

Abstract
Lantibiotics are post-translationally modified peptide antimicrobial agents that are synthesized with an N-terminal leader sequence and a C-terminal propeptide. Their maturation involves enzymatic dehydration of Ser and Thr residues in the precursor peptide to generate unsaturated amino acids, which react intramolecularly with nearby cysteines to form cyclic thioethers termed lanthionines and methyllanthionines. The role of the leader peptide in lantibiotic biosynthesis has been subject to much speculation. In this study, mutations of conserved residues in the leader sequence of the precursor peptide for lacticin 481 (LctA) did not inhibit dehydration and cyclization by lacticin 481 synthetase (LctM) showing that not one specific residue is essential for these transformations. These amino acids may therefore be conserved in the leader sequence of class II lantibiotics to direct other biosynthetic events, such as proteolysis of the leader peptide or transport of the active compound outside the cell. However, introduction of Pro residues into the leader peptide strongly affected the efficiency of dehydration, consistent with recognition of the secondary structure of the leader peptide by the synthetase. Furthermore, the presence of a hydrophobic residue at the position of Leu-7 appears important for activity. Based on the data in this work and previous studies, a model for the interaction of LctM with LctA is proposed. The current study also showcases the ability to prepare other lantibiotics in the class II lacticin 481 family, including nukacin ISK-1, mutacin II, and ruminococcin A using the lacticin 481 synthetase. Surprisingly, a conserved Glu located in a ring that appears conserved in many class II lantibiotics, including those not belonging to the lacticin 481 subgroup, is not essential for antimicrobial activity of lacticin 481.
Keywords: Lantibiotic, leader peptide, lacticin 481, mutacin II, nukacin ISK-1
The problem of multi-drug resistant bacteria has become increasingly apparent in recent years, with several strains posing the threat of becoming immune against all commercially available antibiotics (1, 2). It is evident that in order to prevent potential epidemic outbreaks of infectious diseases, a renewed focus on antibiotic research is highly desired, including the understanding of biosynthetic pathways of natural product antibiotics (3). In this regard, the lantibiotics family of antimicrobial peptides has shown promising properties (4). Nisin, the most studied lantibiotic to date, is produced by Lactococcus lactis and has been used commercially as a preservative in the food industry for over 40 years due to its potent antibacterial properties and non-toxicity to humans (5). The high efficacy of nisin (nM MICs against many Gram-positive bacteria) has been attributed to its dual modes of action: highly efficient formation of pores in the membrane and inhibition of incorporation of lipid II, a membrane-anchored cell wall precursor, into the peptidoglycan (6, 7).
Lantibiotics are a group of peptide antimicrobial agents that are ribosomally synthesized as precursor peptides containing two distinct regions, an N-terminal leader sequence and a C-terminal propeptide (8). Maturation of the propeptide region of these prepeptides results in a family of antibiotics with varying structure, size, and biological activity (4, 8, 9). In the first step of post-translational modification, specific threonine and serine residues undergo dehydration to generate the unsaturated amino acids 2,3-dehydrobutyrine (Dhb) and 2,3-dehydroalanine (Dha). Subsequently, intramolecular Michael-type addition of nearby cysteines to the Dha and Dhb residues form cyclic thioethers termed lanthionine and methyllanthionine (Figure 1A). Two distinct enzymes, a LanB dehydratase and a LanC cyclase, catalyze these processes for class I lantibiotics (eg nisin) while a bifunctional LanM protein performs both reactions for class II members (eg lacticin 481, Figure 1B) (10). The final step of lantibiotic biosynthesis involves proteolytic removal of the leader peptide and transportation out of the cell. For class II lantibiotics, the leader sequences are removed by the protease domain of an ABC-transporter LanT, which is also responsible for secreting the mature product (11-13). A better understanding of the role of the leader peptide in the post-translational modifications is a prerequisite for efficient efforts to reprogram their biosynthetic pathways.
Figure 1.

(A) Dehydroalanine (Dha) and dehydrobutyrine (Dhb) formation during lantibiotic biosynthesis arises from the dehydration of serine and threonine residues, respectively. Cysteine addition to the unsaturated amino acids generates the thioether crosslinks. (B) Lacticin 481 biosynthesis catalyzed by lacticin 481 synthetase (LctM). The enzyme dehydrates specific serine and threonine residues of the substrate LctA and subsequently catalyzes cysteine addition to the dehydrated amino acids, generating two lanthionines and one methyllanthionine. The dual cysteine protease and ABC transport protein LctT is responsible for leader peptide removal and transport of the mature lantibiotic out of the cell.
The substrate specificity of the biosynthetic machinery has been investigated primarily by in vivo experiments with mutant genes that produced variants in the leader and propeptide of the precursor peptides (8, 14, 15). Collectively these studies have shown that the biosynthetic enzymes for both classes of lantibiotics have high tolerance for amino acid changes in their prepeptides. However, when such mutants were not processed to mature lantibiotics, it has been difficult to delineate whether this was the result of incorrect dehydration, cyclization, proteolytic processing/degradation, secretion, or possibly because of breakdown of the cell’s self-resistance system when a lantibiotic analog was produced. Three biosynthetic systems, lacticin 481 (16), haloduracin (17), and nisin (18), have been reconstituted in vitro with purified enzymes and can be utilized to better understand the sequence requirements for substrate recognition by the modification enzymes. We dissected the importance of leader peptide residues for lanthionine formation and proteolysis in vitro, reporting in the accompanying paper on the effects of mutation of the prepeptide of lacticin 481 with respect to proteolysis by LctT, and disclosing in this study the effects on dehydration and cyclization by LctM.
Previous studies suggest that at least part of the function of the leader sequence may involve recognition by the modification enzymes (16, 18-20). However, a recent study demonstrated that the leader sequence of the precursor peptide for lacticin 481 (LctA) is not required for post-translational modification by the bifunctional dehydratase/cyclase LctM but greatly increases the efficiency of modification (21). The current study shows that point mutations of conserved residues of the LctA leader sequence do not impair either catalytic function of the protein, but can result in decreased efficiency of both processes, thereby singling out important residues in the leader peptide. In a different set of experiments probing the importance of the leader peptide, LctM is shown to process chimeras of the LctA leader peptide and propeptides from other members of the lacticin 481 subgroup of lantibiotics including NukA (nukacin ISK-1), MutA (mutacin II), and RumA (ruminococcin A). Surprisingly, mutagenesis of the glutamate that is conserved in the A-ring of the lacticin 481 subgroup and in the B-ring of mersacidin showed that it is not essential for antimicrobial activity of lacticin 481.
MATERIALS AND METHODS
Primers for construction of LctA mutants as well as general methods for expression and purification of peptides are detailed in the Supporting Information.
Construction of pET15b LctNukA expression system
To install the C-terminal mutations, the partial lctA gene was amplified using the primers LctNukA1 FP (5′-TCTCATGACTGTCACATGAATAGCTTCCAATTT-3′) and T7 terminator (5′-GCTAGTTATTGCTCAGCGG-3′) with plasmid pET15b-LctA as the template. The generated mega primer was subsequently used as the terminator primer with the long T7 promoter primer (5′-CGCGAAATTAATACGACTCACTATAGGGGAATTGTGAG-3′) to yield the full lctA gene. The PCR product was digested with NdeI and BamHI restriction enzymes then ligated into the pET15b vector. DNA sequencing revealed the three desired mutations in the C-terminal portion of the propeptide. This plasmid was termed pET15b LctA E13D/N15H/W19F. The partial lctnukA gene was then amplified using the primers LctNukA2 FP (5′-GCAAAAAAAAAAAGTGGAGTTATTCCGACAGTTTCTCAT-3′) and T7 terminator with plasmid pET15b-LctA E13D/N15H/W19F as the template. The generated mega primer was subsequently used as the terminator primer with the long T7 promoter primer to yield the full lctnukA gene. The PCR product was digested with NdeI and BamHI restriction enzymes then ligated into the pET15b vector.
Construction of pET15b NukA expression system
Oligonucleotides corresponding to the full sequence of the NukA leader region were designed, annealed and used as template DNA. The leader sequence of NukA was amplified using a forward primer containing the NdeI restriction site (5′-AGGAATTCCATATGGAAAACTCTAAAGTTATG-3′) followed by 18 bases of NukA leader and a reverse primer consisting of the last 18 bases of the NukA leader sequence followed by the first 18 bases of the NukA propeptide. (3′-AACTCCACTTTTTTTTTTAGCACCCAGAACTTCGTT-5′). This generated a piece of DNA that coded for the NukA leader and a truncated portion of the NukA propeptide. The NukA propeptide was amplified by employing pET15b LctNukA as template DNA. A forward primer consisting of the last 18 bases of the NukA leader sequence as well as the first 18 bases of the NukA propeptide (5′-AACGAAGTTCTGGGTGCTAAAAAAAAAAGTGGAGTT-3′) and a reverse primer complimentary to the C-terminal portion of the propeptide of NukA flanked by the BamHI recognition site (5′-TAGCAGCCGGATCCTTAAGAGCAGCAAGTAAA-3′) amplified the propeptide. With these two pieces in hand, a subsequent PCR reaction was performed where the two pieces primed to each other at the leader-structural interface and the outside primers containing the restriction sites for the endonucleases were employed for amplification. The product was digested with NdeI and BamHI and ligated into the pET15b vector.
Construction of pET15b LctMutA, LctRumA, and MutLctA expression systems
Generation of the LctMutA, LctRumA, and MutLctA peptides was undertaken in a similar manor to the generation of NukA. Oligonucleotides corresponding to the full sequence of the MutA propeptide or RumA propeptide were designed. PCR was conducted on these synthetic oligos as well as the pET15b LctA template for chimera generation. The MutLctA construct was generated by amplifying the MutA leader sequence off genomic DNA isolated from Streptococcus mutans T8 and the LctA propeptide off pET15b LctA. With overlapping fragments in hand, a subsequent PCR reaction was performed such that the two pieces primed to each other at the leader-structural interface. Outside primers were employed for amplification. Digested fragments were ligated into the pET15b vector between XhoI and BamHI. See the Supporting Information for further clarification.
Processivity Assay
The LctA peptide (0.5 μM) was incubated in 1 mL of 50 mM Tris-HCl, pH 7.5, containing 10 mM MgCl2, 25 μg/mL BSA, 2.5 mM ATP, 5 mM TCEP (Fluka) and His6-LctM (1 nM) at 25 °C. At 0, 30, 60, and 90 min, 200 μL of the assay was removed and quenched with 40 μL of 5 % TFA. The aliquot was lyophilized and resuspended in 10 μL of H2O. The assays were analyzed by MALDI-TOF MS after preparing samples utilizing Millipore 10 μL C18 tips and eluting with 4 μL of a saturated solution of α-hydroxyl cinnamic acid dissolved in ACN:H2O (1:1) in 0.5 % TFA. The entire sample was applied to the MALDI target and analyzed.
General Methods for p-Hydroxymercuribenzoic Acid (PHMB) Assays
Assays of His6-LctNukA, His6-LctMutA, His6-LctRumA, or His6-NukA with wt LctM or LctM C781A/C836A were carried out as described above except on a 50 μL scale with 26 μM substrate and 1.7 μM enzyme. A 5 μL aliquot of the assay was removed after mixing prior to enzyme addition, acidified with 2 μL of 5 % TFA and was concentrated to dryness. The reactions were incubated with enzyme at room temperature for 3-4 h. Two 5 μL aliquots were removed and both were acidified with 2 μL of 5 % TFA. One aliquot was zip tipped as described previously to confirm dehydration by MALDI-TOF MS and the second was concentrated to dryness. To the dry 0 and 3 h time point aliquots was added 10 μL of 10 mM TCEP and 4 M guanidine hydrochloride. Subsequently, 10 μL of 10 mM PHMB was added, incubated at 25 °C for 1 h and checked for thiol modification by zip tipping and eluting in 4 μL of α-hydroxyl cinnamic acid to run MALDI-TOF MS as detailed above.
Results
Site-Directed Mutants of Conserved Residues in the LctA Leader Sequence
While the leader sequence is not absolutely required for the LctA propeptide to undergo post-translational modification by LctM in vitro, it provides imperative binding affinity for efficient processing (21). An examination of class II lantibiotic leader peptides reveals a sequence of conserved residues that may be responsible for recognition by their synthetases (Figure 2). These amino acids may provide a secondary structure that is adopted in the presence of LctM, as the use of structure prediction tools expect the C-terminal portion of the leader peptide to be alpha helical (nnPredict (22), residues -17 to -4; GOR4 (23), residues -16 to -4; SSPro (24), residues -18 to -12 and -10 to -3).
Figure 2.

Alignment of class II lantibiotic sequences. Highly conserved sequences in the leader peptide are highlighted in yellow. Ser/Thr that undergo dehydration are highlighted in blue while Cys that undergo cyclization are labeled in red. The conserved negatively charged residue (Glu/Asp) that has been suggested to be important for antimicrobial activity is labeled in blue font. The arrow denotes the proteolytic cleavage site at which the leader peptide is removed from the mature propeptide.
The helix breaking residues Pro and Gly (25) were introduced at positions -8, -6, -5 and -4 to assess whether LctM might require an α-helical structure for substrate binding. All six constructs generated (E-8P, D-6P, D-6G, L-5P, I-4P, and I-4G) were successfully expressed in E. coli and purified (Table 1). In addition, a series of mutants was made of strictly or strongly conserved residues (E-13A, E-8A, L-7E, L-7A, L-7K and L-5Q) to examine the importance of side chain functionality for processing by LctM. The purified LctA mutants were subjected to LctM in the presence of ATP and Mg2+. Interestingly, all mutants in Table 1 were substrates for LctM as demonstrated by the loss of water molecules in the assay products by MALDI-TOF MS (Figure S1). Unfortunately, efforts to obtain reliable kinetic parameters have been hampered by the very low solubility of the His6-LctA peptide (∼ 1.3 μM) as reported previously (26, 27). Qualitatively, most mutant substrates were processed by LctM with similar efficiencies as wild type LctA as exemplified for E-8A in Figure 3A. Like wt LctA, these mutants did not demonstrate significant build-up of intermediates and showed mostly products of fourfold dehydration even after only partial conversion of the substrate at early time points (Figure S1). Upon longer incubation, all substrate was converted to fourfold dehydrated products. On the other hand, incubation of LctM with five mutants of LctA (E-8P, L-7E, L-7K, D-6P, and I-4P) resulted in the observation of significant amounts of incompletely dehydrated products (eg L-7E in Figure 3B). These mutants could not be converted completely to fourfold dehydrated products upon long incubation times. For a complete set of mass spectra, see the Supporting Information.
TABLE 1.
| Entry | His6-Substrate | Antibiotic Activity | Synthetase Activitya (Relative to WT) |
|---|---|---|---|
| 1 | wt | + | ++ |
| 2 | E-13A | + | ++ |
| 3 | E-8P | +b | - |
| 4 | E-8A | ND | ++ |
| 5 | L-7E | +b | - |
| 6 | L-7K | - | - |
| 7 | L-7A | ND | + |
| 8 | D-6G | + | ++ |
| 9 | D-6P | ND | - |
| 10 | L-5P | ND | + |
| 11 | L-5Q | + | ++ |
| 12 | I-4G | + | ND |
| 13 | I-4P | ND | - |
| 14 | G-2V | + | ++ |
| 15 | G-2K | + | ++ |
| 16 | G-2E | + | ND |
| 17 | A-1I | + | ND |
| 18 | A-1K | + | ++ |
| 19 | A-1D | + | ND |
| 20 | A-1G | ND | ++ |
All mutants shown in entries 1-20 underwent four dehydrations when incubated for 12 h except for L-7K. The designations +, ++, and - refer to the following phenotypes of the mutant substrates when monitoring the time-dependence of dehydration by LctM. Mutants that were fully processed to a four-fold dehydrated product in a similar timespan as wt-LctA are indicated with ++. Mutants that were processed by LctM into a mixture of three- and fourfold dehydrated peptides in this time span are indicated with +. These mutants can be converted to fourfold dehydrated products upon longer incubation times. Mutants that were processed to predominantly three-fold dehydrated products without ever going to complete fourfold dehydrated products are indicated with -. ND, biological activity not determined (column 3) or not investigated in a time-dependent fashion (column 4).
Bioactivity was only observed when at least some fourfold dehydrated product was produced.
Figure 3.
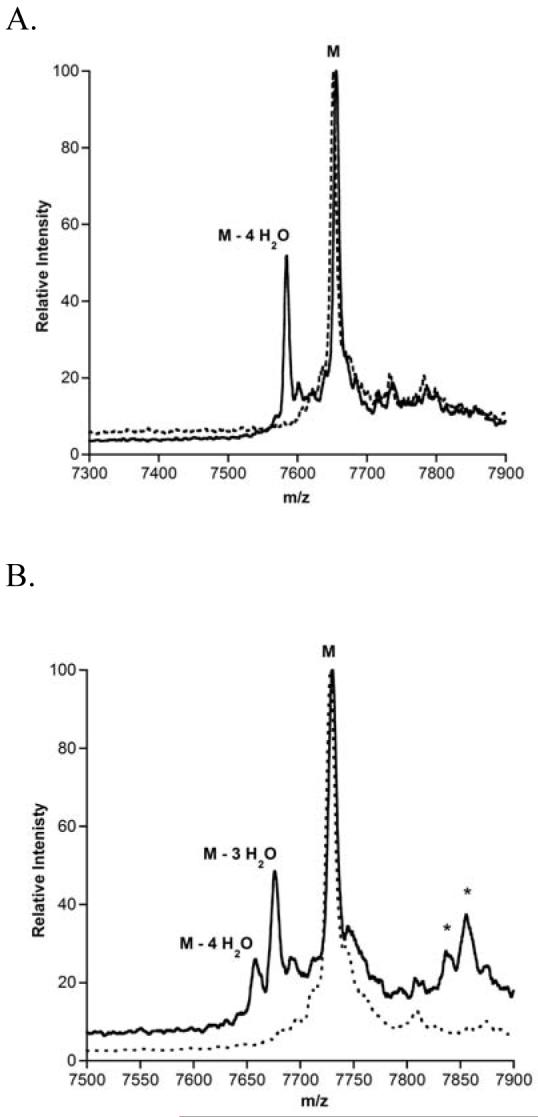
Representative MALDI-TOF mass spectra of LctA leader mutants before (dashed line) and after (solid line) incubation with LctM. Reactions were stopped after 10 min to compare relative efficiencies of product formation, (A) LctA E-8A, (B) LctA L-7E. The (*) indicates phosphorylated peptides (50).
Cyclization Activity with LctA Leader Peptide Mutants
Assessment of correct cyclization activity is more challenging since the cyclized and non-cyclized products have identical masses. The most stringent method to investigate correct cyclization is the use of antimicrobial assays because the thioether rings in lacticin 481 are crucial for its antimicrobial activity (28). Hence, the products of LctM action on a selection of the LctA mutants were subjected to proteolytic removal of the leader peptide with the commercial protease Lys-C (16) and analyzed for antimicrobial activity against the reporter strain Lactococcus lactis CNRZ 117. Only the L-7K mutant failed to show a clear zone of growth inhibition indicating that correct cyclization activity was not abolished for all other mutants tested (Table 1). However, bioactivity for two other substrates that displayed inefficient dehydration (E-8P and L-7E) was weaker than other point mutants. Whereas the qualitative bioassays can be used to assess whether cyclization occurred correctly, these experiments do not report on any uncyclized material if the mutations resulted in reduced cyclization efficiency. To assess the extent of cyclization of a subset of the LctA mutants, the thiol specific modification agent p-hydroxymercuribenzoic acid (PHMB) was used due to its large mass shift of 320 Da upon aryl mercury adduct formation with free cysteines (26, 29). Such adducts would report on incomplete cyclization. LctM-processed LctA point mutants were lyophilized and treated with 4 M guanidine HCl and 10 mM TCEP to disrupt any secondary structure and reduce disulfide bonds. After incubation with PHMB, adduct formation was assessed by MALDI-TOF MS. No peaks corresponding to the loss of four water molecules and a mercury adduct were observed for E-8A, D-6P, and I-4P suggesting they underwent complete cyclization. To confirm that cyclization was catalyzed by LctM, these mutants were treated with a cyclization deficient mutant, LctM-C781A/C836A (26), followed by incubation with PHMB. Arylmercury adduct formation was observed for dehydrated material for all assay products further confirming that wild type LctM catalyzed complete cyclization of E-8A, D-6P, A-1D, and I-4P (Figure S2). On the other hand, substantial adduct formation for dehydrated peptides was observed for the LctM-proceesed products of E-8P, L-7E and L-7K. These findings therefore demonstrate that introducing polar amino acids for hydrophobic Leu-7 drastically affects not only dehydration but also cyclization. Furthermore, for cyclization position -8 is more sensitive with respect to introduction of a Pro than positions -4 and -6.
Mutants of the Double Gly Motif
The leader sequence of the class II lantibiotics ends in the double-glycine motif, which typically consists of either two consecutive Gly residues or a GlyAla sequence (Figure 2) (8, 11). To probe the importance of this motif for LctM processing, the mutants G-2V, G-2K, G-2E, A-1I, A-1K, A-1D, and A-1G were generated. Treatment of these mutant peptides with LctM under the same conditions as described previously resulted in four dehydrations for all analogs (Figure S1). Proteolytic removal of the leader sequences with Lys-C resulted in antimicrobial activity indicating that ring formation had also taken place (Table 1).
Processivity of LctM
The observation of only fourfold dehydrated LctA and no partially dehydrated products even at early time points when the substrate is not completely consumed has been interpreted previously as an example of processive enzyme catalysis (30). On the other hand, partially dehydrated intermediates were observed when the leader peptide and prepeptide were incubated with LctM in trans. These findings suggested that the leader peptide keeps the substrate bound to the enzyme between subsequent dehydration events. During this study on LctA mutants, an alternative explanation for these observations was considered. Given the poor solubility of the substrate, if substrate deaggregation is slow in comparison to dehydration activity, then incompletely dehydrated intermediates would not be observable either. To test this hypothesis, an experiment with low substrate (0.5 μM) and enzyme (1 nM) concentrations was performed in a relatively large volume to allow analysis by MALDI-MS. Intermediate dehydration products were observed under these conditions (Figure 4) suggesting the previously observed processivity of LctM may be a consequence of peptide aggregation.
Figure 4.
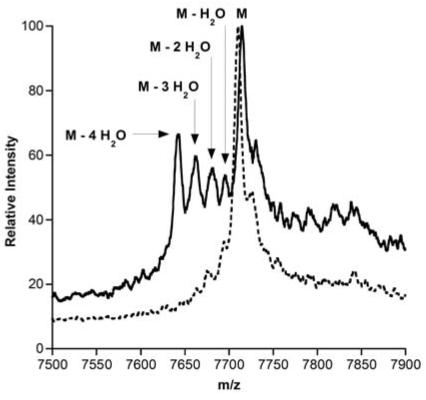
Processivity of LctM with LctA at low substrate concentration (0.5 μM). MALDI-TOF mass spectra of LctA taken before (dashed) and 60 min after LctM addition (solid).
Chimera of the LctA Leader Peptide and the NukA Propeptide
Nukacin ISK-1 is a class II lantibiotic produced by the Gram-positive bacterial strain Staphylococcus warneri ISK-1. It is classified in the lacticin 481 subgroup of lantibiotics (14). The peptide sequences of LctA and NukA have seven amino acid differences in their propeptides (74% identity), but lower sequence identity in the leader peptides with the conservation concentrated in the C-terminal half of the leader peptide (residues -13 to -1 have 61% identity, Figure 2). The similarity in the propeptide includes identical positions of the Ser/Thr and Cys residues resulting in identical topologies of the ring systems of the two lantibiotics. Given the significant similarity between these two prepeptides, a chimeric peptide was constructed consisting of the LctA leader peptide and NukA propeptide (LctNukA) in an attempt to use LctM for the preparation of nukacin ISK-1.
The His6-LctNukA peptide was generated through a series of PCR experiments (Materials & Methods). The peptide was heterologously expressed in E. coli, and purified under denaturing conditions. Incubation of LctNukA with LctM resulted in complete conversion of starting material to a four-fold dehydrated peptide as analyzed by MALDI-MS (Figure 5A). To examine the ability of LctM to catalyze cyclization of this chimeric substrate and generate nukacin ISK-1, PHMB was again used. Incubation of unmodified LctNukA with PHMB resulted in near quantitative derivatization. LctM-processed LctNukA was lyophilized and treated with 4 M guanidine HCl, 10 mM TCEP, and PHMB for 1 h at room temperature. Adduct formation was assessed by MALDI-TOF MS. No peaks corresponding to mercury adducts were observed (Figure 5B) suggesting cyclization had taken place to generate nukacin ISK-1. Treatment of LctNukA with a cyclization deficient mutant, LctM-C781A/C836A (26), followed by incubation with PHMB resulted in arylmercury adduct formation for fully dehydrated material (Figure 5C), confirming that wild type LctM catalyzed complete cyclization of this chimeric substrate.
Figure 5.
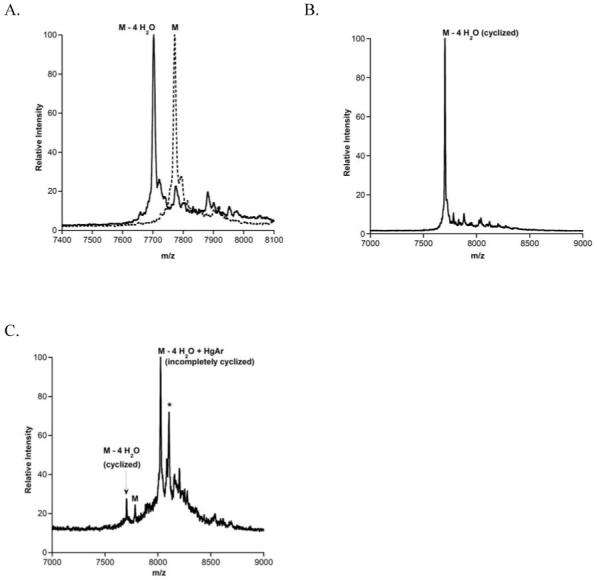
MALDI-TOF mass spectra of (A) LctNukA incubated with LctM (B) LctNukA incubated with LctM and subsequently PHMB, and (C) LctNukA incubated with the cyclization deficient synthetase LctM C781A/C836A and subsequently PHMB. The (*) indicates phosphorylated peptide (50).
Chimera of the LctA Leader Peptide with the MutA or RumA Propeptides
Mutacin II, produced by Streptococcus mutans T8, and ruminococcin A, produced by Ruminococcus gnavus, were chosen to examine the general utility of LctM to generate other class II lantibiotics of the lacticin 481 subgroup. Ruminococcin A is isolated from human fecal matter and has yet to be structurally characterized (31). The peptide sequences of LctA and RumA have only 5 amino acid differences in their propeptides whereas LctA and MutA have 11 variations. The number of amino acids between the Ser/Thr and Cys residues involved in cyclization are identical, but the distances to the leader peptide are quite different in the prepeptides (Figure 2). Furthermore, in ruminoccocin A, the formation of the putative C ring results in a methyllanthionine instead of a lanthionine in lacticin 481. LctM has already been shown to be tolerant of such changes (28). Treatment of both LctRumA and LctMutA with LctM resulted in four dehydrations and analogous analysis for cyclization as described above for nukacin ISK-1 demonstrated that the chimeras were also cyclized (Figures S3 and S4).
Using the MutA leader sequence to generate lacticin 481
The broad substrate specificity of LctM raised the question whether other lantibiotic leader peptides could be appended to the LctA propeptide and promote full processing by LctM. The MutA leader was chosen to replace the LctA leader sequence. This MutLctA chimeric substrate was treated with LctM and reaction progress was monitored by MALDI-MS demonstrating four dehydrations. Furthermore, like wild type LctA, this peptide conveniently contains a lysine residue at position 1 that was used to remove the leader peptide with endoprotease Lys-C. Treatment of L. lactis 117 with the fully processed and leaderless compound resulted in a zone of growth inhibition, demonstrating that the presence of the MutA leader sequence does not impede the cyclization ability of LctM.
Processing of NukA by lacticin 481 synthetase
The gene encoding the precursor peptide of nukacin ISK-1, NukA, was generated via splicing by overlap extension PCR, expressed in E. coli as a His6-tagged peptide, and purified. The peptide was treated with LctM and loss of four water molecules was observed by MALDI-MS (Figure S5A). Removal of the leader peptide and the first three-lysine residues of nukacin ISK-1 using endoprotease Lys-C generated a compound with weak antimicrobial activity against L. lactis 117 (Figure S5B).
The Importance of Glu13 for Lacticin 481 Activity
The mode of action of lacticin 481 is currently unknown. The sequence of its A-ring resembles the C-ring of mersacidin (Figure 6), with the glutamate in this ring being crucial for the antimicrobial properties of the latter (32). In fact, this ring is also conserved in plantaricin C and the α peptides of two-component lantibiotics (Figure 6) (33, 34). The similarities in these ring structures has led to speculation that the conserved glutamate (E13) in the A-ring of the lacticin 481 subgroup may be similarly important for activity (9, 28, 33). To address the importance of this glutamate in lacticin 481, the point mutation E13A was constructed in LctA. Treatment of this mutant with LctM under standard assay conditions resulted in a loss of four waters observed by MALDI-TOF MS (Figure S6A). Removal of the leader peptide by proteolysis with Lys-C and application of the processed compound against a lacticin 481 sensitive bacterial strain resulted in a zone of growth inhibition (Figure S6B).
Figure 6.
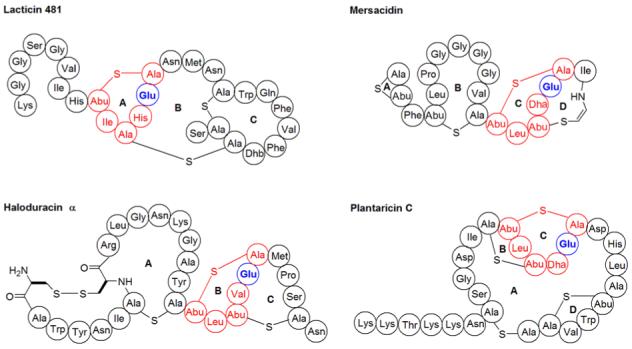
Structures of lantibiotics containing a conserved methyllanthionine ring displayed in red. The glutamate that is essential for both mersacidin and haloduracin antimicrobial activity is bolded.
DISCUSSION
Lantibiotic leader peptides range in length from 23 to 59 amino acids and seem to assist in several processes of lantibiotic biosynthesis. Since the modified propeptide shows no antibiotic activity as long as the leader sequence is attached, the leader peptide keeps the lantibiotic inactive inside the cell, thereby protecting the producing bacterial strain (16, 18, 35). In addition, the leader peptides appear to be important for molecular recognition by the lanthionine incorporation machinery (16, 18-21, 36), by transporter systems that carry the lantibiotic across the cell membrane (37, 38), and by proteases that remove the leader peptide (13, 35, 39). It should be emphasized that the segment of the precursor peptides termed the leader peptide is defined by the protease cleavage site, but that it is currently not known whether it is also this segment that is recognized by biosynthetic enzymes that precede the proteolytic processing step. Similarly, which specific residues in the leader peptide are responsible for constructive interactions with any of the biosynthetic enzymes is still not understood. To date, the sequence requirement of class II lantibiotic leader peptides has only been investigated in vivo. Mutation of the conserved double-glycine motif abolished production of mutacin II and resulted in accumulation of dehydrated premutacin in the cell membrane (39). Whether cyclization had also occurred was not determined. These observations suggested that mutacin II production was disrupted due to the inability of the protease to remove the leader sequence of modified MutA. This hypothesis is corroborated by the in vitro data in this work on mutants of the double-glycine motif that were fully dehydrated and cyclized, and by findings in the accompanying paper that proteolysis by the protease domain of LctT was inhibited by mutations in this motif (13).
Of the remaining MutA leader peptide mutations generated in vivo only L-7K fully disrupted mutacin II biosynthesis, but it could not be determined what step(s) in the overall maturation process had been perturbed (39). The results of the current study suggest that mutacin II production was abolished due to decreased efficiency of dehydration as a result of the incorporation of charge at the Leu-7 position since both L-7E and L-7K mutations in LctA strongly perturbed lacticin 481 synthetase activity (Figures 3B) whereas the L-7A mutant was a better substrate (Figure S1C). The L-7E and L-7K mutants also showed poor cyclization activity as demonstrated by the presence of free thiols in the assay products. Collectively, the MutA leader peptide findings are consistent with the ability of LctM to tolerate almost all point mutations generated in this study, including residues that are fully conserved among class II lantibiotic peptides (Table 1). These findings then pose the conundrum that with the exception of Leu-7 no residues appear to be critically important for activity yet removing the leader peptide altogether strongly affects the efficiency of dehydration (21). The fact that no single mutation alone completely abolished LctM activity suggest that multiple residues are required for binding and raised the possibility that LctM recognizes a secondary structure of the leader peptide.
Circular dichroism experiments on the leader peptides for nisin, subtilin, pep5, epidermin, and gallidermin indicate they adopt an α-helical structure in solutions containing trifluoroethanol (40). Indeed, current structure prediction tools all anticipate helical character for certain stretches of amino acids in the leader peptides, although this has never been demonstrated in aqueous buffer and could also not be confirmed in this study for LctA. Nevertheless, it is possible that LctM induces or captures a helical conformation of its substrate. The importance of a helical conformation for dehydration is supported by the E-8P, D-6P, L-5P, and I-4P mutations that strongly decrease the efficiency of dehydration by LctM. These mutations are in regions that are predicted to be helical by all three bioinformatics tools used. Although the observations with the proline mutants do not unequivocally demonstrate that LctM recognizes a helical conformation of the leader peptide, taken together with the results with the other mutants they are fully consistent with disruption of secondary structure. Interestingly, proline mutations at positions -4, -5, and -6 did not seem to perturb cyclization activity to nearly the same extent as the dehydration activity with only the E-8P mutant showing incomplete cyclization in this set. It should be noted that it is not known whether the binding site for the leader peptide is shared between the dehydration and cyclization activities in the bifunctional LanM proteins. The stand-alone cyclase NisC requires the leader peptide and functions in the absence of the NisB dehydratase suggesting the cyclase has an independent leader peptide binding site (18).
Our current hypothesis for the role of the leader peptide in dehydratase activity is shown in Figure 7 and is based on the results in this study and that of previous reports. LctM is proposed to recognize a certain secondary structure, possibly helical, adopted by the C-terminal segment of the leader peptide. Leader peptide binding then brings the propeptide in close proximity to the active site for dehydration resulting in highly promiscuous dehydration. Ser/Thr residues that are too close to the leader peptide are not dehydrated in the propeptide as they cannot reach into the active site, consistent with previous experimental results (28). Leader peptide binding also results in overall increased efficiency of dehydration because a leaderless substrate is processed much slower than the full length prepeptide (21). However, the observation that the prepeptide without the leader is a substrate demonstrates that the leader peptide is not required for dehydration (21). Therefore, we do not favor a model in which the leader peptide induces a compulsory conformational change to convert inactive LctM to an active dehydratase (Figure 7, pathway B), but rather a mechanism in which the leader stabilizes an active form of LctM that is present as a minor species (pathway A). Figure 7 depicts leader peptide binding as a static event, but a related mechanism in which the leader peptide slides along a binding surface with LctM cannot be ruled out. Furthermore, at present we have insufficient kinetic data to unequivocally determine whether leader peptide binding also contributes energetically to enzymatic catalysis. The use of binding energy of a substrate at non-reactive positions for enzymatic rate acceleration has been discussed previously (41, 42) and has recently received renewed attention (43, 44). The strongly diminished activity with the proline mutants of LctA is consistent with leader peptide binding contributing to catalysis. Similarly the observed higher dehydration activity when the leader peptide is added in trans compared to when only the prepeptide is incubated with LctM is consistent with the leader peptide playing a direct role in rate acceleration. However, both observations can also be explained by a shift in the equilibrium between active and inactive enzyme upon leader peptide binding. In the absence of the leader peptide, this equilibrium will not be shifted toward the active conformation explaining the greatly reduced activity in the absence of the leader peptide. Similarly, the observed reduced activity of some of the proline mutants of LctA can be accounted for by a much reduced affinity of the leader peptide for the enzyme, which would also result in a smaller fraction of active enzyme. Finally, the presence of dehydrated intermediates observed by MALDI for LctM under conditions to limit substrate aggregation suggests that LctM is not processive, in contrast to previous interpretations of the data (30).
Figure 7.
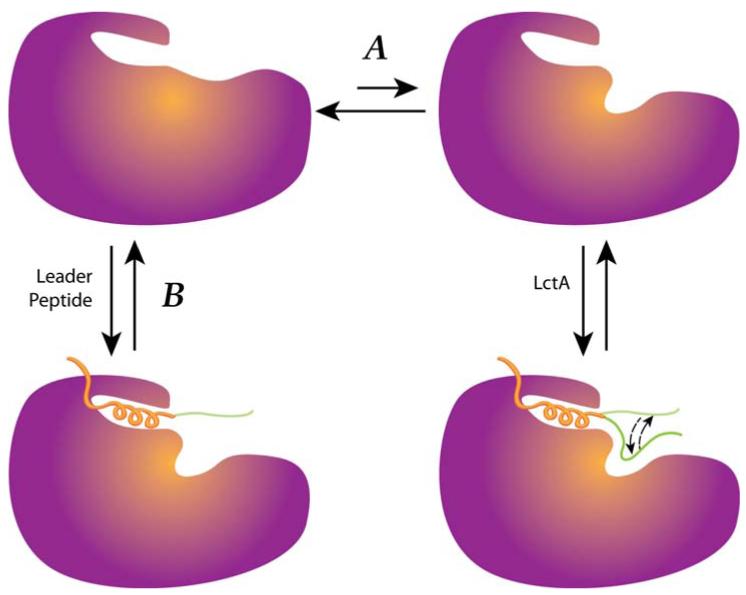
The proposed role of the leader peptide in LctM activity. The leader peptide is proposed to shift the equilibrium between inactive and active enzyme towards the latter.
The observed activity of LctM with LctNukA, LctMutA, and LctRumA displays the promiscuous nature of this modification enzyme. These peptides contain between five and eleven mutations in the prepeptide compared to LctA, but did not affect LctM dehydration activity. These findings with class II substrates are reminiscent of in vivo experiments conducted on the class I lantibiotics nisin and subtilin (45). The activity of LctM on a chimeric substrate containing the leader peptide of another lantibiotic (MutLctA) also demonstrates the overall promiscuous nature of the substrate recognition. Again, these in vitro results with class II lantibiotics parallel in vivo data reported for class I family members as a chimera of the leader sequence of subtilin and the propeptide of nisin was processed in the nisin producer L. lactis (46). However, the ability of LctM to process the NukA substrate highlights one of the advantages of in vitro studies because a recent study reported that LctM could not complement a NukM deficient strain in vivo. It was suggested that this observation resulted from the incapability of LctM to process NukA due to the low sequence identity (42%) between LctM and NukM (47), but our results suggest that the observed lack of nukacin ISK-1 production may more likely result from perturbed protein-protein interactions in a lantibiotic synthetase complex or incorrect folding of LctM. We attribute the weak antimicrobial activity observed against L. lactis 117 for processed NukA to the removal of the first three N-terminal lysine residues after treatment with Lys-C. It was previously shown that removal of these lysines resulted in a 32-fold decrease in antimicrobial activity against several Gram positive strains (48).
Mersacidin (49), plantaricin C (34), and the α peptide of the two component lantibiotic lacticin 3147 (33), which is structurally similar to haloduracin α, all have lipid II as their biological target. For mersacidin (32) and haloduracin (Cooper, L.E., van der Donk, W.A. unpublished results) the conserved Glu highlighted in Figure 6 is critically important for biological activity. The unexpected finding that this Glu in the A-ring of lacticin 481 is not required for antimicrobial activity suggests that lacticin 481 has either a different target or recognizes lipid II by a different molecular mechanism.
In summary, the results presented here provide support for the recognition of a secondary structure in the C-terminal region of the leader peptide of LctA by lacticin 481 synthetase rather than specific residues. The only specific requirement for efficient dehydration appears to be a hydrophobic residue at position -7. Decreased dehydratase activity of helix-disrupting mutants was also observed. Binding studies are required to further illuminate substrate recognition by lantibiotic synthetases, but these have been hampered so far by the poor solubility of the LctA peptide. The higher solubility of the HalA2 substrate for the haloduracin synthetase HalM2 shows promise to extend the current findings and provide quantitative binding data.
Supplementary Material
ACKNOWLEDGEMENTS
We thank L.A. Furgerson Ihnken for cloning and expression of LctA E-13A, LctA L-7A, and LctA A-1G as well as Prof. Caufield for the donation of Streptococcus mutans T8.
Abbreviations
- ATP
adenosine triphosphate
- Dha
2,3-didehydroalanine
- Dhb
2,3-didehydrobutyrine
- DTT
dithiothreitol
- FPLC
fast protein liquid chromatography
- LanB
class I lantibiotic dehydratase enzymes
- LanC
class I lantibiotic cyclase enzymes
- LanM
class II lantibiotic synthetase enzymes
- LanT
lantibiotic transporter/protease
- LctT
class II lantibiotic ABC transporter/protease for lacticin 481
- MALDI-TOF MS
matrix assisted laser desorption ionization - time of flight mass spectrometry
- MIC
minimum inhibitory concentration
- RP-HPLC
reverse phase high performance liquid chromatography
- TFA
trifluoroacetic acid
Footnotes
This work was supported by the National Institutes of Health (GM58822 to WAV) and a Ruth L. Kirschstein National Research Service Award (GM070421 to LEC).
REFERENCES
- 1.Talbot GH, Bradley J, Edwards JE, Jr., Gilbert D, Scheld M, Bartlett JG, Task Force of the Infectious Diseases Society of America Bad bugs need drugs: an update on the development pipeline from the Antimicrobial Availability. Clin. Infect. Dis. 2006;42:657–668. doi: 10.1086/499819. [DOI] [PubMed] [Google Scholar]
- 2.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. Jama. 2007;298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 3.Clardy J, Fischbach MA, Walsh CT. New antibiotics from bacterial natural products. Nat. Biotechnol. 2006;24:1541–1550. doi: 10.1038/nbt1266. [DOI] [PubMed] [Google Scholar]
- 4.Cotter PD, Hill C, Ross RP. Bacterial lantibiotics: strategies to improve therapeutic potential. Curr. Protein Pept. Sci. 2005;6:61–75. doi: 10.2174/1389203053027584. [DOI] [PubMed] [Google Scholar]
- 5.Delves-Broughton J, Blackburn P, Evans RJ, Hugenholtz J. Applications of the bacteriocin, nisin. Antonie van Leeuwenhoek. 1996;69:193–202. doi: 10.1007/BF00399424. [DOI] [PubMed] [Google Scholar]
- 6.Breukink E, Wiedemann I, van Kraaij C, Kuipers OP, Sahl H, de Kruijff B. Use of the cell wall precursor lipid II by a pore-forming peptide antibiotic. Science. 1999;286:2361–2364. doi: 10.1126/science.286.5448.2361. [DOI] [PubMed] [Google Scholar]
- 7.Hasper HE, Kramer NE, Smith JL, Hillman JD, Zachariah C, Kuipers OP, de Kruijff B, Breukink E. A new mechanism of antibiotic action. Science. 2006;313:1636–1637. doi: 10.1126/science.1129818. [DOI] [PubMed] [Google Scholar]
- 8.Chatterjee C, Paul M, Xie L, van der Donk WA. Biosynthesis and Mode of Action of Lantibiotics. Chem. Rev. 2005;105:633–684. doi: 10.1021/cr030105v. [DOI] [PubMed] [Google Scholar]
- 9.Willey JM, van der Donk WA. Lantibiotics: Peptides of Diverse Structure and Function. Annu. Rev. Microbiol. 2007;61:477–501. doi: 10.1146/annurev.micro.61.080706.093501. [DOI] [PubMed] [Google Scholar]
- 10.Xie L, van der Donk WA. Post-Translational Modifications during Lantibiotic Biosynthesis. Curr. Opin. Chem. Biol. 2004;8:498–507. doi: 10.1016/j.cbpa.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 11.Håvarstein LS, Diep DB, Nes IF. A family of bacteriocin ABC transporters carry out proteolytic processing of their substrates concomitant with export. Mol. Microbiol. 1995;16:229–240. doi: 10.1111/j.1365-2958.1995.tb02295.x. [DOI] [PubMed] [Google Scholar]
- 12.Uguen P, Hindré T, Didelot S, Marty C, Haras D, Le Pennec JP, Vallee-Rehel K, Dufour A. Maturation by LctT Is Required for Biosynthesis of Full-Length Lantibiotic Lacticin 481. Appl. Environ. Microbiol. 2005;71:562–565. doi: 10.1128/AEM.71.1.562-565.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Furgerson Ihnken LA, Chatterjee C, van der Donk WA. In vitro Reconstitution and Substrate Specificity of a Lantibiotic Protease. Biochemistry. 2008 doi: 10.1021/bi800278n. Accompanying paper. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dufour A, Hindre T, Haras D, Le Pennec JP. The biology of lantibiotics from the lacticin 481 group is coming of age. FEMS Microbiol. Rev. 2007;31:134–167. doi: 10.1111/j.1574-6976.2006.00045.x. [DOI] [PubMed] [Google Scholar]
- 15.Kuipers OP, Bierbaum G, Ottenwälder B, Dodd HM, Horn N, Metzger J, Kupke T, Gnau V, Bongers R, van den Bogaard P, Kosters H, Rollema HS, de Vos WM, Siezen RJ, Jung G, Götz F, Sahl HG, Gasson MJ. Protein engineering of lantibiotics. Antonie van Leeuwenhoek. 1996;69:161–169. doi: 10.1007/BF00399421. [DOI] [PubMed] [Google Scholar]
- 16.Xie L, Miller LM, Chatterjee C, Averin O, Kelleher NL, van der Donk WA. Lacticin 481: in vitro reconstitution of lantibiotic synthetase activity. Science. 2004;303:679–681. doi: 10.1126/science.1092600. [DOI] [PubMed] [Google Scholar]
- 17.McClerren AL, Cooper LE, Quan C, Thomas PM, Kelleher NL, van der Donk WA. Discovery and in vitro biosynthesis of haloduracin, a two-component lantibiotic. Proc. Natl. Acad. Sci. 2006;103:17243–17248. doi: 10.1073/pnas.0606088103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li B, Yu J-PJ, Brunzelle JS, Moll GN, van der Donk WA, Nair SK. Structure and Mechanism of the Lantibiotic Cyclase Involved in Nisin Biosynthesis. Science. 2006;311:1464–1467. doi: 10.1126/science.1121422. [DOI] [PubMed] [Google Scholar]
- 19.Rink R, Kuipers A, de Boef E, Leenhouts KJ, Driessen AJ, Moll GN, Kuipers OP. Lantibiotic structures as guidelines for the design of peptides that can be modified by lantibiotic enzymes. Biochemistry. 2005;44:8873–8882. doi: 10.1021/bi050081h. [DOI] [PubMed] [Google Scholar]
- 20.Rink R, Wierenga J, Kuipers A, Kluskens LD, Driessen AJM, Kuipers OP, Moll GN. Production of dehydroamino acid-containing peptides by Lactococcus lactis. Appl. Environ. Microbiol. 2007;73:1792–1796. doi: 10.1128/AEM.02350-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levengood MR, Patton GC, van der Donk WA. The Leader Peptide is not Required for Post-translational Modification by Lacticin 481 Synthetase. J. Am. Chem. Soc. 2007;129:10314–10315. doi: 10.1021/ja072967+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kneller DG, Cohen FE, Langridge R. Improvements in protein secondary structure prediction by an enhanced neural network. J. Mol. Biol. 1990;214:171–182. doi: 10.1016/0022-2836(90)90154-E. [DOI] [PubMed] [Google Scholar]
- 23.Garnier J, Gibrat JF, Robson B. GOR method for predicting protein secondary structure from amino acid sequence. Methods Enzymol. 1996;266:540–553. doi: 10.1016/s0076-6879(96)66034-0. [DOI] [PubMed] [Google Scholar]
- 24.Pollastri G, Przybylski D, Rost B, Baldi P. Improving the prediction of protein secondary structure in three and eight classes using recurrent neural networks and profiles. Proteins. 2002;47:228–235. doi: 10.1002/prot.10082. [DOI] [PubMed] [Google Scholar]
- 25.O’Neil KT, DeGrado WF. A thermodynamic scale for the helix-forming tendencies of the commonly occurring amino acids. Science. 1990;250:646–651. doi: 10.1126/science.2237415. [DOI] [PubMed] [Google Scholar]
- 26.Paul M, Patton GC, van der Donk WA. Mutants of the Zinc Ligands of Lacticin 481 Synthetase Retain Dehydration Activity but Have Impaired Cyclization Activity. Biochemistry. 2007;46:6268–6276. doi: 10.1021/bi7000104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.You YO, van der Donk WA. Mechanistic investigations of the dehydration reaction of lacticin 481 synthetase using site-directed mutagenesis. Biochemistry. 2007;46:5991–6000. doi: 10.1021/bi602663x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chatterjee C, Patton GC, Cooper L, Paul M, van der Donk WA. Engineering Dehydro Amino Acids and Thioethers into Peptides using Lacticin 481 Synthetase. Chem. Biol. 2006;13:1109–1117. doi: 10.1016/j.chembiol.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 29.Pitts KE, Summers AO. The roles of thiols in the bacterial organomercurial lyase (MerB) Biochemistry. 2002;41:10287–10296. doi: 10.1021/bi0259148. [DOI] [PubMed] [Google Scholar]
- 30.Miller LM, Chatterjee C, van der Donk WA, Kelleher NL. The Dehydration Activity of Lacticin 481 Synthetase is Highly Processive. J. Am. Chem. Soc. 2006;128:1420–1421. doi: 10.1021/ja057203d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dabard J, Bridonneau C, Phillipe C, Anglade P, Molle D, Nardi M, Ladire M, Girardin H, Marcille F, Gomez A, Fons M. Ruminococcin A, a new lantibiotic produced by a Ruminococcus gnavus strain isolated from human feces. Appl. Environ. Microbiol. 2001;67:4111–4118. doi: 10.1128/AEM.67.9.4111-4118.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Szekat C, Jack RW, Skutlarek D, Farber H, Bierbaum G. Construction of an expression system for site-directed mutagenesis of the lantibiotic mersacidin. Appl. Environ. Microbiol. 2003;69:3777–3783. doi: 10.1128/AEM.69.7.3777-3783.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cotter PD, Deegan LH, Lawton EM, Draper LA, O’Connor PM, Hill C, Ross RP. Complete alanine scanning of the two-component lantibiotic lacticin 3147: generating a blueprint for rational drug design. Mol. Microbiol. 2006;62:735–747. doi: 10.1111/j.1365-2958.2006.05398.x. [DOI] [PubMed] [Google Scholar]
- 34.Wiedemann I, Bottiger T, Bonelli RR, Schneider T, Sahl HG, Martinez B. Lipid II-based antimicrobial activity of the lantibiotic plantaricin C. Appl. Environ. Microbiol. 2006;72:2809–2814. doi: 10.1128/AEM.72.4.2809-2814.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van der Meer JR, Rollema HS, Siezen RJ, Beerthuyzen MM, Kuipers OP, de Vos WM. Influence of amino acid substitutions in the nisin leader peptide on biosynthesis and secretion of nisin by Lactococcus lactis. J. Biol. Chem. 1994;269:3555–3562. [PubMed] [Google Scholar]
- 36.Kluskens LD, Kuipers A, Rink R, de Boef E, Fekken S, Driessen AJ, Kuipers OP, Moll GN. Post-translational Modification of Therapeutic Peptides By NisB, the Dehydratase of the Lantibiotic Nisin. Biochemistry. 2005;44:12827–12834. doi: 10.1021/bi050805p. [DOI] [PubMed] [Google Scholar]
- 37.Izaguirre G, Hansen JN. Use of alkaline phosphatase as a reporter polypeptide to study the role of the subtilin leader segment and the SpaT transporter in the posttranslational modifications and secretion of subtilin in Bacillus subtilis 168. Appl. Environ. Microbiol. 1997;63:3965–3971. doi: 10.1128/aem.63.10.3965-3971.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuipers A, De Boef E, Rink R, Fekken S, Kluskens LD, Driessen AJ, Leenhouts K, Kuipers OP, Moll GN. NisT, the transporter of the lantibiotic nisin, can transport fully modified, dehydrated and unmodified prenisin and fusions of the leader peptide with non-lantibiotic peptides. J. Biol. Chem. 2004;279:22176–22182. doi: 10.1074/jbc.M312789200. [DOI] [PubMed] [Google Scholar]
- 39.Chen P, Qi FX, Novak J, Krull RE, Caufield PW. Effect of amino acid substitutions in conserved residues in the leader peptide on biosynthesis of the lantibiotic mutacin II. FEMS Microbiol. Lett. 2001;195:139–144. doi: 10.1111/j.1574-6968.2001.tb10511.x. [DOI] [PubMed] [Google Scholar]
- 40.Beck-Sickinger AG, Jung G. In: Nisin and Novel Lantibiotics. Jung G, Sahl H-G, editors. ESCOM; Leiden: 1991. pp. 218–230. [Google Scholar]
- 41.Jencks WP. Binding Energy, Specificity, and Enzymic Catalysis: The Circe Effect. Adv. Enzymol. 1975;43:219–410. doi: 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- 42.Ray WJ, Jr., Long JW, Owens JD. An analysis of the substrate-induced rate effect in the phosphoglucomutase system. Biochemistry. 1976;15:4006–4017. doi: 10.1021/bi00663a015. [DOI] [PubMed] [Google Scholar]
- 43.Amyes TL, Richard JP. Enzymatic catalysis of proton transfer at carbon: activation of triosephosphate isomerase by phosphite dianion. Biochemistry. 2007;46:5841–5854. doi: 10.1021/bi700409b. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wicki J, Schloegl J, Tarling CA, Withers SG. Recruitment of both uniform and differential binding energy in enzymatic catalysis: xylanases from families 10 and 11. Biochemistry. 2007;46:6996–7005. doi: 10.1021/bi700359e. [DOI] [PubMed] [Google Scholar]
- 45.Chakicherla A, Hansen JN. Role of the leader and structural regions of prelantibiotic peptides as assessed by expressing nisin-subtilin chimeras in Bacillus subtilis 168, and characterization of their physical, chemical, and antimicrobial properties. J. Biol. Chem. 1995;270:23533–23539. doi: 10.1074/jbc.270.40.23533. [DOI] [PubMed] [Google Scholar]
- 46.Kuipers OP, Rollema HS, de Vos WM, Siezen RJ. Biosynthesis and secretion of a precursor of nisin Z by Lactococcus lactis, directed by the leader peptide of the homologous lantibiotic subtilin from Bacillus subtilis. FEBS Lett. 1993;330:23–27. doi: 10.1016/0014-5793(93)80911-d. [DOI] [PubMed] [Google Scholar]
- 47.Nagao J, Aso Y, Shioya K, Nakayama J, Sonomoto K. Lantibiotic engineering: molecular characterization and exploitation of lantibiotic-synthesizing enzymes for peptide engineering. J. Mol. Microbiol. Biotechnol. 2007;13:235–242. doi: 10.1159/000104749. [DOI] [PubMed] [Google Scholar]
- 48.Asaduzzaman SM, Nagao J, Aso Y, Nakayama J, Sonomoto K. Lysine-oriented charges trigger the membrane binding and activity of nukacin ISK-1. Appl. Environ. Microbiol. 2006;72:6012–6017. doi: 10.1128/AEM.00678-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brötz H, Bierbaum G, Leopold K, Reynolds PE, Sahl HG. The lantibiotic mersacidin inhibits peptidoglycan synthesis by targeting lipid II. Antimicrob. Agents Chemother. 1998;42:154–160. doi: 10.1128/aac.42.1.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chatterjee C, Miller LM, Leung YL, Xie L, Yi M, Kelleher NL, van der Donk WA. Lacticin 481 Synthetase Phosphorylates its Substrate during Lantibiotic Production. J. Am. Chem. Soc. 2005;127:15332–15333. doi: 10.1021/ja0543043. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.