

Abstract
The host cell recognition protein of the Escherichia coli bacteriophage HK620 is a large homotrimeric tailspike that cleaves the O18A1 type O antigen. The crystal structure of HK620 tailspike determined in the apo and substrate-bound form is reported by Barbirz et al. in this issue of Molecular Microbiology. Lacking detectable sequence similarity, the fold and overall organization of the HK620 tailspike are similar to those of the tailspikes of the related phages P22 and Sf6. The substrate-binding site is intra-subunit in P22 and HK620 tailspikes, but inter-subunit in Sf6, demonstrating how phages can adapt the same protein fold to recognize different substrates.
Keywords: tailspike, evolution, polysaccharide, hydrolase, bacteriophage
Conservation of structure in large oligomeric enzymes generally implies that function is conserved and the substrate-binding site is in a similar location, even in the absence of detectable sequence similarity. Two papers from the Seckler and Heinemann laboratories, one in this issue of Molecular Microbiology (Barbirz et al., 2008), refute this generality. Crystal structures of the 500+ residue, trimeric tailspikes from bacteriophages HK620 (Barbirz et al., 2008) and Sf6 (Müller et al., 2008) show that these enzymes, whose overall folds and organizations are conserved, differ markedly in their amino acid sequences, substrate specificities, enzymatic mechanisms, and locations of the substrate binding site. Barbirz et al. present a detailed characterization of the HK620 endo-N-acetylglucosamidase that degrades the O antigen lipopolysaccharide (LPS) of its Escherichia coli host. The crystal structures were determined to a remarkable 1.4 Å resolution for the apoenzyme and 1.6 Å when substrate-bound, allowing the first demonstration that the LPS of E. coli H TD2158 was indeed O18A1. Müller et al. (2008) provide a comparable analysis of the endorhamnosidase of the Shigella flexneri phage Sf6. These papers set a high standard for articles reporting an enzyme structure.
Unlike laboratory strains of E. coli, the LPS of most environmental isolates is complete and is often further extended by a polymer of repeating carbohydrates called O antigen. Some bacteria also display a K antigen, a different capsular polysaccharide (PS) that was first defined as an antigen that masked O antigens. This distinction is no longer clear, as K and O antigens are now known to share more common features than differences (Schnaitman, 2001) Both consist of repeating units of two to seven, often modified, sugars that might be in branched chains. More than 180 O and 80 K antigens have been described for E. coli alone (Schnaitman, 2001). Changes in basal antigen structure by phase variation, or modification of some sugars (e.g., acetylation) within the repeating units can lead to an enormous variation in surface structures. Prophages and plasmids are major contributors to LPS variation.
Phages that infect common laboratory strains of E. coli, which have a truncated LPS, adsorb to cells using tail fibres. The tail fibres are arranged symmetrically around the tail and thus orient the phage particle on the cell surface. Subsequently, the tail recognizes a second receptor, which might be a different molecule or a different part of the same molecule. Only in a very few cases has this second receptor, which can trigger DNA ejection in vitro, or the corresponding phage tail protein been identified. The O and K antigens of natural isolates do not trigger DNA ejection; they form a continuous layer that can prevent the tip of the tail from accessing the second receptor. Barbirz et al. report that the average number of O antigen repeats in the HK620 host is 13; in an extended conformation, this creates a 320 Å layer, about twice the length of the phage tail knob. Some K antigens extend from the cell surface as much as 4000 Å, which clearly prevents direct recognition of the second receptor by the phage tail. In order to infect bacteria displaying O and K antigens, phages like HK620 and K1E have replaced the tail fibre with a tailspike, an enzyme that recognizes and degrades the PS. There are one or two sets of six tailspikes on a tail (Lander et al., 2006; Leiman et al., 2007), where each set is specific for a particular antigen. The tailspikes function to create a crater or tunnel in the PS layer, thereby allowing the tail to approach the cell surface and access its receptor.
Most bacteria do not appear to turn over their own extracellular PS, and phage tailspikes represent a major source of polysaccharide degrading enzymes. Binding and digestion of the cell surface PS by the tailspikes has the normal specificity of an enzyme reaction, meaning that the tailspikes of a specific phage can only degrade one or a few O or K antigens. The bewildering variety of these bacterial antigens is then met by the even greater variety and number of phages in the environment. Although phages have yet to be found for some bacterial species, it is rather unlikely that such phages do not exist somewhere, or could not be created.
Perhaps as a consequence of LPS diversity, phage tail fibre and tailspike genes represent the most diverse part of their already very diverse genomes. In phages P22, Sf6, and HK620, it is possible to detect sequence similarities in all corresponding virion proteins (Casjens et al., 2004). However, although the 114 residue-long, N-terminal, tail-binding domain of the tailspikes show 70% sequence identity, the primary sequences of the catalytic, receptor-binding parts have diverged beyond any detectable similarity. Nevertheless, Barbirz et al. show that the catalytic domains are also homologous because they exhibit very similar structures. The major part of the catalytic domain is formed by a right-handed β-helix found in many proteins that process oligo- and polysaccharides, first described in pectate lyase C (Yoder et al., 1993). In P22, Sf6, and HK620 tailspikes, the β-helix has been enhanced by 5-6 additional rungs (or complete turns of the polypeptide chain around the helix axis), making it longer by 25–30 Å. Three β-helices assemble into an elongated homotrimer with the help of the C-terminal domain.
Phages have evolved two strategies to adapt their tailspikes to the substrate diversity represented by bacterial LPS. First, they use the same robust fold, but change the active site residues. Second, they incorporate foreign domains, already active on a chosen substrate, into the tailspikes. Barbirz et al. argue that the tailspikes of phages P22, Sf6, HK620, and perhaps many others, have evolved using the first strategy. The robust fold is the right-handed β-helix, which can tolerate many mutations as long as stacking interactions between the β-helical rungs are preserved. The amino acid sequence dissimilarity of P22, Sf6, and HK620 tailspikes suggests that a β-helical structure can be as diverse as an α-helix and, thus, just as versatile. The β-helix has a “natural” groove (Fig. 1) that forms a suitable general scaffold for oligosaccharide binding but the actual substrate specificity in the tailspikes is determined by loops that protrude out from the β-helix to form part of the active site.
Figure 1.
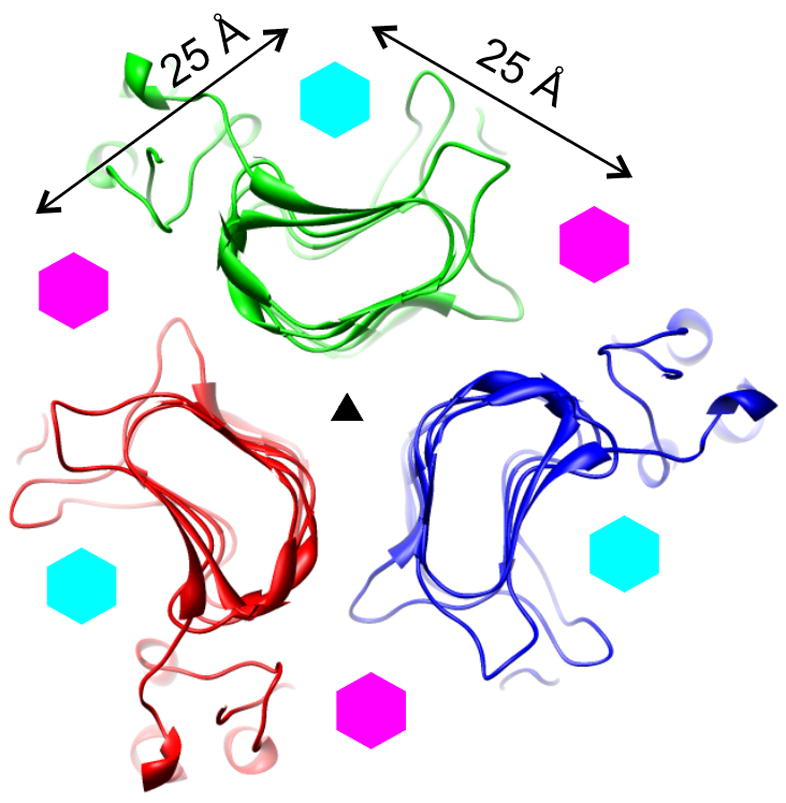
A schematic showing the position of the active sites on the β-helices of three related tailspikes: P22, Sf6, and HK620 (Barbirz et al., 2008), (Müller et al., 2008; Steinbacher et al., 1996). A 20 Å-thick slice through the structure of the P22 tailspike in the region containing the active sites is shown. The slice is perpendicular to the threefold axis of the protein that is indicated with a black triangle. Three polypeptide chains are coloured red, green, and blue. The positions of the P22/HK620 (intrasubunit) and Sf6 (intersubunit) active sites are indicated with cyan and magenta hexagons, respectively.
The active site of the HK620 and P22 tailspikes is on the face of the β-helix, inside its “natural” groove, i.e. it is intra-subunit, whereas the Sf6 tailspike active site lies between two neighbouring β-helices and is thus inter-subunit (Barbirz et al., 2008; Müller et al., 2008; Steinbacher et al., 1996) (Fig. 1). It seems unlikely that a tailspike could lose its capacity to bind substrate intra-molecularly and acquire an ability to bind it inter-molecularly without an intermediate that can bind in both positions. In fact, all three tailspikes in question might possess secondary low affinity binding sites (inter-molecular in P22/HK620 and intra-molecular in Sf6) that are not revealed in standard biochemical experiments. This environment is significantly different to the bacterial cell surface, where the substrate concentration is very high and the tailspikes likely function cooperatively (Leiman et al., 2007). Such secondary binding sites might have evolved to play a more important role as the active site changed from intra- to inter-subunit. As structural investigations of additional tailspikes continue, new β-helical structures might be discovered, and a hypothetical intermediate that has both inter- and intra-molecular active sites might be found.
The second evolutionary strategy, which can be called “cut-and-paste”, is also a very common feature of phage tailspike architecture. There seems to be a conserved set of N-terminal tail-binding domains, and a much greater variety of the C-terminal host cell-binding modules. Phages can exchange their tailspike modules and change their specificity. The N-terminal domain of the CUS-3 tailspike is homologous to the HK620, Sf6 and P22 tailspikes, but the CUS-3 catalytic domain is an endosialidase responsible for degradation of K1 capsular PS (Stummeyer et al., 2006). K1F tailspike contains an endosialidase module fused to an N-terminal domain homologous to the T7 L-shaped fibre (Stummeyer et al., 2006). Cut-and-paste events can also occur within the catalytic domains of tailspikes. For example, the catalytic part of endosialidases is comprised of a classical (exo-)sialidase domain, a small β-barrel domain and an intertwined C-terminal β-helical domain that assembles the entire structure into a homotrimer. The structural location of the active site and even two of the five active site residues in the exosialidase are conserved in the endosialidase. The activity of the endosialidase is many-fold higher than its exo- relative, demonstrating that phages can adjust the properties of “captured” domains to perform new functions.
A novel insight gained from studies of capsule-specific tailspikes is the presence of a secondary substrate-binding site. The crystal structure of K1F endosialidase shows that, in addition to the cleavage site, there is an additional substrate binding site (Stummeyer et al., 2006). The latter might function to hold the phage particle between depolymerization events, resulting in processivity of enzyme action. O antigen degradation by P22, Sf6, and HK620 might occur in a similar manner, and a putative secondary binding site of the tailspikes functions similarly to the K1-specific tailspikes.
Barbirz et al. argue that the three related tailspikes in question (P22, Sf6, and HK620) evolved from a common ancestor (Strategy 1 described above) and that these proteins do not represent gene mosaics (Strategy 2), meaning that they have not recently been pasted together into functional enzymes. This idea is supported by structural similarities in both the β-helices and the C-terminal domains. Although it is likely that the three tailspikes have a common origin, the ancestral enzyme appears to be assembled from three unrelated parts (the N-, catalytic and C-terminal domains), likely a result of several non-homologous recombination events. Furthermore, all three domains might have been exchanged many times with equivalent modules from other tailspikes before reaching the structures we see today. It is, in fact, both the chicken-and-egg and Plutarch’s Ship of Theseus paradoxes.
Tailspike enzymes degrading cell surface polysaccharides have long been known for phages belonging to all three families of tailed phages: Myoviridae, Siphoviridae, Podoviridae (Rieger-Hug and Stirm, 1981). Electron microscopy suggests that many tailspikes have similar elongated structures. The atomic structures of three enzymes from the Podoviridae (Barbirz et al., 2008; Müller et al., 2008; Steinbacher et al., 1996) and one from the Myoviridae (Walter et al., 2008) strongly support the idea that the β-helix is a predominant structural motif in the majority of catalytic domains. These domains must constantly adapt to their ever-changing cell surface PS substrates; they are also frequently exchanged between diverse phages with very different ancestries. The unanswered evolutionary question posed by the results of Barbirz et al. and Müller et al. is then: did Nature switch between an inter- and intra-subunit active site (or vice versa) in generating enzyme diversity or is that diversity a product of different evolutionary pathways converging through exchange of compatible modules? “Cherchez le phage”! Their continued study will undoubtedly reveal more of Nature’s mysterious secrets.
References
- Barbirz S, Müller JJ, Utrecht C, Clark AJ, Heinemann U, Seckler R. Crystal structure of E. coli phage HK620 tailspike reveals that podoviral tailspike endoglycosidase modules are evolutionary related. Molecular Microbiology. 2008 doi: 10.1111/j.1365-2958.2008.06311.x. this issue. [DOI] [PubMed] [Google Scholar]
- Casjens S, Winn-Stapley DA, Gilcrease EB, Morona R, Kuhlewein C, Chua JE, Manning PA, Inwood W, Clark AJ. The chromosome of Shigella flexneri bacteriophage Sf6: complete nucleotide sequence, genetic mosaicism, and DNA packaging. J Mol Biol. 2004;339:379–394. doi: 10.1016/j.jmb.2004.03.068. [DOI] [PubMed] [Google Scholar]
- Lander GC, Tang L, Casjens SR, Gilcrease EB, Prevelige P, Poliakov A, Potter CS, Carragher B, Johnson JE. The structure of an infectious P22 virion shows the signal for headful DNA packaging. Science. 2006;312:1791–1795. doi: 10.1126/science.1127981. [DOI] [PubMed] [Google Scholar]
- Leiman PG, Battisti AJ, Bowman VD, Stummeyer K, Muhlenhoff M, Gerardy-Schahn R, Scholl D, Molineux IJ. The structures of bacteriophages K1E and K1–5 explain processive degradation of polysaccharide capsules and evolution of new host specificities. J Mol Biol. 2007;371:836–849. doi: 10.1016/j.jmb.2007.05.083. [DOI] [PubMed] [Google Scholar]
- Müller JJ, Barbirz S, Heinle K, Freiberg A, Seckler R, Heinemann U. An inter-subunit active site between supercoiled parallel beta-helices in the trimeric tailspike endorhamnosidase of Shigella flexneri phage Sf6. Structure. 2008 doi: 10.1016/j.str.2008.01.019. in the press. [DOI] [PubMed] [Google Scholar]
- Rieger-Hug D, Stirm S. Comparative study of host capsule depolymerases associated with Klebsiella bacteriophages. Virology. 1981;113:363–378. doi: 10.1016/0042-6822(81)90162-8. [DOI] [PubMed] [Google Scholar]
- Schnaitman CA. The genetics and biosynthesis of lipopolysaccharides. In: Sussman M, editor. Molecular Medical Microbiology. Vol. 1. New York: Academic Press; 2001. pp. 93–136. [Google Scholar]
- Steinbacher S, Baxa U, Miller S, Weintraub A, Seckler R, Huber R. Crystal structure of phage P22 tailspike protein complexed with Salmonella sp. O-antigen receptors. Proc Natl Acad Sci U S A. 1996;93:10584–10588. doi: 10.1073/pnas.93.20.10584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stummeyer K, Schwarzer D, Claus H, Vogel U, Gerardy-Schahn R, Muhlenhoff M. Evolution of bacteriophages infecting encapsulated bacteria: lessons from Escherichia coli K1-specific phages. Mol Microbiol. 2006;60:1123–1135. doi: 10.1111/j.1365-2958.2006.05173.x. [DOI] [PubMed] [Google Scholar]
- Walter M, Fiedler C, Grassl R, Biebl M, Rachel R, Hermo-Parrado XL, Llamas-Saiz AL, Seckler R, Miller S, van Raaij MJ. Structure of the receptor-binding protein of bacteriophage Det7: a podoviral tail spike in a myovirus. J Virol. 2008;82:2265–2273. doi: 10.1128/JVI.01641-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder MD, Keen NT, Jurnak F. New domain motif: the structure of pectate lyase C, a secreted plant virulence factor. Science. 1993;260:1503–1507. doi: 10.1126/science.8502994. [DOI] [PubMed] [Google Scholar]