

Abstract
Cholesteryl esters are hydrolyzed by cholesteryl ester hydrolase (CEH) yielding free cholesterol for export from macrophages. Hence, CEH has an important regulatory role in macrophage reverse cholesterol transport (RCT). CEH and human carboxylesterase 1 (CES1) appear to be the same enzyme. CES1 is inhibited by oxons, the bioactive metabolites of organophosphate (OP) pesticides. Here, we show that CES1 protein is robustly expressed in human THP-1 monocytes/macrophages and its biochemical activity inhibited following treatment of cell lysates and intact cells with chlorpyrifos oxon, paraoxon, or methyl paraoxon (with nanomolar IC50 values) or after immunodepletion of CES1 protein. CES1 protein expression in cells is unaffected by 24-h paraoxon treatment, suggesting the reduced hydrolytic activity is due to covalent inhibition of CES1 by oxons and not down-regulation of expression. Most significantly, treatment of cholesterol-loaded macrophages with either paraoxon (a non-specific CES inhibitor) or benzil (a specific CES inhibitor) caused enhanced retention of intracellular cholesteryl esters and a “foamy” phenotype, consistent with reduced cholesteryl ester mobilization. Thus, exposure to OP pesticides, which results in the inhibition of CES1, may also inhibit macrophage RCT, an important process in the regression of atherosclerosis.
Keywords: atherosclerosis, cholesteryl ester hydrolase, carboxylesterase, cholesterol metabolism, organophosphates, benzil
1. Introduction
Cardiovascular disease (CVD) is the leading cause of death in the United States today and has been for most of the last century [1]. Atherosclerosis is the underlying pathologic process responsible for most CVD and was originally thought to result from the passive accumulation of cholesterol in arterial walls. However, it is now recognized that inflammation plays a major role in all stages of atherosclerosis [2]. As part of this inflammatory process, monocytes in blood adhere to the vascular endothelium then migrate to the arterial intima where they differentiate into macrophages and secrete pro-inflammatory cytokines. Intimal macrophages take up free cholesterol from the extracellular matrix and cholesteryl esters from modified and unmodified low-density lipoprotein (LDL) particles via scavenger receptor type A and LDL receptors, respectively [3,4]. Following endocytosis, cholesteryl esters are hydrolyzed within the acidic environment of lysosomes by lysosomal acid lipase (LAL) generating free cholesterol and fatty acids [5]. Excess free cholesterol can be toxic to the cell necessitating that intracellular levels of free cholesterol be tightly controlled [6]. Consequently, free cholesterol is re-esterified by acyl coenzyme A cholesterol acyltransferase 1 (ACAT-1) yielding cholesteryl esters [7], which are stored in the cytoplasm as lipid droplets. Cholesteryl esters are not considered to be as toxic to the cell as free cholesterol and are thought to be a storage form of excess intracellular cholesterol. Macrophages that accumulate excessive amounts cholesteryl esters are transformed into foam cells, which contribute to the progression of atherosclerosis [8]. Mobilization of cholesterol from macrophages is initiated by the hydrolysis of cholesteryl esters stored in cytoplasmic lipid droplets. The liberated free cholesterol is exported to extracellular acceptors (such as high density lipoprotein [HDL] particles), transported to the liver, and secreted in bile. The transport of cholesterol from vessel wall macrophages to the liver is termed macrophage reverse cholesterol transport (RCT) and is proposed to be the process by which regression of atherosclerotic lesions can occur [9].
Hydrolysis of cholesteryl esters stored as lipid droplets in the cytosol of macrophages is catalyzed by a neutral cholesteryl ester hydrolase. This enzymatic reaction is the rate-limiting step in the mobilization of cholesterol from macrophages [10]. Currently, the best candidate for this hydrolase activity in humans is cholesteryl ester hydrolase (CEH), which was originally cloned from a human THP-1 monocyte/macrophage cell line [11]. CEH appears to be the same enzyme as human CES1 [12], a carboxylesterase involved in xenobiotic metabolism [13]. CES1 protein is also called hCE1 [14] and triacylglycerol hydrolase (TGH; [15]), but Ghosh and colleagues use the term CEH to underscore its physiological activity in macrophages. We have chosen to use the term CES1 throughout the remainder of this manuscript when referring to this protein. Zhao et al. [16] reported that over-expression of CES1 in cultured THP-1 macrophages increased the rate of cholesterol efflux. Further, over-expression of CES1 in the macrophages of Ldlr−/− mice was found to reduce atherosclerosis and lesion necrosis and to increase macrophage RCT [17]. These results strongly support the notion that hydrolysis of intracellular cholesteryl esters is the rate-limiting step in macrophage RCT.
Carboxylesterases (CES, EC 3.1.1.1) are serine hydrolases known to metabolize xenobiotics containing ester, amide, and thioester bonds (see [13] for review). CES1 and carboxylesterase 2 (CES2) are the two major human carboxylesterase isoforms and share 48% sequence homology. CES1 is expressed in multiple tissues while CES2 is detected in a more tissue-specific manner, with liver, small intestine, and kidney being major sites of expression [14]. Carboxylesterases, among other serine hydrolases, are inhibited by covalent reaction of the active-site serine residue with organophosphate compounds (oxons) [13]. Oxons are the bioactivated metabolites of organophosphothionate pesticides and are formed mainly in liver following cytochrome P450 monooxygenase-catalyzed desulfuration [18]. Saboori and Newcombe [19] previously showed that a carboxylesterase protein, which was purified from human monocytes, could be inhibited by oxon treatment. If CES1 is responsible for cholesteryl ester hydrolysis, then inhibition of CES1 activity would be predicted to inhibit macrophage RCT thereby increasing the risk of the development of atherosclerosis. In this report, we characterize the inhibitory effects of three oxons, derived from commonly used pesticides, on esterase activities in human THP-1 monocyte/macrophages. Moreover, we show that treatment of cholesterol-loaded THP-1 macrophages with a specific reversible inhibitor of CES1 (benzil), or a bioactive metabolite of an OP insecticide (paraoxon) that broadly targets multiple serine hydrolases including CES1, causes significant retention of cholesteryl esters in cultured macrophages under conditions which promote lipoprotein-dependent cholesterol efflux. Therefore, these chemical treatments appear to promote the formation of foam cells from macrophages.
2. Materials and Methods
2.1. Materials
Human THP-1 monocytes, Hep G2 cells, RPMI-1640 medium, gentamicin sulfate solution (50 mg/ml), and Hanks’ balanced salt solution without calcium, magnesium and phenol red were purchased from the American Type Culture Collection (ATCC) (Manassas, VA). Fetal bovine serum (FBS) was purchased from Invitrogen (Carlsbad, CA). Trypan Blue solution (0.4%), β-mercaptoethanol, 4-methylumbelliferyl acetate (4-MUBA), 4-methylumbelliferyl oleate (4-MUBO), α-naphthyl acetate, β-naphthyl acetate, acetonitrile, monoclonal antibodies against β-actin, horseradish peroxidase (HRP) conjugated to streptavidin, phorbol 12-myristate 13-acetate (PMA), benzil (diphenylethane-1,2-dione), porcine bile-salt stimulated cholesterol esterase [EC 3.1.1.13, also termed carboxyl ester lipase (CEL)], and all components of the buffers were purchased from Sigma (St. Louis, MO). A protein silver staining kit was purchased from Amersham Biosciences (Piscataway, NJ). Polyvinylidine fluoride (PVDF), goat anti-rabbit IgG secondary antibody conjugated to HRP, and goat anti-mouse IgG secondary antibody conjugated to HRP were purchased from Bio-Rad (Hercules, CA). 6-N-biotinylaminohexyl isopropyl phosphorofluoridate hemihydrate (FP-biotin) was purchased from Toronto Research Chemicals (North York, Ontario, Canada). Streptavidin-agarose was from Invitrogen. Cholesteryl [1-14C] oleate was from New England Nuclear (Cambridge, MA). Human acetylated LDL was purchased from Intracel (Frederick, MD). Chlorpyrifos oxon, paraoxon and methyl paraoxon were synthesized and provided as gifts by Dr. Howard Chambers, Department of Entomology, Mississippi State University. Rabbit anti-CES1 was a gift from Dr. M. Hosokawa, Chiba University, Japan. Recombinant CES1 and recombinant CES2 proteins were expressed in baculovirus-infected Spodoptera frugiperda cells and purified [20]. Rabbit anti-CES2 antibodies were generated using the purified protein as an antigen and full details of their properties will be published elsewhere (Hatfield and Potter, manuscript in preparation).
2.2. Culture Conditions
THP-1 monocytes were grown in suspension in RPMI-1640 medium supplemented with 10% FBS, 0.05 mM β-mercaptoethanol, and 50 μg gentamicin/mL (growth medium) at 37°C and 5% CO2. The cells were grown at a density between 0.2x106 and 1x106 cells /ml as recommended by ATCC. THP-1 monocytes were differentiated into macrophages by incubating in growth medium containing 100 nM phorbol 12-myristate 13-acetate (PMA) for 7 days at 37°C and 5% CO2 [11]. Culture medium was replaced every two-to-three days with fresh growth medium containing PMA.
2.3. Preparation of cell lysates
THP-1 cells were collected by centrifugation (200 x g for 7 min) and washed once with phosphate-buffered saline (PBS). The cells were re-suspended in ice-cold 100 mM Tris-HCl (pH 7.4) buffer and lysed by sonication (four 15 second bursts while on ice).
2.4. Protein assay
Protein concentrations of cell lysates were determined using the BCA reagent according to the manufacturer’s instructions (Pierce, Rockford, IL).
2.5. Western blot analysis
Cell lysates were subjected to sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis (PAGE) as described by Laemmli [21]. Proteins were transferred to PVDF by a modification of the method of Towbin et al. [22]. Briefly, PVDF membranes were soaked in methanol for 2 min then soaked in transfer buffer (25 mM Tris base, 192 mM glycine, 15% methanol) for 5 min. Following electrophoretic transfer, PVDF membranes were blocked for 1 hour at room temperature with TBS Tween (100 mM Tris-HCl [pH 7.4], 0.9% NaCl, and 0.1% [v/v] Tween 20) containing 5% (w/v) nonfat dry milk. PVDF membranes were then probed for CES1 or CES2 proteins by incubating them in TBS Tween/5% milk containing either rabbit anti-CES1 diluted 1:4000 or rabbit anti-CES2 diluted 1:4000 for 1 hour at room temperature. The membranes were then washed four times for 15 min each with TBS Tween followed by incubation in TBS Tween/5% milk with goat anti-rabbit secondary antibody conjugated to HRP diluted 1:20,000. The membranes were again washed four times for 15 min each with TBS Tween and the signal developed using the SuperSignal West Pico Chemiluminescent Substrate from Pierce (Rockford, IL). The signal was recorded using X-OMAT photographic film (Eastman Kodak Co., Rochester, NY).
For quantitative analysis of CES1 and β-actin, PVDF membranes were probed with TBS Tween/5% milk containing both rabbit anti-CES1 diluted 1:4000 and mouse anti-β-actin diluted 1:5000 for 1 hour at room temperature and washed as described above. Membranes were then incubated with TBS Tween/5% milk containing goat anti-rabbit HRP and goat anti-mouse HRP secondary antibodies, each diluted 1:20,000. The signal was developed as described above and captured on X-OMAT film. Quantitative analysis was done using 1D-Multi software from Alpha Innotech (San Leandro, CA).
CES1 protein was immunoprecipitated from THP1 cell lysate proteins with anti-CES1 IgG antibody as described previously [23]. Precipitated proteins were analyzed by SDS-PAGE. To control for non-specific binding, lysate proteins were also incubated with pre-immune rabbit IgG antibody (control IgG) prior to SDS-PAGE. Gels were stained with silver according to the manufacturer’s directions to detect separated proteins; alternatively, proteins were transferred to PVDF membranes for subsequent western blotting using anti-CES1 as the primary antibody, as described above.
2.6. Native gel analysis of carboxylesterase activity
Aliquots of THP-1 cell lysate containing 25 μg of protein were subjected to discontinuous native PAGE [24]. Following electrophoresis, the gels were equilibrated in 0.1 M potassium phosphate (pH 6.5) for 15 min followed by esterase staining using either 4-MUBA [25] or a mixture of α- and β-naphthyl acetate [26].
2.7. Treatment of THP-1 cell lysates with oxons
Cell lysates were diluted in 100 mM Tris-HCl (pH 7.4) buffer containing 1 mM EDTA. The indicated oxon was added from a stock solution in ethanol to the diluted cell lysate at the desired concentration. The final concentration of the ethanol in all reactions was 1% (v/v). This mixture was incubated for 10 min at 37ºC before the addition of para-nitrophenyl valerate to measure residual carboxylesterase activity as described below.
2.8. Treatment of intact THP-1 cells with oxons
Five mL of medium, containing 0.6 x 106 cells /ml, were placed in T-25 flasks. The oxon from the ethanol stock solution was diluted 1:10 in a separate aliquot of medium and then added to the medium containing cells to give the desired final concentration. The final concentration of ethanol in the medium in all flasks was always 1% (v/v). The cells were exposed to the various oxons for 24 h. Following oxon exposure, cell viability was assessed using Trypan Blue exclusion. Cells were pelleted and washed 3 times with PBS to remove excess oxon. Cell pellets were re-suspended in 100 mM Tris-HCl (pH 7.4) buffer. The re-suspended cells were lysed by sonication as described above. The resulting cell lysates were used in the carboxylesterase assays.
2.9. Carboxylesterase activity assays
2.9.1. Hydrolysis of para-nitrophenyl valerate
Carboxylesterase assays [27] were performed to measure the activity after THP-1 cell lysates or intact cells were treated with oxons. Briefly, cell lysate was diluted in 100 mM Tris-HCl (pH 7.4) buffer containing 1 mM EDTA. The mixture was incubated for 10 min at 37°C in either the absence (ethanol vehicle only) or presence of 10 μM paraoxon. Para-nitrophenyl valerate (final concentration, 500 μM) in ethanol was added to the mixture and incubated at 37°C for an additional 15 min. A solution of 2% (w/v) SDS, 2% (w/v) Tris base, and 0.044% (w/v) EDTA was added to the reaction mixture to stop the enzymatic reaction. The rate of hydrolysis of para-nitrophenyl valerate was quantified by monitoring the production of para-nitrophenol at 405 nm. The carboxylesterase activity was determined by calculating the difference in the absorbance at 405 nm in the tubes without 10 μM paraoxon and those with paraoxon. The ability of an oxon to inhibit carboxylesterase activity was determined by adding the oxon at the desired concentration from a stock solution in ethanol and incubating for 10 min prior to adding para-nitrophenyl valerate. An extinction coefficient of 13 cm−1 mM−1 [28] was used to calculate enzyme activity (μmol of para-nitrophenol formed/min/mg of protein). The amount of cell lysate was adjusted to be in the linear range with respect to the amount of enzymatic activity present. Data was expressed as percent of control. All reactions were performed in triplicate.
Alternatively, a continuous assay measuring the production of para-nitrophenol was also used [29]. Briefly, hydrolysis reactions were performed in a 96-well plate format with cell lysate diluted to 50–125 μg protein/ml and para-nitrophenyl valerate added at a final concentration of 500 μM. The substrate, dissolved in ethanol, was diluted into 50 mM Tris-HCl (pH 7.4) buffer to 1 mM and 150 μl added to wells in triplicate. After a 5 min pre-incubation at 37° C, the reaction was initiated by the addition of 150 μl of diluted cell lysate (in 50 mM Tris-HCl buffer, pH 7.4). Reaction progress was monitored at 405 nm over 5 min to estimate the rate of formation of para-nitrophenol. The slopes of the activity curves (absorbance units per min) were recorded. Using an extinction coefficient of 13 cm−1 mM−1 [28], data were converted to enzyme activity units (i.e., μmol of para-nitrophenol formed/min/mg protein). Data was expressed as percent of control (no inhibitor added). All reactions were performed in triplicate.
IC50 values were estimated by first determining the linear portion of the dose response curve then running a series of concentrations that were 0.25 log units apart for each oxon. The best line was drawn though the points (r2 values ≥ 0.92) and the IC50 value interpolated. The values reported are the mean ± the standard deviation of three separate determinations. IC50 values for benzil were determined in a similar manner. The final concentrations of benzil used were 0.05, 0.1, 0.5, 1, 5, 10, and 50 μM.
CES1 was immunoprecipitated [23] using protein A-agarose beads [68] to determine the residual hydrolytic activity toward para-nitrophenyl valerate following immunodepletion of CES1 protein. Following immunoprecipitation using pre-immune IgG and anti-CES1 IgG, the resulting supernatants were subjected to western blotting analysis for CES1 and carboxylesterase activity assays.
2.9.2. Hydrolysis of cholesteryl oleate
The cholesteryl ester hydrolase activity in human THP1 cell lysate was determined by incubating lysate proteins (1 mg/ml final concentration) in 10 mM Tris-HCl (pH 7.4) buffer containing 150 mM NaCl and 0.001% (v/v) Brij 78 in the presence of 75 μM cholesteryl [1-14C] oleate (specific activity = 0.75 mCi/mmol; added in acetone, 2.5% v/v final conc.). Reactions were initiated by addition of radioactive cholesteryl oleate. After a 2 h incubation at 37°C, reactions were stopped by addition of 1/10 volume of 1 N NaOH and extracted with an equal volume of heptane:chloroform:methanol (2.73:3.42:3.85 v/v/v). Following brief centrifugation, the aqueous fraction was removed and counted in a scintillation counter to assess the amount of [1-14C]oleate produced. Selected reactions were treated with 10 μM paraoxon for 10 min (37°C) prior to adding cholesteryl oleate. Control reactions contained boiled cell lysate. All reactions were normalized on protein amount and incubation time. Positive control reactions contained porcine bile-salt stimulated cholesterol esterase (EC 3.1.1.13) instead of THP1 cell lysate.
2.9.3. Hydrolysis of 4-methylumbelliferyl oleate
Hydrolysis of the surrogate fluorogenic substrate 4-MUBO by CES1, porcine bile-salt stimulated cholesterol esterase (CEL), and THP-1 cell lysates, in the absence or presence of the inhibitors benzil and paraoxon, was adapted from an assay used for 4-MUBA [29]. In brief, a colloidal suspension of 4-MUBO was prepared at a concentration of 150 μM in 10 mM Tris-HCL (pH 7.4), 150 mM NaCl, and 0.01% Triton X-100. The solution was vortexed vigorously for one min followed by sonication in a bath sonicator for 5 min to disperse the substrate. One-hundred fifty μl of dilute protein or cell lysate in the same buffer was pre-incubated for 5 min at 37°C, with or without chemical inhibitors, in the wells of a black 96-microwell plate. Following this pre-incubation period, 150 μl aliquots of the substrate working solution were then dispensed into the wells to begin the reaction and the fluorescence was monitored using λex 355 nm and λem 460 nm. The activity curves were converted to specific enzymatic activities by the utilization of a standard curve prepared with authentic 4-methylumbelliferone.
2.10. Detection of serine esterases in THP-1 cell lysates by FP-biotin labeling
THP-1 monocytes were cultured under normal conditions, harvested, and lysed in 50 mM Tris-HCl (pH 7.4) buffer. Lysate proteins (1 mg/ml) were treated with increasing concentrations of paraoxon up to 10 nM for 15 min at 37°C. FP-biotin [30] was then added to each sample at a final concentration of 2 μM and incubated further for 1 h at room temperature. To control for non-specific labeling of protein by FP-biotin, lysate samples were boiled for 5 min to denature proteins before adding FP-biotin. Reactions were terminated by the addition an equal volume of 2x SDS-PAGE loading buffer and boiled for 5 min. Equal amounts of protein were loaded and separated on a 10% SDS-PAGE gel. The proteins were transferred to a PVDF membrane, blocked for 1 h in 0.05% Tween buffer containing 3% (w/v) milk, and the membrane incubated with 1:3000 (v/v) Avidin-HRP in 0.05% Tween buffer containing 1% (w/v) milk for 1 h at room temperature. The membrane was washed four times in 0.05% Tween buffer for 15 min each and then incubated with the SuperSignalWestPico chemiluminescent reagent (Pierce) for 5 min. The resulting signal was recorded using X-OMAT film.
To verify specific labeling of CES1 in THP1 cell lysates by FP-biotin, proteins were incubated with FP-biotin as described above. After 1-h reaction, SDS was added to a final concentration of 0.5% (w/v) and heated at 95°C for 5 min. Samples were centrifuged in a spin filter (Pierce, 3,000-Da cutoff) to remove unreacted FP-biotin. The resulting protein concentrate was diluted to 500 μl with 50 mM Tris-HCl (pH 7.4) and 100 μL of streptavidin-agarose beads were added. The mixture was allowed to incubate overnight at 4°C on a rotator. Next morning, the agarose beads were pelleted by brief centrifugation; following aspiration of the supernatant, the beads were washed twice with 1 ml of 50 mM Tris-HCl (pH 7.4) containing 0.2% SDS followed by two washes with 50 mM Tris-HCl (pH 7.4) without SDS. The washed pellet was resuspended in 40 μl of SDS-loading buffer, heated at 95°C for 5 min, cooled to room temp, and briefly centrifuged again. Approximately 30 μl was loaded onto a 10% SDS-PAGE gel and proteins separated. Following electrophoretic transfer of proteins onto a PVDF membrane, the membrane was probed with anti-CES1 antibodies as described above. Alternatively, the PVDF membrane was probed with 1:3000 (v/v) Avidin-HRP as described above to detect all biotin-labeled serine hydrolases after enrichment of the FP-biotin-labeled proteins.
2.11. Cholesteryl ester retention studies
Macrophages were loaded with cholesterol by incubation for 48 h with growth medium containing 50 μg/ml of human acetylated LDL, 1% bovine serum albumin (BSA) and 1% FBS. Following the 48 h loading period, cholesterol efflux from cells was promoted by culturing the cells for another 48 h in growth medium containing 10% FBS as the acceptor for cholesterol. The effect of benzil and paraoxon on the retention of intracellular cholesterol was determined by including either 50 μM benzil or 10 μM paraoxon in the culture medium during the 48-h cholesterol efflux period. The solvent vehicle for benzil and paraoxon was ethanol and its final concentration in the culture medium was 0.1% (v/v). After the 48-h treatment period, the medium was removed and the cells washed twice with Hanks’ balanced salt solution. Cells were scraped from the plates in 50 mM Tris-HCl (pH 7.4) buffer and lysed by sonication (four 15 second bursts while on ice). The protein content of cell lysates was determined using the BCA kit (Pierce, Rockford, IL).
Free and total cholesterol levels in lipid extracts of human THP1 macrophages were determined using gas chromatography (GC)-flame ionization detection as described previously [31] with slight modifications. Cell lysates (~250–300 μl) were transferred to a glass test tube along with internal standard (5α-cholestane, Sigma) to yield a final concentration of 0.02 μg 5α-cholestane/μl of cell lysate. Lipids were extracted with two volumes of 3:2 (v/v) hexane/isopropanol and the extraction was repeated. The pooled organic solvent extracts were evaporated to dryness under nitrogen. Lipid residues were resuspended in 100 μl of 1:1 (v/v) methanol/isopropanol, which was transferred to a GC vial containing a volume-reducing insert. Following injection of four μl onto the GC to determine the amount of free cholesterol (unesterified), the remaining 96 μl of solution was subjected to saponification in 1.0-ml ethanolic KOH (0.57 M KOH in ethanol) for 2 h at 80°C. The head-space of the glass vials was purged with nitrogen gas prior to saponification to prevent auto-oxidation of lipids. Following saponification, the samples were cooled to room temperature and CHCl3 (1 mL) and phosphate-buffered saline (0.9 ml) added; the mixture was vortexed vigorously for one minute. After centrifuging, the aqueous layer was discarded and the organic layer washed by adding water (1 ml) and ethanol (0.5 ml), again followed by vigorous vortexing. Following centrifugation, the organic layer was transferred to a clean glass tube and evaporated to dryness under nitrogen. Residues were resuspended in 100 μl of 1:1 (v/v) methanol/isopropanol and transferred to a GC vial containing a volume-reducing insert. A four μl aliquot was injected into the GC to determine the total cholesterol content (free and esterified). The GC inlet and detector temperatures and carrier gas flows are specified in [30]. The column was ramped from an initial temp of 260°C to 275°C over 15 min and the temp held at 275°C for 5 min before re-establishing the initial column temperature. The tR for 5α-cholestane and cholesterol were 7.5 min and 12 min, respectively. Quantification of cholesterol was done using the internal standard 5α-cholestane as described in [31]. All cholesterol amounts were normalized to the protein content of the cell lysate and expressed as nmol cholesterol/mg protein. The esterified cholesterol levels were determined from the difference between total cholesterol and free cholesterol amounts.
3. Results
3.1. Expression of CES1, but not CES2, in THP-1 monocytes
Western blot analysis of THP-1 cell lysates confirmed the presence of CES1 protein (Fig. 1A), but did not detect CES2 (Fig. 1B). CES1 protein could also be immunoprecipitated from THP-1 cell lysates (Fig. 1C, D). Similarly, when THP-1 cell lysates were analyzed for esterase activity using an in-gel hydrolysis assay [25], the esterase activity migrated on the native PAGE gels with an Rf corresponding to CES1 present in human liver microsomes (Fig. 2 A,B), which was used as a protein marker. However, no esterase activity corresponding to CES2 was observed in the THP1 cell lysate (Fig. 2 A,B). In fact, all carboxylesterase activity detected in the THP-1 cell lysate, using either 4-MUBA or a mixture of α- and β-naphthyl acetate as substrates, was found to migrate in native PAGE gels at the same rate as human liver CES1, which suggests that CES1 is the major carboxylesterase expressed in THP-1 monocytes/macrophages. To confirm this finding, THP-1 cell lysate was depleted of CES1 using anti-CES1 IgG antibodies and its residual carboxylesterase activity measured using para-nitrophenyl valerate (Fig. 3). The immunodepleted cell lysate exhibited only 15% of the carboxylesterase activity present in the lysate treated with non-specific IgG antibodies. This result demonstrates that the para-nitrophenyl valerate hydrolysis activity in THP1 cells is primarily due to CES1.
Fig. 1.
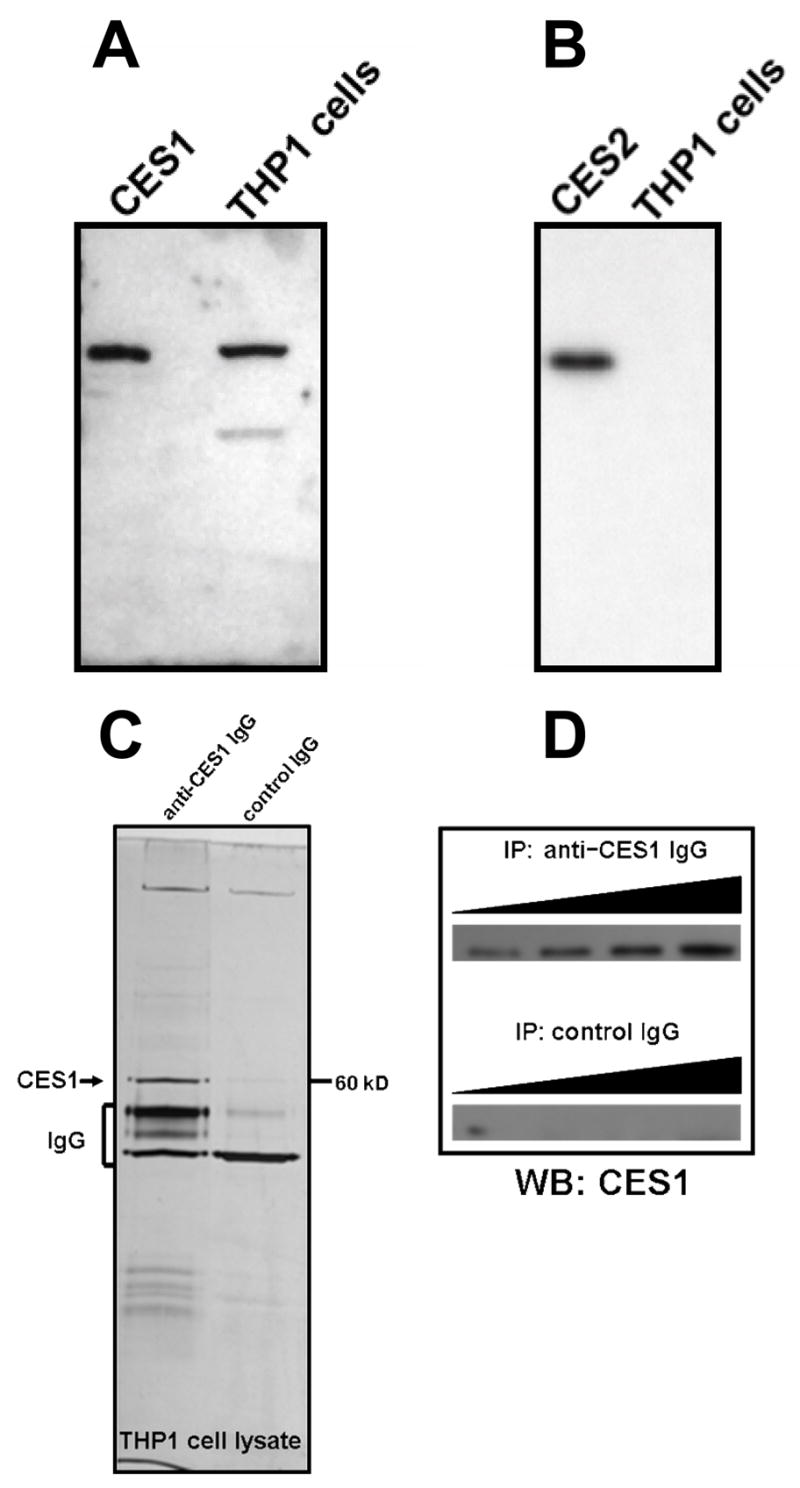
Western blot analysis of THP-1 cell lysates detects CES1 protein, but not CES2. THP-1 cell lysate (25 μg protein) was subjected to SDS-PAGE. The proteins were transferred to a PVDF membrane and probed with anti-CES1 antibody (A) or anti-CES2 antibody (B). Either 0.5 μg of recombinant CES1 (A) or 0.5 μg of recombinant CES2 (B) was included for comparison. CES1 protein can also be immunoprecipitated (IP) from soluble THP1 cell proteomes and is detected on SDS-PAGE gels following silver staining (C) or immunoblotting using anti-CES1 IgG (D). When constant amounts of THP1 cell lysate protein were titrated with increasing levels of rabbit anti-CES1 IgG and incubated, an increased yield of CES1 protein could be detected by immunoblotting in (D), while no CES1 protein was detected in incubations that used control IgG for the IP experiment (D).
Fig. 2.

Esterase activity in THP-1 cell lysates that corresponds to CES1 is detected following native gel electrophoresis. Human liver microsomes were used as a source of CES1 and CES2 proteins. Human liver microsomes (lane 1, 25 μg protein) and THP-1 cell lysate (lane 2, 25 μg protein) were subjected to native gel electrophoresis. Esterase activity in the native gels were detected by staining with either 4-methylumbelliferyl acetate (4-MUBA) (A) or a mixture of α- and β-naphthyl acetate (α,β-NPA) (B).
Fig. 3.

Immunoprecipitation of CES1 from THP1 cell lystates. (A) Western blot showing specific depletion of CES1 protein from cell lysate. (B) Residual carboxylesterase activity of cell lysate toward para-nitrophenyl valerate following immunoprecipitation of CES1 (mean ± SD, n = 3).
3.2. Inhibition of carboxylesterase activity in cell lysates
The ability of three oxons of varying lipophilicity to inhibit carboxylesterase activity, determined with the substrate para-nitrophenyl valerate, in THP-1 cell lysates was evaluated. Chlorpyrifos oxon, paraoxon, and methyl paraoxon all inhibited carboxylesterase activity in cell lysates in a concentration-dependent manner; a representative curve using paraoxon is shown in Fig. 4A. IC50 values were determined for each oxon and are reported in Table 1. Chlorpyrifos oxon and paraoxon were equally potent inhibitors of carboxylesterase activity with no statistically significant difference between them. However, chlorpyrifos oxon and paraoxon were each significantly more potent than methyl paraoxon in inhibiting carboxylesterase activity (p≤ 0.05). The ability of benzil (diphenylethane-1,2-dione), a reversible carboxylesterase inhibitor, to block hydrolytic activity in THP-1 cell lysates was also determined. The IC50 value for benzil using cell lysate as the source of carboxylesterase was estimated to be 160 nM, which is comparable to the IC50 estimated for benzil when using pure recombinant CES1 [32].
Fig. 4.

Carboxylesterase activity in THP-1 cell lysate is inhibited by paraoxon treatments. (A) THP-1 cell lysate was incubated with paraoxon for 30 min at 37°C at various concentrations. Carboxylesterase activity toward para-nitrophenyl valerate is expressed as % of control (mean ± SD, n = 3). (B) THP-1 cell lysate was boiled (control), treated with ethanol vehicle (no inhib), or treated with 10 μM paraoxon (PO) for 10 min before incubation with 75 μM cholesteryl [1-14C]oleate. CEH specific activities are expressed as means ± SEM (n = 9–17 individual reactions per group). As a comparison, porcine bile-salt stimulated cholesterol esterase (0.2 units/reaction), which was used as a positive control for the CEH assay, yielded ~17-fold greater CEH activity than THP-1 cell lysates (no inhib), even without added bile salts (data not shown).
Table 1.
IC50 values for non-specific (oxons) and specific (benzil) inhibitors of carboxylesterase activity following treatments of human THP-1 cell lysates.a
| Inhibitor | Structure | IC50 (nM) |
|---|---|---|
| Chlorpyrifos oxon (CPO) |
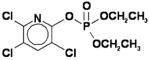
|
0.21 ± 0.03 |
| Paraoxon (PO) |
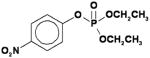
|
0.29 ± 0.12 |
| Methyl paraoxon (MPO) |
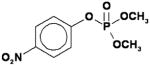
|
49 ± 12 |
| Benzil (diphenylethane-1, 2-dione) |
|
160 ± 10 |
IC50 values are the mean ± SD (n = 3 experiments)
When pure recombinant CES1 protein was incubated with cholesteryl [1-14C] oleate no hydrolytic activity could be observed (data not shown). However, THP1 cell lysates demonstrated cholesteryl ester hydrolase activity that was nearly 2-fold greater than control reactions (Fig. 4B), which had been boiled prior to addition of cholesteryl [1-14C] oleate. The CEH activity of the THP1 cell lysates was reduced by 23% following treatment with 10 μM paraoxon (Fig. 4B).
Despite the difficulty in detecting CEH activity using pure recombinant CES1 protein and cholesteryl [1-14C] oleate (see Discussion), the lipolytic activity of pure CES1 toward a sensitive fluorogenic substrate, 4-methylumbelliferyl oleate (4-MUBO), is clearly evident (Supplementary Fig. 1). However, as can also be seen, the specific activity of CES1 toward the water-soluble substrate pNPV was ~3,400-fold greater than its specific activity toward the water-insoluble 4-MUBO (Supplementary Fig. 1A,B). Thus, based on specific activities, the esterolytic activity of CES1 is clearly more efficient than its lipolytic activity. Nevertheless, CES1 does possess lipase activity, which can be inhibited by both benzil and paraoxon. A representative mammalian lipase, CEL, which can efficiently hydrolyze cholesteryl esters, was also examined to determine its sensitivity to benzil and paraoxon. Porcine pancreatic CEL was shown to have a ~6-fold greater specific activity toward 4-MUBO than CES1 (Supplementary Fig. 1C). This difference would no doubt increase if bile salts had been added to the incubation mixture to stimulate CEL. Importantly, the data presented shows that benzil does not inhibit this representative lipase, while as expected CEL is very sensitive to paraoxon. As will be discussed later, CEL is located in the secretory compartment of macrophages, thus it likely does not contribute to the neutral cytosolic CEH activity found in these cells.
When the lipolytic activity of THP-1 cell lysates toward the surrogate substrate 4-MUBO was determined in the presence of paraoxon (Supplementary Figure 2, right), no significant inhibiton of lipase activity was observed. This lack of inhibition was in contrast to the paraoxon-mediated inhibition observed with the physiological substrate, cholesteryl oleate. For example, while 10 μM paraoxon blunted the hydrolysis rate of cholesteryl oleate (Fig. 4B) the same inhibitor treatment did not reduce the rate of hydrolysis of 4-MUBO (Supplementary Fig. 2). This suggests that 4-MUBO is broadly metabolized in THP-1 cell lysate by enzymes that are insensitive to paraoxon, which possibly masks the action of CES1 since it is capable of hydrolyzing 4-MUBO and is sensitive to the inhibitory effects of paraoxon. In contrast, cholesteryl oleate is hydrolyzed by THP-1 cell lysate at a considerably slower rate than 4-MUBO (~150-fold when normalized on protein amount) by a smaller number of enzymes, including (and perhaps limited to) CES1, in this cell line. Because of the sluggish rate of cholesteryl oleate hydrolysis by THP-1 cell lysates, which may be exacerbated by product inhibition caused by fatty acids liberated during the reaction (Herring et al., unpublished data), the rigorous characterization of inhibitors on CEH activity in THP-1 cells will require development of more sensitive analytical methods, such as LC-MS/MS, to distinguish the properties of these compounds.
3.3. Detection of serine hydrolases in cell lysates
FP-biotin, an activity-based probe employed for labeling enzymatically active serine hydrolases [30], was used to label proteins in THP-1 cell lysates that had been pre-treated with increasing concentrations of paraoxon (Fig. 5A). Without pre-enrichment of biotin-labeled proteins only one protein in the cell lysate (with Mr = 60 kD) was specifically labeled by FP-biotin and visualized. This protein was definitively identified as CES1 by an immunoblotting experiment following its enrichment from FP-biotin-treated cell lysates using streptavidin-agarose beads (Fig. 5B). Pre-treatment of cell lysates with paraoxon concentrations > 1 nM completely inhibited FP-biotin labeling of CES1 (Fig. 5A). Pre-treatment with paraoxon at 0.01 nM, a concentration significantly below the IC50 did not prevent labeling. The upper band present in Fig. 5A is due to non-specific labeling by FP-biotin since it also appears when cell lysates are boiled for 2 min prior to treatment with FP-biotin (data not shown). In order to detect less abundant serine hydrolases, or those which react with FP-biotin less efficiently, we pre-enriched FP-biotin-labeled proteins using streptavidin-agarose beads prior to electrophoresis and subsequent blotting with streptavidin-HRP (Fig. 5C). This procedure resulted in detection of several serine hydrolases besides CES1 in THP1 cells, although it was still the predominant hydrolase detected. For comparison, lysates prepared from HepG2 cells were also analyzed in the same manner as THP1 lysates (Fig. 5C). Numerous hydrolases are detected in the HepG2 cell lysate including CES1, but none are as predominant as CES1 in THP-1 cell lysate. Identification of the labeled proteins besides CES1 in each cell line is the subject of an on-going investigation.
Fig. 5.
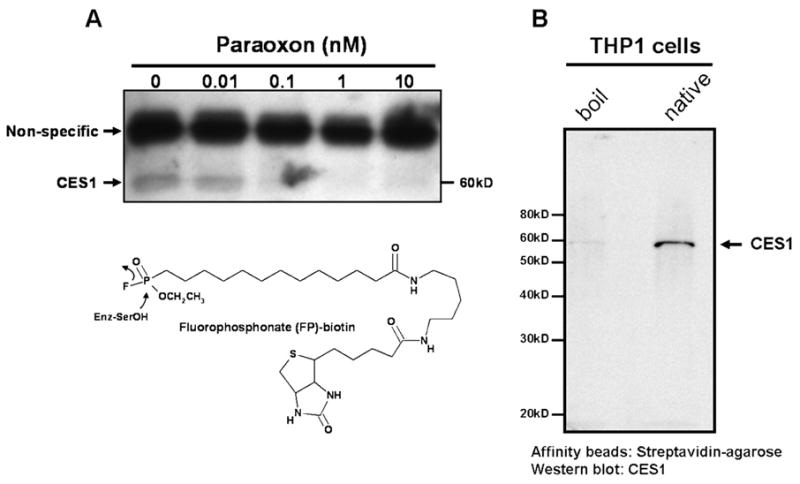

THP-1 cell lysates pre-treated with paraoxon exhibit decreased CES1-specific labeling by FP-biotin (see FP-biotin structure and chemical mechanism below blot in A). (A) Cell lysate proteins (1 mg/ml) were treated with the indicated concentrations of paraoxon for 15 min at 37°C followed by incubation with 2 μM FP-biotin for 1 h at room temperature. The lysates were subjected to SDS-PAGE, transferred to PVDF and probed with streptavidin conjugated to HRP to identify FP-biotin labeled proteins. The non-specific labeled band corresponds to a protein that reacts with FP-biotin after heat denaturation of THP-1 cell lysate proteins. The diminished band intensity of CES1 that corresponds with increasing concentrations of paraoxon indicates that CES1 is targeted by paraoxon. (B and C) Following FP-biotin-treatments of cell lysates, biotinylated proteins were enriched by incubating the lysate with streptavidin-agarose beads. Following SDS-PAGE of enriched proteins, CES1 protein was detected by rabbit anti-CES1 (B) and the biotinylated proteins detected by streptavidin-HRP (C). HepG2 cells were used to compare its suite of serine hydrolases with THP-1 cells. Boil refers to heat-denatured proteins. Native refers to non-denatured ‘active’ proteins.
3.4. Inhibition of carboxylesterase activity in intact cells
When intact cultured THP-1 monocytes were treated with chlorpyrifos oxon, paraoxon, or methyl paraoxon, carboxylesterase activity was inhibited by each oxon in a dose-dependent manner (Fig. 6). The concentrations of oxon used did not affect overall cell viability, as assessed by Trypan Blue exclusion (data not shown). Paraoxon was the most effective at inhibiting carboxylesterase activity, while chlorpyrifos oxon was the least effective. To further investigate the unanticipated difference in inhibition potency observed for chlorpyrifos oxon following treatments of cell lysates versus intact cells, we incubated chlorpyrifos oxon and paraoxon separately in cell-free culture medium for varying lengths of time (0–24 h). When paraoxon was incubated with the culture medium it retained more of its ability to inhibit carboxylesterase activity than chlorpyrifos oxon when the oxon-treated media was added to lysates from untreated cells (data not shown). These results suggest that chlorpyrifos oxon was either being degraded or sequestered by serum proteins in the culture medium to a greater extent than paraoxon, although some of paraoxon’s ability to inhibit carboxylesterase was lost after incubation in the medium.
Fig. 6.
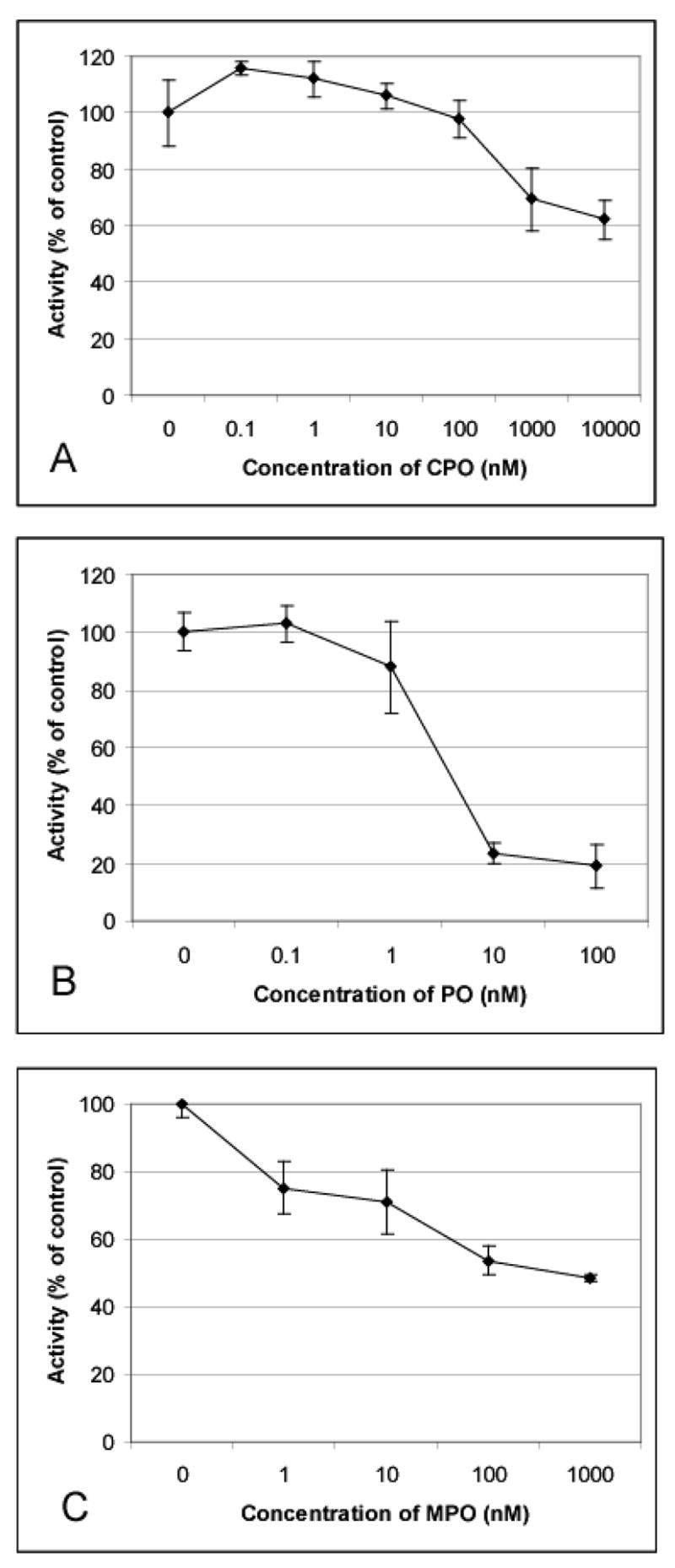
Carboxylesterase activity in intact THP-1 cells is inhibited by chlorpyrifos oxon, paraoxon, or methyl paraoxon. THP-1 cells were incubated with chlorpyrifos oxon (A), paraoxon (B), or methyl paraoxon (C) for 24 h at the indicated concentrations. Following treatments, cells were collected, washed, and lysed. Carboxylesterase activity was measured in cell lysates and is expressed as % of control (mean ± SD, n = 3).
Analysis of cell lysate proteins prepared from either paraoxon-treated or vehicle-treated cells on native PAGE gels confirmed that CES1 activity was markedly inhibited by oxon treatment (Fig. 7). However, Western blot analysis of cell lysate proteins prepared from paraoxon-treated cells showed no difference in the amount of CES1 protein present compared to control (vehicle) treated cells (Fig. 8), indicating that the reduced carboxylesterase activity was due to direct inhibition by paraoxon and not down-regulation of CES1 protein expression.
Fig. 7.
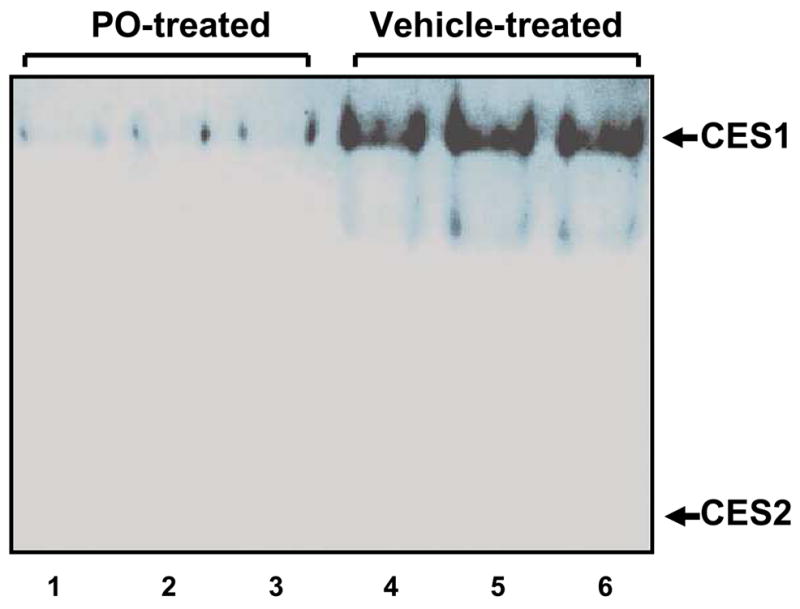
Paraoxon targets CES1 protein in intact THP1 cells. Intact THP-1 cells were exposed to 10 μM paraoxon for 24 h. Control cells were exposed to vehicle only. THP-1 cell lysates were subjected to native gel electrophoresis (25 μg protein in each lane). The gel was stained with a mixture of α,β-naphthyl acetate to detect esterase activity. Lanes 1, 2, and 3 contain THP-1 cell lysate from paraoxon-treated cells and lanes 4, 5, and 6 contain THP-1 cell lysate from control cells. The migration positions of CES1 and CES2 proteins are as indicated.
Fig. 8.

CES1 protein levels are not down-regulated in THP-1 monocytes following exposure to paraoxon. THP-1 cells were incubated with either vehicle (ethanol) or paraoxon (10 μM) for 24 h. Cells were collected, washed, and lysed. Cell lysate proteins were separated by SDS-PAGE and proteins transferred to a PVDF membrane, which was then probed with anti-CES1 and anti-β-actin antibodies. Bar graph represents the pixel density of each gel band (mean ± SD, n = 3).
3.5. Effect of benzil and paraoxon treatments on cholesteryl ester retention in macrophages
THP-1 macrophages were loaded with cholesterol by incubation in culture medium containing human acetylated LDL (50 μg/ml) for 48 h. Subsequently, cholesterol efflux from macrophages was carried out for 48 h using 10% FBS as the extracellular cholesterol acceptor in the presence of either benzil or paraoxon. Benzil, a reversible specific inhibitor of carboxylesterases [32], was previously shown to be cell-permeable, non-cytotoxic, and effective inhibitor of intracellular carboxylesterase activity in cultured U373MG cells transfected with plasmids containing mammalian CES cDNA [33]. The effect of benzil on intracellular cholesterol content is shown in Fig. 9. Exposure of macrophages to 50 μM benzil during the 48-h cholesterol efflux period resulted in a significant and marked increase (>3-fold) in intracellular cholesteryl ester levels. In addition, exposure of macrophages to 10 μM paraoxon during cholesterol efflux also yielded a significant increase in intracellular cholesteryl ester retention (Fig. 9A). Neither benzil nor paraoxon treatments had significant effects on total cellular cholesterol content (Fig. 9B). While free cholesterol levels were similar for the vehicle- and benzil-treated cells, a modest but significant reduction was noted for the paraoxon-treated cells (Fig. 9C). Carboxylesterase activity in cell lysate prepared from 10 μM paraoxon-treated macrophages was significantly inhibited (81% inhibition, p≤0.05) compared to control cells. However, carboxylesterase activity in cell lysate from 50 μM benzil-treated macrophages was not significantly inhibited compared to lysate from control cells due to benzil being a competitive reversible inhibitor of carboxylesterases and its inhibitory activity being extensively diluted during cell lysate preparation [33].
Fig. 9.

Benzil or paraoxon treatments cause significant retention of cholesteryl esters in cholesterol-loaded macrophages. Results are shown for cholesteryl esters (A), total cholesterol (B), and free cholesterol (C). THP-1 monocytes were cultured in growth medium containing 100 nM PMA for 7 days to induce differentiation into macrophages. Adherent cells were then loaded with cholesterol by incubating with 50 μg/ml of human acetylated LDL for 48 h. Cholesterol efflux from the lipid-laden macrophages was accomplished by culturing cells in growth medium containing 10% FBS, which serves as the cholesterol acceptor. Either 50 μM benzil (benzil) or 10 μM paraoxon (PO) was added to the culture media during the cholesterol efflux period to determine their effect on cholesteryl ester retention. Control refers to vehicle-treated cells. Cholesteryl ester, total, and free cholesterol levels are expressed as nmol/mg protein (mean ± SD, n = 4). *, p≤0.05 compared to control (Student’s t-test, n = 4).
4. Discussion
Macrophages contribute significantly to the development and progression of atherosclerosis [2]. Macrophages in arterial walls scavenge free cholesterol and cholesteryl esters in LDL particles and become engorged with lipids. The excessive accumulation of cholesterol in arterial wall macrophages can result in foam cell formation and the progression of atherosclerosis. However, macrophages are also believed to play an important role in the regression of atherosclerosis by the process of macrophage RCT [9]. This mechanism initiates transport of cholesterol from vessel wall macrophages to the liver for secretion in bile. Thus, the relationship between macrophage cholesterol metabolism and atherosclerosis is complex. The rate-limiting step in macrophage RCT is the hydrolysis of cholesteryl esters stored in cytoplasmic droplets, which yields free cholesterol available for export from the cell. A neutral cholesteryl ester hydrolase is believed to catalyze this reaction in macrophages [10], and efflux of stored cholesteryl esters from THP-1 macrophages appears to be regulated by a neutral CEH activity [34].
Several candidate neutral cholesteryl ester hydrolases have been proposed to catalyze the hydrolysis of esterified cholesterol in macrophages. These include hormone-stimulated lipase (HSL) [35], bile salt-stimulated cholesteryl ester hydrolase (also termed carboxyl ester lipase) [36], and a neutral cholesteryl ester hydrolase (CEH; [11]) that is probably the same gene product as human carboxylesterase 1 (CES1; [14]). HSL is found primarily in adipose tissue, but has also been detected in murine macrophages. Further, there are reports indicating that HSL mRNA is expressed in THP-1 monocytes/macrophages [37], although whether or not HSL protein was expressed was not investigated in this particular study. HSL is acutely stimulated by catecholamines and is phosphorylated by a cAMP-dependent kinase, which activates its hydrolytic activity, and triacylglycerol and cholesteryl esters are both substrates [38]. Indeed, overexpression of murine HSL in THP-1 macrophages was shown to increase the rate of cholesterol efflux [39]. In spite of these reports, several lines of evidence argue against HSL being the primary neutral cholesteryl ester hydrolase in macrophages. First, the cholesteryl ester hydrolase activity in primary human monocyte-derived macrophages cannot be stimulated by addition of cAMP [40]. Second, HSL mRNA was not detectable in primary human macrophages [40]. Third, peritoneal macrophages derived from HSL-knockout mice retained the ability to hydrolyze cholesteryl esters [41]. Finally, our own experiments found that HSL protein was not detectable in THP-1 monocyte/macrophage lysates following Western analysis (data not shown). It is further unlikely that the bile salt-stimulated cholesteryl ester hydrolase has a role because this enzyme is secreted by macrophages, thus it would not be available to hydrolyze cholesteryl esters stored in cytoplasmic lipid droplets [36]. Accordingly, it was predicted that another neutral CEH accounts for the catabolism and mobilization of cholesteryl esters in macrophages [42]. Currently, the most likely candidate for this biochemical activity is the neutral CEH that was originally cloned from THP-1 monocyte/macrophages by Ghosh [11]. CEH appears to be the same gene product as the CES1 carboxylesterase expressed abundantly in human liver [12,14]. In support of this, overexpression of CES1 in COS-7 cells yielded demonstrable amounts of cholesteryl ester hydrolytic activity in cell lysates [12]. CES1 was also found to be localized in the cytoplasm of human macrophage cells and to be associated with the cholesteryl ester lipid droplet within the cytoplasm [43]. The most convincing evidence of a role for CES1 in cholesterol metabolism comes from studies in which overexpression of CES1 in THP-1 macrophages was shown to markedly increase the rate of cholesterol efflux in vitro [16]. Moreover, Zhao et al. [17] elegantly demonstrated in vivo that macrophage-specific transgenic over-expression of human CES1 increased macrophage RCT and decreased atherosclerosis in Ldlr−/− mice, a commonly used animal model of atherosclerosis. Thus, both in vitro and in vivo studies point to an essential role for CES1 in cholesteryl ester hydrolysis in macrophages.
Humans are known to have two major carboxylesterases isozymes, CES1 and CES2. Previous reports have shown that CES1 is expressed in human monocytes [44]. In contrast, expression of CES2 mRNA levels in peripheral blood mononuclear cells was reported to be variable with only small amounts of protein being detected [45,46], which is likely due to CES2 expression in lymphocytes but not monocytes. In agreement with this, we detected the presence of CES1 protein, but not CES2 protein, in cell lysates prepared from THP-1 monocytes, and, as mentioned earlier, hormone sensitive lipase (HSL) was not detected in our THP-1 cell lysates. CES1 is known to be stoichiometrically inhibited by oxons, which are the bioactive metabolites of OP pesticides. These pesticides are widely used in agriculture and their metabolites are detected within a significant portion of the U.S. population [47]. Consequently, we have begun investigating the effects of oxons on hydrolytic enzymes in THP-1 cells because of the potential role these compounds may have in the metabolism and mobilization of cholesterol.
Three oxons (chlorpyrifos oxon, paraoxon, and methyl paraoxon) of differing lipophilicity were studied here. These oxons are the active metabolites of three of the most commonly used or well-studied OP pesticides. Chlorpyrifos oxon is the most lipophilic of the three and methyl paraoxon is the least lipophilic [48]. We found that all three oxons were effective inhibitors of carboxylesterase activity in cell lysates prepared from THP-1 monocytes. Consistent with this, we have determined IC50 values for chlorpyrifos oxon, paraoxon, and methyl paraoxon using purified recombinant CES1 protein (Herring et al., unpublished results) that agree well with the inhibition data obtained using THP-1 cell lysates (this study).
We also used FP-biotin, a fluorophosphonate activity-based probe with the same chemotype as OPs, to covalently modify the active-site serine residue of catalytically active serine hydrolases [49]. The goal of this experiment was to detect the suite of serine hydrolases present in THP-1 cell lysates. This probe reacts with a broad spectrum of biochemically-active serine hydrolase enzymes, but not with inactive enzymes such as denaturated or zymogen proteins. Surprisingly, only one protein in THP-1 lysates was reproducibly labeled and detected by FP-biotin without prior enrichment. Further, labeling of this 60-kD protein was inhibited by pre-treatment of cell lysates with paraoxon. It was previously shown that a carboxylesterase in the mouse heart membrane proteome was abundantly labeled by FP-rhodamine [50], and Peeples et al. [51] also demonstrated that FP-biotin could label a serum carboxylesterase in rats. Our immunoblotting results demonstrated that the FP-biotin-labeled 60-kD protein detected in THP-1 cell lysates was specifically identified with anti-CES1 antibodies. Thus, combined with our immunoprecipitation data (Figs. 1 and 3) and native gel data (Fig. 2), we conclude that the 60-kD FP-biotin-labeled protein in Fig. 5 is CES1. Importantly, FP-biotin labeling of CES1 can be inhibited by pre-treatment of the THP-1 cell proteome with paraoxon. The FP-biotin-labeled proteins in cell lysates were also enriched by a pull-down procedure to probe for less abundant serine hydrolases and those less efficiently labeled by FP-biotin as CES1. This allowed the visualization of several other serine hydrolases present in THP-1 cell lysate besides CES1. Although one cannot determine how efficiently each serine hydrolase is labeled by FP-biotin without using normalized quantities of purified proteins, these results suggest that CES1 is one of the more abundantly expressed and/or active serine hydrolases in THP-1 cells and is effectively targeted by oxons in complex proteomes even though oxons are known to react with several classes of serine hydrolases [52].
Inhibition of CES1 activity in THP-1 cells by oxons is due to direct covalent inhibition of the active-site serine residue and not the result of reduced CES1 protein expression (Fig. 6–8). Furthermore, it is important to note that cell viability was not affected by the oxons during the 24-h exposure period. Hence, our data demonstrate that exposure of macrophages to oxons of frequently used pesticides can inhibit an enzyme believed to be responsible for mobilizing free cholesterol from stored cholesteryl ester pools for eventual efflux [16,53]. We therefore postulated that inhibition of CES1 could in turn inhibit macrophage RCT resulting in the excessive accumulation of cholesteryl esters and thus predispose a macrophage to become a foam cell. To investigate this possibility, we loaded THP-1 macrophages with cholesterol and then determined the effect of benzil, a specific reversible inhibitor of carboxylesterases, on the retention of intracellular cholesteryl esters. Consistent with our hypothesis, treatment of macrophages with benzil resulted in an increased retention of intracellular cholesteryl esters (Fig. 9) suggesting reduced cholesteryl ester mobilization and possible impairment of macrophage RCT. This finding suggests that environmental toxicants that inhibit CES1 could also inhibit macrophage cholesterol efflux. In fact, we demonstrated that exposure of cholesterol-loaded macrophages to the toxicant paraoxon also resulted in a significant increase in intracellular cholesteryl ester retention. Importantly, benzil or paraoxon treatments did not significantly change the total cellular cholesterol content of macrophages, rather it increased the ratio of esterified cholesterol:free cholesterol in the cells compared to untreated (control) cells. A consequence of increased retention of cholesteryl esters in cells is that lipid droplets are prone to oxidative damage due to their rich supply of unsaturated fatty acyl chains prone to homolytic cleavage following oxygen radical attack [54]. As a result, marked lipid peroxidation can generate reactive aldehydes (e.g., 4-hydroxynonenal) that damage critical cellular proteins [55], and can upregulate the expression of scavenger receptors further increasing oxidized LDL accumulation [56], thereby exacerbating foam cell formation. Paraoxon is expected to react with other serine hydrolases besides CES1 in THP-1 macrophages. Furthermore, whether benzil can or cannot inhibit lipases besides CES1 in this cell line has not been reported, although our data demonstrates that pure CEL is not inhibited by benzil (Supplementary Figure 1). Accordingly, it is possible that the observed effect of these chemical inhibitors on cholesteryl ester retention is due in part to inhibition of enzymes other than CES1. To address this possibility, we plan to knockdown CES1 expression in THP-1 macrophages using siRNA and determine its effect on cholesterol efflux. Moreover, using activity-based probes such as FP-biotin, it should be possible to determine the inhibitory potency and selectivity of benzil on multiple serine hydrolases in parallel within the THP-1 lysate proteome [50].
The hypothesis that inhibition of a neutral cholesteryl ester hydrolase activity will cause an excess accumulation of intracellular cholesteryl esters is not without precedence. In steroidogenic cells, a neutral cholesteryl ester hydrolase cleaves cholesteryl esters to generate free cholesterol for steroid hormone biosynthesis. A previous report documented the accumulation of cholesteryl esters in steroidogenic cells in the adrenal glands and ovaries following treatment of female rats with either tricresyl phosphate or butylated triphenyl phosphate, both organophosphate compounds [57]. The authors demonstrated that the level of cholesteryl ester accumulation correlated with the degree of inhibition of a neutral cholesteryl ester hydrolase, strongly suggesting that CEH inhibition was responsible for the cholesteryl ester accumulation. Moreover, the hypothesis that reduced cholesteryl ester hydrolase activity can increase atherogenesis also has precedence. Several instances of decreased macrophage cholesteryl ester hydrolase activity have been associated with an increased risk of atherosclerosis and are documented [58–61].
A recent report has demonstrated that CES1 is structurally capable of processing endogenous lipid substrates, and exhibits an alternative channel termed the ‘side-door’ into/from the active site permitting the extrusion of fatty acyl chains of lipid substrates/products [62–64]. Despite repeated attempts by our laboratory to hydrolyze the substrate cholesteryl [1-14C]oleate with purified recombinant CES1 protein, no reproducible enzymatic activity could be detected. Furthermore, addition of detergents, including Triton X-100, sodium cholate, or albumin to the reaction mixture did not improve this outcome. These observations are consistent with the known problems associated with assaying lipase activity in vitro using purified enzymes and water-insoluble substrates [65], particularly since these enzyme-catalyzed reactions are often cell-context dependent [62]. Despite the functional demonstration in cultured macrophages [16] and intact mice [17] that CES1 processes cholesteryl esters for eventual cholesterol efflux, it is likely that undefined co-factors, post-translational modifications, and/or protein-protein interactions, among other possibilities, are required for the correct presentation of the cholesteryl ester substrate to the enzyme active site and its ultimate hydrolysis. In support of this idea, Cravatt and co-workers [66] have shown that a series of N-acyl taurines, which are endogenous lipids found in the central nervous system, were poorly hydrolyzed by pure recombinant fatty-acid amide hydrolase (FAAH) in vitro, while also convincingly demonstrating with FAAH knockout mice that these endogenous lipid compounds were excellent substrates for FAAH in the brain and efficiently catabolized in vivo. Thus, poor substrates for CES1 in vitro such as cholesteryl esters may still be regulated in vivo by CES1, particularly if alternative routes of hydrolysis are absent.
Increasingly, environmental toxicants are being recognized as etiological agents that contribute to atherosclerosis [67]. Our results clearly show that oxons of widely used OP pesticides are able to affect hydrolytic activities in THP-1 monocytes/macrophages catalyzed by CES1. In addition, our results show that exposure of cholesterol-loaded macrophages to benzil (a specific inhibitor of carboxylesterases) and paraoxon (a broad spectrum non-specific inhibitor) caused an increased retention of intracellular cholesteryl esters. This suggests that exposure to oxons may adversely affect macrophage cholesterol efflux and emphasizes the need for a better understanding of the wide range of physiological effects that these compounds may elicit. For example, these findings raise the question of whether exposure to OP pesticides can affect hepatic cholesterol metabolism and the subsequent development of atherosclerosis. Whether or not OPs actually influence the development of atherosclerosis, they should prove to be useful tools for studying fundamental mechanisms of cholesterol metabolism.
Supplementary Material
Acknowledgments
The project was supported by NIH 1R15ES015348 and NIH 5P20RR17661 and the American Lebanese Syrian Associated Charities (ALSAC). Ms. Middleton was supported by NIH T35RR007071. We thank Dr. Howard Chambers, Department of Entomology, Mississippi State University for the gift of chlorpyrifos oxon, paraoxon and methyl paraoxon, Dr. M. Hosokawa, Chiba University, Japan for the gift of rabbit anti-CES1. Kayte Herring and Shuqi Xie are gratefully acknowledged for their help with cell culture and enzyme assays.
List of abbreviations
- ACAT-1
acyl CoA cholesterol acyltransferase
- BSA
bovine serum albumin
- CEH
cholesteryl ester hydrolase
- CES1
carboxylesterase 1
- CES2
carboxylesterase 2
- CPO
chlorpyrifos oxon
- CVD
cardiovascular disease
- EDTA
ethylenediamine tetraacetetic acid
- FP-biotin
6-N-biotinylaminohexyl isopropyl phosphorofluoridate hemihydrate
- HDL
high density lipoprotein
- HRP
horseradish peroxidase
- IC50
50% inhibitory concentration
- LAL
lysosomal acid lipase
- LDL
low density lipoprotein
- MPO
methyl paraoxon
- 4-MUBA
4-methylumbelliferyl acetate
- 4-MUBO
4-methylumbelliferyl oleate
- OP
organophosphorus
- PMA
phorbol 12-myristate 13-acetate
- PMSF
phenylmethylsulfonyl fluoride
- PO
paraoxon
- PVDF
polyvinylidine fluoride
- PBS
phosphate buffered saline
- RCT
reverse cholesterol transport
- SDS PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- 4-MUBA
4-methyl-umbelliferyl acetate
- αβ-NPA
mixture of α- and β-napthylacetate
Footnotes
Conflict of Interest Statement. The authors declare there is no conflict of interest.
References
- 1.American Heart Association. Heart Disease and Stroke Statistics-2008 Update. Dallas, Texas: American Heart Association; 2008. ©2008, American Heart Association. [Google Scholar]
- 2.Libby P, Theroux P. Pathophysiology of coronary artery disease. Circulation. 2005;111:3481–3488. doi: 10.1161/CIRCULATIONAHA.105.537878. [DOI] [PubMed] [Google Scholar]
- 3.Babaev VR, Gleaves LA, Carter KJ, Suzuki H, Kodama T, Fazio S, Linton MF. Reduced atherosclerotic lesions in mice deficient for total or macrophage-specific expression of scavenger receptor-A. Arterioscler Thomb Vasc Biol. 2000;20:2593–2599. doi: 10.1161/01.atv.20.12.2593. [DOI] [PubMed] [Google Scholar]
- 4.Linton MF, Babaev VR, Gleaves LA, Fazio S. A direct role for the macrophage low density lipoprotein receptor in atherosclerotic lesion formation. J Biol Chem. 1999;274:19204–19210. doi: 10.1074/jbc.274.27.19204. [DOI] [PubMed] [Google Scholar]
- 5.Goldstein JL, Dana SE, Faust JR, Beaudet AL, Brown MS. Role of lysosomal acid lipase in the metabolism of plasma low density lipoprotein. J Biol Chem. 1975;250:8487–8795. [PubMed] [Google Scholar]
- 6.Tabas I. Consequences of cellular cholesterol accumulation: basic concepts and physiological implications. J Clin Invest. 2002;110:905–911. doi: 10.1172/JCI16452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang TY, Chang CC, Lin S, Yu C, Li BL, Miyazaki A. Roles of acylcoenzyme A:cholesterol acyltransferase-1 and-2. Curr Opin Lipidol. 2001;12:289–296. doi: 10.1097/00041433-200106000-00008. [DOI] [PubMed] [Google Scholar]
- 8.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 9.Lewis GF, Rader DJ. New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circ Res. 2005;96:1221–1232. doi: 10.1161/01.RES.0000170946.56981.5c. [DOI] [PubMed] [Google Scholar]
- 10.Rothblat GH, Llera-Moya MDL, Favari E, Yancey PG, Kellner-Weibel G. Cellular cholesterol flux studies: methodological considerations. Atherosclerosis. 2002;163:1–8. doi: 10.1016/s0021-9150(01)00713-4. [DOI] [PubMed] [Google Scholar]
- 11.Ghosh S. Cholesteryl ester hydrolase in human monocyte/macrophage: cloning, sequencing and expression of full length cDNA. Physiol Genomics. 2000;2:1–8. doi: 10.1152/physiolgenomics.2000.2.1.1. [DOI] [PubMed] [Google Scholar]
- 12.Zhao B, Natarajan R, Ghosh S. Human liver cholesteryl ester hydrolase: cloning, molecular characterization, and role in cellular cholesterol homeostasis. Physiol Genomics. 2005;23:304–310. doi: 10.1152/physiolgenomics.00187.2005. [DOI] [PubMed] [Google Scholar]
- 13.Satoh T, Hosokawa M. The mammalian carboxylesterases: from molecules to functions. Ann Rev Pharmacol Toxicol. 1998;38:257–288. doi: 10.1146/annurev.pharmtox.38.1.257. [DOI] [PubMed] [Google Scholar]
- 14.Redinbo MR, Potter PM. Mammalian carboxylesterases: from drug targets to protein therapeutics. Drug Discov Today. 2005;10:313–325. doi: 10.1016/S1359-6446(05)03383-0. [DOI] [PubMed] [Google Scholar]
- 15.Dolinsky VW, Gilham D, Alam M, Vance DE, Lehner R. Triacylglycerol hydrolase: Role in intracellular lipid metabolism. Cell Mol Life Sci. 2004;61:1633–1651. doi: 10.1007/s00018-004-3426-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao B, Song J, St Clair R, Ghosh S. Stable over-expression of human macrophage cholesteryl ester hydrolase (CEH) results in enhanced free cholesterol efflux from human THP1-macrophages. Am J Physiol Cell Physiol. 2007;292:C405–C412. doi: 10.1152/ajpcell.00306.2006. [DOI] [PubMed] [Google Scholar]
- 17.Zhao B, Song J, Chow WN, St Clair RW, Rudel LL, Ghosh S. Macrophage-specific transgenic expression of cholesteryl ester hydrolase significantly reduces atherosclerosis and lesion necrosis in Ldlr−/− mice. J Clin Invest. 2007;117:2983–2992. doi: 10.1172/JCI30485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hodgson E, Levi PE. Metabolism of pesticides. In: Krieger RI, Hayes WJ, Laws ER, editors. Handbook of Pesticide Toxicology. I. Academic Press; San Diego, CA: 2001. pp. 531–562. [Google Scholar]
- 19.Saboori AM, Newcombe DS. Human Monocyte Carboxylesterase. J Biol Chem. 1990;265:19792–19799. [PubMed] [Google Scholar]
- 20.Morton CL, Potter PM. Comparison of Escherichia coli, Saccharomyces cerevisiae, Pichia pastoris, Spodoptera frugiperda, and COS7 cells for recombinant gene expression. Application to a rabbit liver carboxylesterase. Mol Biotechnol. 2000;16:193–202. doi: 10.1385/MB:16:3:193. [DOI] [PubMed] [Google Scholar]
- 21.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 22.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ross MK, Borazjani A, Edwards CC, Potter PM. Hydrolytic metabolism of pyrethroids by human and other mammalian carboxylesterases. Biochem Pharmacol. 2006;71:657–669. doi: 10.1016/j.bcp.2005.11.020. [DOI] [PubMed] [Google Scholar]
- 24.Ornstein L. Disc Electrophoresis: background and theory. Ann NY Acad Sci. 1964;121:321–349. doi: 10.1111/j.1749-6632.1964.tb14207.x. [DOI] [PubMed] [Google Scholar]
- 25.Dean RA, Zhang J, Brzezenski MR, Bosron WF. Tissue distribution of cocaine methyl esterase and ethyl transferase activities: Correlation with carboxylesterase protein. J Pharmacol Exp Ther. 1995;275:965–971. [PubMed] [Google Scholar]
- 26.Stok JE, Huang H, Jones PD, Wheelock CE, Morisseau C, Hammock BD. Identification, expression, and purification of a pyrethoid hydrolyzing carboxylesterase from mouse liver microsome. J Biol Chem. 2004;279:29863–29869. doi: 10.1074/jbc.M403673200. [DOI] [PubMed] [Google Scholar]
- 27.Chambers JE, Chambers HW. Time course of inhibition of acetylcholinesterase and aliesterases following parathion and paraoxon exposure in rats. Toxicol Appl Pharmacol. 1990;103:420–429. doi: 10.1016/0041-008x(90)90315-l. [DOI] [PubMed] [Google Scholar]
- 28.Morgan EW, Yan B, Greenway D, Petersen DR, Parkinson A. Purification and characterization of two rat liver microsomal CEs (hydrolase A and B) Arch Biochem Biophys. 1994;315:495–512. doi: 10.1006/abbi.1994.1531. [DOI] [PubMed] [Google Scholar]
- 29.Ross MK, Borazjani A. Enzymatic activity of human carboxylesterases. Curr Protoc Toxicol Unit. 2007;14.24:4.24.1–4.24.14. doi: 10.1002/0471140856.tx0424s33. [DOI] [PubMed] [Google Scholar]
- 30.Liu Y, Patricelli MP, Cravatt BF. Activity-based protein profiling: the serine hydrolases. Proc Natl Acad Sci U S A. 1999;96:14694–14699. doi: 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klansek JJ, Yancey P, St Clair RW, Fischer RT, Johnson WJ, Glick JM. Cholesterol quantitation by GLC: artifactual formation of short-chain steryl esters. 1995;36:2261–2266. [PubMed] [Google Scholar]
- 32.Hyatt JL, Stacy V, Wadkins RM, Yoon KJ, Wierdl M, Edwards CC, Zeller M, Hunter AD, Danks MK, Crundwell G, Potter PM. Inhibition of carboxylesterases by benzil (diphenylethane-1,2-dione) and heterocyclic analogues is dependent upon the aromaticity of the ring and the flexibility of the dione moiety. J Med Chem. 2005;48:5543–5550. doi: 10.1021/jm0504196. [DOI] [PubMed] [Google Scholar]
- 33.Hyatt JL, Tsurkan L, Wierdl M, Edwards CC, Danks MK, Potter PM. Intracellular inhibition of carboxylesterases by benzil: modulation of CPT-11 cytotoxicity. Mol Cancer Ther. 2006;5:2281–2288. doi: 10.1158/1535-7163.MCT-06-0160. [DOI] [PubMed] [Google Scholar]
- 34.Graham A, Angell AD, Jepson CA, Yeaman SJ, Hassall DG. Impaired mobilisation of cholesterol from stored cholesteryl esters in human (THP-1) macrophages. Atherosclerosis. 1996;120:135–145. doi: 10.1016/0021-9150(95)05695-5. [DOI] [PubMed] [Google Scholar]
- 35.Small CA, Goodacre JA, Yeaman SJ. Hormone-sensitive lipase is responsible for the neutral cholesterol ester hydrolase activity in macrophages. FEBS Lett. 1989;247:205–208. doi: 10.1016/0014-5793(89)81335-3. [DOI] [PubMed] [Google Scholar]
- 36.Hui DY, Howles PN. Carboxyl ester lipase: structure-function relationship and physiological role in lipoprotein metabolism and atherosclerosis. J Lipid Res. 2002;43:2017–2030. doi: 10.1194/jlr.r200013-jlr200. [DOI] [PubMed] [Google Scholar]
- 37.Johnson WJ, Jang SY, Bernard DW. Hormone sensitive lipase mRNA in both monocyte and macrophage forms of the human THP-1 cell line. Comp Biochem Physiol B Biochem Mol Biol. 2000;126:543–552. doi: 10.1016/s0305-0491(00)00220-0. [DOI] [PubMed] [Google Scholar]
- 38.Holm C. Molecular mechanisms regulating hormone-sensitive lipase and lipolysis. Biochem Soc Trans. 2003;31:1120–1124. doi: 10.1042/bst0311120. [DOI] [PubMed] [Google Scholar]
- 39.Okazaki H, Osuga J, Tsukamoto K, Isoo N, Kitamine T, Tamura Y, Tomita S, Sekiya M, Yahagi N, Iizuka Y, Ohashi K, Harada K, Gotoda T, Shimano H, Kimura S, Nagai R, Yamada N, Ishibashi S. Elimination of cholesterol ester from macrophage foam cells by adenovirus-mediated gene transfer of hormone-sensitive lipase. J Biol Chem. 2002;277:31893–31899. doi: 10.1074/jbc.M204016200. [DOI] [PubMed] [Google Scholar]
- 40.Contreras JA, Lasuncion MA. Essential differences in cholesteryl ester metabolism between human monocyte-derived and J774 macrophages. Evidence against the presence of hormone-sensitive lipase in human macrophages. Arterioscler Thromb. 1994;14:443–452. doi: 10.1161/01.atv.14.3.443. [DOI] [PubMed] [Google Scholar]
- 41.Osuga J, Ishibashi S, Oka T, Yagyu H, Tozawa R, Fujimoto A, Shionoiri F, Yahagi N, Kraemer FB, Tsutsumi O, Yamada N. Targeted disruption of hormone-sensitive lipase results in male sterility and adipocyte hypertrophy, but not in obesity. Proc Natl Acad Sci USA. 2000;97:787–792. doi: 10.1073/pnas.97.2.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Contreras JA. Hormone-sensitive lipase is not required for cholesteryl ester hydrolysis in macrophages. Biochem Biophys Res Commun. 2002;292:900–903. doi: 10.1006/bbrc.2002.6757. [DOI] [PubMed] [Google Scholar]
- 43.Zhao B, Fisher BJ, St Clair RW, Rudel LL, Ghosh S. Redistribution of macrophage cholesteryl ester hydrolase from cytoplasm to lipid droplets upon lipid loading. J Lipid Res. 2005;46:2114–2121. doi: 10.1194/jlr.M500207-JLR200. [DOI] [PubMed] [Google Scholar]
- 44.Langmann T, Aslanidis C, Schuierer M, Schmitz G. Differentiation-dependant expression of a human carboxylesterase in monocytic cells and transcription factor binding to the promoter. Biochem Biophys Res Commun. 1997;230:215–219. doi: 10.1006/bbrc.1996.5912. [DOI] [PubMed] [Google Scholar]
- 45.Zhang W, Xu G, McLeod HL. Comprehensive evaluation of carboxylesterase-2 expression in normal human tissue using tissue array analysis. Appl Immunohistochem Mol Morphol. 2002;10:374–380. doi: 10.1097/00129039-200212000-00015. [DOI] [PubMed] [Google Scholar]
- 46.Cecchin E, Corona G, Masier S, Biason P, Cattarossi G, Frustaci S, Buonodonna A, Colussi A, Toffoli G. Carboxylesterase isoform 2 mRNA in peripheral blood mononuclear cells is a predictive marker of the irinotecan to SN38 activation step in colorectal cancer patients. Clin Canc Res. 2005;11:6901–6907. doi: 10.1158/1078-0432.CCR-05-0602. [DOI] [PubMed] [Google Scholar]
- 47.Barr DB, Bravo R, Weerasekera G, Caltabiano LM, Whitehead RD, Jr, Olsson AO, Caudill SP, Schober SE, Pirkle JP, Sampson EJ, Jackson RJ, Needham LL. Concentrations of the dialkyl phosphate metabolites of the organphosphorus pesticides in the U.S. population. Environ Health Perspec. 2004;112:186–200. doi: 10.1289/ehp.6503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chambers JE, Carr RL. Inhibition patterns of brain acetylcholinesterase and hepatic and plasma aliesterases following exposures to three phosphorothionate insecticides and their oxons in rats. Fundam Appl Toxicol. 1993;21:111–119. doi: 10.1006/faat.1993.1079. [DOI] [PubMed] [Google Scholar]
- 49.Evans MJ, Cravatt BF. Mechanism-based profiling of enzyme families. Chem Rev. 2006;106:3279–3301. doi: 10.1021/cr050288g. [DOI] [PubMed] [Google Scholar]
- 50.Leung D, Hardouin C, Boger DL, Cravatt BF. Discovering potent and selective reversible inhibitors of enzymes in complex proteomes. Nature Biotechnol. 2003;21:687–691. doi: 10.1038/nbt826. [DOI] [PubMed] [Google Scholar]
- 51.Peeples ES, Schopfer LM, Duysen EG, Spaulding R, Voelker T, Thompson CM, Lockridge O. Albumin, a new biomarker of organophosphorus toxicant exposure, identified by mass spectrometry. Toxicol Sci. 2005;83:303–312. doi: 10.1093/toxsci/kfi023. [DOI] [PubMed] [Google Scholar]
- 52.Casida JE, Quistad GB. Serine hydrolase targets of organophosphorus toxicants. Chem Biol Interact. 2005;157–158:277–283. doi: 10.1016/j.cbi.2005.10.036. [DOI] [PubMed] [Google Scholar]
- 53.Ghosh S, St Clair RW, Rudel LL. Mobilization of cytoplasmic CE droplets by over-expression of human macrophage cholesteryl ester hydrolase. J Lipid Res. 2003;44:1833–1840. doi: 10.1194/jlr.M300162-JLR200. [DOI] [PubMed] [Google Scholar]
- 54.Navab M, Ananthramaiah GM, Reddy ST, Van LentenAnsell BJ, Fonarow GC, Vahabzadeh K, Hama S, Hough G, Kamranpour N, Berliner JA, Lusis AJ, Fogelman AM. The oxidation hypothesis of atherogenesis: the role of oxidized phospholipids and HDL. J Lipid Res. 2004;45:993–1007. doi: 10.1194/jlr.R400001-JLR200. [DOI] [PubMed] [Google Scholar]
- 55.Petersen DR, Doorn JA. Reactions of 4-hydroxynonenal with proteins and cellular targets. Free Radic Biol Med. 2004;37:937–945. doi: 10.1016/j.freeradbiomed.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 56.Yun MR, Im DS, Lee SJ, Woo JW, Hong KW, Bae SS, Kim CD. 4-hydroxynonenal contributes to macrophage foam cell formation through increased expression of class A scavenger receptor at the level of translation. Free Radic Biol Med. 2008;45:177–183. doi: 10.1016/j.freeradbiomed.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 57.Latendresse JR, Azhar S, Brooks CL, Capen CC. Pathogenesis of cholesteryl lipdoisis of adrenocortical and ovarian interstitial cells in F344 rats caused by tricresyl phosphate and butylated triphenyl phosphate. Toxicol Appl Pharmacol. 1993;122:281–289. doi: 10.1006/taap.1993.1197. [DOI] [PubMed] [Google Scholar]
- 58.Mathur SN, Field FJ, Megan MB, Armstrong HL. A defect in mobilization of cholesteryl esters in macrophages. Biochim Biophys Acta. 1985;834:48–57. doi: 10.1016/0005-2760(85)90175-4. [DOI] [PubMed] [Google Scholar]
- 59.Ishii I, Oka M, Katto N, Shirai K, Saito Y, Hirose S. Beta-VLDL-induced cholesterolester deposition in macrophages may be regulated by neutral cholesterol esterase activity. Arterioscler Thomb. 1992;12:1139–1145. doi: 10.1161/01.atv.12.10.1139. [DOI] [PubMed] [Google Scholar]
- 60.Hakamata H, Miyazaki A, Sakai M, Suginohara Y, Sakamoto Y, Horiuchi S. Species differences in cholesteryl ester cycle and HDL-induced cholesterol efflux from macrophage foam cells. Arterioscler Thomb. 1994;14:1860–1865. doi: 10.1161/01.atv.14.11.1860. [DOI] [PubMed] [Google Scholar]
- 61.Yancey PG, St Clair RW. Mechanism of the defect in cholesteryl ester clearance from macrophages of atherosclerosis-susceptible White Carneau pigeons. J Lipid Res. 1994;35:2114–2129. [PubMed] [Google Scholar]
- 62.Bencharit S, Edwards CC, Morton CL, Howard-Williams EL, Kuhn P, Potter PM, Redinbo MR. Multisite Promiscuity in the Processing of Endogenous Substrates by Human Carboxylesterase 1. J Mol Biol. 2006;363:201–214. doi: 10.1016/j.jmb.2006.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wierdl M, Morton CL, Nguyen NK, Redinbo MR, Potter PM. Molecular Modeling of CPT-11 metabolism by carboxylesterases (CEs): Use of pnb CE as a model. Biochemistry. 2004;43:1874–1882. doi: 10.1021/bi035586r. [DOI] [PubMed] [Google Scholar]
- 64.Streit TM, Borazjani A, Lentz S, Wierdl M, Potter PM, Gwaltney SR, Ross MK. Evaluation of the 'side-door' in carboxylesterase-mediated catalysis and inhibition. Biol Chem. 2008;389:149–162. doi: 10.1515/BC.2008.017. [DOI] [PubMed] [Google Scholar]
- 65.Gilham D, Lehner R. Techniques to measure lipase and esterase activity in vitro. Methods. 2005;36:139–147. doi: 10.1016/j.ymeth.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 66.Saghatelian A, Trauger SA, Want EJ, Hawkins EG, Siuzdak G, Cravatt BF. Assignment of endogenous substrates to enzymes by global metabolite profiling. Biochemistry. 2004;43:14332–14339. doi: 10.1021/bi0480335. [DOI] [PubMed] [Google Scholar]
- 67.Brook RD, Franklin B, Cascio W, Hong Y, Howard G, Lipsett M, Luepker R, Mittleman M, Samet J, Smith SC, Tager I. Air Pollution and Cardiovascular Disease (A Statement for Healthcare Professionals From the Expert Panel on Population and Prevention Science of the American Heart Association) Circulation. 2004;109:2655–2671. doi: 10.1161/01.CIR.0000128587.30041.C8. [DOI] [PubMed] [Google Scholar]
- 68.Bonifacino JS, Dell’Angelica EC, Springer TA. Current Protocols in Protein Science. John Wiley and Sons, Inc; Hoboken, NJ: 1999. Unit 9.8 Immunoprecipitation; pp. 9.8.1–9.8.28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.