

Abstract
Many studies have indicated leukocytes as one of the major contributors to brain injuries caused by intracerebral hemorrhage (ICH). Leukocyte-expressed CD18 is important for neutrophil-endothelial interactions in the vasculature and CD18 deficiency protects against ischemia-reperfusion injury. We investigated whether CD18 deficiency provides protection against ICH-induced brain injury. Male wild type (WT) CD18+/+ mice and CD18−/− knockout mice were used in this study. ICH was induced by a collagenase injection. Mortality, neurological function, brain edema and myeloperoxidase (MPO) activity as well as tissue expression of nitrotyrosine and MPO were evaluated at 24 hours after ICH. We discovered a significantly reduced brain edema and diminished mortality with a concomitant decrease in MPO and nitrotyrosine immunoreactivities in brains of CD18 knockout mice.
Keywords: CD18−/− mice, intracerebral hemorrhage, collagenase, inflammation, brain edema
INTRODUCTION
Intracerebral hemorrhage (ICH) remains a major medical problem, which so far does not have an effective treatment. Mechanisms, underlying brain injury after ICH, have been extensively studied with the inflammatory response playing an extremely important role in ICH-induced brain injury. This response includes the infiltration of neutrophils and macrophages into and around the hematoma, which peaks at 48 to 72 hours after ICH onset (Gong et al., 2000; Wu et al., 2006; Xi et al., 2004).
Many studies attribute a significant involvement of leukocytes in ICH-induced brain injury (Huang et al., 2006; Power et al., 2003; Wang et al., 2007b). Interactions between β2-integrins (CD18) and intercellular adhesion molecule-1 (ICAM-1) are responsible for firm adhesion of neutrophils to the endothelium in the acute stage of inflammation (Winn et al., 1998). Importantly, β2 integrins (CD11/CD18) are exclusively expressed in leukocytes hence their importance for immune responses. Following their recruitment, the activated leukocytes infiltrate the brain and may contribute to its injury via nitration of tyrosine residues (Wang et al., 2007a). The leukocyte myeloperoxidase (MPO) is believed to be involved in this process (Eiserich et al., 1998). Although it has been proven that the deficiency of CD18 plays a great role in lessening of ischemia-reperfusion injury (Huang et al., 2006; Jean et al., 1998; Soriano et al., 1999) it is still unknown whether neutrophil-endothelial cell interactions contribute in ICH-induced brain injury.
The genetically engineered mice with targeted mutation of CD18, the common β2 subunit of CD11/CD18 integrins, were used in our study. Neutrophils from CD18−/− mice do not express CD11/CD18 adhesion complexes (Horwitz et al., 2001; Wong et al., 2007). Our hypothesis was that CD18 genetargeted deficiency, via reducing inflammatory response will ameliorate brain injury, neurological function and mortality after ICH.
MATERIALS AND METHODS
Experimental animals
All procedures for this study were approved by the Animal Care and Use Committee at Loma Linda University and complied with the NIH Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publication No. 85-23, revised 1985) and with Guidelines for the Use of Animals in Neuroscience Research by the Society for Neuroscience. Fifty seven 19–22 weeks old mice were used in our study: 36 wild type C57BL/6J (including 2 sham-operated and 2 naïve control mice) and 21 CD18 knockout mice (C57BL/6J-Itgb2tm1bay).
Mice were housed in a 12-h light/dark cycle in a specific pathogen free facility with controlled temperature and humidity and were allowed free access to food and water. All neurological tests were performed during the light cycle.
Experimental design
Mice were divided into wild type and CD 18−/− knock-out groups. The additional control groups included WT sham-operated mice and naïve control mice. All animals were neurologically tested and sacrificed at 24 hours after ICH induction. Brain samples were collected for measurements of brain edema and hemorrhage volume. The evaluation of neurological deficit was carried out by investigators blinded to mice type. Mortality was examined during 24 hr after stroke onset.
ICH induction
We performed the collagenase-induced ICH model as previously described (Rosenberg et al., 1990; Tang et al., 2004; Tang et al., 2005). Briefly, mice were anesthetized with ketamine (100 mg/kg, i.p.) and xylazine (5 mg/kg, i.p.) and positioned prone in a stereotaxic head frame (Kopf Instruments, Tujunga, CA). An electronic thermostat-controlled warming blanket was used to maintain the core temperature at 37±0.5°C. A cranial burr hole (1 mm) was drilled near the right coronal suture 1 mm lateral to the midline. A 27-gauge needle was inserted stereotaxically into the right basal ganglia (coordinates: 0.9mm posterior to the bregma, 1 mm lateral to the midline, and 4 mm below the dura). The collagenase (VII-S, Sigma; 0.075 U in 0.5 μL of Saline) was infused into the brain over 2 minutes at a rate 0.25 μL/min with a micro-infusion pump (Harvard Apparatus, Holliston, MA). Sham operated mice were subjected to the needle insertion only. The needle was left in place for additional 10 min after injection to prevent the possible leakage of collagenase solution. After removal of the needle, the skull hole was closed with bone wax, the incision was closed with sutures and the mice were allowed to recover. To avoid post surgical dehydration, normal saline was given to each mouse by subcutaneous injection in the amount of 2% of body weight, immediately after surgery.
Neurological deficit
Neurological evaluation was conducted at 24 hours after ICH, using a 28-point neurological scoring system developed by Clark et al. (Clark et al., 1998). The examiner did not have any knowledge of the procedure and mice type. The neurobehavioral study consisted of seven tests with score 0–4 for each test. These seven tests are: 1- Body symmetry (open bench top), 2- Gait (open bench top), 3- Climbing (gripping surface, 45° angle), 4- Circling behavior (open bench top), 5- Front limb symmetry (mouse suspended by its tail), 6- Compulsory circling (front limbs on bench, rear suspended by tail), and 7- Whisker response (light touch from behind). The score given to each mouse at the completion of the evaluation is the sum of all seven individual test scores. The minimum neurological score is “0” for healthy mouse and the maximum is “28” for mouse with the most severe focal deficit.
Brain water content
The brain water content was measured as previously described (Tang et al., 2004; Tang et al., 2005). Briefly, mice were decapitated under deep anesthesia. Brains were immediately removed and divided into 3 parts: ipsilateral hemisphere, contralateral hemisphere, and cerebellum. Cerebellum was used as an internal control for brain water content. Tissue samples were weighed on an electronic analytical balance (APX-60, Denver instrument) to the nearest 0.1 mg to obtain the wet weight (WW). The tissue was then dried at 100°C for 24 hours to determine the dry weight (DW). Brain water content (%) was calculated as [(WW-DW)/WW] X 100.
Hemorrhage volume
Hemoglobin assay was conducted as described previously (Tang et al., 2005). Briefly, mice were sacrificed with overdose of isoflurane at 24 hours after ICH and transcardially perfused with 200 ml of ice cold phosphate-buffered saline (PBS). Brains were extracted and dissected free of olfactory bulbs and cerebellum. Ipsilateral hemisphere was homogenized (Tissue Miser Homogenizer, Fisher scientific, Pittsburgh, PA, USA) for 60 seconds in a test tube containing distilled water (total volume of 3 mL). After centrifugation (15,800g for 30 minutes; model 5417 R centrifuge; Eppendorf AG, Hamburg, Germany), Drabkin’s reagent (400 μL; Sigma-Aldrich) was added to 100 μL aliquots of the supernatant (four samples per brain) and allowed to react for 15 minutes. The absorbance of this solution was read using a spectrophotometer (540 nm; model Spectronic Genesis 5; Thermo Electron Corporation; USA) and the amount of blood in each brain was calculated using a standard curve generated with known blood volumes.
CD18 immunohistochemistry
Brains from euthanized wild type and knockout mice were frozen in OCT media and 5-μm sections were cut and then fixed in 95% ethanol and 5% glacial acetic acid at −20°C for 30 minutes, as previously described (Barlow et al., 2004). Next, sections were incubated in 0.3% H2O2 (30 min) followed by phosphate-buffered saline (PBS) with 4% rabbit serum plus 4 drops/ml avidin blocking solution (Vector Laboratories, Burlingame, CA) for 30 minutes. The rat antimouse primary antibody (eBiosciences, San Diego, CA), anti-CD18 (M18/2) was applied at 1:200 dilution in PBS with 1% rabbit serum and 4 drops/ml biotin solution (Vector Laboratories) for 1 hour in a humidified chamber. Next, sections were incubated with biotinylated rabbit anti-rat IgG in PBS with 0.1% rabbit serum for 1 hour in a humidified chamber, followed by VectaStain ABC (Vector Laboratories) for 1 hour, and then coverslipped (Barlow et al., 2004).
NT and MPO double immunofluorescence staining
Under excessive narcosis mice were perfused with 10% buffered formalin, brains collected and postfixed overnight. Brains were then stored in 30% sucrose in PBS for 4 days. Cryoprotected brains were cut into tissue blocks, embedded in OCT medium, frozen and mounted in the cryostat. Coronal brain sections, 10 μm thick, were cut and mounted on glass slides. The following primary antibodies (from Santa Cruz Biotechnology, Inc.) were used: mouse anti-nitrotyrosine and goat anti-MPO, both at 1:100 dilutions. Sections were pretreated with 3% donkey serum/PBS then incubated with goat anti-MPO antibody followed by donkey anti-goat Texas red-conjugated antibody (1:200, Jackson Immunoresearch Laboratories) and washed extensively. For the next step sections were pretreated with 3% goat serum/PBS then incubated with mouse anti-tyrosine antibody, followed by incubation with goat anti-mouse IgG2b FITC-conjugated antibody (1:200, Santa Cruz). After final washing in PBS (5min, 4 times), sections were mounted with anti-fade mounting medium (Molecular Probes) under glass coverslip. All incubations were carried out at room temperature, for 1 hour. We performed both single and double stains for NT and MPO following the above procedures. For a double IF, the microphotographs were taken separately for each stain within the same visual fields by means of a digital camera connected to the fluorescent microscope (OLYMPUS BX51). Merged images were generated by using Image ProPlus software (Yatsushige et al., 2007). Six high power fields were evaluated in the region immediately surrounding the hematoma, known for a high abundance of dying cells after ICH (Felberg et al., 2002). Representative microphotographs were selected in control, WT ICH and CD18 −/− ICH groups.
MPO activity
The ipsilateral hemispheres were weighed and homogenized at 4 °C in 1ml 50mM Tris-HCl, pH 7.4. Samples were added to 3 ml 50mM sodium buffer pH 6.0, homogenized and centrifuged at 30000g for 30mins at 4 °C. The pellet was resuspended in 0.5% hexadecyltrimethylammonium bromide (Sigma, St Louis, MO, USA) in 50mM potassium phosphate buffer at 25 °C for 2 min. The samples were frozen in liquid nitrogen. Freezing and thawing were carried out 3 times with 10s sonications between each cycles. After the last sonication, the samples were incubated at 4 °C for 15 min and centrifuged at 12,500g for 15 min. The supernatant was mixed with the same volume of 50mM phosphate buffer containing 0.167 mg/ml o-dianisidine dihydrochloride (Sigma) and hydrogen peroxide (0.005%), pH 6.0. The rate of change of absorbance was measured spectrophotometrically at 460nm. MPO activity was expressed in units per gram weight of wet tissue (Vita et al., 2004).
Statistical analysis
Quantitative data were expressed as mean ±SEM. Inter-group comparisons were analyzed by using ANOVA (MPO activity, brain water content), Chi-square test (mortality) and Student t-test (hemorrhage volume, neurological scores). Differences with p<0.05 were considered statistically significant.
RESULTS
Neurological evaluation
Neurological function was examined at 24 hours after surgery by investigators blinded to experimental groups and mice genotype (Fig. 1B). It was found that intracerebral collagenase injection induced significant neurological deficit in both wild type and CD18 null knockout mice. The examination of neurological function showed a tendency towards a decrease in neurological deficit in CD18−/− mice when compared with WT mice at 24 hr after ICH onset, but the difference was not significant (p>0.05, Student t-test).
Figure 1.

(A) Brain water content was assayed at 24 h after ICH by using wet/dry method. Brain edema was significantly reduced in CD18-knockout mice compared with wild type mice (*p<0.05, ANOVA). (B) The Garcia neuroscoring system was used for neurological deficit evaluation. CD18 null mice showed a noticeable but insignificant amelioration of neurological function (C) Mortality was calculated by dividing the number of dead animals by the number of total animals used in each group at 24 hrs after ICH (Ostrowski et al., 2006). Mortality rate was significantly reduced in the CD18-knockout mice (*p<0.05, Chi square test). (D) In MPO activity assay we used whole hemispheres ipsilateral to hemorrhage. CD18 deficiency was associated with a reduced MPO activity, by 70%, although statistical significance was not reached (p>0.05, t-test).
Mortality
Mortality was reduced in CD 18−/− mice (28.6%) compared with wild type mice (46.9%). This reduction was significant (p<0.05, Chi-square test) (Figure 1C).
Brain water content
Brain water content increased after collagenase injection in all ICH mice especially in the ipsilateral hemisphere as shown in the Figure 1A. Brain water content was significantly reduced in CD18 null mice when compared with wild type mice at 24 hr after ICH onset (ANOVA, p<0.05). No water content changes were observed in cerebellum in all groups (p>0.05, ANOVA).
Hemorrhage volume
There was no statistically significant difference in hemorrhage volume between wild type and CD18 null mice. This confirms that our hemorrhagic model is consistent in size for both compared groups of mice (Fig. 2) (n=5 in wild type group and n=3 in CD18 knockout group of mice).
Figure 2.
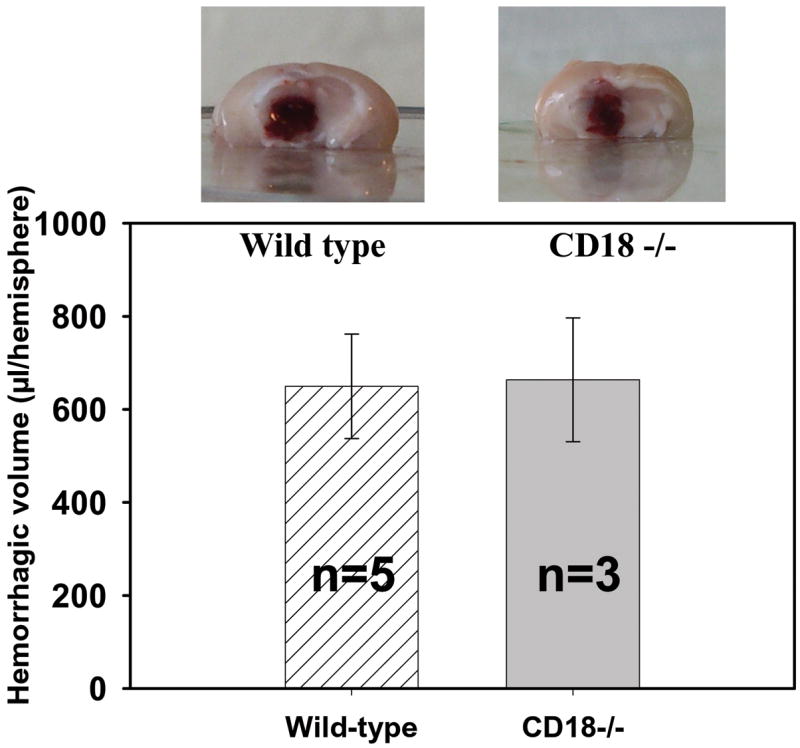
Hemorrhage volumes and representative photographs of hemorrhagic brains of WT and CD18-knockout mice. The hemorrhage volume is consistent in size in both WT and KO mice. Supernatant obtained from homogenates of cerebral hemispheres, containing blood, clots were used. Product of reaction between hemoglobin and Drabkin reagent was measured spectrophotometrically.
CD18 immunohistochemistry
The immunostaining of wild-type brain sections with anti-CD18 antibody demonstrated increased CD18 expression in the cerebral tissues after ICH (Fig. A1&A2). CD18 staining was markedly concentrated in the perivascular regions within hematoma-affected cerebral tissue (Fig. A3), indicating the presence of infiltrating leukocytes. In the WT normal brains the stain was negative (Fig. 3B).
Figure 3.
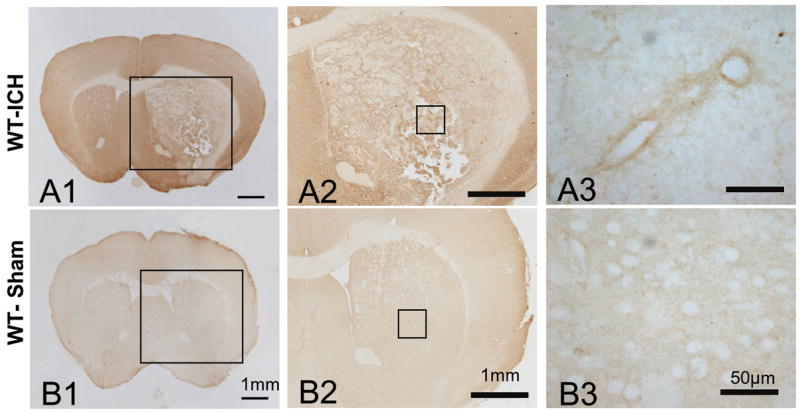
Immunohistochemical stain with anti-CD18 antibody (brown color). Intracerebral hemorrhage increases CD18 expression in brains of wild-type mice with ICH. Rat anti-CD18 primary antibody, anti-rat biotinylated secondary antibody and DAB (ABC kit) were used. Bar = 50μm.
Nitrotyrosine and myeloperoxidase immunofluorescence
As seen in Figure 4B, ICH induced nitrotyrosine immunoreactivity in the pyramidal cells of the CA1 zone. Relatively weaker staining was observed for MPO in this region (Fig. 4E). Both stains were reduced in the CD18 deficient mice (Fig. 4C&F). In the cerebral cortex double immunofluorescence demonstrated a colocalization of MPO with nitrotyrosine epitopes after ICH in WT mice (Fig. 4I). Similarly to CA1, CD18 deficiency reduced tissue expression of NT and MPO after ICH (Fig. 4J-L).
Figure 4.
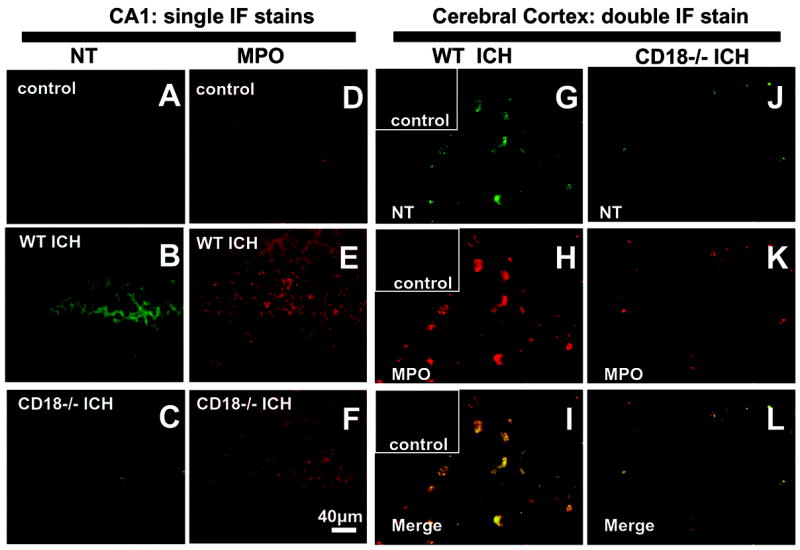
Anti-MPO and anti-NT antibodies bound to their respective epitopes were detected by Texas Red and FITC-conjugated secondary antibodies, respectively. Hemorrhage increased NT (Fig. 4B&G) and MPO (Fig. 4E&H) tissue expression in brains of WT mice but less so in CD18−/− mice (Fig. 4C, 4F&4L). Double immunofluorescence stain for NT and MPO epitopes in the perihematomal region of cerebral cortex after ICH revealed their colocalization in the WT mice (Figure 4I). Control: nonhemorrhagic mouse brain; bar = 40μm.
MPO activity
There were no statistically significant differences between WT and CD18−/− groups in MPO activity, however a trend towards a lesser activity in the CD18−/− mice can be clearly seen (Fig. 1 D).
DISCUSSION
Our study demonstrates that CD18 deficiency reduces brain edema and mortality after ICH. Tendencies in reduction of neurological deficit and MPO activity were also found. In mouse brains with ICH we observed colocalization of MPO and nitrotyrosine immuno reactivities, both reduced in CD18 deficient mice. All above findings speak in favor of CD18 integrin subunit playing an important role in the development of ICH-induced brain injury. Even though 70% reduction of MPO activity, suggesting lessened leukocyte infiltration, did not reach statistical significance, there was an evident reduction in immunofluorescent stain for MPO. Additionally, difference in mortality might impact our study through a negative selection that eliminated WT animals with severe neutrophil accumulation and neurologic deficit. It is possible that significance would have been achieved if very large numbers of mice were included in each group or if samples were collected from a peak time point at 2 days after ICH; however, it is known that our model is notorious for posing difficulties in terms of statistically significant outcome measures (Clark et al., 1998; Soriano et al., 1999; Titova et al., 2007)
In one of the previous studies with CD18 null mice subjected to ischemic stroke the authors observed reduced numbers of infiltrating leukocytes, however leukocyte counts did not reach statistical significance (Soriano et al., 1999). Nonetheless authors’ results suggested that CD11b/CD18 has a role in mediating neutrophil extravasation and infarct development (Soriano et al., 1999).
It has been well documented that β2 integrins regulate adhesion-dependent injury responses on venular endothelium after ischemia reperfusion through adhesion events involving activated endothelial cells and leukocytes (Hakkert et al., 1991; Huang et al., 2006; Kakkar and Lefer, 2004; Kurose et al., 1997). Countering these processes could lead to a reduction of blood brain barrier permeability and brain edema accordingly and might alleviate hematoma expansion in our study (Hu et al., 1999; Wu et al., 2006; Xi et al., 2004).
The reduced edema and mortality may indicate that deficiency of CD18 reduced brain injury via several mechanisms. As demonstrated by double immunofluorescence stain for MPO and NT, the mechanism underlying brain protection in this present study may involve reduced nitration of cell proteins. Consistent with the notion that leukocyte MPO provides a major mechanism for protein nitration (Eiserich et al., 1998), we observed a reduced tissue expression of MPO and NT in the cerebral tissues of CD18 knockout mice.
CD18 deficiency could also decrease leukocyte free radical production and reduce release of inflammatory cytokines and pro-inflammatory proteases, thereby protecting neurovascular unit (Huang et al., 2006; Wang and Dore, 2007). Besides, lessened interactions between CD18 and ICAM might result in less accumulation of neutrophils in microcapillary bed and thusly could improve rCBF (Soriano et al., 1999). Prevention of microvascular plugging in CD18−/−mice would create a great relief for microvessels constricted by edema and compressed by blood clots. Improved microcirculation additive to edema relief could lead to a significant reduction of mortality, observed in our study. This particular endpoint has a great clinical relevance in ICH, associated with higher that in ischemic stroke mortality rates (Sahni and Weinberger, 2007).
In conclusion, our study provided evidence that deficiency of CD18 β2 integrin subunit reduces mortality and the acute brain injury after ICH although further studies are required for better understanding of the exact mechanism of protection. Based on our data, targeting CD11/CD18 integrin-regulated adhesion appears to be a favorable direction for the development of novel therapies of acute ICH.
Acknowledgments
This work was supported by NIH NS052492, to JT, and by NIH NS53407 to JHZ.
References
- Barlow SC, Langston W, Matthews KM, Chidlow JH, Jr, Kevil CG. CD18 deficiency protects against multiple low-dose streptozotocin-induced diabetes. Am J Pathol. 2004;165:1849–1852. doi: 10.1016/S0002-9440(10)63237-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark W, Gunion-Rinker L, Lessov N, Hazel K. Citicoline treatment for experimental intracerebral hemorrhage in mice. Stroke. 1998;29:2136–2140. doi: 10.1161/01.str.29.10.2136. [DOI] [PubMed] [Google Scholar]
- Eiserich JP, Hristova M, Cross CE, Jones AD, Freeman BA, Halliwell B, van d V. Formation of nitric oxide-derived inflammatory oxidants by myeloperoxidase in neutrophils. Nature. 1998;391:393–397. doi: 10.1038/34923. [DOI] [PubMed] [Google Scholar]
- Felberg RA, Grotta JC, Shirzadi AL, Strong R, Narayana P, Hill-Felberg SJ, Aronowski J. Cell death in experimental intracerebral hemorrhage: the “black hole” model of hemorrhagic damage. Ann Neurol. 2002;51:517–524. doi: 10.1002/ana.10160. [DOI] [PubMed] [Google Scholar]
- Gong C, Hoff JT, Keep RF. Acute inflammatory reaction following experimental intracerebral hemorrhage in rat. Brain Res. 2000;871:57–65. doi: 10.1016/s0006-8993(00)02427-6. [DOI] [PubMed] [Google Scholar]
- Hakkert BC, Kuijpers TW, Leeuwenberg JF, van Mourik JA, Roos D. Neutrophil and monocyte adherence to and migration across monolayers of cytokine-activated endothelial cells: the contribution of CD18, ELAM-1, and VLA-4. Blood. 1991;78:2721–2726. [PubMed] [Google Scholar]
- Horwitz BH, Mizgerd JP, Scott ML, Doerschuk CM. Mechanisms of granulocytosis in the absence of CD18. Blood. 2001;97:1578–1583. doi: 10.1182/blood.v97.6.1578. [DOI] [PubMed] [Google Scholar]
- Hu B, Liu C, Zivin JA. Reduction of intracerebral hemorrhaging in a rabbit embolic stroke model. Neurology. 1999;53:2140–2145. doi: 10.1212/wnl.53.9.2140. [DOI] [PubMed] [Google Scholar]
- Huang J, Upadhyay UM, Tamargo RJ. Inflammation in stroke and focal cerebral ischemia. Surg Neurol. 2006;66:232–245. doi: 10.1016/j.surneu.2005.12.028. [DOI] [PubMed] [Google Scholar]
- Jean WC, Spellman SR, Nussbaum ES, Low WC. Reperfusion injury after focal cerebral ischemia: the role of inflammation and the therapeutic horizon. Neurosurgery. 1998;43:1382–1396. doi: 10.1097/00006123-199812000-00076. [DOI] [PubMed] [Google Scholar]
- Kakkar AK, Lefer DJ. Leukocyte and endothelial adhesion molecule studies in knockout mice. Curr Opin Pharmacol. 2004;4:154–158. doi: 10.1016/j.coph.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Kurose I, Argenbright LW, Anderson DC, Tolley J, Miyasaka M, Harris N, Granger DN. Reperfusion-induced leukocyte adhesion and vascular protein leakage in normal and hypercholesterolemic rats. Am J Physiol. 1997;273:H854–H860. doi: 10.1152/ajpheart.1997.273.2.H854. [DOI] [PubMed] [Google Scholar]
- Ostrowski RP, Tang J, Zhang JH. Hyperbaric oxygen suppresses NADPH oxidase in a rat subarachnoid hemorrhage model. Stroke. 2006;37:1314–1318. doi: 10.1161/01.STR.0000217310.88450.c3. [DOI] [PubMed] [Google Scholar]
- Power C, Henry S, Del Bigio MR, Larsen PH, Corbett D, Imai Y, Yong VW, Peeling J. Intracerebral hemorrhage induces macrophage activation and matrix metalloproteinases. Ann Neurol. 2003;53:731–742. doi: 10.1002/ana.10553. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Mun-Bryce S, Wesley M, Kornfeld M. Collagenase-induced intracerebral hemorrhage in rats. Stroke. 1990;21:801–807. doi: 10.1161/01.str.21.5.801. [DOI] [PubMed] [Google Scholar]
- Sahni R, Weinberger J. Management of intracerebral hemorrhage. Vasc Health Risk Manag. 2007;3:701–709. [PMC free article] [PubMed] [Google Scholar]
- Soriano SG, Coxon A, Wang YF, Frosch MP, Lipton SA, Hickey PR, Mayadas TN. Mice deficient in Mac-1 (CD11b/CD18) are less susceptible to cerebral ischemia/reperfusion injury. Stroke. 1999;30:134–139. doi: 10.1161/01.str.30.1.134. [DOI] [PubMed] [Google Scholar]
- Tang J, Liu J, Zhou C, Alexander JS, Nanda A, Granger DN, Zhang JH. Mmp-9 deficiency enhances collagenase-induced intracerebral hemorrhage and brain injury in mutant mice. J Cereb Blood Flow Metab. 2004;24:1133–1145. doi: 10.1097/01.WCB.0000135593.05952.DE. [DOI] [PubMed] [Google Scholar]
- Tang J, Liu J, Zhou C, Ostanin D, Grisham MB, Neil GD, Zhang JH. Role of NADPH oxidase in the brain injury of intracerebral hemorrhage. J Neurochem. 2005;94:1342–1350. doi: 10.1111/j.1471-4159.2005.03292.x. [DOI] [PubMed] [Google Scholar]
- Titova E, Ostrowski RP, Sowers LC, Zhang JH, Tang J. Effects of apocynin and ethanol on intracerebral haemorrhage-induced brain injury in rats. Clin Exp Pharmacol Physiol. 2007;34:845–850. doi: 10.1111/j.1440-1681.2007.04664.x. [DOI] [PubMed] [Google Scholar]
- Vita JA, Brennan ML, Gokce N, Mann SA, Goormastic M, Shishehbor MH, Penn MS, Keaney JF, Jr, Hazen SL. Serum myeloperoxidase levels independently predict endothelial dysfunction in humans. Circulation. 2004;110:1134–1139. doi: 10.1161/01.CIR.0000140262.20831.8F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Dore S. Inflammation after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2007;27:894–908. doi: 10.1038/sj.jcbfm.9600403. [DOI] [PubMed] [Google Scholar]
- Wang J, Fields J, Zhao C, Langer J, Thimmulappa RK, Kensler TW, Yamamoto M, Biswal S, Dore S. Role of Nrf2 in protection against intracerebral hemorrhage injury in mice. Free Radic Biol Med. 2007a;43:408–414. doi: 10.1016/j.freeradbiomed.2007.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Tang XN, Yenari MA. The inflammatory response in stroke. J Neuroimmunol. 2007b;184:53–68. doi: 10.1016/j.jneuroim.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn R, Vedder N, Ramamoorthy C, Sharar S, Harlan J. Endothelial and leukocyte adhesion molecules in inflammation and disease. Blood Coagul Fibrinolysis. 1998;9(Suppl 2):S17–S23. [PubMed] [Google Scholar]
- Wong D, Prameya R, Dorovini-Zis K. Adhesion and migration of polymorphonuclear leukocytes across human brain microvessel endothelial cells are differentially regulated by endothelial cell adhesion molecules and modulate monolayer permeability. J Neuroimmunol. 2007;184:136–148. doi: 10.1016/j.jneuroim.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Wu G, Xi G, Huang F. Spontaneous intracerebral hemorrhage in humans: hematoma enlargement, clot lysis, and brain edema. Acta Neurochir Suppl. 2006;96:78–80. doi: 10.1007/3-211-30714-1_19. [DOI] [PubMed] [Google Scholar]
- Xi G, Fewel ME, Hua Y, Thompson BG, Jr, Hoff JT, Keep RF. Intracerebral hemorrhage: pathophysiology and therapy. Neurocrit Care. 2004;1:5–18. doi: 10.1385/ncc:1:1:5. [DOI] [PubMed] [Google Scholar]
- Yatsushige H, Ostrowski RP, Tsubokawa T, Colohan A, Zhang JH. Role of c-Jun N-terminal kinase in early brain injury after subarachnoid hemorrhage. J Neurosci Res. 2007;85:1436–1448. doi: 10.1002/jnr.21281. [DOI] [PubMed] [Google Scholar]