

Abstract
Objective
Collagen antibody-induced arthritis (CAIA) in mice exhibits a requirement for amplification by the alternative pathway (AP) of complement. Although the AP is activated by spontaneous hydrolysis, it is not known whether the AP can also be initiated directly by IgG antibodies in immune complexes (IC). IgG lacking terminal sialic acid and galactose (G0-IgG) can activate the lectin pathway (LP) of complement, but it is not known if G0-IgG can also activate the classical pathway (CP) or AP.
Methods
To examine initiation of the AP by IC, we used adherent IC containing bovine collagen type II (CII) and four monoclonal Ab (mAb) to CII, adCII-IC. C3 activation was measured in the presence of sera from wild type C57BL/6 mice or from mice deficient in informative complement components. The mAb were used intact or after enzyme digestion to create G0-IgG or to completely remove the N-glycan.
Results
Both the CP and the AP, but not the LP, mediated C3 activation induced by the adCII-IC. Mannose inhibited the AP-mediated C3 activation but had no effect on the CP, and N-glycans in IgG were required by the AP but not the CP. The CP and AP both mediated C3 activation by G0-IgG. MBL bound avidly to G0-IgG, but LP-mediated C3 activation was only slightly increased by G0-IgG.
Conclusion
The AP is capable of initiating C3 activation induced by adCII-IC and requires N-glycans on the IgG. G0-IgG activates both the CP and AP more strongly than the LP.
Keywords: Complement, immune complexes, rheumatoid arthritis
Immune complex (IC)5 diseases are caused by the deposition in vessel walls, or in the basement membrane of the kidneys, of preformed soluble antigen-antibody complexes, or the in situ formation of adherent IC (adIC) from the binding of antibodies (Ab) to tissue antigens. Tissue damage in IC diseases is mediated in large part by activation of the complement system resulting in the release of complement fragments such as C5a (1).
The complement system consists of three major activation pathways that all converge on C3 with the enzymatic generation of C3b by the classical pathway (CP) and alternative pathway (AP) convertases (2,3). The CP is initiated by IgG or IgM Ab binding C1q, followed by proteolysis of C1r and C1s, cleavage of C4 and C2 by activated C1s, and generation of the CP C3 convertase (C4b2a) that cleaves C3 into C3a and C3b. The AP may be continually activated by a “tickover” mechanism characterized by spontaneous hydrolysis of the thioester bond in native C3 to generate a C3b-like molecule, C3(H2O) (4). Factor B binds this C3b-like molecule in solution and is then cleaved by Factor D, generating an AP C3 convertase (C3(H2O)Bb) that cleaves further C3. The newly formed C3b has a very short half-life and quickly binds to nearby surfaces, including adherent IgG. Both properdin and factor H bind to this adherent C3b, either enhancing or inhibiting the AP activity, respectively (5,6). The AP may function primarily as an amplification loop of C3b after initiation by the CP and the lectin pathway (LP). Whether the AP is capable of primarily initiating complement activation remains unclear. The LP is mediated by a complex of mannose-binding lectin (MBL) and MBL-associated proteases (MASP-1, MASP-2, and MASP-3) binding to terminal fucose, glucose, mannose or N-acetylglucosamine (GlcNAc) residues on the surface of microorganisms or other targets (7). The proteases in the LP resemble C1r and C1s in cleaving C2 and C4 to generate the CP convertase C4b2a. MBL is also involved in an additional mechanism of C3 activation called the C2/C4 bypass pathway where, in the absence of C2 or C4, MBL may directly activate C3 and the AP in a MASP-independent fashion (8).
IgG molecules, either alone or in IC, possess complex biantennary N-glycans linked to Asn 297 on the Fc portion of the heavy chain (Ch2 domain) (9). IgG molecules with N-glycans containing two non-reducing terminal galactose residues are termed G2, with G1-IgG containing one terminal galactose residue, and G0-IgG possessing no terminal galactose residues (10). MBL binds to initiating residues through its carbohydrate recognition domains when both galactose residues are removed, but does not bind to galactose residues. G0-IgG levels are increased in the sera of patients with rheumatoid arthritis with the exposed terminal GlcNAc residues able to bind MBL and activate the LP (11). In addition, IgM and IgA molecules lacking terminal sialic acid and galactose are also capable of binding MBL with activation of the LP (12,13). The relative ability of G0-IgG to activate all 3 complement activation pathways is not known.
Enzymatic removal of all N-glycans from the IgG molecule results in a variable change in the ability to bind C1q, with a loss in C1q binding probably secondary to conformational changes in the Fc portion of the IgG molecule (14–16). A mouse-human chimeric IgG1 molecule that expressed high-mannose intermediate N-glycans, but lacked the terminal glycosylation residues of galactose and sialic acid, exhibited decreased but not absent C1q binding (17). In a more recent study, the N-glycans linked to Asn 297 on a pair of murine IgG2a monoclonal Ab (mAb) either enhanced or inhibited C1q binding, while no effect was observed on the AP (18).
The results of recent studies indicated that the AP of the complement system was required in two experimental animal models of arthritis induced by adherent immune complexes, the K/BxN serum transfer model and passive collagen antibody-induced arthritis (CAIA) (18–21). Our data in mice genetically deficient in factor B showed a near absence of clinical disease in CAIA (20,21). However, the CP and LP generated detectable C3b bound to the synovium and cartilage in factor-B-deficient mice, although presumably at levels too low to induce clinical disease in the absence of amplification by the AP (20). The AP alone mediated robust CAIA in vivo as seen with studies in mice deficient in both C1q and MBL. The AP alone also led to high levels of C3 activation using an in vitro system of adIC containing bovine collagen type II (CII) and a mixture of four murine mAb reactive with CII (adCII-IC) (21). The results of these in vivo and in vitro experiments suggested that the AP alone could initiate complement activation induced by adIC, in addition to its important in vivo role in amplification of bound C3b. The results of our in vitro studies indicated that the adCII-IC initiated C3 activation but this system did not fully display all of the characteristics of amplification by the AP (21). In the experiments described herein, we used this in vitro system to explore the possible mechanisms whereby the AP initiates complement activation induced by IC. Our results indicate an important role for N-glycans on the IgG molecules in this process.
Materials and Methods
Sera from WT and complement-deficient mice
Sera from C57BL/6 mice deficient in genes for specific complement components were obtained from the following sources: MBL−/− (deficient in both MBL-A and MBL-C), MBL−/−/Df−/− (factor D), and C1q−/−/Df−/− mice from Drs. Takahashi and Stahl, or from a colony at UCDHSC; and C1q−/− and C4−/− mice from breeding colonies at UCD. Control sera were obtained from wild type (WT) C57BL/6 mice (Jackson Laboratories). Fresh sera were used in all experiments. All animals were kept in a barrier animal facility at UCD with a climate-controlled environment having 12-hour light/dark cycles. Filter top cages were used with three mice in each cage. During the course of this study, all experimental mice were fed breeder’s chow provided by the Center for Laboratory Animal Care, UCD.
C3 activation induced by adherent immune complexes of collagen and anti-collagen antibodies
The levels of C3 activation induced by adCII-IC in vitro were measured by ELISA. Preparation of the in vitro adCII-IC and analysis of C3 deposition on the IC were carried out as described using veronal buffered saline (0.14 M NaCl, 1.8 mM Sodium Barbital, 1 mM MgCl2, and 2 mM CaCl2) (21). In all experiments, C3 activation was also measured under identical conditions by using adherent CII alone without anti-collagen mAb. Data were expressed by using the following formula: mean OD of adCII-IC minus OD of CII alone. C3 activation by the adherent adCII-IC in the presence or absence of an intact AP was examined by using a specific mAb to murine factor B (1379) (22) incubated at 40 μg/ml with 1:10 dilutions of sera from WT or complement-deficient mice for 10 min prior to addition to the adCII-IC.
Effect of mannose on C3 activation by immune complexes of collagen and anti-collagen antibodies
Soluble mannose inhibits the binding of MBL to mannose-containing carbohydrates in plasma membranes or adherent substrates. To examine the effects of mannose on C3 activation induced by adCII-IC, serially increasing amounts of mannose (0 – 200 mM) were added to the adCII-II before incubation with sera from WT mice or mice deficient in various complement components. C3 deposition on the IC was measured as described (21). All fragments of C3 were detected.
MBL binding to anti-collagen Ab
A 96-well Nunc ELISA plate was coated with a cocktail of four anti-CII mAb at 25 ug/ml in 0.1M sodium carbonate buffer pH 9.5 followed by incubation at room temperature for 24 h. The plates were then washed 7× with PBS and 0.05% Tween-20 followed by blocking with 200 ul/well of PBS with 1% BSA and 0.05% Tween-20 during incubation at RT for another 1 h. After washing the ELISA plate 7×, serum samples (100 ul/well) diluted 1:10 in sodium barbital buffer (0.14 M NaCl, 4 mM Sodium Barbital, 1 mM MgCl2, 2 mM CaCl2, and 7.5 mM NaN3) were added and the plate was incubated for 2 h at room temperature. After washing 7× with PBS and 0.5% Tween-20, a mixture of biotinylated rat IgG anti-MBL-A Ab (11 μg/ml) and anti-MBL-C Ab (15 μg/ml) in PBS with 1% BSA (Sigma ELISA grade) was added to the wells. Both of these Ab were provided by Dr. J.C. Jensenius (Aarhus, Denmark). The ELISA plate was incubated at 4°C for 24 h. HRP-conjugated streptavidin (R & D Systems) diluted 1:250 in PBS with 1% BSA and 0.05% Tween-20 was added, followed by incubation for 90 min at room temperature. After seven more washings, the color reaction was developed for 30 min by adding 100 ul/well of tetramethylbenzidine substrate reagent mix (1:1). The reaction was stopped by adding 50 ul/well of a 2N H2SO4 solution followed by absorbance at 450 nm corrected for background by absorbance at 550 nm. MBL demonstrated binding to plates coated with CII alone, so these experiments were carried out with plates coated with IgG anti-collagen mAb alone and not with adCII-IC.
Enrichment in G0-IgG or removal of all N-glycans from anti-collagen mAb
The G2, G1, and G0 content in three different murine IgG preparations was determined by HPLC combined with exoglycosidase array digestions (23). The Arthrogen preparation possessed 19.6% of the glycans as G0 (Table 1). Removal of N-glycans from the anti-collagen mAb (25 ug/ml) was performed by incubation with glycosidases (Sigma-Aldrich) in 50 mM sodium acetate buffer pH 5.5 in an Eppendorf tube at 37°C for 30 h., according to the standard protocol provided by the manufacturer with modifications. To create G0-IgG, sialic acid and β-galactose were cleaved from the anti-collagen monoclonal Ab using a mixture (10 mUnits each) of neuraminidase (EC 3.2.1.18, from Streptococcus pneumoniae) and β-D-galactosidase galactohydrolase (EC 3.2.1.23, from Saccharomyces fragilis). To remove all the N-glycans, anti-collagen mAb were digested with 5 mUnits N-glycosidase (PNGase F) (EC 3.5.1.52, from Chryseobacterium (Flavobacterium) meningsepticum). After digesting the mAb with glycosidases, enzymatic activity was stopped by heating at 65°C for 5 min. Control anti-collagen mAb were incubated with sodium acetate buffer, adding the enzyme buffer without any enzymes. The IgG digested with PNGase F showed the expected decrease in size, as detected by Western blot analyses (data not shown), with no N-glycans detected by matrix-assisted laser-desorption ionization time-of-flight mass spectrometry (MALDI/TOF-MS) (Complex Carbohydrate Research Center, Athens GA). The control IgG mAb and samples after each enzyme digestion were used to prepare adCII-IC with subsequent C3 activation measured as described (21). Exposure of the adherent CII alone to enzymes led to no detectable C3 deposition over CII alone. All 3 IgG preparations bound to the CII-coated plates to the same degree as determined by detection with goat anti-mouse IgG. The cleaved residues and inactive enzyme were removed from the adherent IgG by repeated washings.
Statistics
Statistical analyses were carried using Student’s t test used to determine levels of significance.
Results
C3 activation by adCII-IC and effects of mannose
C3 activation was measured after induction by adCII-II in the presence of sera from WT mice or from mice deficient in various complement components. Equivalent levels of C3 deposition on IC were observed using sera from WT, C4−/−, C1q−/−, or MBL−/−/Df−/− mice (Fig. 1). The C4−/− sera possess only the AP as C4 is necessary for both the CP and LP. C1q−/− sera possess intact AP and LP, and MBL−/−/Df−/− sera possess only an intact CP. Sera from C1q−/−/Df−/− mice, possessing only the LP, failed to exhibit any C3 activation induced by the adCII-II. These results indicated that both the CP and AP, but not the LP, were capable of mediating C3 activation induced by adCII-IC.
Figure 1.

C3 activation by adCII-IC incubated with WT or complement deficient sera and inhibition by mannose. AdCII-IC were incubated in the indicated concentrations of mannose prior to addition of mouse sera and measurement of C3 deposition on the IC by ELISA. The data are expressed as C3 deposition in OD values at 450 nm, mean ± SEM based on n = 3. Comparing C3 deposition in the presence of mannose vs. no added mannose: * p < 0.01, ** p < 0.001.
To assess the role of lectin interactions, serial amounts of mannose (0 – 200 mM) were added to the adCII-II before addition of the various sera and assessment of C3 activation. Increasing amounts of mannose exhibited progressive inhibition of adCII-IC-induced C3 activation mediated by the AP (using sera from C4−/− or C1q−/− mice) (Fig. 1). Mannose exhibited a partial inhibition of adCII-IC-induced C3 activation using WT sera but no inhibition was observed with sera from MBL−/−/Df−/− mice where only the CP was intact. The results of C3 activation induced by adherent mannan showed that the LP was active in sera from C1q−/−/Df−/− mice and was progressively inhibited by mannose (data not shown). These results indicated that mannose inhibited adCII-IC-induced C3 activation mediated by the AP but not by the CP and suggested a possible role for N-glycans in initiation of complement activation by the AP.
MBL binding to anti-collagen Ab
Further experiments were carried out to explore a possible role for N-glycans on the IgG mAb in the adCII-IC in C3 activation mediated by the three complement activation pathways. To confirm that MBL was binding to G0-IgG in the mixture of mAb to CII, binding studies were performed with the IgG anti-CII mAb alone bound to wells of microtiter plates. AdCII-IC were not examined in these studies because the MBL bound to the CII alone in vitro. Sera from WT, C1q−/−/Df−/−, and C4−/− mice displayed high levels of MBL binding to the untreated IgG anti-CII mAb whereas sera from MBL−/− mice exhibited no binding (Fig. 2).
Figure 2.
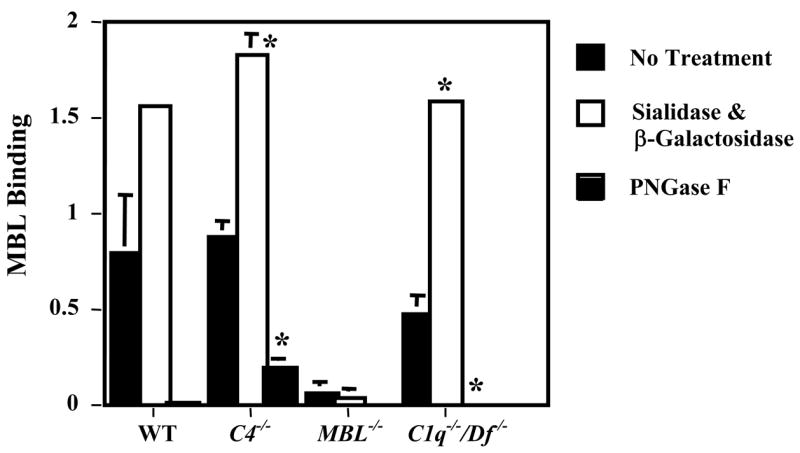
MBL binding to plates coated with IgG anti-collagen antibodies. MBL binding was determined in the presence of one to ten dilutions of sera from WT, C4−/−, MBL−/− or C1q−/−/Df−/− mice before and after treatment with a mixture of sialadase and β-galactosidase, to remove terminal sialac acid and galactose from the N-glycans, or with PNGase F, to remove all of the N-glycans. The data are expressed as MBL binding in OD units at 450 nm, mean ± SEM based on n = 3 for all sera. * p = 0.003 compared with no enzyme treatment. This experiment was repeated 3 times with identical results.
To further explore the carbohydrate specificity of MBL binding, IgG anti-CII mAb were enzymatically treated to remove the terminal sialic acid and galactose residues from the N-glycans or to completely remove all of the N-glycans. The IgG anti-CII mAb lacking sialic acid and galactose (G0-IgG) exhibited increased binding of MBL using sera from WT or C4−/− mice but removal of all N-glycans from the IgG eliminated most MBL binding (Fig. 2). Similar results were obtained using sera from C1q−/−/Df−/− mice where only the LP is active. The sera from MBL−/− mice exhibited no MBL binding using control (untreated) or enzymatically-treated IgG. These results suggested that MBL bound to G0-IgG mAb to CII in the adCII-IC.
C3 activation by adCII-IC
To examine for a role of N-glycans on the IgG mAb present in anti-CII-IC to activate complement, C3 activation was examined using sera from WT, C4−/−, MBL−/−, and C1q−/−/Df−/− mice. The IgG anti-CII mAb in adCII-IC were untreated, treated with specific enzymes to remove terminal sialic acid and galactose residues, or treated with PNGase F to remove all N-glycans. The results showed high levels of C3 activation by the untreated IgG, IgG lacking terminal sialic acid or galactose (G0-IgG), or IgG lacking all N-glycans in the presence of sera from WT or MBL−/− mice (Fig. 3A). The sera from C4−/− mice (AP only) exhibited no change in C3 activation when stimulated by G0-IgG; however, a near absence in C3 activation was observed with IgG lacking any N-glycans. Lastly, the sera from C1q−/−/Df−/− mice (LP only) exhibited a low level of C3 activation only after enrichment for G0-IgG mAb in the adCII-IC.
Figure 3.
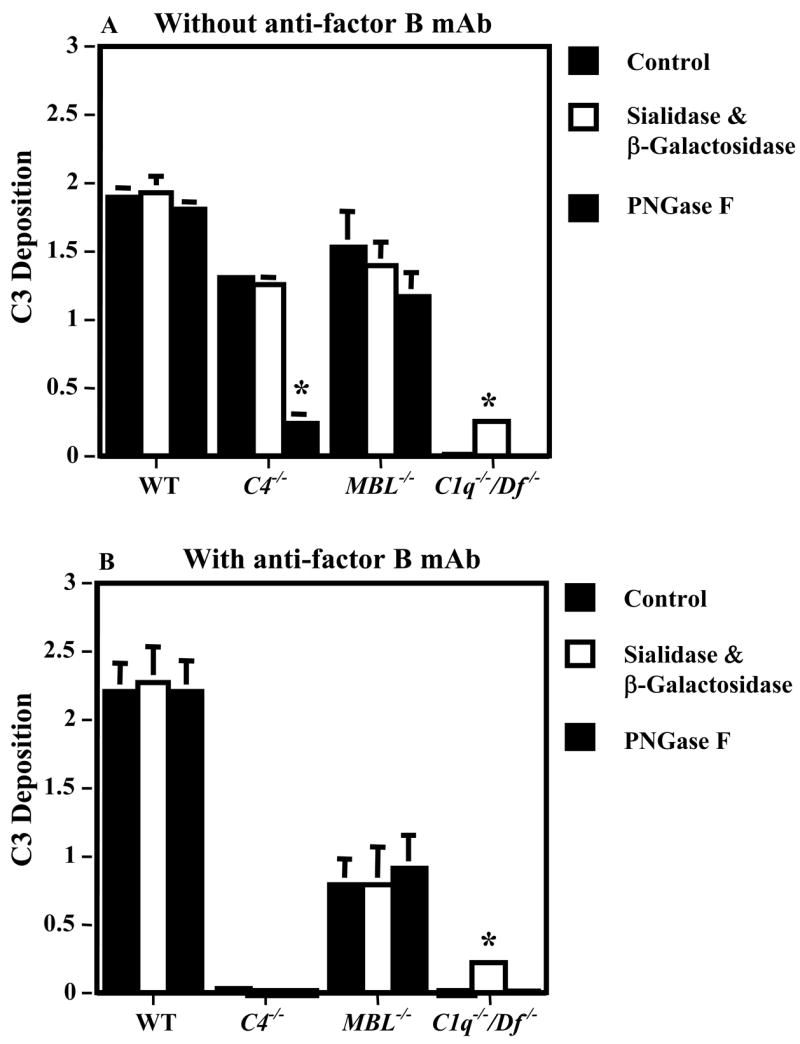
Effect of blockade of the AP on C3 activation induced by plates coated with adCII-IC. Plates coated with adCII-IC were incubated with sera from WT, C4−/−, MBL−/− or C1q−/−/Df−/− mice. The IgG mAb preparations in the IC were untreated, treated with a mixture of sialadase and β-galactosidase, to remove terminal sialac acid and galactose from the N-glycans, or treated with PNGase F, to remove all of the N-glycans. C3 deposition was determined by ELISA and the data are expressed as OD values at 450 nm, mean ± SEM based on n = 3 for all sera. A. The sera were not pre-incubated with anti-factor B mAb and the AP was intact. B. The sera were pre-incubated with a neutralizing mAb to murine factor B to block the AP. * p < 0.001 compared with no enzyme treatment. This experiment was repeated 3 times with different preparations of enzyme-treated IgG with identical results.
To further explore a possible influence of N-glycans in the IgG mAb on initiation of complement activation by the AP, this experiment was repeated using an anti-factor B mAb. The results showed that inhibition of the AP by the anti-factor B mAb completely suppressed any adCII-IC-induced C3 activation mediated by the C4−/− sera (AP only) before enzyme treatment of the IgG mAb in the IC, with G0-IgG, and after removal of all N-glycans (Fig. 3B). Depletion of factor B in the WT sera led to no change in C3 activation using all three forms of IgG mAb in the IC. However, MBL−/− sera (intact CP and AP), in the presence of the mAb to factor B, exhibited a 65% decrease in C3 activation induced by all three forms of IgG mAb in the IC. The sera from C1q−/−/Df−/− mice, possessing only an intact LP, exhibited a low level of C3 activation after enrichment for G0-IgG mAb to CII in the adCII-IC.
These results indicated that the AP alone was capable of initiating C3 activation induced by adCII-IC. Initiation of complement activation by the AP was dependent on N-glycans on IgG in the adCII-IC, but terminal sialic acid and galactose were not required. The AP was solely responsible for initiation of adCII-IC-induced C3 activation in the absence of the CP or the traditional LP (i.e. using C4−/− sera); thus, the MBL-dependent C4 bypass pathway of C3 activation played no role. In the absence of the LP (MBL−/− sera), the AP mediated twice the level of adCII-IC-induced C3 activation in comparison to the CP. In contrast to the AP, N-glycans played no role in IgG activation of the CP. Lastly, the LP (C1q−/−/Df−/− sera) exhibited a low level of adCII-IC-induced C3 activation only after enrichment for G0-IgG in the mAb. The relative ability of G0-IgG mAb to CII in adCII-IC to induce C3 activation by the three pathways of complement activation appeared to be AP > CP > LP.
Discussion
The experiments reported herein indicate that the AP is fully capable of initiating C3 activation induced by adCII-IC in vitro. When both the CP and AP are intact, C3 activation appears to be initiated by the AP at twice the level observed with the CP. Initiation of the complement system by the AP, but not the CP, requires the presence of N-glycan on the IgG molecule. Generation of G0-IgG leads to a low level of C3 activation using the LP. However, both the CP and AP are also activated by G0-IgG generating considerably more C3 activation than seen with the LP.
Amplification of C3b deposition by the AP is required to produce synovitis in CAIA, and in other models of adherent IC disease, after initiation of the complement system potentially by all three pathways. However, whether the AP is capable of primarily initiating complement activation as opposed to amplification, has remained unclear. The AP is thought to exhibit low-grade continuous activation by spontaneous hydrolysis, termed the “tickover” mechanism (3). The C3b generated by this mechanism binds via covalent interactions to amino or hydroxyl groups on nearby surfaces as well as to soluble or adherent IgG. Amplification by the AP results in further C3 cleavage induced by factor B in the presence of factor D.
The role of antibody in activation of the AP has been reviewed (24) with the best studied example being the solubilization of IC (25). These experiments indicated that the AP could both primarily initiate and amplify C3b deposition in immune precipitates, leading to solubilization. However, initiation of C3b deposition by the CP greatly accelerated the rate of solubilization (26). IC-induced activation by the CP has been assumed to be of primary importance in human diseases, until recent evidence has shown the importance of the AP (2).
The AP exists in a dynamic state of equilibrium with activation by factor B enhanced by properdin and inhibited by factors H and I (4,5). Disruption of a balance between activation and inhibition of the AP may lead to disease as exemplified by the association of factor H deficiency with age-related macular degeneration and atypical hemolytic uremic syndrome (27,28). The mechanism of IgG induction of the AP, and the necessity for N-glycans in this process, remains unknown. Possible mechanisms for N-glycans in IgG influencing the initiation of complement by the AP may occur at four different points: binding of C3b to IgG, binding of properdin to C3b, binding of factor B to C3b, or binding of factor H to C3b.
Multiple binding sites for C3b exist on the heavy chains of the Fc portion of IgG, in both the Ch1 and Ch2 domains (29). Binding of C3b to IgG occurs via both ester and amide linkages, with a serial dimer of two C3b molecule favored. Although suggested by the results of early studies (30), C3b appears not to bind directly to the N-glycans linked to Asn 297 in the Ch2 domain of the Fc portion. In contrast, C3b binds avidly to certain terminal sugars in polysaccharides on the surface of bacteria (31). C3b-C3b dimers bound to IgG are protected from inactivation by factor I possibly through strong binding of properdin to the dimers and steric hindrance of one of the factor H binding sites (32). The effect of N-glycans in IgG on C3b binding is not known although the possibility exists that N-glycans are necessary to maintain proper conformation of the IgG. The results of one study suggest that N-glycans can influence the CP but not the AP (18). However, variations in experimental conditions may show differences in dependency of the AP on N-glycans. In contrast, removal of N-glycans from IgG markedly inhibits Fc binding to FcR (14–16). The mechanisms of oligosaccharide interactions with Ch2 residues and of the effects on Fc functions have been discussed (33).
Studies on the X-ray structure of C3b reveal marked conformational changes after enzymatic cleavage of C3 to C3b with exposure of the internal thioester bond and of binding sites for properdin, factor B, and factor H (34). Properdin is a positive regulator of complement activation that stabilizes the AP convertases (C3bBb). Binding of properdin to C3b on a RBC surface occurs before the binding of factor B to C3b and greatly enhances this interaction (35). Furthermore, properdin inhibits factor I binding to and action on cell-bound C3b but does not compete with binding of factor B or factor H (35,36). Properdin binds to C3b on a single site located within residues 1402–1435 on the C-terminus of the α-chain (37). Furthermore, properdin binds avidly to sulfated glycoconjugates, theoretically increasing the affinity of properdin for C3b if appropriate sulfated glycans are located nearby (38). These glycans could be on C3b itself, or could originate from the IgG or surface to which the C3b was bound. Hypothetically, the requirement for N-glycans in AP initiation by IgG observed in our studies could be due to enhancement of properdin binding to C3b. Lastly, the results of recent studies indicate that properdin may initiate complement activation by primarily binding to microbial surfaces through C3b, iC3b, or other ligands, then binding more C3b or C3bBb through its unoccupied site with further in situ assembly of AP convertases (39,40). It is not known if IgG could offer such a site for primary binding of properdin, or what might be the role of N-glycans on IgG in this proposed initiation mechanism for the AP.
Factor B may bind to C3b in the region between residues 727 and 768, although other sites on C3b may influence this binding (34,41). The possible influence of N-glycans on Asn 297 of IgG on factor B binding are unknown although these polysaccharides linked to IgG may enhance the binding of factor B to C3b through secondary interactions.
Factor H regulates the complement system by acting as a cofactor for factor I-mediated cleavage of C3b and by accelerating the decay of the AP C3 and 5 convertases. Factor H is present at a high concentration in plasma, ~ 500 μg/ml, and binds to multiple polyanions on cell surfaces to protect them from attack by the AP (42). Although factor H possesses three binding sites for C3b, the C-terminal domains 19–20 offer the most critical binding site (43). The presence of polyanions on cells greatly enhances the binding of factor H to C3b through domains 19–20 (43). Three sites exist on C3b for binding of factor H, with two sites partially overlapping with factor B, factor H domains 19–20, and CR1 where all 3 molecules may bind to a site on C3d (34,44). Although factor H binds to glycosaminoglycans (45), whether factor H binding to C3b could be sterically inhibited by N-glycans on IgG is unknown.
The results of our in vitro studies clearly show that G0-IgG in adherent anti-CII mAb can activate C3 through both the CP and AP. RA is associated with an increased prevalence of G0-IgG molecules (11,46). G0-IgG was concluded to be pathogenic in an experimental model of inflammatory arthritis as passive transfer of agalactosyl isoforms of polyclonal anti-CII Ab to mice primed to CII induced more disease than did transfer of the untreated Ab (47). IgG RF from patients with RA demonstrated self-association with formation of cyclic dimers (48,49). It has been hypothesized that increased amounts of G0-IgG in IgG RF may predispose to more self-association with the potential to increase pathogenicity (50).
The AP plays an important pathophysiologic role in multiple immunologic diseases, some involving adherent immune complexes (3). Although anti-CII antibodies may not play a primary role in the pathogenesis of RA, studies on adCII-IC may provide important information on pathogenic mechanisms of adherent IC in general. The overall results of our studies indicate that in addition to the key role played by the amplification loop of the AP in experimental models of arthritis, the AP may primarily initiate complement activation. Our observations that this process is dependent on N-glycans in IgG provide a foundation for further studies on the involved mechanism. Additional studies are in progress evaluating all three pathways of complement in the initiation of activation in vivo.
Table I.
Galactose content of the N-glycans from various murine IgG preparations
| G0 | G1 | G2 | |
|---|---|---|---|
| (% of total glycans)* | |||
| Normal mouse IgG | 35.6 | 45.6 | 19.1 |
| mAb to human CR2 | 33.8 | 39.3 | 19.3 |
| mAb to collagen | 19.6 | 47.9 | 31.5 |
Calculated after all samples were digested with sialadase.
Acknowledgments
We thank Drs. Yuanyuan Ma (University of Alabama at Birmingham) and Marina Botto (Imperial College, London) for the original colonies of factor Df−/− and C1q−/− mice.
Supported by NIH grant AR51749.
References
- 1.Guo R-F, Ward PA. Role of C5a in inflammatory responses. Annu Rev Immunol. 2005;23:821–52. doi: 10.1146/annurev.immunol.23.021704.115835. [DOI] [PubMed] [Google Scholar]
- 2.Walport MJ. Complement. New Engl J Med. 2001;244:1058–66. 1140–4. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 3.Thurman JM, Holers VM. The central role of the alterative complement pathway in human diseases. J Immunol. 2006;176:1305–10. doi: 10.4049/jimmunol.176.3.1305. [DOI] [PubMed] [Google Scholar]
- 4.Pangburn MK, Schreiber RD, Müller-Eberhard HJ. Formation of the initial C3 convertase of the alternative complement pathway. Acquisition of the C3b-like activities by spontaneous hydrolysis of the putative thioester in native C3. J Exp Med. 1981;154:856–67. doi: 10.1084/jem.154.3.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fearon DT, Austen KF. Properdin: Initiation of alternative complement pathway. Proc Natl Acad Sci USA. 1975;72:3220–4. doi: 10.1073/pnas.72.8.3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whaley K, Ruddy S. Modulation of the alternative complement pathway by β1H globulin. J Exp Med. 1976;144:1147–63. doi: 10.1084/jem.144.5.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fujita T. Evolution of the lectin – complement pathway and its role in innate immunity. Nature Rev Immunol. 2002;2:346–53. doi: 10.1038/nri800. [DOI] [PubMed] [Google Scholar]
- 8.Selander B, Mårtensson U, Weintraub A, Holström E, Matshshita M, Thiel S, et al. Mannan-binding lectin activates C3 and the alternative complement pathway without involvement of C2. J Clin Invest. 2006;116:1425–34. doi: 10.1172/JCI25982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arnold JN, Wormald MR, Sim RB, Rudd PM, Dwek RA. The impact of glycosylation on the biological structure of human immunoglobulins. Annu Rev Immunol. 2007;25:21–50. doi: 10.1146/annurev.immunol.25.022106.141702. [DOI] [PubMed] [Google Scholar]
- 10.Dwek EA, Lellouch AC, Wormald MR. Glycobiology: The function of the sugar in the IgG molecule. J Anat. 1995;187:279–92. [PMC free article] [PubMed] [Google Scholar]
- 11.Malhotra R, Wormald MR, Rudd PM, Fischer PB, Dwek RA, Sim RB. Glycosylation changes of IgG associated with rheumatoid arthritis can activate complement via the mannose-binding protein. Nature Medicine. 1995;1:237–43. doi: 10.1038/nm0395-237. [DOI] [PubMed] [Google Scholar]
- 12.Arnold JN, Wormald MR, Suter DM, Radcliffe CM, Harvey DJ, Dwek RA, et al. Human serum IgM glycosylation. Identification of glycoforms that can bind to mannan-binding lectins. 2005;280:29080–29087. doi: 10.1074/jbc.M504528200. [DOI] [PubMed] [Google Scholar]
- 13.Terai I, Kobayashi K, Vaerman J-P, Mafune N. Degalactosylated and/or denatured IgA, but not native IgA in any form, bind to mannose-binding lectin. 2006;177:1737–45. doi: 10.4049/jimmunol.177.3.1737. [DOI] [PubMed] [Google Scholar]
- 14.Nose M, Wigzell H. Biological significance of carbohydrate chains on monoclonal antibodies. Proc Natl Acad Sci USA. 1983;80:6632–36. doi: 10.1073/pnas.80.21.6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leatherbarrow RJ, Rademacher TW, Dwek RA, Woof JM, Clark A, Burton DR, et al. Effector functions of a monoclonal aglycosylated mouse IgG2a: binding and activation of complement component C1 and interaction with human monocyte Fc receptor. Molecular Immunol. 19895;22:407–15. doi: 10.1016/0161-5890(85)90125-7. [DOI] [PubMed] [Google Scholar]
- 16.Tao M-H, Morrison SL. Studies of aglycosylated chimeric mouse-human IgG. Role of carbohydrate ion the structure and effector functions mediated by the human IgG constant region. J Immunol. 1989;143:2595–601. [PubMed] [Google Scholar]
- 17.Wright A, Morrison SL. Effect of altered CH2-associated carbohydrate structure on the functional properties and in vivo fate of chimeric mouse-human immunoglobulin G1. J Exp Med. 1994;189:1087–96. doi: 10.1084/jem.180.3.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.White KD, Cummings RD, Waxman FJ. Ig N-glycan orientation can influence interactions with the complement system. J Immunol. 1997;158:426–35. [PubMed] [Google Scholar]
- 19.Hong J, Ohmura K, Mahmood U, Lee DM, Hofhuis FMA, Boackle SA, et al. Arthritis critically dependent on innate immune system players. Immunity. 2002;16:157–68. doi: 10.1016/s1074-7613(02)00275-3. [DOI] [PubMed] [Google Scholar]
- 20.Banda NK, Thurman JM, Kraus D, Wood A, Carroll MC, Arend WP, et al. Alternative complement pathway activation is essential for inflammation and joint destruction in the passive transfer model of collagen-induced arthritis. J Immunol. 2006;177:1904–12. doi: 10.4049/jimmunol.177.3.1904. [DOI] [PubMed] [Google Scholar]
- 21.Banda NK, Takahashi K, Wood AK, Holers VM, Arend WP. Pathogenic complement activation in collagen antibody induced arthritis requires amplification by the alternative pathway. J Immunol. 2007;179:4101–9. doi: 10.4049/jimmunol.179.6.4101. [DOI] [PubMed] [Google Scholar]
- 22.Thurman JM, Kraus DM, Girardi G, Hourcade D, Kang HJ, Royer PM, et al. A novel inhibitor of the alternative complement pathway prevents antiphospholipid antibody-induced pregnancy loss in mice. Molecular Immunol. 2005;42:87–97. doi: 10.1016/j.molimm.2004.07.043. [DOI] [PubMed] [Google Scholar]
- 23.Royle L, Radcliffe CM, Dwek RA, Rudd PM. Detailed structural analysis of N-glycans released from glycoproteins in SDS-PAGE gel bands using HPLC combined with endoglycosidase array digestions. In: Brockhausen I, editor. Methods in Molecular Biology, vol 7, Glycobiology Protocols. Vol. 47. Humana Press; Totowa, NJ: 2006. pp. 125–43. [DOI] [PubMed] [Google Scholar]
- 24.Ratnoff WD, Fearon DT, Austen KF. The role of antibody in the activation of the alternative complement pathway. Springer Semin Immunopathol. 1983;6:361–71. doi: 10.1007/BF02116280. [DOI] [PubMed] [Google Scholar]
- 25.Takahashi M, Tack BF, Nussenzweig V. Requirements for the solubilization of immune aggregates by complement. Assembly of a factor B-dependent C3-convertase on the immune complexes. J Exp Med. 1977;145:87–100. doi: 10.1084/jem.145.1.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takahashi M, Takahashi S, Brade V, Nussenzweig V. Requirements for the solubilization of immune aggregates be complement. The role of the classical pathway. J Clin Invest. 1978;62:349–58. doi: 10.1172/JCI109135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prosser BE, Johnson S, Roversi P, Herbert AP, Blaum BS, Tyrrell J, et al. Structural basis for complement factor H-linked age-related macular degeneration. J Exp Med. 2007;204:2277–83. doi: 10.1084/jem.20071069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pickering MC, Goicoechea de Jorge E, Martinez-Barricarte R, Recalde S, Garcia-Layana A, Rose KL, et al. Spontaneous hemolytic uremic syndrome triggered by complement factor H lacking surface recognition domains. J Exp Med. 2007;204:1249–56. doi: 10.1084/jem.20070301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vivanco F, Muñoz E, Vidarte L, Pastor C. The covalent interaction of C3 with IgG immune complexes. Molecular Immunol. 1999;36:843–52. doi: 10.1016/s0161-5890(99)00105-4. [DOI] [PubMed] [Google Scholar]
- 30.Capel PJA, Groeneboer O, Grosveld G, Pondman KW. The binding of activated C3 to polysaccharides and immunoglobulins. J Immunol. 1978;121:2596–72. [PubMed] [Google Scholar]
- 31.Sahu A, Kozel TR, Pangburn MK. Specificity of the thioester-containing reactive site of human C3 and its significance to complement activation. Biochem J. 1994;302:429–36. doi: 10.1042/bj3020429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jelezarova E, Lutz HU. Assembly and recognition of the complement amplification loop in blood; the role of C3b-C3b-IgG aggregates. Molecular Immunol. 1999;36:837–42. doi: 10.1016/s0161-5890(99)00104-2. [DOI] [PubMed] [Google Scholar]
- 33.Jefferis R, Lund J, Pound JD. IgG-Fc-mediated effector functions: molecular definition of interaction sites for effector ligands and the role of glycosylation. Immunological Reviews. 1998;163:59–76. doi: 10.1111/j.1600-065x.1998.tb01188.x. [DOI] [PubMed] [Google Scholar]
- 34.Janssen BJC, Christodoulidou A, McCarthy A, Lambris JD, Gros P. Structure of C3b reveals conformational changes that underlie complement activity. Nature. 2006;444:213–16. doi: 10.1038/nature05172. [DOI] [PubMed] [Google Scholar]
- 35.Farries TC, Lachmann PJ, Harrison RA. Analysis of the interactions between properdin, the third component of complement (C3), and its physiological activation products. Biochem J. 1988;252:47–54. doi: 10.1042/bj2520047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jelezarova E, Vogt A, Lutz HU. Interaction of C3b2-IgG complexes with complement proteins properdin, factor B and factor H: implications for amplification. Biochem J. 2000;349:217–23. doi: 10.1042/0264-6021:3490217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Daoudaki ME, Becherer JD, Lambris JD. A 34-amino acid peptide of the third component of complement mediates properdin binding. J Immunol. 1988;140:1577–80. [PubMed] [Google Scholar]
- 38.Holt GD, Pangburn MK, Ginsburg V. Properdin binds to sulfatide [Gal(3-SO4)β1-1Cer] and has a sequence homology with other peptides that bid sulfated glycoconjugates. J Biol Chem. 1990;265:2852–55. [PubMed] [Google Scholar]
- 39.Hourcade DE. The role of peoperdin in the assembly of the alternative pathway C3 convertase of complement. J Biol Chem. 2006;281:2128–32. doi: 10.1074/jbc.M508928200. [DOI] [PubMed] [Google Scholar]
- 40.Spitzer D, Mitchell LM, Atkinson JP, Hourcade DE. Properdin can initiate complement activation by binding specific target surfaces and providing a platform for de novo convertase assembly. J Immunol. 2007;179:2600–08. doi: 10.4049/jimmunol.179.4.2600. [DOI] [PubMed] [Google Scholar]
- 41.Lambris JD, Lao Z, Oglesby TJ, Atkinson JP, Hack CE, Becherer JD. Dissection of CR1, factor H, membrane cofactor protein, and factor B binding and functional sites in the third complement component. J Immunol. 1996;156:4821–32. [PubMed] [Google Scholar]
- 42.Rodriquez de Córdoba S, Esparza-Gordillo J, Goicoechea de Jorge E, Lopez-Trascasa M, Sánchez-Corral P. The human complement factor H: functional roles, genetic variations and disease states. Molecular Immunol. 2004;41:355–67. doi: 10.1016/j.molimm.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 43.Ferreira VP, Herbert AP, Hocking HG, Barlow PN, Pangburn ML. Critical role of the C-terminal domains of factor H in regulating complement activation at cell surfaces. J Immunol. 2006;177:6308–16. doi: 10.4049/jimmunol.177.9.6308. [DOI] [PubMed] [Google Scholar]
- 44.Jokiranta TS, Hellwage J, Koistinen V, Zipfel PF, Meri S. Each of the three binding sites on complement factor H interacts with a distinct site on C3b. J Biol Chem. 2000;275:27657–62. doi: 10.1074/jbc.M002903200. [DOI] [PubMed] [Google Scholar]
- 45.DiScipio RG, Daffern PJ, Schraufstätter IU, Sriramarao P. Human polymorphonuclear leukocytes adhere to complement factor H through an interaction that involves αMβ2 (CD11b/CD18) J Immunol. 1998;160:4057–66. [PubMed] [Google Scholar]
- 46.Parekh RB, Dwek RA, Sutton BJ, Fernades DL, Leung A, Stanworth D, et al. Association of rheumatoid arthritis and primary osteoarthritis with changes in the glycosylation pattern of total serum IgG. Nature. 1985;316:452–7. doi: 10.1038/316452a0. [DOI] [PubMed] [Google Scholar]
- 47.Rademacher TW, Williams P, Dwek RA. Agalactosyl glycoforms of IgG antibodies are pathogenic. Proc Natl Acad Sci USA. 1994;91:6123–7. doi: 10.1073/pnas.91.13.6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pope RM, Teller DC, Mannik M. The molecular basis of self-association of antibodies to IgG (rheumatoid factors) in rheumatoid arthritis. Proc Natl Acad Sci USA. 1974;71:517–21. doi: 10.1073/pnas.71.2.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pope RM, Teller DC, Mannik M. The molecular basis of self-association of IgG-rheumatoid factors. J Immunol. 1975;115:365–73. [PubMed] [Google Scholar]
- 50.Rademacher TW, Parekh RB, Dwek RA, Isenberg D, Rook G, Axford JS, et al. The role of IgG glycoforms in the pathogenesis of rheumatoid arthritis. Springer Sem Immunopathol. 1988;10:231–49. doi: 10.1007/BF01857227. [DOI] [PubMed] [Google Scholar]