionic current properties. J Physiol. 2003;551:801–813. doi: 10.1113/jphysiol.2003.046417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gaspo R, Bosch RF, Bou-Abboud E, et al. Tachycardia-induced changes in Na+ current in a chronic dog model of atrial fibrillation. Circ Res. 1997;81:1045–1052. doi: 10.1161/01.res.81.6.1045. [DOI] [PubMed] [Google Scholar]
- 45.Wijffels MC, Kirchhof CJ, Dorland R, et al. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation. 1995;92:1954–1968. doi: 10.1161/01.cir.92.7.1954. [DOI] [PubMed] [Google Scholar]
- 46.Bosch RF, Zeng X, Grammer JB, et al. Ionic mechanisms of electrical remodeling in human atrial fibrillation. Cardiovasc Res. 1999;44:121–131. doi: 10.1016/s0008-6363(99)00178-9. [DOI] [PubMed] [Google Scholar]
- 47.Eijsbouts S, Ausma J, Blaauw Y, et al. Serial cardioversion by Class IC drugs during 4 months of persistent atrial fibrillation in the goat. J Cardiovasc Electrophysiol. 2006;17:648–654. doi: 10.1111/j.1540-8167.2006.00407.x. [DOI] [PubMed] [Google Scholar]