

Abstract
Background
Myocardin (Myocd) is a strong coactivator that binds the serum response factor (SRF) transcription factor over CArG elements embedded within smooth muscle cell (SMC) and cardiac muscle cyto-contractile genes. Here, we sought to ascertain whether Myocd-mediated gene expression confers a structural and physiological cardiac or SMC phenotype.
Methods and Results
Adenoviral-mediated expression of Myocd in the BC3H1 cell line induces cardiac and SMC genes while suppressing both skeletal muscle markers and cell growth. Immunofluorescence microscopy shows that SRF and a SMC-like cyto-contractile apparatus are elevated with Myocd overexpression. A short hairpin RNA to Srf impairs BC3H1 cyto-architecture; however, co-transduction with Myocd results in complete restoration of the cyto-architecture. Electron microscopic studies demonstrate a SMC ultrastructural phenotype with no evidence for cardiac sarcomerogenesis. Biochemical and time-lapsed videomicroscopy assays reveal clear evidence for Myocd-induced SMC-like contraction.
Conclusion
Myocd is sufficient for the establishment of a SMC-like contractile phenotype.
Condensed Abstract
Though Myocd activates cardiac and smooth muscle genes, which cell type is conferred physiologically is unclear. We show Myocd overexpression is sufficient for structural and functional attributes of the smooth muscle contractile phenotype. Such studies have implications for understanding and treating a variety of smooth muscle-associated diseases where the normal contractile phenotype is destabilized.
Keywords: smooth muscle, serum response factor, myocardin, contraction, knockdown
Differentiated vascular smooth muscle cells (SMC) have two important phenotypic characteristics. First, they replicate infrequently within the normal vessel wall 1. Second, they express a unique cyto-contractile gene program encoding a sub-proteome necessary for the principal function of these cells, namely contraction. Following physical or chemical injury to the vessel wall however, vascular SMC increase their replication rate and reduce the expression of many cyto-contractile genes including the smooth muscle isoforms of alpha actin, myosin heavy chain, and calponin 2. Such changes in SMC-restricted genes are thought to contribute to the pathogenesis of atherosclerosis, transplant arteriopathy, hypertension, bypass-graft failure, and the malignant phenotype 2,3. Although the majority of vascular diseases correlate with reductions in SMC contractile proteins, at least one example exists in which increases in SMC contractile proteins are associated with a disorder 4. Elucidating the intrinsic and extrinsic cues that specify one vascular SMC phenotype over another has therefore been the subject of intense study, with myriad proteins and signal transduction pathways identified 2.
A muscle cell’s differentiated phenotype is largely determined by the expression of both ubiquitous and cell-specific transcription factors (TF). The latter are exemplified by members of the MyoD family of basic helix-loop-helix TF which can convert a variety of cells, including cultured SMC, into skeletal muscle cells 5. Widely expressed TF such as serum response factor (SRF), are critical for normal skeletal muscle, cardiomyocyte, and SMC differentiated phenotypes 6. Ubiquitous TF such as SRF orchestrate specific programs of gene expression through combinatorial associations with coregulators, some of which display cell-specific patterns of expression 7. Among SRF coregulators, myocardin (Myocd) has emerged as one of central importance for the establishment of SMC identity. First cloned in a bioinformatic screen for unknown cardiac-specific genes, Myocd was shown initially to stimulate a battery of SRF target genes associated with cardiac muscle differentiation 8. A subsequent series of complementary reports demonstrated Myocd’s pivotal role in directing a genetic program of SMC differentiation 9–13. Levels of Myocd mRNA are reduced concurrently with SMC contractile genes upon SMC phenotypic change in culture 9 and in vivo following arterial injury 14,15. These results and the growing number of studies showing Myocd induces SMC contractile gene expression in non-muscle cell types suggest that, similar to MyoD in skeletal muscle, Myocd is a nodal point for the specification of SMC. Interestingly, Myocd can repress MyoD family members and re-direct cells fated for skeletal muscle to adopt a SMC-like phenotype, suggesting Myocd is dominant over the MyoD program of skeletal muscle cell differentiation 16.
Although a growing number of studies have documented Myocd’s ability to stimulate SMC-restricted gene expression, to what extent this program of gene expression recapitulates aspects of a structural and functional vascular SMC phenotype is unclear 17. Here, we show that despite the activation of both cardiac and SMC genes, Myocd confers an ultrastructural and contractile phenotype that most closely resembles that seen in SMC; no evidence for structural or physiological signs of cardiac muscle differentiation are manifest. These data demonstrate that Myocd overexpression is sufficient for structural and functional attributes of mature SMC, implicating it as a potential master regulator for the SMC contractile phenotype.
Materials and Methods
An expanded Materials and Methods section is available in the online data supplement.
Adenoviral Transduction
Adenoviral transductions were carried out as described 4 using multiplicity of infections (moi) from 10 to 100.
Microscopy
Bright field, immunofluorescence, and electron microscopic analyses of Myocd vs control expressing BC3H1 cells were performed without knowledge of the experimental condition.
Contractile Competence Assays
Contractile competence was carried out in control and Myocd-transduced cells (BC3H1 and human airway SMC) using a standard cell shortening assay and a 3-dimensional collagen gel system.
Results
Myocd inhibits cell growth and cyclin D1 expression
A hallmark of mature, differentiated vascular SMC is a low replication rate 1. To assess the effects of Myocd on BC3H1 cell proliferation, cells were transduced with either a control adenovirus or one carrying the short form of Myocd (amino acids 128–935). Overexpression of Myocd changes BC3H1 cell morphology from a polygonal to more spindle-like shape, reminiscent of mature SMC (Fig. 1A). Data in Figure 1A further demonstrate a reduction in cell density with Myocd over-expression. A temporal study of cell proliferation reveals decreases in Myocd-transduced cell number beginning 2 days following adenoviral transduction and persisting over 5 days (Fig. 1B). Consistent with growth attenuation, Myocd transduction leads to lower level expression of cyclin D1 (Fig. 1C). Transient promoter transfection studies suggest that Myocd directly represses cyclin D1 transcription (Fig. 1D). These results suggest that Myocd promotes a more SMC-like morphology and suppresses growth in BC3H1 cells, in part, through direct repression of a key cell cycle-associated gene.
Figure 1. Myocd inhibits proliferation of BC3H1 cells.
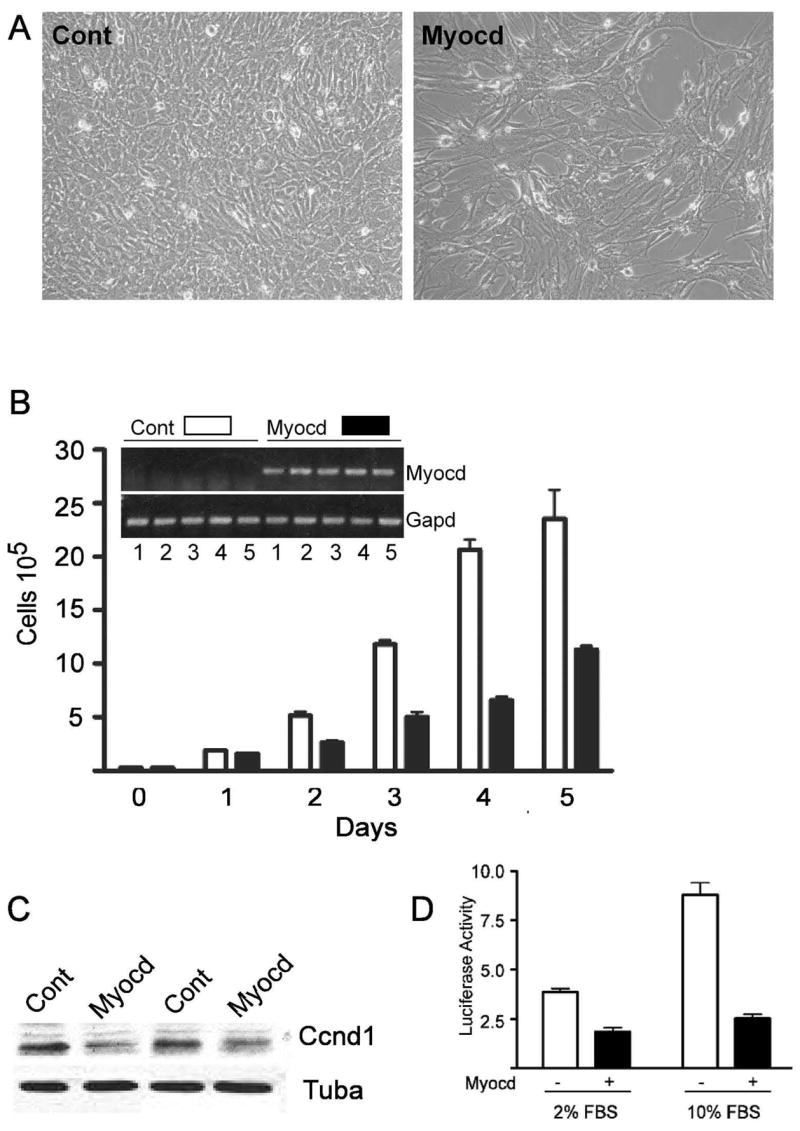
A. Phase contrast micrographs of control- and Myocd-transduced cells 5 days post-transduction. B. BC3H1 cell growth over 5 days following control (open bars) or Myocd (closed bars) adenoviral transduction. Bars here and below indicate standard error of the mean. Inset verifies daily Myocd mRNA expression. C. Cyclin D1 protein expression in BC3H1 cells ± Myocd. D. Luciferase activity (ratio of luciferase to renilla) in BC3H1 cells ± Myocd using a cyclin D1 luciferase reporter in low or high serum.
Reciprocal changes in SMC versus skeletal muscle markers in BC3H1 cells
BC3H1 cells can adopt a skeletal muscle fate upon serum withdrawal 18. Consistent with this concept, BC3H1 cells induced to differentiate to skeletal muscle with low serum exhibit robust expression of myogenin (Myog) mRNA, which encodes for one of four myogenic regulatory factors that orchestrate skeletal muscle cell differentiation (Fig. 2A, compare WT lanes). In contrast, endogenous Myocd mRNA levels are seen to diminish upon BC3H1 cell differentiation as are levels of the Myocd target gene, SM calponin (Cnn1, Fig. 2A, compare WT lanes). Interestingly, we observe a similar decrease in Srf mRNA with BC3H1 cell differentiation (Fig. 2B). To determine whether the changes in mouse Cnn1 expression apply to the human ortholog as well, we generated three BC3H1 cell lines stably-expressing the human CNN1 gene harbored within a bacterial artificial chromosome 19 (lanes labeled BAC in Fig. 2A). As with endogenous mouse Cnn1, human CNN1 mRNA levels are higher in growing versus differentiated BC3H1 cells (Fig. 2A), and immunocytochemistry confirms that similar changes apply to human CNN1 protein as well (Fig. 2C). Taken together, results reveal reciprocal expression profiles between key SMC and skeletal muscle markers in cells that transition from a SMC- to skeletal muscle cell-like phenotype in vitro.
Figure 2. Modulated muscle marker profile in growing versus differentiated BC3H1 cells.

A. RT-PCR analysis of endogenous mRNAs in growing versus differentiated (sarcomeric) BC3H1 cells. B. Northern blot of SRF in growing vs differentiated BC3H1 cells. Hprt is a house-keeping gene. C. Immunostaining of human CNN1 (in red) in growing (left) vs differentiated (right) BC3H1 cells carrying a BAC with human CNN1. Nuclei are stained blue with DAPI. Final magnifications are 400x.
Disparate effects of Myocd on the three muscle cell programs of differentiation
Myocd is a potent inducer of SMC and cardiac muscle genes that contain SRF-binding CArG elements 20. To determine the effects of Myocd on these muscle cell programs as well as skeletal myogenic regulatory factors, we transduced BC3H1 cells that had undergone initial differentiation to the skeletal muscle phenotype with control or Myocd adenovirus. Myocd induces endogenous SMC markers such as Acta2, Cnn1, Myh11, and Tagln (Fig. 3A) as well as human CNN1 mRNA (data not shown). Similarly, several cardiac muscle transcripts are elevated upon Myocd over-expression (Fig. 3A). Consistent with a recent report in C2C12 myoblasts 16, skeletal myogenic regulatory factors are attenuated with Myocd expression in BC3H1 cells (Fig. 3A). We see similar trends with respect to SMC and skeletal muscle proteins (Fig. 3B). These results show that Myocd induces SMC and cardiac muscle target genes, but represses skeletal muscle markers of differentiation in the BC3H1 cell line.
Figure 3. Myocd regulated muscle markers in BC3H1 cells.
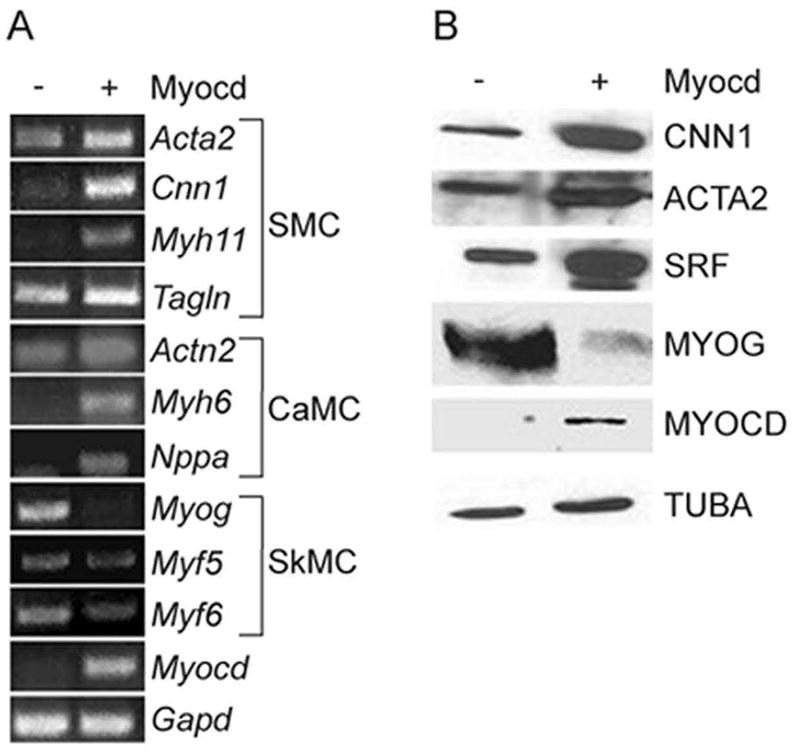
A. BC3H1 cell mRNA expression of smooth muscle cell (SMC), cardiac muscle cell (CaMC), and skeletal muscle cell (SkMC) markers ± 100 moi Myocd. B. Muscle marker proteins ± 100 moi Myocd. Alpha tubulin (TUBA) serves as an internal loading control.
Myocd induces structural attributes of a contractile SMC phenotype
A structural hallmark of mature, differentiated SMCis the presence of a rich array of myofilaments. Immunofluorescence microscopy shows Myocd-mediated increases in cyto-contractile fibers and SRF expression in BC3H1 (Fig. 4, panels a versus c). Further studies demonstrate that the increase in cyto-contractile fibers is attributable to elevations in SM α-actin filaments (Supplemental Fig. I, please see http://atvb.ahajournals.com). The cyto-contractile apparatus in BC3H1 cells is disrupted with a short hairpin RNA to Srf (Fig. 4, panels a versus b). Co-transduction with Myocd results in complete restoration of the cyto-contractile phenotype (Fig. 4, panels b versus d). Western blotting results indicate that co-transduction of Myocd and shSRF normalizes SRF levels (Supplemental Figure II) though we cannot rule out an SRF-independent effect of Myocd 21,22 in rescuing the cyto-contractile phenotype. Importantly, staining with an antibody to cardiac alpha actinin in Myocd-transduced BC3H1 cells does not show the typical periodic staining indicative of cardiac sarcomeres (Supplemental Figure II).
Figure 4. Myocd induces structural attributes of differentiated SMC.

BC3H1 cells transduced (100 moi) with control (a), shSRF (b), Myocd (c) or Myocd + shSRF (d) adenovirus for 72 hr and stained for SRF (green) or cyto-contractile fibers using phalloidin (red). All photomicrographs were taken at same exposure time.
To probe the structure of Myocd-transduced cells deeper, we performed transmission electron microscopy. Mature SMC of the adult mouse aorta show characteristic myofilaments throughout the cytosol, punctuated with focal densities (Fig. 5A). Control-transduced BC3H1 cells show little, if any, indication of myofilament arrays (Fig. 5B). Myocd-transduced BC3H1 cells, however, exhibit bands of myofilaments that appear similar to those in mature vascular SMC in vivo (Fig. 5C and Supplemental Figure III). In agreement with the cardiac alpha actinin staining, we found no evidence for cardiac myofilaments organized as repeating sarcomeres. A blind, quantitative analysis of more than 80 cells each from control- and Myocd-transduced cultures reveals, respectively, 2.3% and 58.8% cells exhibiting ultrastructural evidence of smooth myofilaments (Fig. 5D). These results establish Myocd as a mediator of the SMC ultrastructure phenotype.
Figure 5. Myocd induces SMC-like myofilaments in BC3H1 cells.
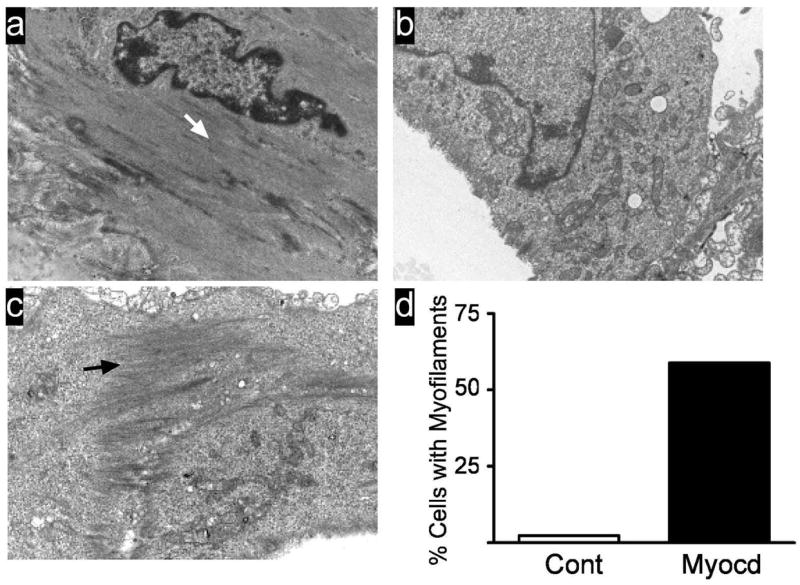
A. Ultrastructure of in vivo mouse aortic SMC showing myofilament array (white arrow). B. BC3H1 cells transduced with control adenovirus. C. BC3H1 cells transduced with Myocd. Note bundle of smooth myofilaments (black arrow). Magnifications, 15,000x . D. Quantitative measure of smooth myofilaments. Data represent semi-quantitative scoring (see Supplemental Methods at http://atvb.ahajournals.com) of more than 80 cells from two independent experiments.
Myocd induces SMC-like contractile competence in two distinct cell types
The preceding structural data suggest that Myocd facilitates bonafide SMC contractions in BC3H1 cells. To explore this novel concept, we first assessed expression of additional proteins necessary for SMC contractile competence. As shown in Figure 6A, Myocd dose-dependently increases the SM isoform of myosin light chain kinase (SM-MLCK), a known SRF target gene 23. We also see Myocd-induced increases in phosphorylated myosin light chain 20 (Fig. 6B), an essential mediator of SMC contractile activity 24. No spontaneous contractions are seen in Myocd-transduced BC3H1 cells. However, time-lapsed videomicroscopy shows slow, SMC-like contractions in Myocd-transduced BC3H1 cells stimulated with 75 mM KCl (Fig. 6C and Supplemental Movies). Quantitative measures of percent cell shortening indicate that Myocd-transduced cells display >3-fold increases over control cells (Fig. 6D). Human bronchial SMC transduced with Myocd also show a dramatic contractile response following histamine stimulation, suggesting Myocd evokes contractile competence in visceral SMC with inherent deficits in contractile activity in vitro (Supplemental Figure IV). Taken in aggregate, the results of this report support the notion that Myocd is sufficient for a SMC-like contractile phenotype.
Figure 6. Myocd-induces SMC-like contraction.
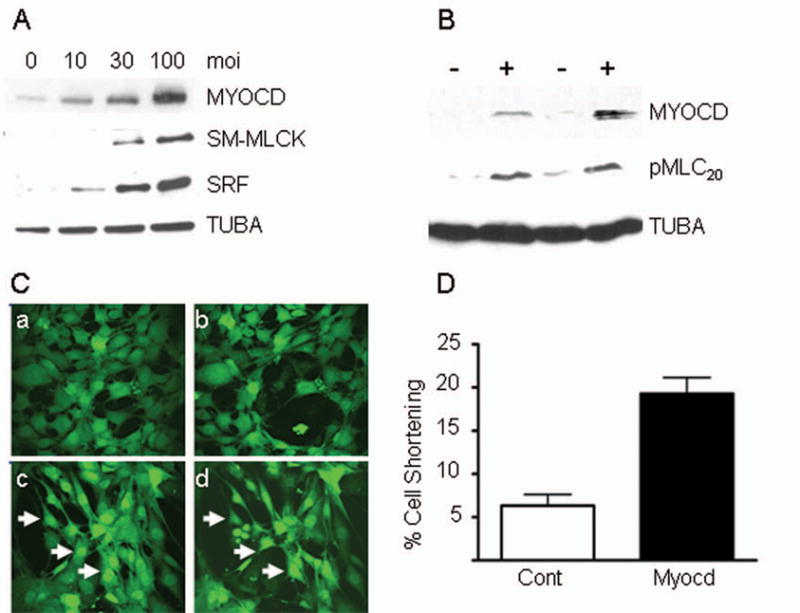
A. Expression of SM proteins in BC3H1 ± increasing amounts of Myocd adenovirus. B. Expression of pMLC20 in BC3H1 ± Myocd. C. Control-transduced BC3H1 cells (a, b) and Myocd-transduced cells (c, d) before (a, c) and after (b, d) 8 minute stimulation with 75 mM KCl. Arrows point to cells exhibiting changes in cell size. D. Quantitative measureof contractility in contro l- vs Myocd-transduced BC3H1 cells.
Discussion
Designating a transcription factor a master regulator of differentiation implies an intrinsic ability of the factor to auto-regulate its expression while eliciting biochemical and physiological attributes of the differentiated cell. MyoD was among the first master regulators of differentiation defined by virtue of its capacity to induce contractile proteins unique to skeletal muscle, organizing proteins into repeating sarcomeres, and eliciting contractile activity 25 as well as auto-regulating its expression 26. Might we similarly consider Myocd a master regulator of SMC differentiation? Clearly, Myocd can activate SMC genes in a variety of cell contexts, and there is some evidence for Myocd auto-regulating its own expression 27. However, until now, nothing was known about Myocd’s ability to orchestrate both ultrastructural and physiological attributes of the SMC differentiated phenotype. Here, we show expected changes in SMC, cardiac muscle, and skeletal muscle marker expression upon Myocd over-expression in BC3H1 cells. Despite the activation of some cardiac-restricted genes, Myocd stimulates smooth myofilaments with no evidence for cardiac sarcomerogenesis. Consistent with ultrastructure studies, Myocd provokes SMC-like contractions in two distinct cell types that otherwise are weakly responsive to contractile agonists. We conclude, therefore, that while Myocd may not stimulate every SMC-associated gene 28, the ability to auto-regulate its own expression 27 combined with the structural and functional data reported here, support Myocd’s designation as a master regulator of the SMC differentiated phenotype.
Despite a literature replete with studies showing Myocd-mediated SMC contractile gene/protein expression, little is known as to this cofactor’s ability to mediate contractile competence. Wamhoff and colleagues showed that voltage-gated calcium channel activation stimulated Myocd mRNA expression in rat SMC 29. The same study used embryoid bodies to show spontaneous SMC-like contractions were dependent upon voltage-gated channel activity. Husain and colleagues recently demonstrated defective Myocd expression and reduced SMC-like contractions in embryoid bodies null for the c-Myb transcription factor 30. It will be important to determine whether this model of SMC contractile competence is dependent upon Myocd expression and whether Myocd is a direct target of c-Myb. Moreover, effects of Myocd modulation on SMC-restricted ion channel expression and activity should be assessed to gain further insight into Myocd’s role in mediating SMC contractile competence. While the present work was underway, parallel studies uncovered Myocd as a marker for Alzheimer’s angiopathy, and gain-of-function studies demonstrated increases in human cerebral SMC shortening 4. Thus, there may be a number of SMC-associated diseases where exaggerated Myocd expression influences disease progression (e.g., Alzheimer’s disease, asthma, intestinal pseudo-obstruction).
It is important to point out that because Myocd levels are low in cultured SMC where contractile competence is rarely seen and most SMC markers are severely down-regulated, we were unable to address effects of loss of Myocd on SMC ultrastructure and contractile activity. However, ultrastructural analysis of the mouse ductus arteriosis where Myocd is conditionally ablated, reveals loss in SMC myofilaments and an abundance of rough endoplasmic reticulum indicative of a SMC synthetic phenotype 31. These results are congruent with a Myocd pan-knockout study where aortic SM alpha actin expression was essentially absent in day 10.5 embryos 13. In both cases, loss in SMC Myocd is apparently uncompensated for by the myocardin-related transcription factors 32.
The results of this study and others clearly show that cardiac and SMC genes are co-activated with ectopic Myocd expression. The initial report of Myocd 8 proposed it playing a critical role in cardiac muscle differentiation. Subsequent studies in Xenopus showed ectopic Myocd to activate cardiac muscle (and SMC) genes 33. Importantly, the latter article as well as a more recent study where Myocd was ectopically expressed in human cardiac fibroblasts 34, found no evidence for striated structures or spontaneous contractions that would support a structural and functional cardiac state. Similarly, we have never seen any evidence for cardiac sarcomerogenesis in cells over-expressing Myocd. The absence of cardiac sarcomerogenesis with Myocd overexpression may not be surprising given the sarcomere’s highly organized pattern of construction involving dozens of proteins 35. Nevertheless, there is some evidence for Myocd-mediated cardiac channel induction and the restoration of cardiac electrical conduction in vitro 34 suggesting some contexts exist where the milieu and signaling input facilitates the coexistence of both cardiac and SMC markers of differentiation. Indeed, a number of SMC restricted genes are known to be expressed in early cardiac muscle 36 and some of these are redeployed during pathological remodeling of cardiac muscle where Myocd levels are also elevated 37,38.
One of the hallmarks of a mature, differentiated SMC phenotype is growth cessation. We first reported that Myocd could reduce cell growth 9; however, the mechanisms underlying such growth attenuation were not addressed. Here, we provide evidence for Myocd directly repressing cyclin D1 promoter activity. Down-regulated Myocd and SMC contractile markers has been associated with human malignant transformation 3,39. Thus, it is tempting to speculate that in addition to activating genes involved in SMC contraction, Myocd represses cell cycle activity thereby contributing to the quiescent phenotype of differentiated SMC in the vessel wall and perhaps other cell types as well. In this context, Myocd represses gene expression in skeletal muscle 16, and a more recent study 40 demonstrated Myocd-mediated inhibition of NF-κB/p65 transcriptional activity and cell cycle protein levels in SMC. Myocd therefore appears to be a multi-functional protein with transcriptional coactivator and repressor activities.
In summary, we demonstrate that while ectopic Myocd expression induces both cardiac and SMC genes, structural and physiological data support its role in directing a SMC-like contractile phenotype. In no instance have we ever observed evidence for the manifestation of cardiac muscle structure or function. The results of the present study therefore support Myocd’s designation as a master regulator of SMC differentiation. The fact that Myocd levels decrease in parallel with contractile genes both in vitro and in vivo following vascular injury implies that the nearly 40 year-old phenomenon of SMC phenotypic modulation 41, likely stems from defective Myocd expression. A critical goal for future research will be elucidating the transcriptional regulation of Myocd under normal and pathological conditions.
Supplementary Material
Acknowledgments
We thank Dr. Burns C. Blaxall for the generous gift of neonatal cardiomyocytes.
Sources of Funding This work was supported by grants from the National Institutes of Health (HL62572 and AG026950 to JMM; AG023084 to BVZ; and HL077726 to WTG) and the American Heart Association (0625938T to XL).
Footnotes
Disclosures The authors have no disclosures.
This PDF receipt will only be used as the basis for generating PubMed Central (PMC) documents. PMC documents will be made available for review after conversion (approx. 2-3 weeks time). Any corrections that need to be made will be done at that time. No materials will be released to PMC without the approval of an author. Only the PMC documents will appear on PubMed Central -- this PDF Receipt will not appear on PubMed Central.
Bibliography
- 1.Spaet TH, Lejnieks I. Mitotic activity of rabbit blood vessels. Proc Soc Exp Biol Med. 1967;125:1197–1201. doi: 10.3181/00379727-125-32312. [DOI] [PubMed] [Google Scholar]
- 2.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 3.Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003;33:49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- 4.Chow N, Bell RD, Deane R, Streb JW, Chen J, Brooks A, Van Nostrand W, Miano JM, Zlokovic BV. Serum response factor and myocardin mediate arterial hypercontractility and cerebral blood flow dysregulation in Alzheimer’s phenotype. Proc Natl Acad Sci, USA. 2007;104:823–828. doi: 10.1073/pnas.0608251104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weintraub H, Tapscott SJ, Davis RL, Thayer MJ, Adam MA, Lassar AB, Miller AD. Activation of muscle-specific genes in pigment, nerve, fat, liver, and fibroblast cell lines by forced expression of MyoD. Proc Natl Acad Sci, USA. 1989;86:5434–5438. doi: 10.1073/pnas.86.14.5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miano JM, Long X, Fujiwara K. Serum response factor: master regulator of the actin cytoskeleton and contractile apparatus. Am J Physiol Cell Physiol. 2007;292:C70–C81. doi: 10.1152/ajpcell.00386.2006. [DOI] [PubMed] [Google Scholar]
- 7.Spiegelman BM, Heinrich R. Biological control through regulated transcriptional coactivators. Cell. 2004;119:157–167. doi: 10.1016/j.cell.2004.09.037. [DOI] [PubMed] [Google Scholar]
- 8.Wang D-Z, Chang PS, Wang Z, Sutherland L, Richardson JA, Small E, Krieg PA, Olson EN. Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell. 2001;105:851–862. doi: 10.1016/s0092-8674(01)00404-4. [DOI] [PubMed] [Google Scholar]
- 9.Chen J, Kitchen CM, Streb JW, Miano JM. Myocardin: a component of a molecular switch for smooth muscle differentiation. J Mol Cell Cardiol. 2002;34:1345–1356. doi: 10.1006/jmcc.2002.2086. [DOI] [PubMed] [Google Scholar]
- 10.Du K, Ip HS, Li J, Chen M, Dandre F, Yu W, Lu MM, Owens GK, Parmacek MS. Myocardin is a critical serum response factor cofactor in the transcriptional program regulating smooth muscle cell differentiation. Mol Cell Biol. 2003;23:2425–2437. doi: 10.1128/MCB.23.7.2425-2437.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoshida T, Sinha S, Dandre F, Wamhoff BR, Hoofnagle MH, Kremer BE, Wang D-Z, Olson EN, Owens GK. Myocardin is a key regulator of CArG-dependent transcription of multiple smooth muscle marker genes. Circ Res. 2003;92:856–864. doi: 10.1161/01.RES.0000068405.49081.09. [DOI] [PubMed] [Google Scholar]
- 12.Wang Z, Wang D-Z, Pipes GCT, Olson EN. Myocardin is a master regulator of smooth muscle gene expression. Proc Natl Acad Sci, USA. 2003;100:7129–7134. doi: 10.1073/pnas.1232341100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li S, Wang D-Z, Richardson JA, Olson EN. The serum response factor coactivator myocardin is required for vascular smooth muscle development. Proc Natl Acad Sci, USA. 2003;100:9366–9370. doi: 10.1073/pnas.1233635100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hendrix JA, Wamhoff BR, McDonald OG, Sinha S, Yoshida T, Owens GK. 5′ CArG degeneracy in smooth muscle α-actin is required for injury-induced gene suppression in vivo. J Clin Invest. 2005;115:418–427. doi: 10.1172/JCI22648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tharp DL, Wamhoff BR, Turk JR, Bowles DK. Upregulation of intermediate-conductance Ca2+-activated K+ channel (IKCa1) mediates phenotypic modulation of coronary smooth muscle. Am J Physiol Heart Circ Physiol. 2006;291:H2493–H2503. doi: 10.1152/ajpheart.01254.2005. [DOI] [PubMed] [Google Scholar]
- 16.Long X, Creemers EE, Wang D-Z, Olson EN, Miano JM. Myocardin is a bifunctional switch for smooth versus skeletal muscle differentiation. Proc Natl Acad Sci, USA. 2007;104:16570–16575. doi: 10.1073/pnas.0708253104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miano JM. Channeling to myocardin. Circ Res. 2004;95:340–342. doi: 10.1161/01.RES.0000140893.16465.2d. [DOI] [PubMed] [Google Scholar]
- 18.Olson EN, Caldwell KL, Gordon JI, Glaser L. Regulation of creatine phosphokinase expression during differentiation of BC3H1 cells. J Biol Chem. 1983;258:2644–2652. [PubMed] [Google Scholar]
- 19.Miano JM, Kitchen CM, Chen J, Maltby KM, Kelly LA, Weiler H, Krahe R, Ashworth LK, Garcia E. Expression of human smooth muscle calponin in transgenic mice revealed with a bacterial artificial chromosome. Am J Physiol Heart Circ Physiol. 2002;282:H1793–H1803. doi: 10.1152/ajpheart.00875.2001. [DOI] [PubMed] [Google Scholar]
- 20.Pipes GCT, Creemers EE, Olson EN. The myocardin family of transcriptional coactivators: versatile regulators of cell growth, migration, and myogenesis. Genes Dev. 2006;20:1545–1556. doi: 10.1101/gad.1428006. [DOI] [PubMed] [Google Scholar]
- 21.Qiu P, Ritchie RP, Fu Z, Cao D, Cumming J, Miano JM, Wang D-Z, Li HJ, Li L. Myocardin enhances Smad3-mediated transforming growth factor-β1 signaling in a CArG box-independent manner: Smad-binding element is an important cis element for SM22α transcription in vivo. Circ Res. 2005;97:983–991. doi: 10.1161/01.RES.0000190604.90049.71. [DOI] [PubMed] [Google Scholar]
- 22.Lockman K, Taylor JM, Mack CP. The histone demethylase, jmjd1a, interacts with the myocardin factors to regulate SMC differentiation marker gene expression. Circ Res. 2008;101:e115–e123. doi: 10.1161/CIRCRESAHA.107.164178. [DOI] [PubMed] [Google Scholar]
- 23.Yin F, Hoggatt AM, Zhou J, Herring BP. The 130kDa smooth muscle myosin light chain kinase is transcribed from a CArG-dependent, internal promoter within the mouse MYLK gene. Am J Physiol Cell Physiol. 2006;290:C1599–C1609. doi: 10.1152/ajpcell.00289.2005. [DOI] [PubMed] [Google Scholar]
- 24.Somlyo AP, Somlyo AV. Signal transduction and regulation in smooth muscle. Nature. 1994;372:231–236. doi: 10.1038/372231a0. [DOI] [PubMed] [Google Scholar]
- 25.Lattanzi L, Salvatori G, Coletta M, Sonnino C, Cusella De Angelis MG, Gioglio L, Murry CE, Kelly R, Ferrari G, Molinaro M, Crescenzi M, Mavilio F, Cossu G. High efficiency myogenic conversion of human fibroblasts by adenoviral vector-mediated MyoD gene transfer. An alternative strategy for ex vivo gene therapy of primary myopathies. J Clin Invest. 1998;101:2119–2128. doi: 10.1172/JCI1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thayer MJ, Tapscott SJ, Davis RL, Wright WE, Lassar AB, Weintraub H. Positive autoregulation of the myogenic determination gene MyoD1. Cell. 1989;58:241–248. doi: 10.1016/0092-8674(89)90838-6. [DOI] [PubMed] [Google Scholar]
- 27.Creemers EE, Sutherland LB, McNally J, Richardson JA, Olson EN. Myocardin is a direct transcriptional target of Mef2, Tead and Foxo proteins during cardiovascular development. Development. 2006;133:4245–4256. doi: 10.1242/dev.02610. [DOI] [PubMed] [Google Scholar]
- 28.Yoshida T, Kawai-Kowase K, Owens GK. Forced expression of myocardin is not sufficient for induction of smooth muscle differentiation in multipotential cells. Arterioscler Thromb Vasc Biol. 2004;24:1596–1601. doi: 10.1161/01.ATV.0000137190.63214.c5. [DOI] [PubMed] [Google Scholar]
- 29.Wamhoff BR, Bowles DK, McDonald OG, Sinha S, Somlyo AP, Somlyo AV, Owens GK. L-type voltage-gated Ca2+ channels modulate expression of smooth muscle differentiation marker genes via a Rho kinase/myocardin/SRF-dependent mechanism. Circ Res. 2004;95:406–414. doi: 10.1161/01.RES.0000138582.36921.9e. [DOI] [PubMed] [Google Scholar]
- 30.Kolodziejska KM, Ashraf HN, Nagy A, Bacon A, Frampton J, Xin H-B, Kotlikoff MI, Husain M. c-Myb-dependent smooth muscle cell differentiation. Circ Res. 2008;102:554–561. doi: 10.1161/CIRCRESAHA.105.162628. [DOI] [PubMed] [Google Scholar]
- 31.Huang J, Cheng L, Li J, Chen M, Zhou D, Lu MM, Proweller A, Epstein JA, Parmacek MS. Myocardin regulates expression of contractile genes in smooth muscle cells and is required for closure of the ductus arteriosus in mice. J Clin Invest. 2008;118:515–525. doi: 10.1172/JCI33304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang DZ, Li S, Hockemeyer D, Sutherland L, Wang Z, Schratt G, Richardson JA, Nordheim A, Olson EN. Potentiation of serum response factor activity by a family of myocardin- related transcription factors. Proc Natl Acad Sci, USA. 2002;99:14855–14860. doi: 10.1073/pnas.222561499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Small EM, Warkman AS, Wang D-Z, Sutherland LB, Olson EN, Krieg PA. Myocardin is sufficient and necessary for cardiac gene expression in Xenopus. Development. 2005;132:987–997. doi: 10.1242/dev.01684. [DOI] [PubMed] [Google Scholar]
- 34.van Tuyn J, Pijnappels DA, de Vries AAF, de Vries I, van der Velde-van Dijke I, Knaan-Shanzer S, van der Laarse A, Schalij MJ, Atsma DE. Fibroblasts from human postmyocardial infarction scars acquire properties of cardiomyocytes after transduction with a recombinant myocardin gene. FASEB J. 2007;21:3369–3379. doi: 10.1096/fj.07-8211com. [DOI] [PubMed] [Google Scholar]
- 35.Hoshijima M. Mechanical stress-strain sensors embedded in cardiac cytoskeleton: Z disk, titin, and associated structures. Am J Physiol Heart Circ Physiol. 2006;290:H1313–H1325. doi: 10.1152/ajpheart.00816.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miano JM. Serum response factor: toggling between disparate programs of gene expression. J Mol Cell Cardiol. 2003;35:577–593. doi: 10.1016/s0022-2828(03)00110-x. [DOI] [PubMed] [Google Scholar]
- 37.Black FM, Packer SE, Parker TG, Michael LH, Roberts R, Schwartz RJ, Schneider MD. The vascular smooth muscle α-actin gene is reactivated during cardiac hypertrophy provoked by load. J Clin Invest. 1991;88:1581–1588. doi: 10.1172/JCI115470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Torrado M, López E, Centeno A, Medrano C, Castro-Beiras A, Mikhailov AT. Myocardin mRNA is augmented in the failing myocardium: expression profiling in the porcine model and human dilated cardiomyopathy. J Mol Med. 2003;81:566–577. doi: 10.1007/s00109-003-0470-7. [DOI] [PubMed] [Google Scholar]
- 39.Milyavsky M, Shats I, Cholostoy A, Brosh R, Buganim Y, Weisz L, Kogan I, Cohen M, Shatz M, Madar S, Kalo E, Goldfinger N, Yuan J, Ron S, Mackenzie K, Eden A, Rotter V. Inactivation of myocardin and p16 during malignant transformation contributes to a differentiation defect. Cancer Cell. 2007;11:133–146. doi: 10.1016/j.ccr.2006.11.022. [DOI] [PubMed] [Google Scholar]
- 40.Tang R, Zheng X-L, Callis TE, Stansfield WE, He J, Baldwin AS, Wang D-Z, Selzman CH. Myocardin inhibits cellular proliferation by inhibiting NF-κB(p65)-dependent cell cycle progression. Proc Natl Acad Sci, USA. 2008;105:3362–3367. doi: 10.1073/pnas.0705842105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fritz KE, Jarmolych J, Daoud AS. Association of DNA synthesis and apparent dedifferentiation of aortic smooth muscle cells in vitro. Exp Mol Pathol. 1970;12:354–362. doi: 10.1016/0014-4800(70)90066-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.