

Abstract
Scurfy mice have a deletion in the forkhead domain of Foxp3, fail to develop thymic-derived Foxp3+ regulatory T cells (nTreg), and develop a fatal lymphoproliferative syndrome with multi-organ inflammation. Transfer of thymic-derived Foxp3+ nTreg into neonatal Scurfy mice prevents the development of disease. Stimulation of conventional CD4+Foxp3− via the TCR in the presence of TGFβ and IL-2 induces the expression of Foxp3 and an anergic/suppressive phenotype. To determine whether the TGFβ-induced Treg (iTR) were capable of suppressing disease in the Scurfy mouse, we reconstituted newborn Scurfy mice with polyclonal iTR. iTR-treated Scurfy mice do not show any signs of disease and have drastically reduced cell numbers in peripheral lymph nodes and spleen in comparison to untreated Scurfy controls. The iTR retained their expression of FoxP3 in vivo for 21 days, migrated into the skin, and prevented the development of inflammation in skin, liver and lung. Thus, TGFβ-differentiated Foxp3+ Treg appear to possess all of the functional properties of thymic-derived nTreg and represent a potent population for the cellular immunotherapy of autoimmune and inflammatory diseases.
Keywords: TGFβ, regulatory T cells, Scurfy mice, autoimmune disease
Introduction
FoxP3+ naturally occurring regulatory T cells are potent suppressors of autoreactive CD4+ T cells which escape negative selection in the thymus [1–3]. The forkhead transcription factor FoxP3 is indispensable for the differentiation, maintenance, and function of nTreg [4]. Mutations in FoxP3 result in the fatal “Immunedysregulation, Polyendocrinopathy, Enteropathy, X-linked syndrome” (IPEX) autoimmune syndrome [5] in man, while a 2-bp insertion in the FoxP3 gene leads to production of a truncated non-functional protein, resulting in the lymphoproliferative syndrome of the Scurfy mouse [6, 7]. Scurfy mice suffer from an aggressive CD4+ and CD8+ T cell-mediated autoimmune disease with multi-organ inflammation and die at the age of 3 to 5 weeks. The nature of the cell type in the Scurfy mouse responsible for disease induction is unknown. It has been proposed that due to the lack of FoxP3, nTreg may have altered their program from protective to autoaggressive. Alternatively, disease in the Scurfy mice may be induced by the normal repertoire of autoreactive T cells secondary to their uncontrolled activation in the absence of functional nTreg. As ablation of FoxP3+ cells in adult and young mice rapidly results in a scurfy-like phenotype, it is likely that uncontrolled autoreactive effector T cells are responsible for disease in the Scurfy mouse [8, 9].
Adoptive transfer of nTreg into neonatal Scurfy mice prevents the development of disease [4] and transfer of both polyclonal and antigen-specific nTreg have proven effective in the prevention and treatment of autoimmune disease in animals models [10–12]. Cellular biotherapy with nTreg represents a promising approach for the treatment of autoimmune disease in man. Major efforts have been made to expand both polyclonal and antigen-specific nTreg in vitro. Nevertheless, isolation/identification of antigen-specific nTreg and expansion of substantial cell numbers of nTreg without outgrowth of contaminating effector T cells during long-term culture periods still remain challenging. An alternative approach for the potential therapeutic use of Treg is based on the demonstration that TCR stimulation of conventional CD4+ T cells in the presence of TGFβ and IL-2 results in the induction of FoxP3 expression [13]. These TGFβ-induced FoxP3+ T cells are identical to nTreg in that they are anergic and suppressive in vitro. Both polyclonal and antigen-specific iTR have demonstrated anti-inflammatory potential in animal models of organ-specific autoimmune disease, such as EAE, autoimmune diabetes, gastritis and colitis [10–12]. However, some studies have suggested that iTR lack regulatory function even though they express FoxP3 [14] and that the iTR rapidly lose Foxp3 expression following transfer in vivo [15]. The use of polyclonal iTR is further complicated by potential differences between nTreg and iTR both in their homing capabilities and in their T cell repertoires. nTreg are thought to express a TCR repertoire with a bias for self, and several studies [16, 17] have suggested that there is limited (15–20%) overlap between the nTreg TCR repertoire and the TCR repertoire of conventional T cells. Thus, iTR derived from a polyclonal population of CD4+CD25− T cells may be restricted in their ability to effectively suppress autoimmune disease secondary to a lack of T cells with anti-self TCRs.
As both antigen-specific and polyclonal iTR can be easily generated in large numbers in vitro, they represent a potent population for the cellular immunotherapy of autoimmune and inflammatory diseases. Therefore we think that it is important to evaluate their potential to suppress autoreactive effector T cells in vivo in a mouse model of severe muti-organ autoimmune disease, like the Scurfy mouse model.
In the present study we have addressed whether polyclonal iTR derived from a healthy adult mouse can protect Scurfy mice from the development of disease. We demonstrate that neonatal transfer of iTR, derived from polyclonal CD4+CD25− T cells, prevents development of disease in Scurfy mice. The transferred iTR could be identified 21 days after adoptive transfer in the peripheral lymph nodes and skin and maintain high expression levels of FoxP3.
Results
Induction of FoxP3+ T cells by TGFβ
CD4+CD25− T cells from WT C57BL/6 mice were FACS-sorted and cultured on plate-bound anti-CD3 and anti-CD28 in the presence of TGFβ and IL-2. On days 3 and 7, the cells were analyzed for FoxP3 expression by intracellular staining. More than 90% of the cells were FoxP3+ on day 3 and the high level of FoxP3 expression was maintained until day 7 (Fig. 1A). As a control for the FoxP3+ iTR, we stimulated C57BL/6 CD4+CD25− T cells under the same conditions, but in the presence of anti-TGFβ instead of TGFβ. The expanded cell population contained only 2–3% Foxp3+ T cells (Fig. 1A, lower panel). Similar results were observed when we activated sorted CD4+GFP− T cells from the Foxp3-GFP ‘knock-in’ mice ruling out the possibility that the high percentage of FoxP3+ T cells generated in the presence of TGFβ is due to expansion of a very small number of residual FoxP3+ T cells in the starting population of sorted CD4+CD25− T cells (Fig. 1B).
Figure 1.

FoxP3 expression of TGFβ-induced regulatory T cells. (A) CD4+ CD25− T cells were sorted from single cell suspensions and stimulated in vitro in the presence of rhTGFβ (5ng/ml) or anti-TGFβ (10μg/ml). Directly postsort and at day 3 and 7 the cells were stained for FoxP3 (black open histogram) and plotted against isotype control (grey filled histogram). (B) GFP− cells were sorted from single cell suspensions of Foxp3-GFP ‘knock-in’ mice and stimulated in vitro in the presence of rhTGFβ (5ng/ml). GFP was measured after sorting and on day 7. Numbers represent the percentage of cells in the gate. One representative experiment of at least 10 experiments with similar results is shown.
Neonatal transfer of FoxP3+ iTR prevents the development of disease in Scurfy mice
In vitro differentiated FoxP3+ iTR or FoxP3− cells (10 × 106) were injected i.p. into male Scurfy mice on day 1 or 2 of life. By day 12 to 15 of life, untreated Scurfy mice and Scurfy mice that received the FoxP3− cells showed overt signs of disease such as failure to thrive, hunched posture, scruffy fur, and scaly erythematous skin on the ears and tails, whereas the Scurfy mice that received the iTR developed normally. By day 15 to 20 of life, the untreated Scurfy mice and Scurfy mice that received Foxp3− cells showed signs of terminal autoimmune disease and became moribund between day 20 and 30 of life. Therefore, we chose day 21 after birth for all further analyses.
Macroscopic evaluation on day 21 after transfer did not reveal any clinical signs of disease in the iTR-treated Scurfy mice. The size of the body and the skin of the ears, tail and eyelids was comparable to untreated WT controls, whereas Scurfy mice treated with FoxP3− control cells showed the same phenotype as the untreated Scurfy control (Fig. 2A). A closer view of the tail skin revealed no visible lesions in the Scurfy mouse, if iTR had been transferred (Fig. 2B). Transfer of 5 × 106 iTR into Scurfy mice also prevented disease as measured by macroscopic analysis, whereas transfer of 1 × 106 iTR did not (data not shown). Histologic evaluation of various organs (Fig. 2C) demonstrated that untreated Scurfy mice had severe tissue inflammation consisting of massive lymphocytic and mononuclear infiltration in the liver parenchyma, lung interstitium, and the dermis of the skin. In contrast, no lesions were observed in either the treated WT or Scurfy mice. Importantly WT mice did not show any lesions by macroscopic or microscopic examination at day 21 after transfer of iTR, ruling out the concern that the iTR themselves might cause disease.
Figure 2.

Transfer of TGFβ-induced iTR prevents the development of disease in Scurfy mice. (A) FoxP3+ iTR (10 × 106) or FoxP3− control cells (ctrl) were transferred i.p. into newborn Scurfy or WT mice on day 1 of life. Untreated Scurfy mice and male WT littermates served as controls. Macroscopic appearance 21 days after transfer. (B) Closer view of the skin of the tail. (C) H&E staining of representative sections of ear, lung and liver (10X) 21days after transfer. One representative experiment of 3 experiments with similar results is shown.
Transfer of FoxP3+ iTR reduces lymphadenopathy in Scurfy mice
We next analyzed the cellularity of the inguinal lymph nodes and spleen of either WT or Scurfy mice 21 days after transfer of iTR. Scurfy mice that received 10 × 106 iTR showed significantly reduced cell numbers in the inguinal lymph node and in the spleen (Fig. 3), while transfer of 5 × 106 iTR resulted in only slight reduction in cell numbers which was not statistically significant (data not shown).
Figure 3.
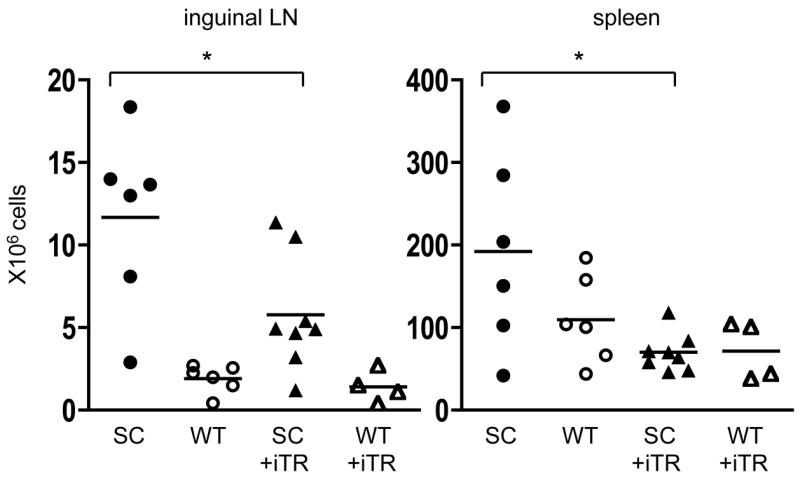
Transfer of TGFβ-induced iTR reduces the cell number in the peripheral lymph nodes and spleen of Scurfy mice. Cellularity of the inguinal lymph nodes and spleen 21 days after transfer of 10 × 106 iTR into newborn Scurfy mice (▲) or male WT littermates (△). Untreated Scurfy mice (●) and male WT littermates (○) served as controls. Each symbol represents a single mouse from 3 different experiments; * p =0.0283 (left), * p = 0.0147 (right), unpaired Student’s t-test.
iTR preferentially expand in the peripheral lymph nodes and migrate into the skin
The use of the congenic markers CD45.1 and CD45.2 allowed us to distinguish the transferred iTR from the Scurfy or WT host cells 21 days after transfer. The percentage of transferred iTR in the LN was higher in the Scurfy animals than in the WT, indicating expansion or preferential survival of the iTR only in the sick animal (Fig. 4A). The absolute numbers of transferred iTR in the inguinal LN were also significantly higher in the Scurfy animals than in the WT (Fig. 4B). After transfer of CFSE-labeled iTR, a higher percentage of iTR diluted CFSE in the Scurfy mouse than in the WT animal indicating more expansion in the sick animal than in a non-inflammatory environment (Fig. 4C).
Figure 4.

TGFβ-induced iTR preferentially expand in the Scurfy host and migrate into the skin. (A) 21days after transfer of 10 × 106 CD45.1+ iTR, inguinal lymph node (LN) cells were analyzed by flow cytometry, gating on CD4+ T cells. Numbers represent the percentage of cells in the gate. (B) The absolute numbers of iTR in the inguinal LN were calculated by multiplying the percentages of CD45.1+ cells by the total cell number of each node, (n = 4), * p = 0.0286, Mann-Whitney test. (C) 7 days after transfer of CFSE-labeled CD45.1+ iTR, cells from the inguinal LN were analyzed by flow cytometry, gating on CD45.1+ cells. (D) 21days after transfer of 10 × 106 CD45.1+ iTR, cells from the skin of the ear were isolated and analyzed by flow cytometry.
To determine if iTR were also present in tissues, we isolated cells from the skin of the ear from WT and “rescued” Scurfy mice 21 days after transfer. In the WT animals, very few iTR were detectable in the skin, whereas in the treated Scurfy mice there was a substantial number of iTR present in the skin (Fig. 4D).
The transferred iTR maintain their FoxP3+ phenotype in vivo
The use of congenically-labeled iTR allowed us to analyze FoxP3-expression of the transferred iTR after 21 days in vivo. The iTR in the inguinal LN of the Scurfy mouse maintained a high expression level of FoxP3, whereas in the WT animal only 50% of the transferred cells were still FoxP3+ (Fig. 5A, upper panel). The iTR also maintained their level of FoxP3 in the skin of the Scurfy mouse, but not in the WT mouse (Fig. 5A, lower panel). Although the iTR prevented all manifestations of disease upon transfer to neonatal Scurfy mice, T cells isolated from treated Scurfy animals still expressed an activated phenotype as measured by the low expression of the activation marker CD62L suggesting that some level of T cell activation was present (Fig. 5B).
Figure 5.

The transferred iTR maintain their FoxP3+ phenotype for 21 in vivo. (A) 21 days after transfer of 10 × 106 CD45.1+ iTR, inguinal lymph node cells (upper panel) and the cells isolated from the skin of the ear (lower panel) were analyzed by flow cytometry. Gating on the transferred CD45.1+ cells, the black histogram shows the expression of FoxP3, the grey histogram represents the corresponding isotype control. Numbers represent the percentage of cells in the gate. (B) CD62L expression in the iLN of a WT mouse (left panel, black histogramm, the grey histogramm is the corresponding isotype control), of a untreated Scurfy control (middle panel) and in a Scurfy mouse 21 days after transfer of 10 × 106 CD45.1+ iTR (right panel), here gating on the host CD45.2+ cells.
iTR efficiently suppress disease after co-transfer of Scurfy T cells into RAG−/− mice
One problem with the design of these studies is that the iTR are only injected once at day 1 of life, while there is a constant thymic output of newly generated autoreactive T cells in the Scurfy mice. To correct for this imbalance of effector T cells and iTR, we developed a model in which male RAG−/− mice were reconstituted with 5 × 106 total lymph node and spleen cells from 7 day-old Scurfy mice alone or co-transferred with either 1 × 106 iTR or FoxP3− control cells. The RAG−/− mice 4 weeks after transfer of Scurfy cells alone or Scurfy cells and FoxP3− control cells exhibited skin inflammation with hair loss, marked erythematous lesions and scaly skin (data not shown). Histologic analysis of the skin, lung and liver showed severe lesions in recipients that were reconstituted with Scurfy cells only or co-transferred with FoxP3− control cells (Fig. 6A). In contrast, mice that received iTR did not show skin inflammation on macroscopic analysis (data not shown) and the histopathology scores in skin, lung and liver sections were significantly lower (Fig. 6A). The cellularity of the inguinal lymph node was also significantly reduced by co-transfer of iTR, but not by transfer of the FoxP3− control cells (Fig. 6B). The majority of the transferred iTR were still FoxP3+ at 4 weeks after transfer (data not shown). Most importantly, in the presence of iTR, the Scurfy T cells maintained higher levels of CD62L and CD45RB expression than Scurfy mice transferred with control cells (Fig. 6C).
Figure 6.
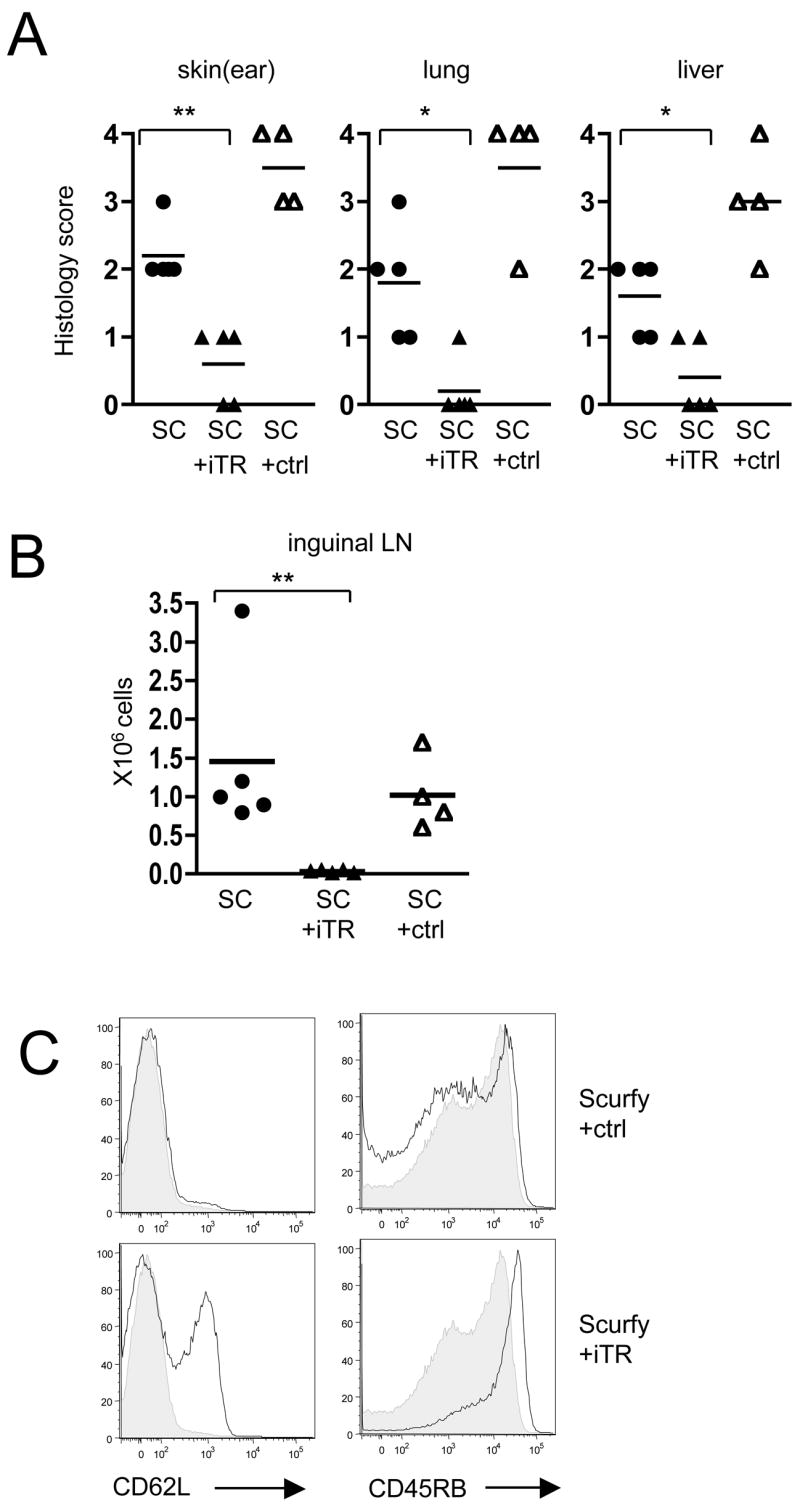
Cotransfer of TGFβ-induced iTR prevents activation of Scurfy effector T cells in RAG−/− mice. (A) Peripheral lymph node and spleen cells (5 × 106) from 7 day-old Scurfy mice (SC) were either transferred alone (●) or co-transferred with 1 × 106 CD45.1+ iTR (▲) or 1 × 106 CD45.1+ FoxP3− control cells (ctrl) (△) into male RAG−/− mice. 28 days after transfer the ears, lungs and the livers were evaluated histologically; ** p = 0.0079 (ear), * p = 0.0159 (lung), * p = 0.0317 (liver), Mann Whitney test. (B) Cellularity of the inguinal lymph nodes 28 days after transfer; ** p = 0.0079, Mann Whitney test. (C) Flow cytometric analysis of CD62L and CD45RB expression in the inguinal lymph node, gating on the CD45.2+ Scurfy cells. The grey histogram shows the expression levels of the CD45.2+ Scurfy cells transferred alone.
Discussion
In summary, our data indicate that TGFβ-iTR can suppress all the pathologic manifestations of the severe autoimmune disease that develops in Scurfy mice in both lymphoid sites and in tissues. When used in cell transfer studies with limiting numbers of cells from the Scurfy mice, they were even capable of significantly suppressing the earliest changes that occur during cell activation such as the modulation of CD62L and CD45RB expression. It should be noted that the potency of the iTR is significantly less than what has been reported with nTreg, where 0.5 × 106 nTreg prevented disease in Scurfy mice [4], while 5–10 × 106 iTR were needed in our studies. The high number of iTR injected to rescue Scurfy mice might in fact not represent the actual number of actively suppressing iTR. After their in vitro activation iTR are mostly CD62L low (data not shown) and accordingly only a certain percentage might be able to home to the lymph nodes. Moreover, the activation and expansion of iTR in vivo as shown by CFSE dilution might also be accompanied by cell death. The greater potency of the nTreg could be secondary to their superior ability to home to sites where inflammation is initiated or due to differences in the mechanisms of suppression used by different Treg populations. Nevertheless, the TCR repertoire of the iTR generated from normal adult mice appears to be sufficient to prevent autoimmune disease in the Scurfy mouse which is consistent with recent studies demonstrating that disease in the Scurfy mouse is not mediated by nTreg precursors that have been converted into effector cells. One important finding is that the iTR maintained high levels of FoxP3+ expression in the inflammatory environment, whereas the same cells when transferred to normal mice lost expression of FoxP3, suggesting that certain factors such as proinflammatory cytokines in the sick Scurfy mouse might act as survival or expansion signals for the iTR. The higher CFSE dilution of the iTR in the Scurfy mouse than in the WT animal supports this view.
Taken together, the results of these studies and others [10–12] strongly suggest that mouse polyclonal and autoantigen-specific iTR can be easily generated in large numbers in vitro and can exhibit potent inhibitory effects on the development of both systemic and organ-specific autoimmune diseases. However, in the human setting several issues still impair the use of iTR for the treatment of autoimmune disease in the clinic. It remains controversial whether one can generate human iTR [18] using protocols similar to the one that is highly effective in the mouse. It is may also be difficult to generate large numbers of iTR of sufficient purity needed for the treatment of organ-specific autoimmune diseases in man. Therefore more studies are necessary to further elucidate the mechanism and suppressive activity in vivo for improving the potential future treatment of autoimmune disease, the prevention of transplant rejection, and graft versus host disease in man.
Materials and Methods
Mice
Female heterozygous B6.Cg-Foxp3sf/J (Scurfy) mice were purchased from Jackson Laboratories and bred to C57BL/6 wild-type male mice to generate hemizygous male B6 (Cg-Foxp3sf/Y [Scurfy] offspring); the C57BL/6 wild-type (WT) male littermates were used as controls. C57BL/6 CD45.1+ mice were obtained from the National Institute of Health. The Foxp3-GFP ‘knock-in’ mice (FoxP3-GPF-KI mice) [19] were a kind gift of Dr. Y. Belkaid and Dr. V. Kuchroo. All mice were held under specific pathogen-free conditions and cared for in accordance with institutional guidelines and regulations. All animal experimental protocols used in this study were approved by the NIAID ACUC (Animal Care and Use Committee).
Flow Cytometry
Anti-CD25-PE (PC61), anti-CD4-FITC (L3T4), anti-CD62L-FITC (MEL-14) anti-CD45RB-PE (16A), anti-CD45.1-APC (A20) were purchased from BD. Anti-CD45.2-PE (104), APC-, FITC-, or PacificBlue-labeled anti-FoxP3 (FJK-16s) were purchased from eBioscience.
For FACS-analysis the data were acquired either on a FACSCalibur (BD Biosciences) or a LSRII (BD Biosciences). The data were analyzed using the FLOWJo Software (Tree Star).
Cell Isolation
CD4+CD25− T cells and CD4+CD25+ T cells or CD4+GFP− and CD4+GFP+ T cells were FACS-sorted from single-cell suspensions from peripheral lymph nodes and spleen. In some experiments CD4+CD25− T cells were isolated using magnetic bead sorting after depletion of CD25+ cells.
For isolation of cells from the skin, ears were split and incubated with the dermis down in DMEM containing Liberase enzyme blend (ROCHE) for 45 minutes at 37°C. Then the ears were chopped, transferred into 1ml of DMEM 10%FBS with 0.05% DNAse I (Sigma) and ground in a medicon (50mm, BD) for 6 minutes using the Medimaschine (BD). The cells were rinsed in PBS, filtered through a 70μm pore-size filter and used for further analysis.
In vitro differentiation by TGFβ and adoptive transfer
CD4+CD25− T cells were stimulated with plate-bound anti-CD3 (1μg/well) and anti-CD28 (1μg/well) in the presence of rhTGFβ1 (5ng/ml, R&D systems) in complete RPMI (RPMI 1640 with 5% heat-inactivated FCS (Atlanta Biologicals), penicillin/streptomycin, 2 mM L-glutamine, 1 mM HEPES, 0.1 mM nonessential amino acids and 1 mM sodium pyruvate (all from BioSource International)) containing 100U/ml rhIL-2. On day 2, the cells were split in complete RPMI containing rhIL-2 (100U/ml). On day 3 to 7, cells were harvested, washed twice in PBS and resuspended in 50μl PBS for i.p. injection into newborn Scurfy mice on day 1 or 2 of life. Expression of FoxP3 was measured by flow cytometry on the day of injection and only populations with 80% to 99% FoxP3+ T cells were used.
Histology
For histopathologic evaluation 21 days after reconstitution, routine necropsies were performed, and selected tissues (skin, liver, lung) fixed in 10% neutral buffered formalin. Fixed tissues were embedded in paraffin, stained with H&E by American Histolabs (Gaithersburg, MD) and evaluated by two blinded veterinary pathologists. Inflammation was scored on a grading scale of: normal (0) with no evidence of inflammation; minimal (1) inflammation indicating few scattered lymphocytes, plasma cells perivascularly around portal and centrilobular veins in the liver, bronchioles in the lung, and multifocally in the pinnae; mild (2) inflammation indicating slightly increased numbers of primarily lymphocytes, plasma cells and occasional neutrophils perivascularly around portal and centrilobular veins in the liver, bronchioles in the lung, and multifocally in the pinnae; moderate (3) inflammation characterized by increased numbers of lymphocytes, neutrophils, plasma cells and scattered areas of more severe inflammation or necrosis bordering central veins in the liver, perivascularly around bronchioles and smaller airways in the lung, and multifocal areas of shallow abrasion or few ulcerations in the pinnae; marked (4) inflammation characterized by multifocal to coalescing or diffuse infiltrates of abundant degenerate and nondegenerate neutrophils, lymphocytes, plasma cells and cellular debris with variable areas of necrosis in the liver or bronchioles and multifocal abrasions to locally extensive ulcerations covered by a serocellular crust in the pinnae.
Statistical analysis
The data are expressed as mean ± SD. Significance was analysed using Prism and InStat (GraphPad Software) and p values <0.05 were considered significant.
Acknowledgments
The authors thank C. Henry and T. Moyer for cell sorting and the staff of the animal facilities for their work maintaining the mice. This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Allergy and Infectious Diseases. Lily I. Cheng and Jerrold M. Ward are in part supported by SoBran, Inc. (Fairfax, VA).
Abbreviations
- FoxP3
forkhead transcription factor p3
- iTR
TGFβ-induced regulatory T cells
References
- 1.Shevach EM. CD4+ CD25+ suppressor T cells: more questions than answers. Nat Rev Immunol. 2002;2:389–400. doi: 10.1038/nri821. [DOI] [PubMed] [Google Scholar]
- 2.Maloy KJ, Powrie F. Regulatory T cells in the control of immune pathology. Nat Immunol. 2001;2:816–822. doi: 10.1038/ni0901-816. [DOI] [PubMed] [Google Scholar]
- 3.Sakaguchi S, Sakaguchi N, Shimizu J, Yamazaki S, Sakihama T, Itoh M, Kuniyasu Y, et al. Immunologic tolerance maintained by CD25+ CD4+ regulatory T cells: their common role in controlling autoimmunity, tumor immunity, and transplantation tolerance. Immunol Rev. 2001;182:18–32. doi: 10.1034/j.1600-065x.2001.1820102.x. [DOI] [PubMed] [Google Scholar]
- 4.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 5.Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27:20–21. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 6.Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, Levy-Lahad E, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. 2001;27:18–20. doi: 10.1038/83707. [DOI] [PubMed] [Google Scholar]
- 7.Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27:68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- 8.Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8:191–197. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- 9.Lahl K, Loddenkemper C, Drouin C, Freyer J, Arnason J, Eberl G, Hamann A, et al. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J Exp Med. 2007;204:57–63. doi: 10.1084/jem.20061852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DiPaolo RJ, Brinster C, Davidson TS, Andersson J, Glass D, Shevach EM. Autoantigen-specific TGFbeta-induced Foxp3+ regulatory T cells prevent autoimmunity by inhibiting dendritic cells from activating autoreactive T cells. J Immunol. 2007;179:4685–4693. doi: 10.4049/jimmunol.179.7.4685. [DOI] [PubMed] [Google Scholar]
- 11.Weber SE, Harbertson J, Godebu E, Mros GA, Padrick RC, Carson BD, Ziegler SF, Bradley LM. Adaptive islet-specific regulatory CD4 T cells control autoimmune diabetes and mediate the disappearance of pathogenic Th1 cells in vivo. J Immunol. 2006;176:4730–4739. doi: 10.4049/jimmunol.176.8.4730. [DOI] [PubMed] [Google Scholar]
- 12.Fantini MC, Becker C, Tubbe I, Nikolaev A, Lehr HA, Galle P, Neurath MF. Transforming growth factor beta induced FoxP3+ regulatory T cells suppress Th1 mediated experimental colitis. Gut. 2006;55:671–680. doi: 10.1136/gut.2005.072801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hill JA, Feuerer M, Tash K, Haxhinasto S, Perez J, Melamed R, Mathis D, Benoist C. Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity. 2007;27:786–800. doi: 10.1016/j.immuni.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 15.Selvaraj RK, Geiger TL. A kinetic and dynamic analysis of Foxp3 induced in T cells by TGF-beta. J Immunol. 2007;179:11, 1390. [PubMed] [Google Scholar]
- 16.Hsieh CS, Zheng Y, Liang Y, Fontenot JD, Rudensky AY. An intersection between the self-reactive regulatory and nonregulatory T cell receptor repertoires. Nat Immunol. 2006;7:401–410. doi: 10.1038/ni1318. [DOI] [PubMed] [Google Scholar]
- 17.Pacholczyk R, Kern J, Singh N, Iwashima M, Kraj P, Ignatowicz L. Nonself-antigens are the cognate specificities of Foxp3+ regulatory T cells. Immunity. 2007;27:493–504. doi: 10.1016/j.immuni.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tran DQ, Shevach EM. Response: Anti human FOXP3 mAb PCH101 stains activated human naive T cells nonspecifically. Blood. 2008;111:464–466. [Google Scholar]
- 19.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]