

Abstract
Metallo-β-lactamases are responsible for conferring antibiotic resistance on certain pathogenic bacteria. In consequence, the search for inhibitors that may be useful in combating antibiotic resistance has fueled much study of the active sites of these enzymes. There exists circumstantial evidence that the binding of substrates and inhibitors to metallo-β-lactamases may involve binding to the organic part of the molecule, in addition to or prior to binding to one or more active site metal ions. It has also been postulated that a conformational change may accompany this putative binding. In the present study, electron paramagnetic resonance spectrokinetic study of a spin-labeled variant of the class B2 metallo-β-lactamase ImiS identified movement of a component residue on a conserved α-helix in a catalytically competent time upon formation of a transient reaction intermediate with the substrate imipenem. In a significant subpopulation of ImiS, this conformational change was not associated with substrate binding to the active site metal ion but, rather, represents a distinct step in the reaction with ImiS. This observation has implications regarding the determinants of substrate specificity in metallo-β-lactamases and the design of potentially clinically useful inhibitors.
Keywords: Lactamase, metallo-β-lactamase, cobalt, zinc, EPR, rapid-freeze-quench, conformation
INTRODUCTION
Antibiotic resistance to β-lactam-containing antibiotics is most often a consequence of bacterial production of β-lactamases. β-Lactamases hydrolyze the amide bond in the β-lactam ring of the antibiotic, rendering it ineffective in treating bacterial infection. β-Lactamases are categorized into four classes, A, B, C, and D, based on amino acid sequence, mechanism of action, sensitivity towards clinical inhibitors, and other biochemical characteristics.1 There are over 400 β-lactamases known and ca. 40 of these β-lactamases are known to require Zn(II) for catalytic activity. Metallo-β-lactamases (MβLs) are in class B and inactivate all penicillin-class antibiotics. MβLs are produced by a number of pathogenic bacteria, including Aeromonas sobria, Bacillus anthracis, and Stenotrophomonas maltophilia, and render these bacteria resistant to the largest class of antibiotics available in the clinic.2 MβLs have been shown to contain an αββα structural motif that is characteristic of the ‘β-lactamase fold’ superfamily to which they belong.3 Zn(II) binds to MβLs at the interface of the β-sheets in the enzymes. The class B MβLs have been further separated into 3 subclasses: B1, B2, and B3. Early crystallographic studies on several B1 and B3 MβLs identified a position-conserved loop that extends over the metal binding site and indicated that this loop was highly-disordered.4–8 Analogous loops have been observed in other enzymes belonging to the β-lactamase fold superfamily, which suggests a role for these loops in all of the proteins.9,10 X-ray crystallographically-derived structural models of MβL complexes with inhibitors have shown that the loop region is less disordered and is positioned closer to the metal center in these complexes than in the naked proteins.5–7,11 NMR studies of the MβL CcrA, site-directed substitution of an albeit non-conserved loop residue, and kinetic analyses12 indicated that the loop of CcrA may play a role in inhibitor (and, by analogy, substrate) binding and thus be integral to the mechanism of the catalytic reaction. Deletion of the entire loop region of CcrA resulted in 150 – 5000-fold decreases in kcat/Km.13 Analogous kinetic studies have been performed with variants of the MβLs IMP-1 and BcII, in which deletions or substitutions in the loop region were made.14 These studies highlighted effects on microscopic rate constants, steady-state kinetic constants, and the characteristics of formation of a reaction intermediate when using nitrocefin as substrate. Time-dependent spectrofluorometric studies on a tryptophan residue incorporated into a loop that covers the active site in the MβL L1 showed that changes in the tryptophan fluorescence occurred at catalytically competent rates.15 There exists, therefore, a body of evidence that implicates elements of protein structure other than the catalytic metal-containing site in substrate recognition and binding, and in the catalytic mechanism. Indeed, the observation that the mechanism of action of the MβL BcII appears to differ from those of the otherwise similar MβLs L1, IMP-1 and CcrA has been rationalized as being consistent with a shorter loop in BcII.16
ImiS is a B2 MβL that was identified in a clinical isolate of Aeromonas veronii bv. sobria and, like other B2 MβLs from aeromonads such as CphA and AsbM1, readily hydrolyzes carbapenems.17 The enzyme has been kinetically and biochemically characterized;18 ImiS requires one Zn(II) ion for catalysis and addition of more Zn(II) is inhibitory. EPR and NMR spectroscopic studies have shown that the catalytically required Zn(II) (or Co(II) in substituted ImiS) is bound by a cysteine and a histidine residue and can adopt four- or fivefold coordination, likely depending on the number of solvent ligands.19 This site corresponds to the B2 MβL consensus ‘Zn2’ site whose structure is known from X-ray crystallographic characterization of the very closely related B2 MβL CphA.20 A catalytically- competent five-coordinate Co(II) reaction intermediate species was characterized by EPR spectrokinetic studies of Co-substituted ImiS and confirmed the direct involvement of the metal ion in catalysis.21 These studies also clearly identified a product complex of ImiS, and supporting kinetic studies indicated that the product of imipenem hydrolysis inhibited catalysis by ImiS, with Ki ~ 55 μM.
ImiS has not been structurally characterized by X-ray crystallography. However, sequence comparison of ImiS with the related B2 MβL CphA indicated a very high level (98 %) of sequence homology between ImiS and CphA,17 and a high level of sequence similarity among the B2 MβLs. X-ray crystallographic studies on CphA20 indicated that B2 MβLs do not contain a loop over their active sites, in contrast to the B1 and B3 MβLs; instead, there is a position-conserved α-helix in B2 MβLs, the role of which has not been probed to date. Crystal structures of the class B2 MβL CphA in the naked state, as a complex with the product of biapenem hydrolysis, and as a complex with the CphA inhibitor pyridine-2,4-dicarboxylate (PDB ID: 2GKL) have been presented.20 Taken together, these suggest that the side-chain of the residue Glu152, which is solvent-accessible and resides on the conserved α-helix, moves when CphA is complexed with product or inhibitor. However, these complexes may not be representative of species that arise during the catalytic reaction. Further, the propensity of ImiS to form complexes with products and, perhaps, the natures of the complexes themselves, differ depending on the nature of the product.21 Direct information on the nature of catalytic intermediates is, therefore, desirable.
In the present study, a combination of rapid-freeze-quench (RFQ) EPR and site-directed spin-labeling (SDSL) techniques have been used to investigate the dynamics of an α-helix, spanning residues 150–160 and corresponding to the position-conserved helix in other B2 MβLs, in Co(II)-substituted ImiS that was spin-labeled at a substituted Cys152 residue with (1-oxyl-2,2,5,5-tetramethylpyrroline-3-methyl)methanethiosulfonate (MTSL). RFQ-EPR has been utilized for over 45 years for the investigation of enzyme reaction intermediates22,23 and the use of spin labeling combined with EPR and computer simulation is a rapidly growing area for the study of protein structure and dynamics.24,25 The use of metal ion-spin label interactions to probe interspin distances in proteins has been described,26 and full matrix diagonalization simulation suites that explicitly treat S > ½ and multispin systems are available.27 In this study, we combined these approaches in order to study conformational changes in ImiS during the catalytic reaction by monitoring changes in the Co(II)-MTSL distances in samples of ImiS before, during, and after reaction with the substrate imipenem.
MATERIALS AND METHODS
An ImiS variant in which glutamate 152 was substituted with cysteine (‘ImiS[E152C]’) was generated by mutating the imiS gene in pET26bimiS using the Stratagene Quikchange mutagenesis kit. The following oligonucleotides were used for the preparation of the mutant: E152C forward, gAAgAgCgACTgggCCTgCATTgTCgCCTTTACC and E152C reverse, ggTAAAggCgACAATgCAggCCCAgTCgCTCTTC. DNA sequence verification of the mutated imiS gene using T7forward (TAATACgACTCACTATAgg) and T7reverse (CgATCAATAACGAgTCgCC) as the primers was performed (PerkinElmer Life Sciences ABI 5300 Genetic Analyzer). To test for over-expression of the mutant enzyme, E. coli BL21(DE3) cells were transformed with the mutated over-expression plasmid, and small scale cultures were used. ImiS[E152C] was purified according to a protocol described earlier.18 ImiS[E152C] (20 μL of 30μM) was incubated with a 10-fold molar excess of MTSL spin-label dissolved in 300 μL dimethylsulfoxide. The coupling reaction was allowed to proceed overnight in the dark at 4 °C. The resulting solution was centrifuged to remove any precipitates. The mixture was passed through a Sephadex G-25 column to separate any excess, unbound spin label from spin-labeled ImiS (ImiS*). Zn-ImiS* and Co-ImiS*, containing one equivalent of Zn(II) and Co(II), respectively, were prepared and characterized as described for wild-type Zn- and Co-ImiS.19 Steady state kinetics with imipenem as substrate were carried out at 25 °C in 50 mM Tris buffer, pH 7.0.
Co-ImiS* (0.5 mM in 50 mM Tris buffer, pH 7.0) was reacted with imipenem (0.5 mM) at 2 °C, and the reaction mixture was freeze-quenched for EPR spectroscopy, using a system described elsewhere;21,28 the calibrated reaction time was 12 ms. Following EPR data collection, samples were thawed by agitation of the sample tubes in water at 25 °C for 1 min and refrozen in liquid nitrogen. Samples were thawed again and incubated with an additional 4.5 mM imipenem for 10 min at 25 °C. A separate sample (1 mM) of Co-ImiS* was incubated with 5 mM imipenem for 10 min at 25 °C and frozen for EPR in the absence of isopentane, in order to investigate any dependence of the EPR spectrum upon isopentane.
Low temperature EPR spectroscopy was carried out using a Bruker EleXsys E600 spectrometer equipped with an Oxford Instruments ITC4 liquid helium flow system and a 90 dB dynamic range microwave bridge for low power measurements. Ambient temperature EPR spectroscopy (25 ± 1 °C) was carried out using a Bruker EMX spectrometer equipped with a flat cell and nitrogen flow temperature control. Recording parameters for individual spectra are given in the figure legends. In each case, great care was taken to ensure that spectra were recorded under non-saturating conditions, including the characterization of conditions under which ‘pseudo-absorption’ (second harmonic modulation quadrature detection) signals were observable due to rapid passage effects. EPR simulations were carried out using the matrix diagonalization program XSophe (Version 1.1.3).27 Spins were quantified by double integration (IGORPro 6.02, Wavemetrics) of simulated spectra; this approach better captures the intensity in the ‘wings’ of the spectra than does integration of inherently noisy experimental spectra. Differences between the spectra of MTSL in Co-ImiS* and Zn-ImiS* were used as the models to which theoretical spectra invoking spin-spin interaction between Co(II) and MTSL in Co-MTSL were fitted. Contributions due to individual components were systematically varied and the resulting model spectra, normalized for the total spin density, were compared to the experimental data. Proportions of each species were changed in order to minimize the integral of the square of the difference spectrum, generated by subtracting the simulation from the experimental spectrum.
RESULTS
Kinetic analyses showed that ImiS[E152C] and ImiS* exhibited very similar kinetic behavior to wild-type ImiS.18 Metal analyses indicated that as-prepared ImiS[E152C] contained 0.80 equivalents of Zn, which is similar to the metal content of wild-type ImiS, and displayed 85 % of the catalytic activity of wild-type ImiS. Steady state kinetic parameters for ImiS[E152C] were kcat = 292 ± 28 s-1 and KM = 106 ± 20 μM, and for ImiS* were kcat = 295 ± 20 s−1 and KM = 104 ± 15 μM. Co-ImiS, containing 1.0 eq. Co, exhibited kinetic parameters kcat = 255 ± 16 s−1 and KM = 99 ± 12 μM;19 the steady state rates of hydrolysis of imipenem by Co-ImiS[E152C] and by Co-ImiS* were indistinguishable from that by Co-ImiS.
EPR spectra of Zn-ImiS* and Co-ImiS* recorded at ambient temperature (25 ± 1 °C) are shown in Figure 1 and low temperature (12 K) EPR spectra of Zn-ImiS* and of Co-ImiS* in the resting state and upon incubation with imipenem are presented in Figure 2.
Figure 1.
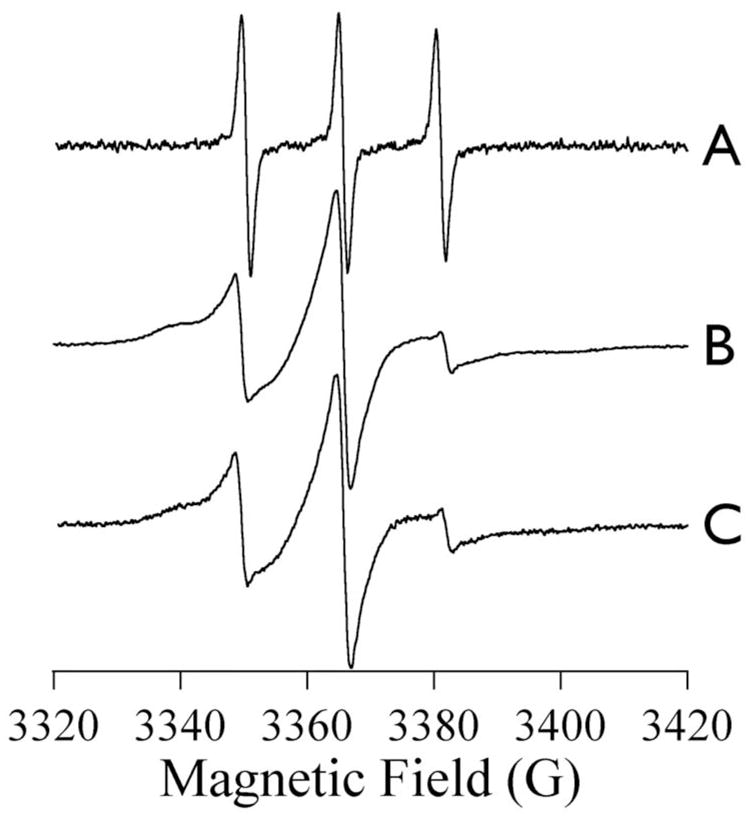
EPR spectra of (A) MTSL, (B) Zn-ImiS* and (C) Co-ImiS* recorded at 25 ± 1 °C, 9.395 GHz, 1 mW microwave power and 0.1 mT (1 G) field modulation at 100 kHz.
Figure 2.
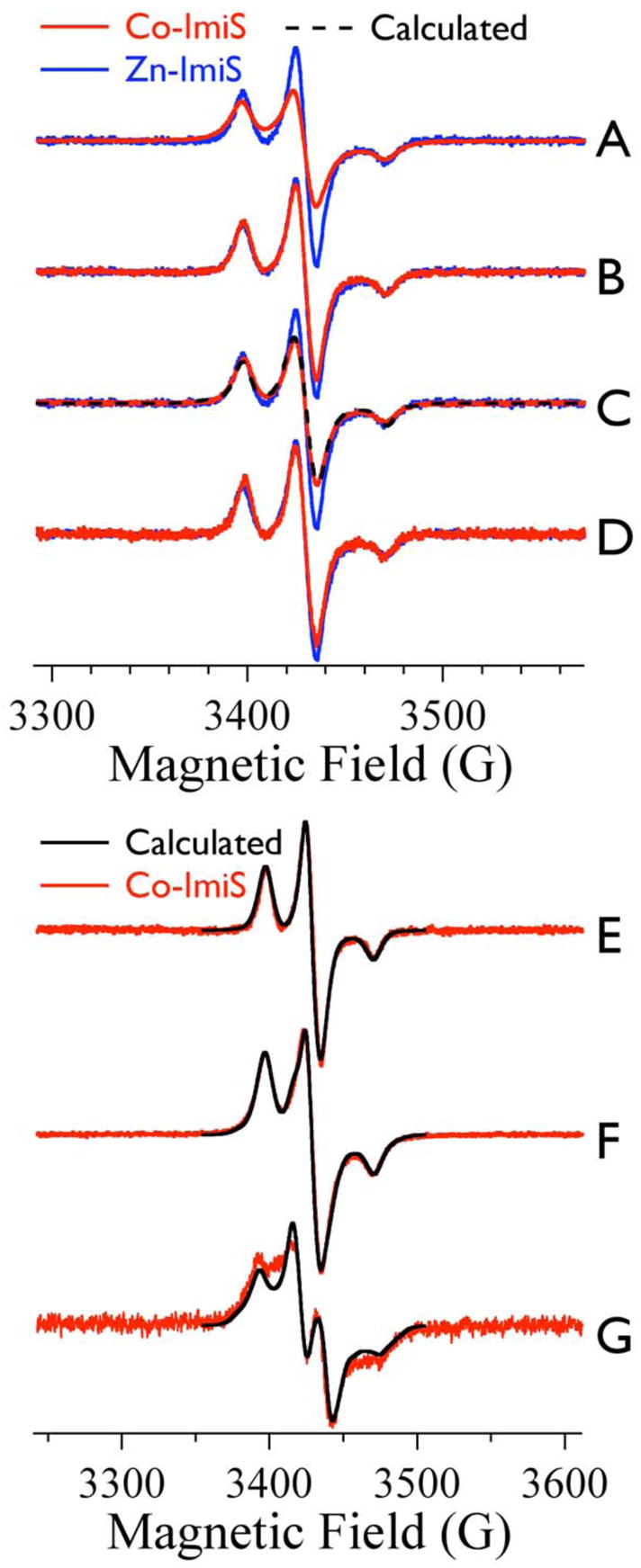
The top panel shows experimental EPR spectra of Co-ImiS* species (A – D, red lines) overlaid with the EPR signal from resting Zn-ImiS* (blue) in each case. The bottom panel (E – G) shows computer simulations, with computed spectra in black and experimental spectra in red. The dashed black trace of C is also a computer simulation. The experimental Co-ImiS* spectra are from (A, F) resting Co-ImiS*, (B, E) 0.5 mM Co-ImiS* after reaction with 0.5 mM imipenem for 12 ms at 2 °C, (C) 0.5 Co-ImiS* after reaction with 0.5 mM imipenem for 1 min at 2–25 °C and (D) 0.5 mM Co-ImiS* after reaction with 5 mM imipenem for 10 min at 25 °C; Trace G (red) is the difference spectrum obtained by subtraction of the experimental spectrum of Co-ImiS* after reaction with 0.5 mM imipenem for 12 ms from that of resting Co-ImiS*. The intensities of all traces in A – D correspond to the same number of total spins; the traces in E – G are shown with arbitrarily adjusted intensities. The black Traces E and G are computed spectra representing (E) an isolated MTSL radical, and (G) an MTSL radical experiencing a dipolar interaction with a Co(II) ion at a distance of 1.6 nm (16 Å). The simulated spectrum F (black) was generated by adding the computed spectra of Trace E (55 % of total spin density) and Trace G (45 % of total spin density). The simulated spectrum C (dashed, black) was generated by adding the computed spectra of Trace E (70 % of total spin density) and Trace G (30 % of total spin density). Experimental spectra were recorded at 9.634 GHz, 12 K, 0.05 mW microwave power, with 0.1 mT (1.0 G) magnetic field modulation at 100 kHz. Simulations were carried out by matrix diagonalization (XSophe) assuming a spin Hamiltonian ℋ = (βg.B.S)MTSL + (S.A[14N].I[14N])MTSL + (βg.B.S)Co(II) + (S.D.S)Co(II) + (S.J.S)MTSL-Co(II) and parameters are given in Table 1.
The EPR spectra of Zn-ImiS* and Co-ImiS* recorded at ambient temperature were indistinguishable from each other but clearly very different from that of MTSL in solution (Figure 1). The 0.1 mT (1 G) field-modulated spectra of Zn- and Co-ImiS* each exhibited three relatively narrow derivative features with an average peak-to-trough linewidth of 0.20 mT (2.0 G) and split by 1.62 mT (16.2 G). Comparison with the linewidth (0.14 mT; 1.4 G) and splitting (1.54 mT; 15.4 G) in the spectrum of MTSL indicated that the derivative features in the ImiS* spectra are due to a system in which the anisotropy in the 14N hyperfine tensor, A, is largely averaged on the EPR timescale (~ 10 ns); they arise from a population of MTSL that is mobile. In addition, there were other features in the spectra of ImiS* with very different properties. Two broad shoulders were evident at 334 mT (3340 G) and 340 mT (3400 G), with an approximate average full-width-at-half-height linewidth of 1.3 mT (13 G). These features exhibited, therefore, an effective AI = 1(14N) of 3.0 mT (30 G), much closer to the rigid limit of 3.6 mT (36 G) for Az than to the rotationally averaged Aav. of 1.5 mT (15 G). The splitting and linewidth of these features indicate that they arise from a second, distinct population of MTSL that is rotamerically constrained on the 10 ns timescale. The relative amounts of the two species in the ambient-temperature spectra were estimated by modeling the MI = +1 region of the spectrum (3330–3360 G) as being due to an absorption feature due to the immobile component and a derivative due to the more mobile one. This approach is relatively crude but does not rely on a detailed knowledge of nitroxide dynamics; it is estimated that the EPR spectra contain roughly equal proportions of the two species.
The 12 K EPR signal from resting Zn-ImiS* is shown as the blue line in each of Traces A – D of Figure 2, overlaid, in each case, with the experimental EPR signals from the g ~ 2 region of the EPR spectrum from various Co-ImiS* samples (Fig. 2A–D, red lines); note that the signal intensities shown in A – D of Figure 2 are corrected for the total spin density corresponding to each spectrum. The EPR signal due to the nitroxide spin label in resting Co-ImiS* clearly differs from the signal from Zn-ImiS* (Fig. 2A). There is additional intensity in the ‘wings’ (3365–3390 G and 3478–3490 G) and in the region 3408–3418 G of the spectrum of resting Co-ImiS*, and the peak-to-trough intensity of the center line is only 50 % of that from Zn-ImiS*. In contrast, the spectrum from Co-ImiS* after reaction with imipenem for 12 ms at 2 °C (the ‘12 ms’ signal) is very similar to that of Zn-ImiS* (Fig. 2B); the only noticeable differences are an approximately 7 % diminution of the height of the center line and some very weak additional absorption at the extremities of the Co-ImiS* spectrum. After complete reaction (1 min) of 0.5 mM Co-ImiS* with 0.5 mM imipenem, the observed Co-ImiS* ‘1:1 product’ EPR signal (Fig. 2C) was line-broadened compared to the Zn-ImiS* and Co-ImiS* ‘12 ms’ EPR signals but not as broadened as resting Co-ImiS*. After complete reaction of 0.5 mM Co-ImiS* with 5 mM imipenem, the observed Co-ImiS* ‘1:10 product’ EPR signal was very similar to the Zn-ImiS* and Co-ImiS* ‘12 ms’ signals (Fig. 2D).
Computer simulations were carried out in order to better understand the origins of the various EPR signals; spin Hamiltonian parameters for the individual species are given in Table 1 and the contributions of the individual components to the simulations of experimental spectra are summarized in Figure 3. The Co-ImiS* ‘12 ms’ signal could be readily simulated (Fig. 2E) as a single species using parameters typical for a nitroxide and invoking no spin-spin interaction. A distance of 2.5 nm is considered to be around the effective limit for the use of continuous wave EPR for distance determination,29 and therefore the MTSL-Co(II) interspin distance (rMTSL-Co(II)) is likely ≥ 2.5 nm (25 Å) in most molecules. (In fact, computed spectra were surprisingly discriminating among distances even as long as ~ 2.5 nm; significant differences between spectra were observed with changes in the interspin distance and the Euler angle χ - see Table 1 for definition - of ΔrMTSL-Co(II) = 0.05 nm and Δχ = 5°, respectively.) The small differences between the center line heights of the spectra of Zn-ImiS* and the Co-ImiS* ‘12 ms’ signal suggested a small proportion, ~ 5 %, of some additional species in the ‘12 ms’ signal and the very weak extra absorption in the spectrum’s wings suggested that this species may be due to dipolar splitting or broadening of the nitroxide signal in a small proportion of the molecules in the sample.
Table 1.
Spin Hamiltonian parameters and interspin distances from EPR simulations.
| Species | gz [Az, (mT)] | gy [Ay, (mT)] | gx [Ax, (mT)] | rCo-MTSL(nm) [χ]a |
|---|---|---|---|---|
| 2Eb | 2.0028 [3.66] | 2.0064 [0.53]c | 2.0084 [0.58]c | ∞c |
| 2Gb | 2.0035 [3.66] | 2.0080 [0.55] | 2.0080 [0.55] | 1.6e [40 °] |
χ is the angle between the z axis of the MTSL g tensor and the interspin vector between MTSL and Co(II).
Species labeled as in Figure 2.
No MTSL-Co(II) interaction was assumed for this species.
The interaction between MTSL and Co(II) was calculated assuming Co(II) spin Hamiltonian parameters of S = 3/2, MS = ± 1/2, g(x,y,z) = 2.29, 2.29, 2.50, D = 50 cm−1 and E/D = 0.09.
Figure 3.
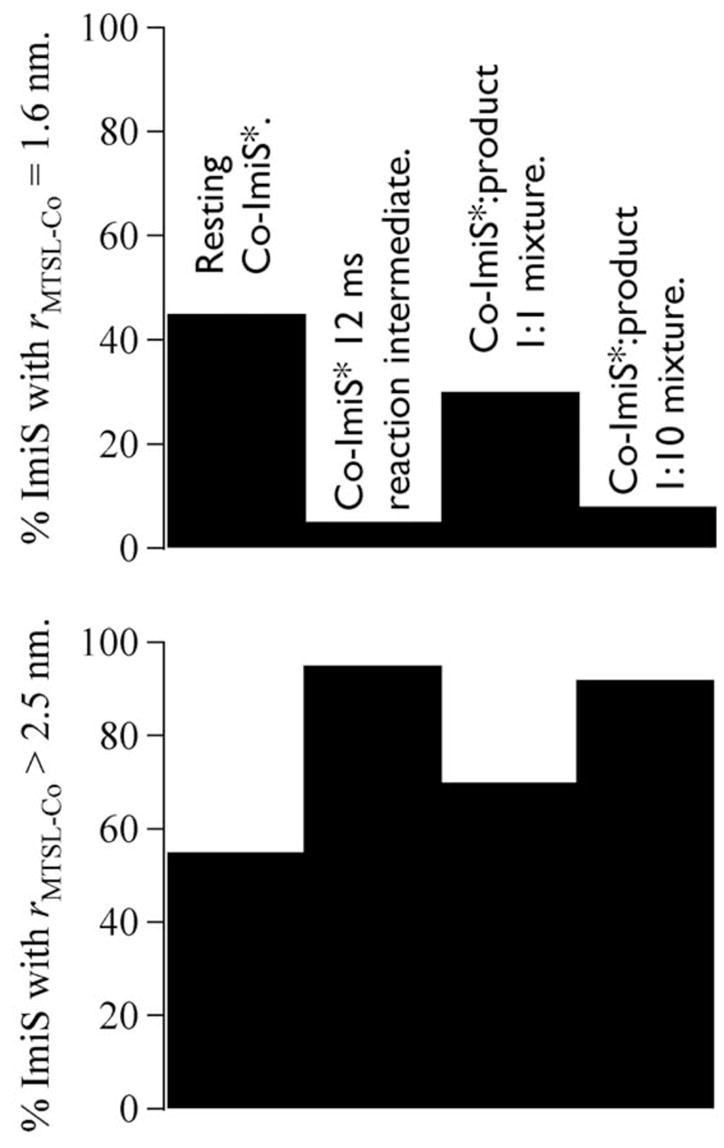
Fractional intensities of the two classes of EPR signals that contribute to the spectra of Co-ImiS* species. The top panel shows the percentage of total spins arising from signals due to Co-ImiS* species with a MTSL-Co(II) interspin distance of 1.6 nm (16 Å). The bottom panel shows the percentage of spins from species in which the MTSL-Co(II) interspin distance appears to be greater than 2.5 nm (25 Å).
In contrast to the Co-ImiS* ‘12 ms’ signal, the EPR signal from resting Co-ImiS* was markedly different from that of Zn-ImiS* (Fig. 2A). The Co-ImiS* resting signal was readily resolved into two components. One species was similar to those of the Zn-ImiS* and Co-ImiS* ‘12 ms’ signals, and was modeled using identical parameters to those for the Co-ImiS* ’12 ms’ signal. A second ‘split’ species, shown as the red trace of Fig. 2G, was generated by subtraction of the experimental ‘12 ms’ spectrum from the resting signal. This ‘split’ species was modeled (black trace of Fig. 2G) invoking an MTSL-Co(II) distance of 1.6 nm ± 0.05 nm (16 Å) and accounting for 45 % ± 5 % of the spins. These two species provided a very good overall simulation of the Co-ImiS* resting species (Fig. 2F). Interestingly, these data are also consistent with the estimate of equal populations of two species, one relatively mobile and the other almost immobile, in the ambient temperature spectra.
The Co-ImiS* ‘1:1 product’ species had the appearance of being comprised of contributions from the resting species and the narrower ‘12 ms’ species and was simulated (Fig. 2C) using 70 % (by spins) of the unsplit (rMTSL-Co(II) > 2.5 nm) ‘12 ms’ simulation (2E), and 30 % of the ‘split’ (rMTSL-Co(II) = 1.6 nm) species (2G).
Incubation of either 0.5 mM Co-ImiS* with 5 mM imipenem in the presence of isopentane or 1 mM ImiS with 5 mM imipenem in the absence of isopentane gave samples upon freezing that exhibited EPR spectra essentially indistinguishable from each other and from that of Zn-ImiS*. It is estimated that up to about 7 % of the signal may be due to MTSL experiencing a significant interaction with Co(II) based on the height of the center line. It is noteworthy that in the course of the present work indistinguishable rMTSL-Co(II) = 1.6 nm (16 Å) spectral components were observed in samples in the absence (2A) and presence (2C) of isopentane, and were also seen to disappear in an imipenem-dependent fashion in both the absence and presence of isopentane; isopentane did not, therefore, interfere with either the reaction of Co-ImiS* with imipenem or with the response of the spin label under the conditions employed.
RFQ-EPR was also used to probe the environment of the Co(II) ion in Co-ImiS*. The direct involvement of the Co(II) ion in wild-type Co-ImiS catalysis has already been well characterized by RFQ-EPR and stopped-flow electronic absorption spectroscopies.21 EPR spectra of Co(II) in Co-ImiS* are presented in Figure 4. The resting signal (Fig. 4A) was devoid of resolved 59Co hyperfine structure and did not exhibit, from 3.6 to 20 K, any sharp feature at geff. > 6 that would signify a population of Co(II) with MS = |±3/2〉. After 12 ms reaction with imipenem, a clear indication of 59Co hyperfine structure from the rhombic signal that was observed in the earlier work was observed (Fig 4C). The intensity of these features diminished upon further incubation for 1 min (Fig 4D). The fractional intensity of the rhombic, hyperfine split species was estimated to be ~ 10 % of total Co(II) based on comparison with the data presented in the earlier study.21
Figure 4.
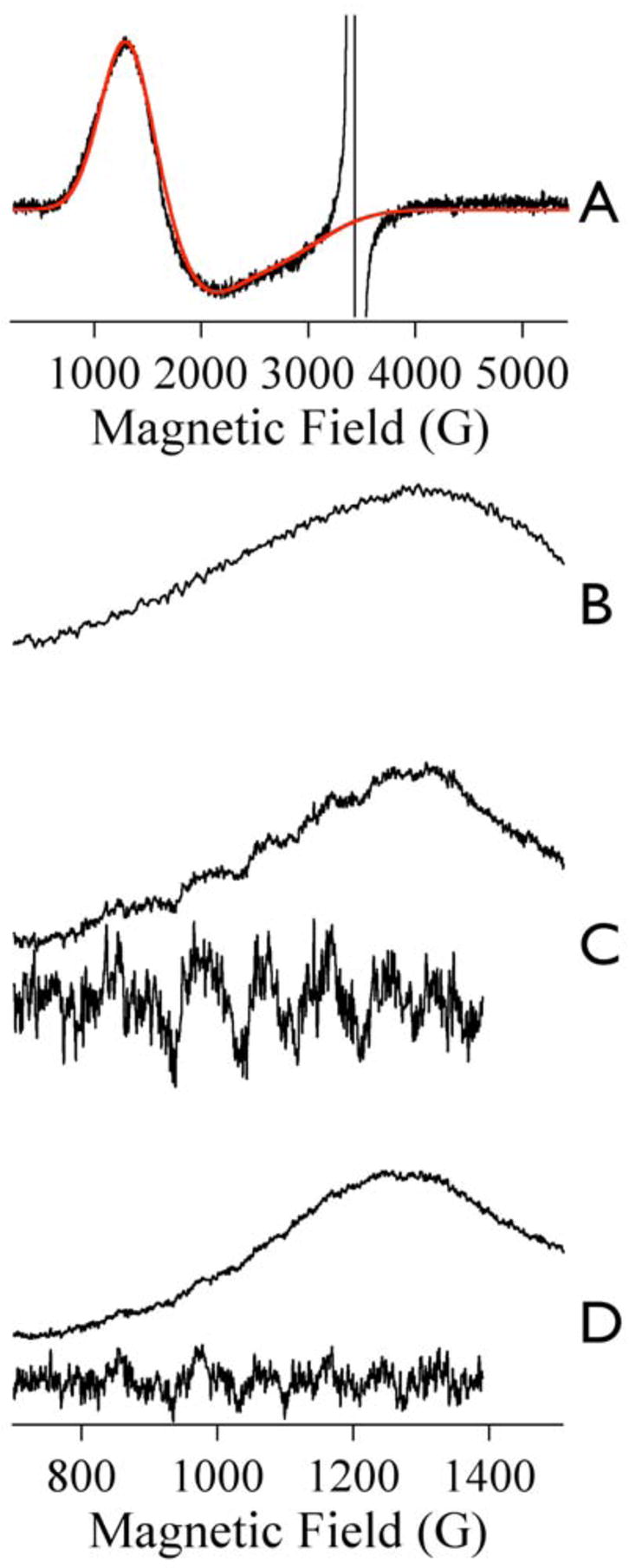
(A) Experimental (black line) and simulated (red line) EPR spectra of resting Co-ImiS*; the simulation was calculated using ℋ = βg.B.S + S.D.S with spin Hamiltonian parameters of S =3/2, MS = ±½, g(x,y,z) = 2.29, 2.29, 2.50, D = 50 cm−1 and E/D = 0.09. Traces B – D show partial EPR spectra from Co(II) in Co-ImiS*. (B) Resting Co-ImiS*. (C) Co-ImiS* after reaction with 1 mM imipenem for 12 ms at 2 °C. (D) Co-ImiS* after reaction with 1 mM imipenem for 1 min at 2–25 °C. The insets in C and D show the 59Co hyperfine region (× 4 in each case). Spectra were recorded at 9.634 GHz, 12 K, 1 mW microwave power, with 1.4 mT (14 G) magnetic field modulation at 100 kHz.
In order to obtain further information on the nature of the broadening of the MTSL signals observed in some of the samples of Co-ImiS*, MTSL spectra were recorded at 12 K, where the signal from Co(II) is optimum, and at 40 K, where the Co(II) signal is undetectable due to relaxation broadening. At 12 K the spectrum of resting Co-ImiS* (Fig 5A, black line) is clearly broadened extensively compared to that of Zn-ImiS* (Fig 5A, blue line). At 40 K, however, the signals (Fig 5B) from Co- and Zn-ImiS* are indistinguishable. This behavior is entirely analogous to the temperature-dependent splitting of the signal from slow-relaxing Mo(V) in xanthine oxidase due to dipolar spin-spin interaction with a fast-relaxing reduced iron-sulfur cluster.30,31 In marked contrast, spectral splittings due to dipolar interaction between two nitroxide spin labels do not exhibit this temperature dependence.29
Figure 5.
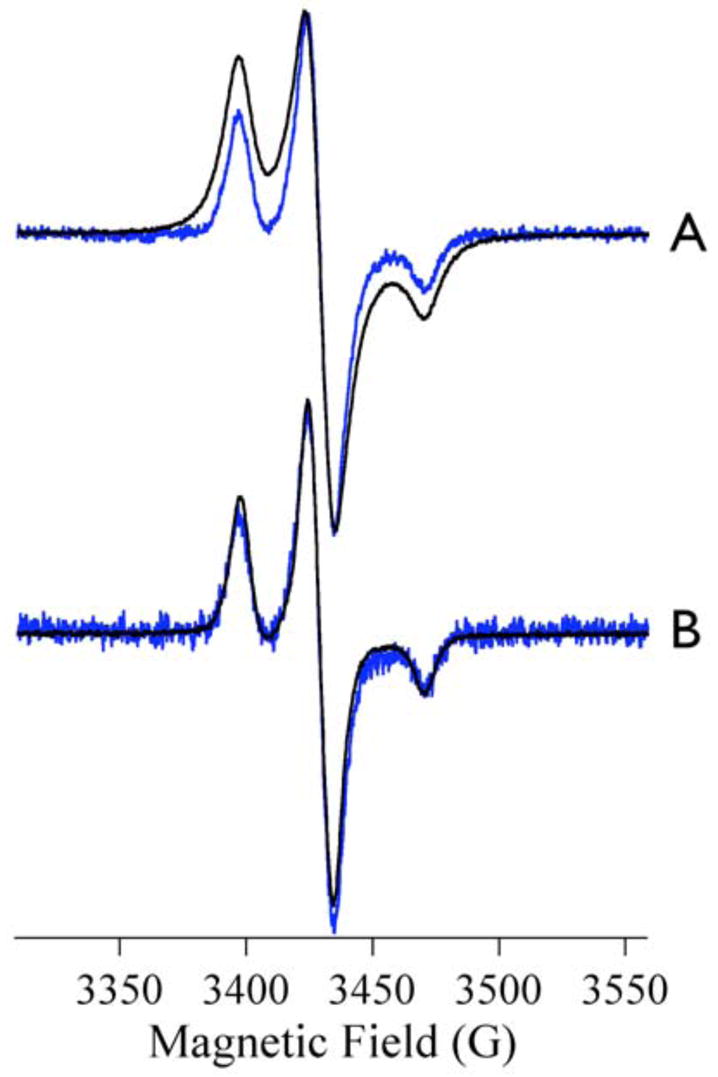
EPR spectra of resting Co-ImiS* (black lines) and Zn-ImiS* (blue lines) recorded at 12 K (A) and 40 K (B). Other recording parameters were as for Figure 1. Spectra are shown normalized for the intensity of the center line.
DISCUSSION
The EPR spectrum from the resting species of Co-ImiS* is clearly significantly broadened with respect to that of Zn-ImiS*. The obvious conclusion to draw is that the MTSL in Co-ImiS* experiences a magnetic (dipolar) interaction with Co(II) due to the proximity of the two paramagnets in Co-ImiS*. In support of this conclusion, we have observed that the broadening of the MTSL signal from Co-ImiS* (i) is only exhibited at temperatures where the Co(II) signal is detectable, (ii) changes upon incubation of Co-ImiS* with imipenem both in a time that is catalytically competent and concomitant with changes in the signal from Co(II), and (iii) can be modeled extremely well by computer simulations invoking MTSL-Co(II) distances that are realistic given structural models of ImiS. The EPR spectrum due to MTSL in Co-ImiS* is sensitive, then, to the proximity of MTSL to the active site Co(II); thus the nature of the EPR signal from MTSL in Co-ImiS* provides a probe of conformational changes in ImiS during the catalytic reaction.
As in earlier work on ImiS,21 the EPR spectrum of Co(II) bound to ImiS* in Tris buffer, pH 7.0, indicated a single Co(II) environment in which the metal ion is five-coordinate, with non-proteinous ligands provided by solvent water. This species has been well characterized19 and is optimally detected at 12 K; above 12 K the spectrum is broadened by relaxation and below 12 K rapid passage effects are observed that result in contamination of the field-modulated first-derivative spectrum with absorption-like components. The rhombic intermediate species observed upon reaction with imipenem has a similar temperature dependence to the resting signal,21 and the temperature dependences signify much faster relaxation times for Co(II) than for MTSL. In addition, the g tensors of the Co(II) signals do not overlap with that of MTSL.19,21 Therefore, a spin system due to MTSL and Co(II) experiencing weak spin-spin coupling will not give rise to spectra that are distorted due to mixing of the wavefunctions; rather, the individual spin systems of Co(II) and MTSL retain their identity.32,33 The slower relaxing species, MTSL, will give rise to an unsplit spectrum at high temperature (where Co(II) is unobservable and T1CoII very short) that is characterized by a linewidth (T2MTSL)−1 + J2T1CoII.30 Comparison of the spectra of Co-ImiS* and Zn-ImiS* at 40 K clearly shows that the latter term is negligible. At low temperature (where T1CoII is long and Co(II) is observable), the MTSL signal is expected to exhibit a distance-dependent dipolar splitting and a linewidth (T2MTSL)−1 + ½(T1CoII)−1; again, in practice, the latter term is expected to be small and the linewidths comparable to those of the high temperature signal. This is, in fact, precisely what is observed for the ‘split’ species at 12 K; the signal exhibits a dipolar splitting but no significant broadening of the individual lines. The quality of the simulations, which do not take into account relaxation-dependent line broadening, support the assignment of the ‘split’ signal to a dipolar-coupled system in the low temperature limit. As the splittings are clearly resolved, they can be used as a probe of interspin, i.e. Co(II)-MTSL, distances in ImiS.
The RFQ-EPR data indicated that resting ImiS appears to be in equilibrium between two conformations, one in which the catalytic metal ion is 1.6 nm (16 Å) from the MTSL label on Cys152, and another in which the paramagnets are > 2.5 nm (> 25 Å) distant. Consistent with this observation, the ambient temperature EPR spectra of ImiS* also indicated two distinct populations of MTSL, one mobile on the EPR timescale and the other approaching the rigid limit. Upon binding of the substrate imipenem, a conformation predominates in which the catalytic metal ion and the paramagnetic nitroxide of the MTSL spin label, specifically the nitrogen atom on which most of the spin density is resident, are distant. The partial restoration of the Co-ImiS* MTSL signal toward the resting signal upon exhaustion of substrate suggests interference of full return to the resting equilibrium by product, perhaps by the formation of a product complex with KD > 0.5 mM. At higher product concentrations (5 mM), the product complex predominates, again favoring a longer MTSL-Co(II) distance, suggesting 5 mM > KD > 0.5 mM.
Insight into the interaction of imipenem and ImiS during the reaction can be obtained by consideration of the possible mechanisms for the imipenem-dependent change of > 0.9 nm (> 9 Å) in the Co(II)-MTSL distance. In the absence of a crystal structure of ImiS, inspection of the structure of the very closely related (98 % homology) CphA is instructive. Figure 6 shows the structure of the CphA complex with the hydrolysis product of biapenem (blue), and into which imipenem was modeled (red). Glu152 was replaced with Cys-MTSL in two orientations, A and B. The five dihedral angles along the nitroxide tether are generally referred to, sequentially, as X1 – X5, with X1 being the dihedral angle around the Cα-Cβ bond.
Figure 6.
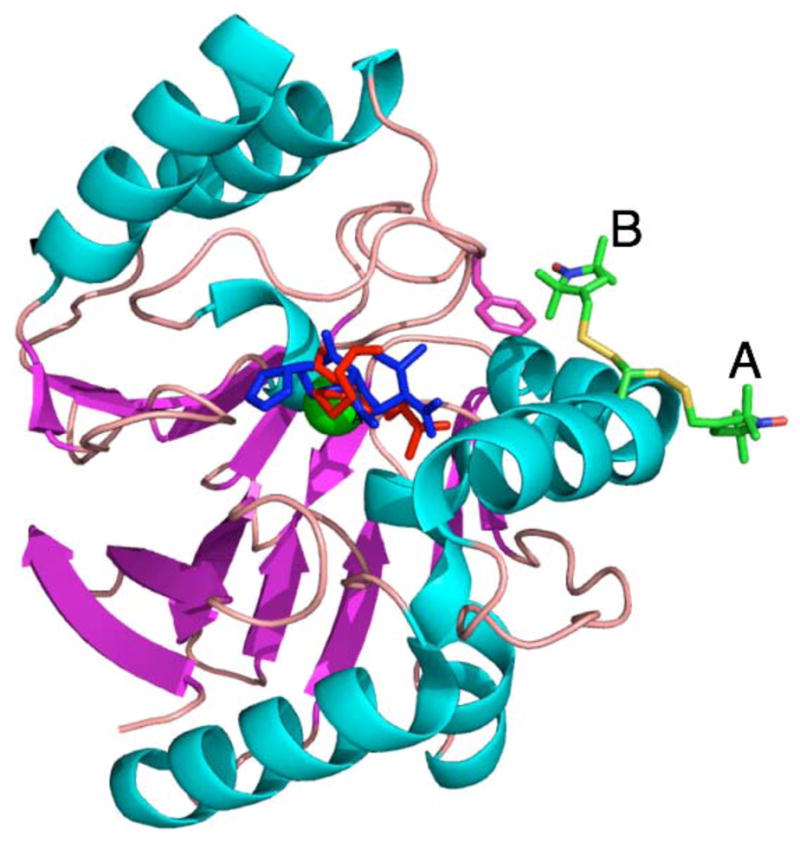
A model for ImiS* based on the structure of the CphA complex with the hydrolysis product of biapenem. A Glu152Cys in silico mutation (MacPyMol v.0.99) was carried out on the CphA structure (PDB-ID 1X8I) and the Cys152 side chain was replaced by a Cys-MTSL fragment from the structure of spin-labeled T4 lysozyme (PDB-ID 1ZYT). Two conformations of Cys-MTSL are shown, A and B, that correspond to rotation around the X1 (Cα-Cβ) dihedral axis. Imipenem (red) was manually modeled onto biapenem (blue) to preserve, as far as possible, both the Zn binding and the presumed orientation of the lactam ring. The Zn(II) ion is shown in green in the center of the figure and the side chain of Phe236 is shown in magenta.
Using the model shown in Figure 6, some distances can be compared with those from EPR. The orientation, A, of the nitroxide represents the longest possible distance between the nitrogen atom and the Zn(II) ion, 2.2 nm (22 Å). Fast motion of surface nitroxides is common, though fast rotation is usually restricted to the X4 and X5 dihedral axes.34,35 In the model of Figure 6, this would correspond to a continuum of Co(II)-NMTSL distances from 1.85–2.20 nm. Allowing for additional steric hindrance between the helix and nitroxide ring, from the model it may be expected that the range of distances observed would more likely be 2.1 – 2.2 nm. The dipolar broadening that would result from these distances may be difficult to detect in the EPR spectrum of resting Co-ImiS*, which is dominated by an even broader signal due to the species with rMTSL-Co(II) = 1.6 nm. It is clear, however, that in more than 90% of the molecules responsible for the ‘12 ms’ species, rMTSL-Co(II) > 2.5 nm. Upon reaction with imipenem, then, ImiS adopts a conformation in which the observed Co(II)-MTSL distance is greater than can be explained by rotational flexibility of the spin label superimposed upon the static model provided by the 98 % homologous CphA. This observation suggests that the helix itself moves relative to the active site upon addition of imipenem to ImiS.
Further insight comes from consideration of the resting enzyme. The major species in resting ImiS exhibited a Co(II)-MTSL distance of 1.6 nm. This can be modeled by the orientation B shown in Figure 6, which differs from orientation A principally in a rotation around the X1 dihedral angle. The coexistence of such rotameric pairs is relatively common34,35 and is supported in the case of ImiS by the two-component ambient temperature EPR spectrum. In that spectrum, the major species is relatively immobile and in the MTSL conformation B, the nitroxide is stabilized by numerous hydrophobic interactions with the side chain of Phe236 (interresidue C-C distances of 0.26, 0.26, 0.32 and 0.34 nm and a C-S distance of 0.30 nm are present in the model). The model is consistent with a well-defined Co(II)-MTSL distance (1.6 ± 0.5 nm), as indicated by EPR, rather than a wide distribution.
The EPR and structural information available suggest models, (i) for the ‘12 ms’ species, in which the helix must move at least 0.3 nm away from the active site relative to resting enzyme to be consistent with the EPR, and, (ii) for the resting state, in which the B rotamer is stabilized by Phe236. One possible explanation for the almost total loss of an rMTSL-Co(II) = 1.6 nm component in the ‘12 ms’ species, then, is the loss of stabilization of the B rotameric conformation upon a ≥ 0.3 nm displacement of the Cys152-containing helix due to binding of imipenem. The cause of this proposed helix motion is, however, unclear. Both the crystallographically observed biapenem hydrolysis product and the modeled imipenem molecule approach side chain residues of the helix to ~ 0.3 nm in the structure of CphA. However, there are no interactions between modeled imipenem and the helix that would suggest a necessity that the helix move further away. On the other hand, a small rotation of the helix about its axis in response to imipenem binding would be sufficient to both increase the Co(II)-MTSL distance in conformation A to the range indicated by EPR of the ‘12 ms’ species and to prevent stabilization of conformation B by Phe236. This would imply a role for substrate recognition by the helix, consistent with its conservation in this class of metallo-β-lactamases. The level of sophistication of the modeling of imipenem into the structure is too low to make more detailed structural predictions and further work will be necessary to more thoroughly interrogate the models proposed herein.
An alternative explanation for the changes in the EPR upon adding imipenem to ImiS can be considered if the spin label is assumed to be immobilized relative to the helix. This model would suggest two conformations of the resting enzyme in slow equilibrium, one with the helix and nitroxide arranged as in B of Figure 6, and a second with the helix either translated some further 1 nm distant, which is difficult to envisage, or else rotated through a large angle. The distance between the center of the paramagnetic electron density of MTSL and the α-carbon of the peptide backbone is 0.7 nm (7 Å), and the radius of the helix itself is 0.5 nm (5 Å). A change in the MTSL-Co(II) distance of 1 nm could be obtained purely by rotation around the axis of the helix; the minimum angle of rotation for which this would be possible is ~ 50 °. The addition of translational components would require correspondingly smaller minimum angles of rotation to achieve the necessary change in interspin distance. Again, further studies will be required to address the detailed nature of the conformational change.
The models for the imipenem-induced structural changes in ImiS, although very different, each require movement, rotation, or both of the 140–152 helix in response to imipenem and indicate a role for the 140–152 helix in the substrate binding process. Intriguingly, strong evidence for the importance of the protein itself in substrate binding, rather than the metal center alone, comes from comparison of the changes of the EPR signals from MTSL in Co-ImiS* with those of Co(II). As in earlier work, the catalytically-competent intermediate Co(II) species accounted for only ~ 10 % of the Co(II) in Co-ImiS* after reaction with imipenem for 12 ms. In contrast, EPR of the spin label showed that over 90 % of the molecules in this sample had adopted the ‘bound’ conformation, in which Co(II) and MTSL are distant from each other; more than 35 % of the molecules had shifted conformation. These data provide solid evidence for a binding mechanism of imipenem by ImiS that does not involve the catalytic metal center but involves a significant conformational change of the enzyme molecule.
These studies have implications for the design of inhibitors of MβLs with possible use in the clinic to combat antibiotic strains of bacteria. While spectrokinetic studies do not yield the kind of high-resolution structural information available from X-ray diffraction, they are applicable to trapped reaction intermediates and allow direct investigation of these species. The present work has identified an event in substrate binding that is associated with a conformational change in the ImiS enzyme molecule and that is distinct from any binding to the active site metal ion. The active sites of metallohydrolases in general and the MβLs in particular share common features and are unlikely to be able to confer the level of substrate specificity observed in these enzymes. The present work suggests that substrate binding by the organic part of the protein molecule, that is reported by a catalytically competent conformational change involving the 140–152 conserved helix, is the first step in the catalytic reaction; it is perhaps at this point that substrate specificity is conferred upon the reaction. One can speculate that the binding of a recognized substrate displaces the conserved helix from a position that hitherto prevented access to the active site; the data presented here are certainly consistent with that scenario. In that case, then, in the search for a rationale for the design of anti-MβL inhibitors, the present work suggests that more attention should be placed on the nature of the interaction of substrates and inhibitors with the organic part of the protein, in addition to the continuing studies of interactions with the active sites in MβLs. A combined approach may ultimately be the most successful one.36
Acknowledgments
This work was supported by the National Institutes of Health (EB001980, AI056231 to B.B., GM40052 to M.W.C.)
References
- 1.(a) Crowder MW, Spencer J, Vila A. Acc Chem Res. 2006;10:721–728. doi: 10.1021/ar0400241. [DOI] [PubMed] [Google Scholar]; (b) Heinz U, Adolph HW. Cell Mol Life Sci. 2004;61:2827–39. doi: 10.1007/s00018-004-4214-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walsh TR, Toleman MA, Poirel L, Nordmann P. Clin Microbiol Rev. 2005;18:306–25. doi: 10.1128/CMR.18.2.306-325.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Daiyasu H, Osaka K, Ishino Y, Toh H. FEBS Lett. 2001;503:1–6. doi: 10.1016/s0014-5793(01)02686-2. [DOI] [PubMed] [Google Scholar]
- 4.Ullah JH, Walsh TR, Taylor IA, Emery DC, Verma CS, Gamblin SJ, Spencer J. J Mol Biol. 1998;284:125–136. doi: 10.1006/jmbi.1998.2148. [DOI] [PubMed] [Google Scholar]
- 5.Concha NO, Janson CA, Rowling P, Pearson S, Cheever CA, Clarke BP, Lewis C, Galleni M, Frere JM, Payne DJ, Bateson JH, Abdel-Meguid SS. Biochemistry. 2000;39:4288–4298. doi: 10.1021/bi992569m. [DOI] [PubMed] [Google Scholar]
- 6.Fitzgerald PMD, Wu JK, Toney JH. Biochemistry. 1998;37:6791–6800. doi: 10.1021/bi9730339. [DOI] [PubMed] [Google Scholar]
- 7.Toney JH, Fitzgerald PMD, Grover-Sharma N, Olson SH, May WJ, Sundelof JG, Vanderwall DE, Cleary KA, Grant SK, Wu JK, Kozarich JW, Pompliano DL, Hammond GG. Chemistry and Biology. 1998;5:185–196. doi: 10.1016/s1074-5521(98)90632-9. [DOI] [PubMed] [Google Scholar]
- 8.Scrofani SDB, Chung J, Huntley JJA, Benkovic SJ, Wright PE, Dyson HJ. Biochemistry. 1999;38:14507–14514. doi: 10.1021/bi990986t. [DOI] [PubMed] [Google Scholar]
- 9.Cameron AD, Ridderstrom M, Olin B, Mannervik B. Structure. 1999;7:1067–1078. doi: 10.1016/s0969-2126(99)80174-9. [DOI] [PubMed] [Google Scholar]
- 10.de la Sierra-Gallay IL, Pellegrini O, Condon C. Nature. 2005;433:657–661. doi: 10.1038/nature03284. [DOI] [PubMed] [Google Scholar]
- 11.Toney JH, Hammond GG, Fitzgerald PMD, Sharma N, Balkovec JM, Rouen GP, Olson SH, Hammond ML, Greenlee ML, Gao Y-D. J Biol Chem. 2001;276:31913–31501. doi: 10.1074/jbc.M104742200. [DOI] [PubMed] [Google Scholar]
- 12.Huntley JJA, Fast W, Benkovic SJ, Wright PE, Dyson HJ. Protein Sci. 2003;12:1368–1375. doi: 10.1110/ps.0305303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Y, Keeney D, Tang XJ, Canfield N, Rasmussen BA. J Biol Chem. 1999;274:15706–15711. doi: 10.1074/jbc.274.22.15706. [DOI] [PubMed] [Google Scholar]
- 14.Moali C, Anne C, Lamotte-Brasseur J, Groslambert S, Devreese B, Van Beeumen J, Galleni M, Frere JM. Chemistry & Biology. 2003;10:319–329. doi: 10.1016/s1074-5521(03)00070-x. [DOI] [PubMed] [Google Scholar]
- 15.Garrity JD, Pauff JM, Crowder MW. J Biol Chem. 2004;279:39663–39670. doi: 10.1074/jbc.M406826200. [DOI] [PubMed] [Google Scholar]
- 16.Dal Peraro M, Carloni P, Vila AJ. Biophys J. 2002;82:2141. [Google Scholar]
- 17.Walsh TR, Neville WA, Haran MH, Tolson D, Payne DJ, Bateson JH, MacGowan AP, Bennett PM. Antimicrob Agents Chemother. 1998;42:436–439. doi: 10.1128/aac.42.2.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crawford PA, Sharma N, Chandrasekar S, Sigdel T, Walsh TR, Spencer J, Crowder MW. Prot Express Purif. 2004;36:272–279. doi: 10.1016/j.pep.2004.04.017. [DOI] [PubMed] [Google Scholar]
- 19.Crawford PA, Yang K-W, Narayan S, Bennett B, Crowder MW. Biochemistry. 2005;44:5168–5176. doi: 10.1021/bi047463s. [DOI] [PubMed] [Google Scholar]
- 20.Garau G, Bebrone C, Anne C, Galleni M, Frere JM, Dideberg O. J Mol Biol. 2005;345:785–795. doi: 10.1016/j.jmb.2004.10.070. [DOI] [PubMed] [Google Scholar]
- 21.Sharma NP, Hajdin C, Chandrasekar S, Bennett B, Yang K-W, Crowder MW. Biochemistry. 2006;45:10729–10738. doi: 10.1021/bi060893t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bray RC. Biochem J. 1961;81:189–195. doi: 10.1042/bj0810189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar A, Periyannan GR, Narayanan B, Kittell AW, Kim J-J, Bennett B. Biochem J. 2007;403:527–536. doi: 10.1042/BJ20061591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hubbell WL, Cafiso DS, Altenbach C. Nat Struct Biol. 2007;7:735–739. doi: 10.1038/78956. [DOI] [PubMed] [Google Scholar]
- 25.Hustedt EJ, Smirnov AI, Laub CF, Cobb CE, Beth AH. Biophys J. 1997;74:1861–1877. doi: 10.1016/S0006-3495(97)78832-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eaton SS, Eaton GR. Biol Magn Reson. 2001;19:1–28. [Google Scholar]
- 27.Hanson GR, Gates KE, Noble CJ, Mitchell A, Benson S, Griffin M, Burrage K. XSophe-Sophe-XeprView: A computer simulation software suite for the analysis of continuous wave EPR spectra. In: Shiotani M, Lund A, editors. EPR of Free Radicals in Solids: Trends in Methods and Applications. Kluwer Press; Dordrecht: 2003. pp. 197–237. [Google Scholar]
- 28.Garrity JD, Bennett B, Crowder MW. Biochemistry. 2005;44:1078–1087. doi: 10.1021/bi048385b. [DOI] [PubMed] [Google Scholar]
- 29.Rabenstein MD, Shin Y-K. Proc Natl Acad Sci USA. 1995;92:8239–8243. doi: 10.1073/pnas.92.18.8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lowe DJ, Lynden-Bell RM, Bray RC. Biochem J. 1972;130:239–249. doi: 10.1042/bj1300239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bertrand P, More C, Guigliarelli B, Fournel A, Bennett B, Howes B. J Am Chem Soc. 1994;116:3078–3086. [Google Scholar]
- 32.Lynden-Bell RM, Harris RK. Nuclear Magnetic Resonance Spectroscopy. Nelson; London: 1969. [Google Scholar]
- 33.Abragam A, Bleaney B. Electron Paramagnetic Resonance of Transition Ions. Clarendon; Oxford: 1970. [Google Scholar]
- 34.Langen R, Oh KJ, Cascio D, Hubbell WL. Biochemistry. 2000;39:8396–8405. doi: 10.1021/bi000604f. [DOI] [PubMed] [Google Scholar]
- 35.Altenbach C, Oh K-J, Trabanino RJ, Hideg K, Hubbell WL. Biochemistry. 2001;40:15471–15482. doi: 10.1021/bi011544w. [DOI] [PubMed] [Google Scholar]
- 36.Kurosaki H, Yamaguchi Y, Higashi T, Soga K, Matsueda S, Yumoto H, Misumi S, Yamagata Y, Arakawa Y, Goto M. Angew Chem Int Ed Engl. 2005;44:3861–3864. doi: 10.1002/anie.200500835. [DOI] [PubMed] [Google Scholar]