

Abstract
Proteasome inhibitors are known to suppress the proteasome-mediated degradation of IκBα in stimulated cells. This results in the cytoplasmic retention of NFκB and its reduced nuclear transcriptional activity. In this study, we show that in the metastatic prostate cancer cells, the proteasome inhibitors exhibit a novel, previously unrecognized effect: they increase the cellular levels of IκBα, which then translocates to the nucleus, associates with the nuclear p65 NFκB, thus inhibiting the constitutive NFκB DNA binding activity and inducing apoptosis. The proteasome inhibition-induced nuclear translocation of IκBα is dependent on de novo protein synthesis, occurs also in other cell types, and does not require IκBα phosphorylation on Ser-32. Since NFκB activity is constitutively increased in many human cancers as well as in inflammatory disorders, the proteasome inhibition-induced nuclear translocation of IκBα could thus provide a new therapeutic strategy aimed at the specific inhibition of NFκB activity by the nuclear IκBα.
Keywords: Proteasome inhibition, IκBα, nuclear translocation, prostate cancer, NFκB, apoptosis
INTRODUCTION
NFκB is a key regulator of genes involved in immune and inflammatory responses, as well as genes regulating cell proliferation and survival [1–4]. In most unstimulated resting cells, NFκB is localized in the cytoplasm bound to the inhibitory protein IκBα. Following cell stimulation by extracellular stimuli, including apoptotic signals, inflammatory cytokines, and other forms of cellular stress, IκBα is phosphorylated on Ser-32 and Ser-36 by the enzymes of IκB kinase complex (IKK), ubiquitinated, and selectively degraded by the 26S proteasome [5]. This releases NFκB proteins from the inhibitory complex, and NFκB then translocates to the nucleus and stimulates transcription of NFκB-dependent anti-apoptotic and pro-inflammatory genes [6, 7].
In addition to many inflammatory disorders, NFκB is constitutively activated in a variety of human cancers, including prostate cancer [8–11]. Prostate cancer is the third most common cause of death from cancer in men of all ages [12, 13]. It proceeds from a localized, curable, androgen dependent disease to an invasive, metastatic, and always fatal, androgen-independent disease [14, 15]. The metastatic, androgen-independent prostate cancer is characterized by a constitutive activation of NFκB that induces synthesis of pro-survival genes Bcl-2 and Bcl-xL [16–18]. Thus, inhibition of NFκB activity and induction of apoptosis represent an attractive therapeutic approach for prostate cancer as well as other cancers characterized by the high constitutive NFκB activation [19–22].
We have previously shown that NFκB activity can be inhibited by nuclear accumulation of IκBα, and this is associated with the induction of apoptosis [23, 24]. We have searched for experimental treatment, or conditions, that would induce the nuclear translocation of IκBα, thus inhibiting NFκB activity and inducing apoptosis. The 26S proteasome inhibitors such as MG132 and MG115 are known to suppress NFκB activation and induce apoptosis by inhibiting the inducible degradation of phosphorylated IκBα in stimulated cells [3, 4]. However, several studies indicated that proteasome inhibitors can also induce nuclear translocation and accumulation of transcriptional regulators such as aryl hydrocarbon receptor (ACH)[25], glucocorticoid receptor (GR)[26] and varicella-zoster virus DNA binding protein ORF29p [27] in unstimulated resting cells.
In this study, we considered the possibility that proteasome inhibitors may increase the cellular levels of IκBα, which may then translocate to the nucleus and inhibit the NFκB DNA binding activity, thus inducing apoptosis. To this end, we selected the androgen independent metastatic prostate cancer PC-3 cells that are characterized by nuclear localization of NFκB proteins and high levels of constitutive NFκB DNA binding activity [28–30]. We found that in these cancer cells, as well as in HeLa cells, proteasome inhibition by MG132 and MG115 leads to the nuclear translocation of IκBα. The nuclear accumulation of IκBα then results in the nuclear IκBα-p65 NFκB association, inhibition of NFκB DNA binding activity and induction of apoptosis. Our data show that proteasome inhibitors suppress NFκB activity, and induce apoptosis, by a previously unrecognized mechanism that consists of inducing the nuclear translocation and accumulation of IκBα.
MATERIALS AND METHODS
Antibodies and Reagents
Purified polyclonal antibodies against human IκBα (sc-371), NFκB-p65 (sc-372), NFκB-p50 (sc-7178), Bcl-2 (sc-492), Bcl-xL (sc-7195), and lamin B (sc-6216), as well as monoclonal antibody against Ser-32 phosphorylated IκBα (sc-8404) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Purified polyclonal antibody against lactate dehydrogenase (LDH; 20-LG22) was from Fitzgerald Industries International (Concord, MA, USA), and actin antibody was from Sigma (St Louis, MO). Horseradish peroxidase (HRP)-conjugated anti-rabbit, anti-mouse and anti-goat secondary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA).
T4 polynucleotide kinase, poly (dI-dC), and Sephadex G25 spin columns were purchased from Pharmacia (Piscataway, NJ). CREB (sc-2504, sc-2517) and NFκB (sc-2505, sc-2511) gel shift oligonucleotides were from Santa Cruz Biotechnology (Santa Cruz, CA). [32P]-γ-ATP was purchased from Perkin Elmer (Boston, MA). Proteasome inhibitors MG132, MG115 and PSI, and IκB kinase inhibitor VII (401486) were purchased from EMD Chemicals (San Diego, CA). Cycloheximide, recombinant human tumor necrosis factor-α (TNF) and all other reagents were molecular biology grade and were purchased from Sigma (St Louis, MO).
Cell Culture
All cell lines were obtained from American Type Culture Collection (ATCC; Rockville, MD). Human prostate cancer PC-3 cells were cultured in Ham’s F12K medium (ATCC; Rockville, MD) supplemented with 2 mM L-glutamine, 1% penicillin and streptomycin, and 10% fetal bovine serum (Invitrogen, Grand Island, NY). Epithelial cancer HeLa cells and leukemia HL-60 cells were cultured in Dulbecco’s modified Eagle’s medium and RPMI 1640 medium, respectively, supplemented with 10% fetal bovine serum and antibiotics. Prior to cell treatment, cells were seeded (106 cells/ml) for 24 hours in 6-well plates and grown at 37°C with 5% CO2.
Transfection with siRNA and Proteasome Inhibition
Human IκBα (sc-29360) and control (sc-37007) small interfering RNAs (siRNAs) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The day before transfection, PC-3 cells were seeded into a 24-well plate and incubated in a humidified 5% CO2 incubator at 37°C in antibiotic-free F12K medium supplement with 10% FBS for 24 h to 80% confluence. For each transfection, 60 μmol of either control siRNA-A (sc-37007) or IκBα siRNA (sc-29360) were used. The cells were transfected for 6 hours in transfection medium with siRNA-transfection reagent according to manufacturer’s instructions (Santa Cruz Biotechnology; Santa Cruz, CA). After transfection, fresh F12K medium supplemented with FBS and antibiotics was added, and the cells were treated with proteasome inhibitors for 24 hours.
Proteasome inhibitors MG132, MG115 and PSI were dissolved in DMSO and stored at −20°C. An equivalent volume of DMSO was used in all experiments as a solvent control.
Preparation of Cytoplasmic and Nuclear Extracts
Nuclear (NE) and cytoplasmic extracts (CE) were prepared as described previously [23, 24]. Contamination of nuclear and cytoplasmic fractions by cytoplasmic and nuclear proteins, respectively, was determined by Western analysis using LDH and lamin B as specific markers as described [23, 24].
Electrophoretic Mobility Shift Assay (EMSA)
DNA binding activity of NFκB and CREB was measured by using specific NFκB and CREB double-stranded oligonucleotides as described [23, 31].
Immunoprecipitation
Nuclear extracts were prepared by using the Active Motif’s Nuclear Complex Co-IP Kit (Active Motif, 54001; Carlsbad, CA). The nuclear extracts were incubated (4°C, overnight) with IκBα antibody (sc-371) or control rabbit pre-immune IgG (sc-2027). The immune complexes were immunoprecipitated on A/G Plus Agarose (sc-2003), washed four times with PBS buffer, resolved on 10% SDS gel and detected with IκBα and p65 antibodies as described [32].
Proteasome Activity Assay
Activity of the 20S catalytic proteasomal core unit was measured in whole cell extracts by using the 20S Proteasome Activity Assay Kit (Chemicon, APT280; Temecula, CA) as described by the manufacturer. Briefly, cells were lysed in a lysis buffer (50 mM Hepes, pH 7.5; 5 mM EDTA; 150 mM NaCl; 1% Triton X-100) for 30 minutes on ice, and the lysates were collected by centrifugation (10,000g, 15 min, 4°C). The cell lysates were incubated (2h, 37°C) with a labeled substrate, LLVY-7-amino-4-methylcoumarin, and the cleavage activity was monitored by detection of the free fluorophore 7-amino-4-methylcoumarin using a fluorescence plate reader (Berthold Mithras LB940, Berthold Technologies) at 355/460 nm.
Apoptosis Assay
Apoptosis was quantified with a cell death detection ELISA kit that quantifies release of nucleosomes into the cytoplasm (Cell Death Detection ELISAPLUS, Roche, Indianapolis, IN). The assay was performed at the indicated time points as per the manufacturer’s instructions.
Statistical Analysis
The results represent at least three independent experiments. Numerical results are presented as means ± S.E. Data were analyzed by using an InStat software package (GraphPAD, San Diego, CA). Statistical significance was evaluated by using Mann-Whitney U test with Bonferroni correction for multiple comparisons, and p<0.05 was considered significant.
RESULTS
Proteasome Inhibitor MG132 Induces Nuclear Translocation of IκBα in Prostate Cancer Cells
The metastatic androgen-independent prostate cancer PC-3 cells are characterized by high levels of constitutive NFκB activity that consists of the NFκB p50 and p65 subunits [16, 17]. To analyze the nucleo-cytoplasmic distribution of IκBα and NFκB proteins in these cells, and determine whether proteasome inhibition induces IκBα translocation to the nucleus, PC-3 cells were treated with increasing concentrations of MG132 for 24 hours, and the cytoplasmic and nuclear fractions were prepared and analyzed by western blotting. As a control of the purity of cytoplasmic and nuclear extract preparations, we used lactate dehydrogenase (LDH) and lamin B as specific cytoplasmic and nuclear markers, respectively. Interestingly, in the resting unstimulated PC-3 cells, IκBα was localized predominantly in the cytoplasm, while NFκB p65 and p50 proteins were found mainly in the nuclear fraction (Fig. 1A). Cell treatment with MG132 remarkably decreased IκBα levels in the cytoplasm and induced its dose-dependent translocation to the nucleus. The nuclear translocation of IκBα was induced by 10 μM MG132, and 25 μM and higher concentrations of MG132 resulted in almost complete nuclear localization of IκBα. The nucleo-cytoplasmic distribution of NFκB p50 and p65 proteins was not changed by 1–100 μM MG132, and there was no pronounced effect on the nuclear levels of p50 and p65 NFκB proteins within these MG132 concentrations (Fig. 1A).
Figure 1. Proteasome inhibitor MG132 induces nuclear translocation of IκBα in prostate cancer PC-3 cells.
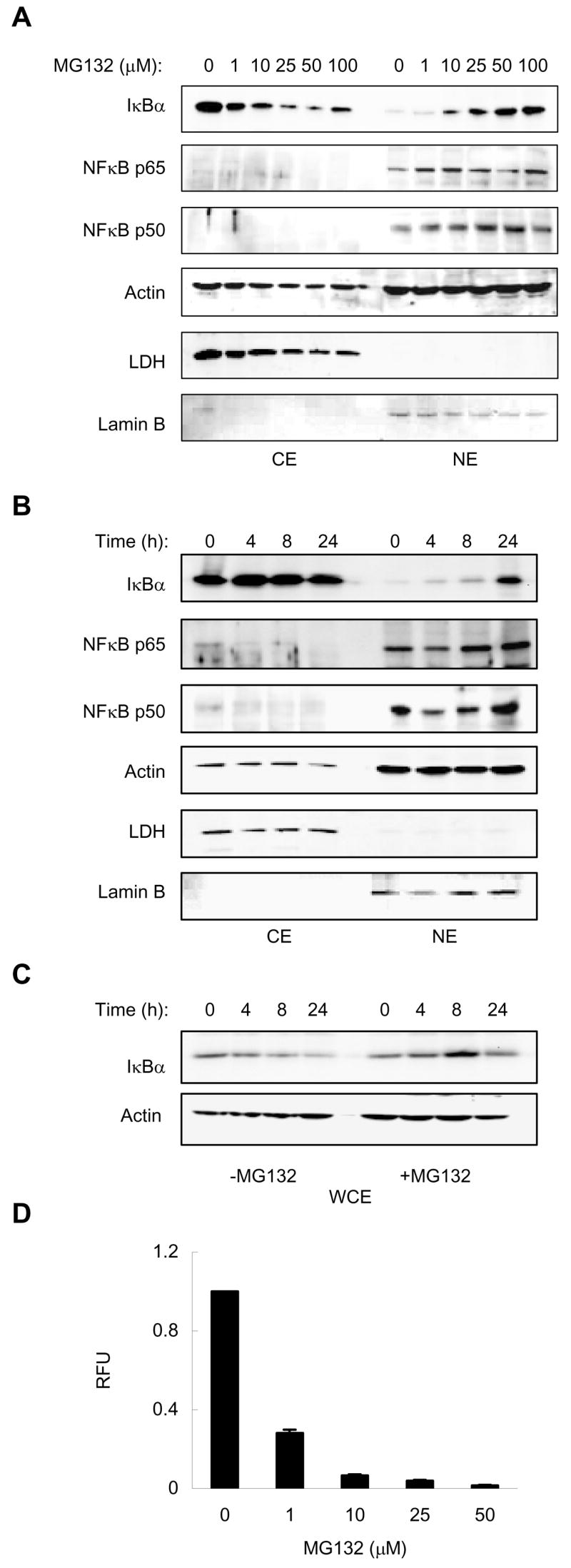
(A) Western blotting of cytoplasmic (CE) and nuclear extracts (NE) prepared from PC-3 cells treated with increasing concentrations of MG132 for 24 hours, and analyzed by using IκBα, p65 and p50 NFκB antibodies. To confirm equal protein loading, the membranes were stripped and re-probed with actin antibody. The presence of cytoplasmic proteins in nuclear fraction was evaluated by re-probing the membrane with LDH antibody. Nuclear contamination in the cytoplasmic fraction was assessed by using lamin B specific antibody. Each lane corresponds to approximately 5×104 cells. (B) Western blotting of CE and NE performed as described above, prepared from PC-3 cells treated with 50 μM MG132 for 0–24 hours. (C) Western blotting of whole cell extracts (WCE) prepared from unstimulated cells and cells treated with 50 μM MG132 for 0–24 hours. Each lane corresponds to approximately 2×104 cells. (D) Proteasome activity measured in WCE prepared from PC-3 cells treated with increasing concentrations of MG132 for 24 hours. The 20S proteasomal activity is expressed as relative fluorescence units (RFU) of MG132-treated cells compared to untreated cells. The values represent the mean +/−SE of three independent experiments measured in duplicates.
To determine whether the nuclear translocation of IκBα in response to MG132 is time dependent, we analyzed IκBα levels in cytoplasmic and nuclear extracts of PC-3 cells treated with 50 μM MG132 for 0 to 24 hours. As shown in Fig. 1B, while the nuclear IκBα translocation occurred 4 to 8h after treatment with MG132, substantial accumulation of IκBα in the nucleus was observed after 24 hour treatment with MG132.
However, since we observed that in addition to inducing the nuclear translocation of IκBα, 4 and 8 h treatment with MG132 also somewhat increased the cytoplasmic IκBα levels (Fig. 1B), and because IκBα in PC-3 cells was shown to have a rapid metabolic turnover attributed to its phosphorylation on Ser-32 and subsequent degradation by the proteasome [16, 17], we investigated whether MG132 treatment increases the total cellular levels of IκBα in PC-3 cells. As shown in Fig. 1C, in untreated cells, the total cellular levels of IκBα, analyzed in whole cell extracts, gradually decreased during 24 hour incubation. In contrast, 8 and 24 h treatment with 50 μM MG132 increased the total cellular levels of IκBα compared to the whole cell extracts of untreated cells (Fig. 1C). These data suggested that the nuclear translocation of IκBα induced by proteasome inhibition in PC-3 cells is associated with an increase of the total cellular IκBα levels, and indicated that the proteasome is constitutively active in these cells.
To demonstrate the proteasome activity in PC-3 cells, and to verify that MG132 indeed inhibits the proteasome activity, we measured activity of the 20S proteasome catalytic subunit in the whole cells extracts of PC-3 cells incubated 24 hours with increasing concentrations of MG132. As shown in Fig. 1D, 1 μM MG132 inhibited about 70% of the proteasome activity in PC-3 cells, and about 90% inhibition was achieved by 10 μM MG132. Thus, these data indicate that a substantial inhibition of the proteasome is required to induce the nuclear accumulation of IκBα, and suggest that even a small residual proteasomal activity in PC-3 cells is sufficient to control the level of IκBα in the cytoplasm.
The Nuclear IκBα, Induced upon Proteasome Inhibition, Associates with p65 NFκB in the Nucleus, Thus Inhibiting the Constitutive NFκB DNA Binding Activity
To determine whether the nuclear translocation of IκBα, induced by proteasome inhibition, is associated with the inhibition of NFκB activity, we measured NFκB DNA binding activity in nuclear extracts prepared from PC-3 cells treated with 0–50 μM MG132. As shown in Fig. 2A, the constitutive nuclear NFκB DNA-binding activity in PC-3 cells was drastically reduced by 24 hour treatment with 10 μM MG132, which also induced the nuclear translocation of IκBα (Fig. 1A), and completely inhibited by 25 and 50 μM MG132 concentrations, which resulted in the predominantly nuclear localization of IκBα (Fig. 1A, 2A). Using supershift characterization, we confirmed that this activity consisted of p50/p65 NFκB heterodimers (Fig. 2B). In contrast to NFκB, DNA binding activity of another, IκBα-independent transcription factor, CREB, was not affected by 1–50 μM concentrations of MG132, indicating specificity for NFκB (Fig. 2A). Fig. 2C shows a time course of 50 μM MG132 effect on the NFκB and CREB DNA binding activities. While the CREB activity was not markedly changed, 24 hour treatment with 50 μM MG132 resulted in a pronounced inhibition of NFκB DNA binding (Fig. 2C), consistent with the increased nuclear levels of IκBα (Fig. 1B).
Figure 2. The MG132-induced nuclear translocation of IκBα is associated with the inhibition of NFκB activity.
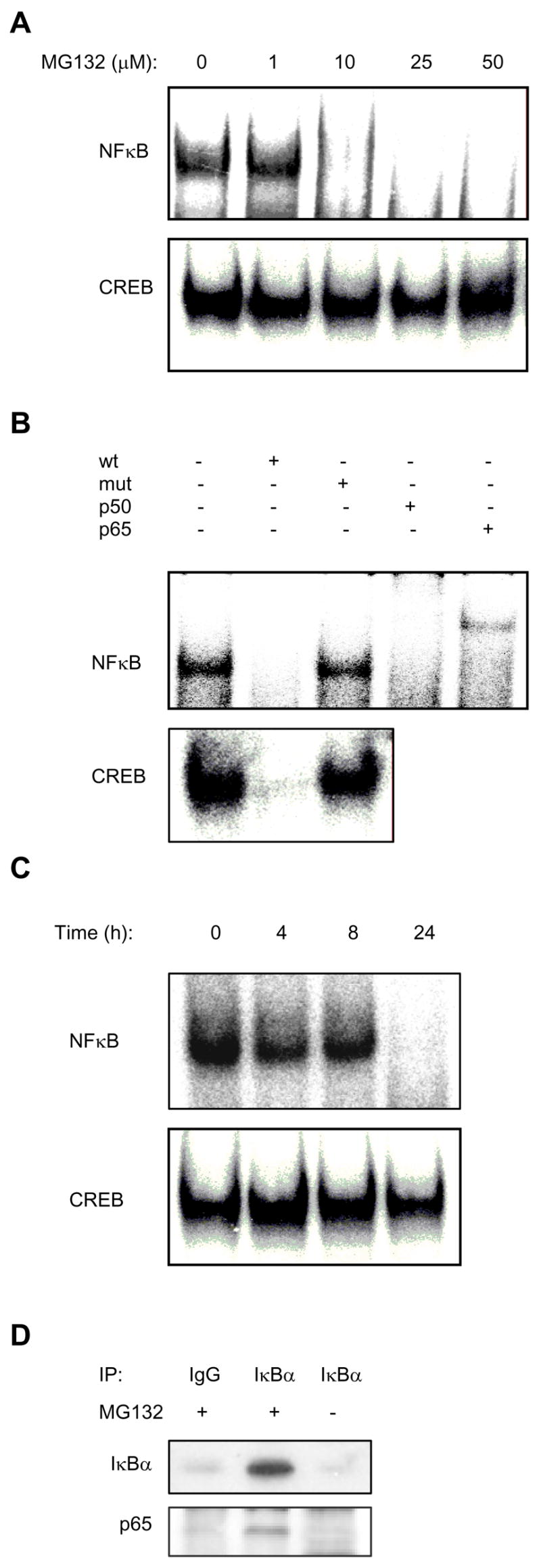
(A) EMSA of NFκB and CREB DNA binding activities analyzed in nuclear extracts from PC-3 cells treated for 24 hours with increasing concentrations of MG132. (B) Specificity analysis of the constitutive NFκB and CREB activities in PC-3 cells. Nuclear extracts from untreated PC-3 cells were incubated with NFκB or CREB specific DNA probes alone or in the presence (+) of 30 molar excess of unlabeled wild type (wt) or mutant (mut) oligonucleotides. Antibodies used in supershift NFκB assays included antibodies to p50 and p65. (C) EMSA of NFκB and CREB activities analyzed in nuclear extracts from PC-3 cells treated with 50 μM MG132 for 0–24 hours. (D) Co-immunoprecipitation experiment from nuclear extracts of untreated or MG132-treated (50 μM, 24h) cells by using pre-immune IgG or IκBα-specific antibody. The western blots were analyzed with IκBα and p65 NFκB antibodies.
To support the data that the NFκB DNA binding activity is inhibited by IκBα that translocates to the nucleus as a result of the proteasome inhibition, we performed a co-immunoprecipitation experiment using IκBα antibody and nuclear extracts prepared from untreated as well as MG132-treated (50 μM, 24h) PC-3 cells. As shown in Fig. 2D (top panel), IκBα was immunoprecipitated from the nuclear extracts of MG132-treated cells, but not from the nuclear extracts of untreated cells, or from MG132-treated cells immunoprecipitated by using control pre-immune IgG. Immunoblotting by using p65 NFκB antibody revealed the presence of p65 NFκB in the nuclear extracts of MG132-treated cells immunoprecipitated with IκBα antibody, but not with pre-immune IgG (Fig. 2D, lower panel). Importantly, no p65 NFκB signal was detected in the IκBα-immunoprecipitates prepared from the nuclear extracts of untreated cells (Fig. 2D, lower panel), even though p65 NFκB is present in the nucleus of untreated PC-3 cell (Fig.1 A, B), demonstrating specificity for the IκBα-binding proteins. Together, these data show that IκBα that translocates to the nucleus after the proteasome inhibition, associates with p65 NFκB in the nucleus, and provide the mechanism by which the nuclear IκBα inhibits NFκB DNA binding activity.
The Nuclear Translocation of IκBα Is Induced by Other Proteasome Inhibitors As Well
To ascertain that the nuclear translocation of IκBα is not restricted only to MG132, but can be induced by other proteasome inhibitors as well, we tested effects of other reversible proteasome inhibitors, MG115 (Z-Leu-Leu-Nva-CHO) and PSI (Z-Ile-Glu(OtBu)-Ala-Leu-CHO). Both MG115 and PSI induced a dose-dependent translocation of IκBα from the cytoplasm to the nucleus, similarly as in the cells treated with MG132, and this was associated with the inhibition of NFκB DNA binding activity (data not shown).
The MG132-Induced Nuclear Translocation of IκBα Results in the Induction of Apoptosis
To determine whether the MG132-induced nuclear translocation of IκBα and inhibition of NFκB activity are associated with the induction of apoptosis, we analyzed the protein levels of two NFκB-regulated anti-apoptotic proteins, Bcl-2 and Bcl-xL, and measured apoptosis by a quantitative ELISA assay based on the detection of nucleosome release into the cytoplasm. As shown in Fig. 3A and B, Bcl-2 and Bcl-xL expression were pronouncedly reduced by 25 and 50 μM MG132, and the cytoplasmic nucleosome release was increased by 10–50 μM MG132. Altogether, these data indicated that the 26S proteasome inhibitors suppress NFκB activity and induce apoptosis, at least partly, by inducing translocation of IκBα from the cytoplasm to the nucleus.
Figure 3. The MG132-Induced Nuclear Translocation of IκBα Results in the Induction of Apoptosis.
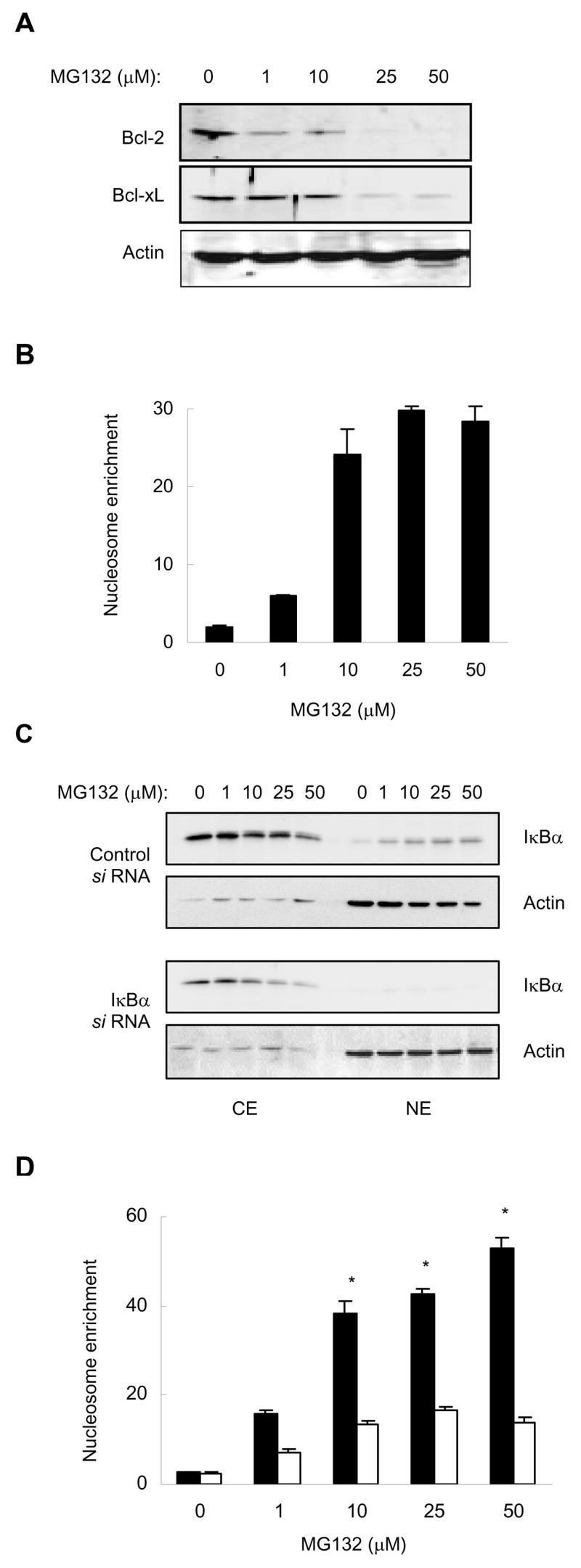
(A) Western blotting of WCE prepared from PC-3 cells treated with increasing concentrations of MG132 for 24 hours, and analyzed by using Bcl-2, Bcl-xL, and actin antibodies. Each lane corresponds approximately to 2×104 cells. (B) Apoptosis measured by the quantitative nucleosome enrichment assay in PC-3 cells treated 24 hours with increasing concentrations of MG132. (C) Western blotting of CE and NE prepared from PC-3 cells transfected with control and IκBα si RNA, followed by treatment with increasing concentrations of MG132 for 24 hours. The membranes were analyzed by using IκBα and actin antibodies. Each lane corresponds approximately to 4×104 cells. (D) Apoptosis measured by the nucleosome enrichment assay in PC-3 cells transfected with control (full columns) and IκBα (empty columns) si RNA, followed by treatment with increasing concentrations of MG132 for 24 hours. The values represent the mean +/−SE of four independent experiments. Asterisks denote a statistically significant (P<0.01) inhibition.
To determine whether the increased apoptosis observed in MG132-treated cells is caused by the increased nuclear levels of IκBα, rather than by some side effects of MG132, we hypothesized that suppression of IκBα nuclear levels should inhibit apoptosis in MG132-treated cells. To test this hypothesis, we silenced IκBα expression by IκBα-specific si RNA, and then treated cells with increasing concentrations of MG132 for 24 hours. As expected, IκBα si RNA greatly reduced the cellular protein levels of IκBα compared to cells transfected with control si RNA, resulting in barely detectable IκBα in the nucleus of MG132-treated cells (Fig. 3C). The decreased nuclear expression of IκBα in cells transfected with IκBα si RNA, compared to cells transfected with control RNA, resulted in a significantly reduced apoptosis in response to treatment with 10–50 μM MG132 (P<0.01) (Fig. 3D), demonstrating that the increased nuclear IκBα levels induce apoptosis.
New Protein Synthesis Is Required for the MG132-Induced Nuclear Translocation of IκBα
The induced nuclear accumulation of IκBα in response to proteasome inhibition raised a question whether de novo protein synthesis is required for the IκBα nuclear translocation. To investigate this, PC-3 cells were treated with cycloheximide (CHX, 10 μg/ml, 1h) to inhibit new protein synthesis, before 24 h incubation with increasing concentrations of MG132. Interestingly, CHX treatment completely prevented the MG132-induced nuclear IκBα translocation, resulting in exclusively cytoplasmic localization of IκBα (Fig. 4). These results suggest that de novo protein synthesis is required for the proteasome inhibition-induced nuclear translocation of IκBα.
Figure 4. De novo protein synthesis is required for the MG132-induced nuclear translocation of IκBα in PC-3 cells.
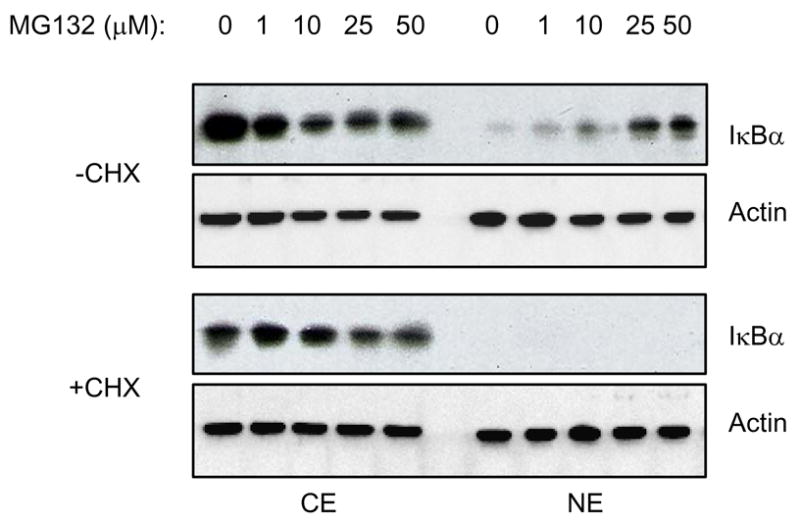
Western blotting of CE and NE prepared from PC-3 cells treated with increasing concentrations of MG132 for 24 hours, in the absence and presence of 1h pre-treatment with CHX (10 μg/ml). The membranes were analyzed by using IκBα and actin antibodies. Each lane corresponds approximately to 5×104 cells.
The MG132-Induced Nuclear Translocation of IκBα Is Not Dependent on IκBα Phosphorylation by IκB Kinase and Occurs Also in Other Cell Types
Since the constitutive NFκB activity in PC-3 cells is associated with high level of IκBα that is phosphorylated by IκB kinase (IKK) on Ser-32 [16, 17], we investigated whether the phosphorylated IκBα translocates to the nucleus after MG132 treatment. PC-3 cells were treated with control vehicle (DMSO) or 50 μM MG132 for 0–24 hours, and the cytoplasmic and nuclear extracts were analyzed by immunoblotting using monoclonal antibody specific against IκBα phosphorylated on Ser-32. As expected, in unstimulated cells and cells treated only with DMSO, IκBα was phosphorylated, and was localized in the cytoplasm (Fig. 5A). However, during 4–24 h treatment with MG132, the phosphorylated form of IκBα translocated from the cytoplasm to the nucleus (Fig. 5A). These results suggested that phosphorylation by IKK may be required for the proteasome inhibition-induced nuclear translocation of IκBα.
Figure 5. The MG132-induced nuclear translocation of IκBα is not dependent on IκBα phosphorylation by IκB kinase.
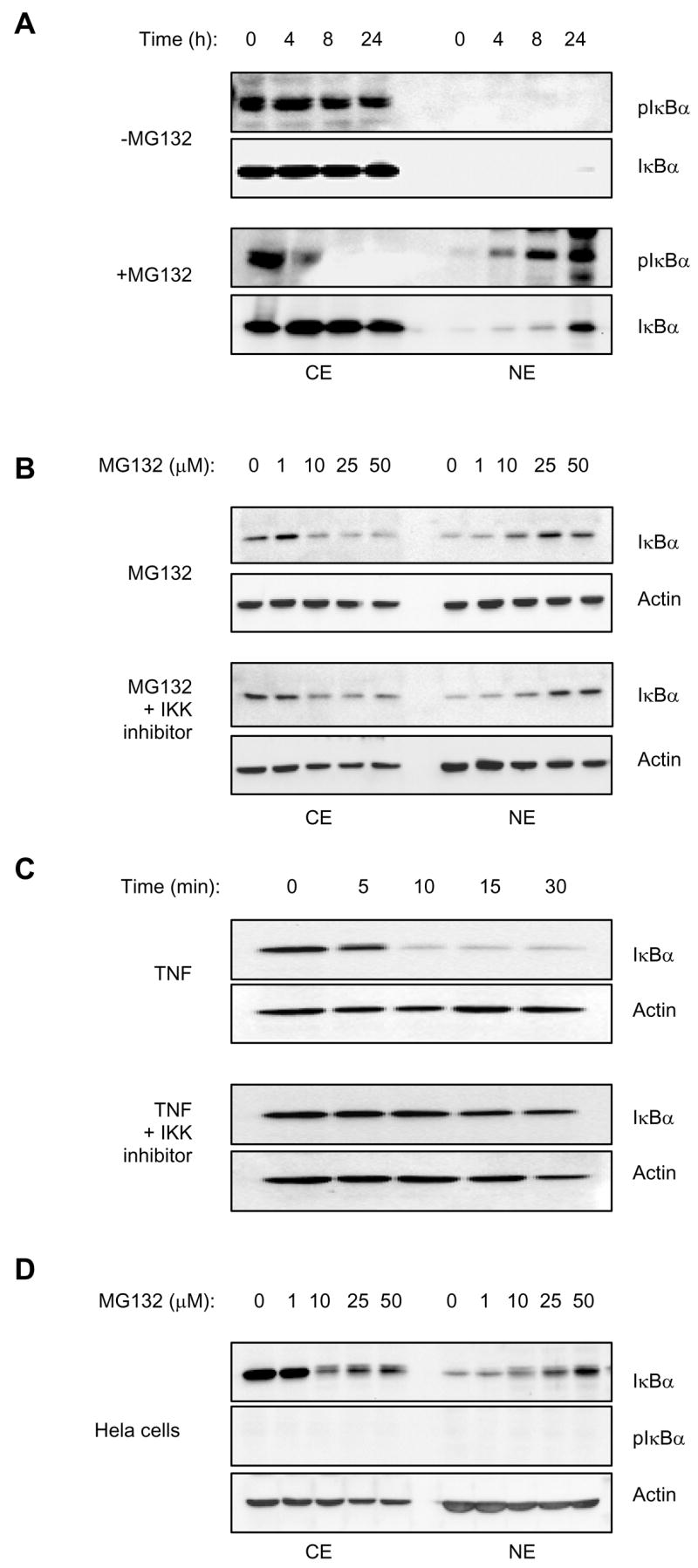
(A) Western blotting of CE and NE prepared from PC-3 cells treated for 0–24 hours with control DMSO or 50 μM MG132, and analyzed by using Ser-32-phosho-IκBα (pIκBα) and IκBα antibodies. Each lane corresponds approximately to 5×104 cells. (B) Western analysis of CE and NE prepared from PC-3 cells pre-treated for 1 hour with 10 μM IKK inhibitor VII (Calbiochem, 401486) or control DMSO, followed by 24 hour incubation with increasing concentrations of MG132. The samples were analyzed by using IκBα and actin antibodies; each lane corresponds approximately to 5×104 cells. (C) Western analysis of cytoplasmic extracts prepared from PC-3 cells treated for 1 hour with 10 μM IKK inhibitor VII or control DMSO, followed by 0 – 30 minutes stimulation with 10 ng/ml TNF. The samples were analyzed by using IκBα and actin antibodies, and each lane corresponds approximately to 5×104 cells. (D) Western analysis of CE and NE prepared from HeLa cells treated for 24 hours with increasing concentrations of MG132, and analyzed by using IκBα, Ser-32-phosho-IκBα (pIκBα) and actin antibodies. Each lane corresponds approximately to 5×104 cells.
To further examine the role of phosphorylation by IKK in the MG132-induced nuclear translocation of the endogenous IκBα, we analyzed the nucleo-cytoplasmic localization of IκBα in PC-3 cells treated with MG132 in the absence and presence of a specific inhibitor of IKK. As shown in Fig. 5B, 10 μM IKK inhibitor did not prevent the MG132-induced nuclear translocation of IκBα, even though it did inhibit the TNF-induced degradation of IκBα in the cytoplasmic extracts of PC-3 cells (Fig. 5C), thus verifying the inhibitor’s functionality.
To validate these results to be independent of a specific cancer cell line, we used additional well-characterized cancer cell lines, epithelial HeLa cells and leukemia HL-60 cells. Even though IκBα is not constitutively phosphorylated on Ser-32 in these cells [33, 34], MG132 induced a dose-dependent translocation of IκBα from the cytoplasm to the nucleus (Fig. 5D). Thus, even though the Ser-32 phosphorylated IκBα translocates to the nucleus in response to proteasome inhibition in prostate cancer cells, this phosphorylation does not seem to be required for the nuclear translocation of IκBα. Together, these data show that proteasome inhibition suppresses NFκB activity by a novel mechanism that consists of inducing the nuclear translocation and accumulation of IκBα.
DISCUSSION
Proteasome inhibitors are known to suppress NFκB activity and induce apoptosis by inhibiting the 26S proteasome-mediated degradation of IκBα in stimulated cells [1–4]. Thus, modulation of proteasome function has emerged as a novel target for the treatment of multiple cancers as well as inflammatory and immune disorders [35–37]. In this study, we have shown that proteasome inhibitors suppress the constitutive NFκB DNA binding activity in metastatic prostate cancer cells by a novel mechanism, which consists of increasing the cellular levels of IκBα, which then translocates from the cytoplasm to the nucleus (Fig. 6). The nuclear accumulation of IκBα, induced by proteasome inhibition, results in the nuclear IκBα-p65 NFκB association, inhibition of NFκB DNA binding activity and induction of apoptosis of prostate cancer cells. Since NFκB activity is constitutively increased in many human cancers as well as in inflammatory disorders, the proteasome inhibition-induced nuclear accumulation of IκBα could thus provide a new therapeutic strategy aimed at the specific inhibition of NFκB activity by the nuclear IκBα.
Figure 6. Current model of the regulation of nuclear IκBα translocation by proteasome inhibition.
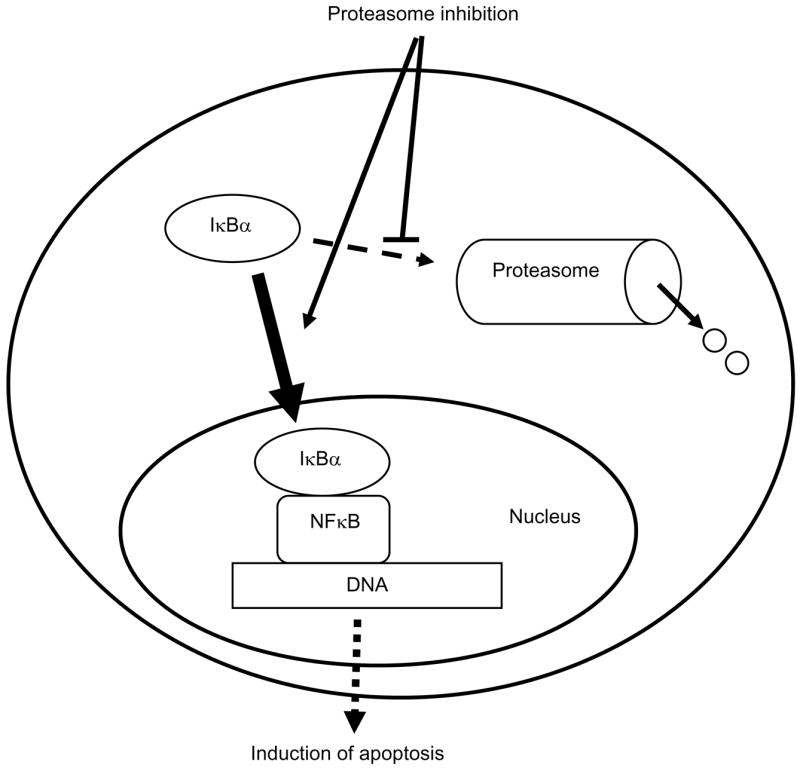
The 26S proteasome is a large, multi-subunit protein complex that selectively degrades cellular proteins, present both in the cytoplasm and in the nucleus. Our data show that although proteasome inhibition resulted in the translocation of IκBα from the cytoplasm to the nucleus, it did not change the cellular localization on NFκB p50 and p65 proteins, nor induced nuclear translocation of LDH (Fig. 1). Interestingly, however, proteasome inhibition has been also shown to induce the nuclear translocation of GR [26], aryl hydrocarbon receptor [25], and varicella zoster DNA-binding protein ORF29p [27]. All these proteins are transcriptional regulators that can be specifically degraded by the 26S proteasome. Although the exact mechanisms by which the proteasome inhibition induces their nuclear translocation have not been fully elucidated, studies suggest an involvement of protein stabilization resulting in increased total cellular levels, and post-translational modification by phosphorylation by protein kinase C [25–27, 38].
Our data demonstrate that the nuclear translocation of IκBα induced by proteasome inhibition in metastatic prostate cancer cells correlates with the stabilization of IκBα, resulting in the increased total cellular levels of IκBα (Fig. 1). Even though in prostate cancer cells, the proteasome inhibition induces nuclear translocation of IκBα that is phosphorylated by IKK on Ser-32 (Fig. 5A), this phosphorylation does not seem to be necessary for the IκBα nuclear translocation in response to proteasome inhibition. First, the nuclear translocation of the endogenous IκBα is not prevented by the inhibition of IKK (Fig. 5B and C), and second, the nuclear translocation of IκBα occurs also in cells that exhibit low levels of constitutive IKK activity and IκBα Ser-32 phosphorylation: HeLa cells (Fig. 5D) and HL-60 cells (not shown). These data suggest that there might be an IKK-independent mechanism that targets IκBα to the proteasome in these cells. In this context, several mechanisms have been described that lead to ubiquitin-mediated proteasome-dependent NFκB activation that occurs in the absence of IκBα phosphorylation on Ser-32 and Ser-36 [39–44]. These IKK-independent mechanisms include the UV-induced NFκB activation [39–41], NFκB activation induced by hepatitis B virus X protein [42], and the IκBα degradation and NFκB activation induced by prolonged cell treatment with a chemotherapeutic agent, doxorubicin [43]. Although the precise mechanisms by which these slow-activating agents induce the proteasomal IκBα degradation have not been identified, they seem to involve phosphatidylinositol 3-kinases, a serine-threonine kinase Akt, and the extracellular signal regulated kinase ERK [43, 44].
Since IκBα is a protein that has a high rate of metabolic turnover, our data indicate that proteasome inhibition prevents degradation of the cytoplasmic IκBα, which then translocates to the nucleus, and by binding to p65 NFκB inhibits NFκB DNA binding activity. These data are also consistent with our results showing that new protein synthesis is required in order for IκBα to translocate to the nucleus (Fig. 4). The lack of IκBα nuclear translocation in response to proteasome inhibition in CHX-treated cells could be explained by two mutually non-exclusive mechanisms. In the first model, treatment with CHX might prevent resynthesis of a protein that is otherwise necessary for the nuclear translocation of IκBα in MG132-treated cells, but has a short half-life; thus, treatment with CHX would significantly decrease its level. Alternatively, the nuclear translocation of IκBα in MG132-treated cells may require that the cellular (cytoplasmic) level of IκBα increases above certain threshold level. When the cells are treated with CHX, de-novo synthesis of IκBα is inhibited, and IκBα never reaches this threshold level, even after the degradation of IκBα is blocked by MG132. Similar mechanism has been suggested to account for the nuclear accumulation of GR [26] and the varicella-zoster virus DNA binding protein ORF29p [27]. In this model, the nuclear translocation of IκBα, triggered by the increased cytoplasmic level of IκBα, could be explained by saturating a cytoplasmic protein, or a cellular structure, which binds and anchors IκBα in the cytoplasm. This model is also supported by previous studies that used cells transfected with constructs expressing wild type (wt) IκBα and demonstrated that when the wt IκBα protein is over-expressed, it localizes in the nucleus [45–48]. These studies indicated that IκBα is retained in the cytoplasm through its association with the NFκB proteins; however, when not bound to NFκB, the free IκBα translocates to the nucleus [45–48].
The nuclear translocation of IκBα is mediated by the second ankyrin repeat of IκBα that also mediates interaction with NFκB proteins [49, 50]. As a result, in most unstimulated cells, IκBα is localized in the cytoplasm bound to NFκB proteins, which can translocate to the nucleus only after IκBα had been degraded by proteasome upon stimulation [1–4]. However, during post-induction repression in continuously stimulated cells, since the NFκB proteins are in the nucleus, the newly synthesized IκBα is not bound to NFκB in the cytoplasm, resulting in IκBα translocation to the nucleus and terminating NFκB-dependent transcription [51]. Interestingly, however, we have found that in the metastatic prostate cancer PC-3 cells, IκBα is localized in the cytoplasm, while the NFκB proteins are predominantly in the nucleus (Fig. 1). Thus, the mechanisms that regulate the cytoplasmic localization of IκBα in PC-3 cells, and its MG132-induced nuclear translocation, are clearly different from the IκBα nuclear shuttling during post-induction repression.
In addition, the cytoplasmic localization of IκBα in PC-3 cells raises a question of the mechanisms responsible for its cytoplasmic retention in these cells. Since IκBα contains nuclear export sequences [52–54], one possibility might be that IκBα continuously shuttles in the prostate cancer cells, and the nuclear export is dominant over the nuclear import, as has been described in most unstimulated cells [46, 47]. However, this does not seem very likely, since our data indicate that once in the nucleus, IκBα binds to p65 NFκB, inhibits NFκB DNA binding activity and induces apoptosis of the androgen-independent PC-3 cells (Figs. 2 and 3). Alternatively, the cytoplasmic retention of IκBα in untreated PC-3 cells can be explained by binding of IκBα to cytoplasmic proteins or components. This hypothesis seems to be supported by our CHX data (Fig. 4), suggesting that the nuclear translocation of IκBα is regulated by its total cytoplasmic levels, as well as by previous study by Prigent et al demonstrating that in HeLa cells, IκBα is retained in the cytoplasm by binding to protein G3BP2 [48]. In addition, even though the cytoplasmic level of p65 NFκB in PC-3 cells is relatively low, it might be sufficient to retain IκBα in the cytoplasm. However, if the concentration of free cytoplasmic IκBα is increased by blocking the proteasome-dependent IκBα degradation, IκBα translocates and accumulates in the nucleus.
Inhibitors of the 26S proteasome have been used in the treatment of patients with multiple myeloma, and are undergoing evaluation in clinical trials in a variety of malignancies, including the metastatic, androgen independent prostate cancer [55, 56]. In summary, our results show that the proteasome inhibitors have a novel, previously unrecognized effect: they induce the nuclear translocation and accumulation of IκBα. In the metastatic prostate cancer cells, this results in the inhibition of the constitutive NFκB activity and induction of apoptosis. Future studies should address the specific mechanisms by which the increased stability of IκBα leads to its translocation to the nucleus. The induction of nuclear accumulation of IκBα could provide a basis for novel therapies in disorders characterized by high levels of constitutive NFκB activity, such as certain cancers and inflammatory disorders.
Acknowledgments
This work was supported by the National Institutes of Health (NIH) research grant GM079581 and by St. John’s University Faculty Research Award to I. Vancurova.
References
- 1.Ghosh S, Karin M. Cell. 2002;109:S81–S96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 2.Gilmore TD. Oncogene. 2006;25:6680–6684. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- 3.Baeuerle PA, Baltimore D. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 4.Baldwin AS. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 5.Verma IM, Stevenson J. Proc Natl Acad Sci USA. 1997;94:11758–11760. doi: 10.1073/pnas.94.22.11758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Basseres DS, Baldwin AS. Oncogene. 2006;25:6817–6830. doi: 10.1038/sj.onc.1209942. [DOI] [PubMed] [Google Scholar]
- 7.Hayden MS, West AP, Ghosh S. Oncogene. 2006;25:6758–6780. doi: 10.1038/sj.onc.1209943. [DOI] [PubMed] [Google Scholar]
- 8.Yamamoto Y, Gaynor RB. Curr Mol Med. 2001;1:287–296. doi: 10.2174/1566524013363816. [DOI] [PubMed] [Google Scholar]
- 9.Bharti AC, Aggarwal BB. Biochem Pharmacol. 2002;64:883–888. doi: 10.1016/s0006-2952(02)01154-1. [DOI] [PubMed] [Google Scholar]
- 10.Dutta J, Fan Y, Gupta N, Fan G, Gelinas C. Oncogene. 2006;25:6800–6816. doi: 10.1038/sj.onc.1209938. [DOI] [PubMed] [Google Scholar]
- 11.Karin M. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 12.Nelson WG, De Marzo AM, Isaacs WB. N Engl J Med. 2003;349:366–381. doi: 10.1056/NEJMra021562. [DOI] [PubMed] [Google Scholar]
- 13.De Marzo AM, Nakai Y, Nelson WG. Urol Oncol. 2007;25:398–400. doi: 10.1016/j.urolonc.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 14.Catz SD, Johnson JL. Oncogene. 2001;20:7342–7351. doi: 10.1038/sj.onc.1204926. [DOI] [PubMed] [Google Scholar]
- 15.Suh J, Payvandi F, Edelstein LC, Amenta PS, Zong WX, Gélinas C, Rabson AB. Prostate. 2002;52:183–200. doi: 10.1002/pros.10082. [DOI] [PubMed] [Google Scholar]
- 16.Palayoor ST, Youmell MY, Calderwood SK, Coleman CN, Price BD. Oncogene. 1999;18:7389–7394. doi: 10.1038/sj.onc.1203160. [DOI] [PubMed] [Google Scholar]
- 17.Gasparian AV, Yao YJ, Kowalczyk D, Lyakh LA, Karseladze A, Slaga TJ, Budunova IV. J Cell Sci. 2002;115:141–151. doi: 10.1242/jcs.115.1.141. [DOI] [PubMed] [Google Scholar]
- 18.Yemelyanov A, Gasparian A, Lindholm P, Dang L, Pierce JW, Kisseljov F, Karseladze A, Budunova I. Oncogene. 2006;25:387–398. doi: 10.1038/sj.onc.1209066. [DOI] [PubMed] [Google Scholar]
- 19.Huang S, Pettaway CA, Uehara H, Bucana CD, Fidler IJ. Oncogene. 2001;20:4188–4197. doi: 10.1038/sj.onc.1204535. [DOI] [PubMed] [Google Scholar]
- 20.Shukla S, Gupta S. Clin Cancer Res. 2004;10:3169–3178. doi: 10.1158/1078-0432.ccr-03-0586. [DOI] [PubMed] [Google Scholar]
- 21.Xu C, Shen G, Chen C, Gelinas C, Kong AN. Oncogene. 2005;24:4486–4495. doi: 10.1038/sj.onc.1208656. [DOI] [PubMed] [Google Scholar]
- 22.O’Connor S, Shumway S, Miyamoto S. Mol Cancer Res. 2005;3:42–49. [PubMed] [Google Scholar]
- 23.Vancurova I, Miskolci V, Davidson D. J Biol Chem. 2001;276:19746–19752. doi: 10.1074/jbc.M100234200. [DOI] [PubMed] [Google Scholar]
- 24.Castro-Alcaraz S, Miskolci V, Kalasapudi B, Davidson D, Vancurova I. J Immunol. 2002;169:3947–3953. doi: 10.4049/jimmunol.169.7.3947. [DOI] [PubMed] [Google Scholar]
- 25.Santiago-Josefat B, Pozo-Guisado E, Mulero-Navarro S, Fernandez-Salguero PM. Mol Cell Biol. 2001;21:1700–1709. doi: 10.1128/MCB.21.5.1700-1709.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deroo BJ, Rentsch C, Sampath S, Young J, DeFranco DB, Archer TK. Mol Cell Biol. 2002;22:4113–4123. doi: 10.1128/MCB.22.12.4113-4123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stallings CL, Duigou GJ, Gershon AA, Gershon MD, Silverstein SJ. J Virol. 2006;80:1497–1512. doi: 10.1128/JVI.80.3.1497-1512.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shukla S, MacLennan GT, Fu P, Patel JS, Marengo SR, Resnick MI, Gupta S. Neoplasia. 2004;6:390–400. doi: 10.1593/neo.04112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Domingo-Domenech J, Mellado B, Ferrer B, Truan D, Codony-Servat J, Sauleda S, Alcover J, Campo E, Gascon P, Rovira A, Ross JS, Fernández PL, Albanell J. Br J Cancer. 2005;93:1285–1294. doi: 10.1038/sj.bjc.6602851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lessard L, Begin LR, Gleave ME, Mes-Masson AM, Saad F. Br J Cancer. 2005;93:1019–1023. doi: 10.1038/sj.bjc.6602796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vancurova I, Wu R, Miskolci V, Sun S. J Virol. 2002;76:1533–1536. doi: 10.1128/JVI.76.3.1533-1536.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miskolci V, Rollins J, Vu HY, Ghosh CC, Davidson D, Vancurova I. Mol Med. 2007;13:134–142. doi: 10.2119/2006-00072.Miskolci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miyamoto S, Maki M, Schmitt MJ, Hatanaka M, Verma IM. Proc Natl Acad Sci USA. 1994;91:12740–12744. doi: 10.1073/pnas.91.26.12740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DiDonato JA, Mercurio F, Karin M. Mol Cell Biol. 1995;15:1302–1311. doi: 10.1128/mcb.15.3.1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lawrence T, Gilroy DW, Colville-Nash PR, Willoughby DA. Nat Med. 2001;12:1291–1297. doi: 10.1038/nm1201-1291. [DOI] [PubMed] [Google Scholar]
- 36.Hideshima T, Chauhan D, Richardson P, Mitsiades C, Mitsiades N, Hayashi T, Munshi N, Dang L, Castro A, Palombella V, Adams J, Anderson KC. J Biol Chem. 2002;277:16639–16647. doi: 10.1074/jbc.M200360200. [DOI] [PubMed] [Google Scholar]
- 37.Mitchell BS. N Engl J Med. 2003;348:2597–2598. doi: 10.1056/NEJMp030092. [DOI] [PubMed] [Google Scholar]
- 38.Santiago-Josefat B, Fernandez-Salguero PM. J Mol Biol. 2003;333:249–260. doi: 10.1016/j.jmb.2003.08.020. [DOI] [PubMed] [Google Scholar]
- 39.Li N, Karin M. Proc Natl Acad Sci USA. 1998;95:13012–13017. doi: 10.1073/pnas.95.22.13012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bender K, Göttlicher M, Whiteside S, Rahmsdorf HJ, Herrlich P. EMBO J. 1998;17:5170–5181. doi: 10.1093/emboj/17.17.5170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang TT, Feinberg SL, Suryanarayanan S, Miyamoto S. Mol Cell Biol. 2002;22:5813–5825. doi: 10.1128/MCB.22.16.5813-5825.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Purcell NH, Yu C, He D, Xiang J, Paran N, DiDonato JA, Yamaoka S, Shaul Y, Lin A. Am J Physiol Gastrointest Liver Physiol. 2001;280:G669–677. doi: 10.1152/ajpgi.2001.280.4.G669. [DOI] [PubMed] [Google Scholar]
- 43.Tergaonkar V, Bottero V, Ikawa M, Li Q, Verma IM. Mol Cell Biol. 2003;23:8070–8083. doi: 10.1128/MCB.23.22.8070-8083.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sathe SS, Sizemore N, Li X, Vithalani K, Commane M, Swiatkowski SM, Stark GR. Proc Natl Acad Sci USA. 2004;101:192–107. doi: 10.1073/pnas.0306812101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zabel U, Henkel T, Silva MS, Baeuerle PA. EMBO J. 1993;12:201–211. doi: 10.1002/j.1460-2075.1993.tb05646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang TT, Miyamoto S. Mol Cell Biol. 2001;21:4737–4747. doi: 10.1128/MCB.21.14.4737-4747.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee SH, Hannink M. J Biol Chem. 2001;276:23599–23606. doi: 10.1074/jbc.M011197200. [DOI] [PubMed] [Google Scholar]
- 48.Prigent M, Barlat I, Langen H, Dargemont C. J Biol Chem. 2000;275:36441–36449. doi: 10.1074/jbc.M004751200. [DOI] [PubMed] [Google Scholar]
- 49.Sachdev S, Hoffmann A, Hannink M. Mol Cell Biol. 1998;18:2524–2534. doi: 10.1128/mcb.18.5.2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Turpin P, Hay RT, Dargemont C. J Biol Chem. 1999;274:6804–6812. doi: 10.1074/jbc.274.10.6804. [DOI] [PubMed] [Google Scholar]
- 51.Rodriguez MS, Thompson J, Hay RT, Dargemont C. J Biol Chem. 1999;274:9108–9015. doi: 10.1074/jbc.274.13.9108. [DOI] [PubMed] [Google Scholar]
- 52.Huang TT, Kudo N, Yoshida M, Miyamoto S. Proc Natl Acad Sci USA. 2000;97:1014–1019. doi: 10.1073/pnas.97.3.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Johnson C, Van Antwerp D, Hope TJ. EMBO J. 1999;18:6682–6693. doi: 10.1093/emboj/18.23.6682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tam WF, Lee LH, Davis L, Sen R. Mol Cell Biol. 2000;20:2269–2284. doi: 10.1128/mcb.20.6.2269-2284.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Papandreou CN, Logothetis CJ. Cancer Res. 2004;64:5036–5043. doi: 10.1158/0008-5472.CAN-03-2707. [DOI] [PubMed] [Google Scholar]
- 56.Canfield SE, Zhu K, Williams SA, McConkey DJ. Mol Cancer Ther. 2006;5:2043–2050. doi: 10.1158/1535-7163.MCT-05-0437. [DOI] [PubMed] [Google Scholar]