

Abstract
OBJECTIVE
To determine if patients with advanced prostate cancer carrying a polymorphism that codes for a more active testosterone transporter have less durable responses to androgen-deprivation therapy (ADT) than patients not carrying this polymorphism.
PATIENTS AND METHODS
We previously determined that a polymorphism in SLCO1B3 affects testosterone transport and that those men who have at least one wild-type T allele at the 334 T > G polymorphism in this gene have a shorter survival. We hypothesized that the T allele which increases testosterone transport would be associated with a shorter interval from ADT to androgen independence. We examined the association between this SLCO1B3 polymorphism and time from ADT to androgen independence, ADT to prostate-specific antigen (PSA) nadir and PSA nadir to androgen independence in 68 Caucasian patients with advanced prostate cancer who were treated with ADT with metastatic disease (D2) or biochemical failure with no metastatic disease (D0).
RESULTS
When examined separately, patients in the individual stages tended to have a shorter time to androgen independence with the T allele in the D0 (P = 0.11) and D2 (P = 0.18) groups. Combining these groups and stratifying by stage yielded a statistically significant shorter time to androgen independence with the T allele (P = 0.048).
CONCLUSION
A polymorphism in a transporter that increases testosterone import is associated with a shorter time to androgen independence in patients with prostate cancer who are treated with ADT.
Keywords: prostate cancer, androgen independence, testosterone, androgen deprivation, hormonal therapy, transporter
INTRODUCTION
Androgen deprivation therapy (ADT) is standard first-line therapy for metastatic prostate cancer. Although most patients will respond, the disease eventually progresses to androgen-independent prostate cancer (AIPC), despite castrate levels of testosterone [1]. ADT is also used in the setting of biochemical failure with no metastatic disease after local therapy, although treatment in this setting is controversial. AIPC also occurs in this setting, and is also termed ‘castrate nonmetastatic’ prostate cancer. The mechanisms of AI are many and varied; they include androgen receptor (AR) gene amplification, mutations in AR, ligand-independent AR reactivation with growth factors or cytokines with phosphorylation-dependent mechanisms and increased synthesis of androgens local to the tumour, leading to an increase in the local concentration of androgens [2–4]. Many of these mechanisms are due to somatic genetic perturbations. However, using the knowledge of these mechanisms to predict a priori which patients are more prone to develop AI disease and which patients will achieve durable responses is problematic.
We previously showed that the organic anion transporter polypeptide family member 1B3 (OATP1B3), encoded by the polymorphic SLCO1B3 gene, is overexpressed in prostate cancer, and transports testosterone with varying efficiency based on two polymorphic variants. These polymorphic variants (SLCO1B3 334 T > G and 699 G > A) are in complete linkage disequilibrium, and encode for the amino acid changes OATP1B3 S112A and M233I, respectively [5]. The OATP1B3 protein carrying the 112S/233M amino acid combination provides nearly twice the transport of testosterone than the protein carrying the 112 A/233I combination. Furthermore, in a group of 180 patients with prostate cancer, the median survival from diagnosis for those who had one or two copies of the wild-type SLCO1B3 334T/699G haplotype (hereafter referred to as the T allele) at 6.4 years, was significantly shorter than in those with two copies of the mutant 334G/699 A allele (hereafter referred to as the G allele), at 8.5 years. AI still depends on AR reactivation and we hypothesized that the shorter survival associated with the wild-type T allele might be directly due to increased testosterone import, leading to AR reactivation and the earlier emergence of AI disease. We previously characterized the natural history of 80 patients treated with ADT at the time of biochemical recurrence (D0) or with radiographic evidence of metastatic disease (D2) [6]. Although there were both Caucasian and African-American patients in that group, there were few African-Americans, which precluded a meaningful analysis in that subset. In addition, SLCO1B3 haplotype frequencies vary with ethnicity, so comparing all ethnic groups in such a study might confound a possible difference in the natural history of prostate cancer attributable to ethnicity with an association with haplotype frequency [7]. We characterized the SLCO1B3 genotypes of those patients and correlated the natural history with respect to the presence of at least one T allele or two G alleles in SLCO1B3 and found that Caucasian patients who carried one or two copies of the T allele have a significantly shorter time from ADT to AI than those who have two copies of the G allele. Furthermore, we found no definitive evidence that the shorter time from ADT to AI in patients with the T allele is necessarily attributable to a shorter time from ADT to PSA nadir, or a shorter time from PSA nadir to AI.
PATIENTS AND METHODS
Genomic DNA was extracted from serum or white blood cell buffy coat layers of whole blood using either the QiAamp Ultrasens Viral DNA kit or the QIAamp DNA Blood Kit, as described by the manufacturer’s instructions (Qiagen, Valencia, CA, USA). Primers and assay conditions for determining the SLCO1B3 haplotype were as described previously [5]. Given that the SLCO1B3 334T > G and 699 G > A polymorphisms are in complete linkage disequilibrium, any individual carrying the 334T allele was considered to be carrying the 334T/699G haplotype, and any individual carrying the 334G allele was considered to be also carrying the 334G/699 A haplotype. We therefore only assessed the genotype at the 334 T > G locus.
Clinical outcomes were obtained for 68 Caucasian patients who had enrolled in a clinical trial using ketoconazole with or without alendronate, or a clinical trial of docetaxel with or without thalidomide [6]. We characterized the genotypes of these patients in two groups. The first, referred to as D2, comprised patients who were treated with ADT after being diagnosed with metastatic disease; the second, referred to as D0, comprised those treated with ADT after therapy for localized disease, who had an increasing PSA level and no evidence of metastatic disease. AI was defined as the first increase in PSA level in a series from the PSA nadir after starting ADT with medical or surgical castration. We assessed these groups for Gleason score, and if there was a difference in high Gleason grade tumours with genotype for the individual stages (D0 and D2) and for the whole group of patients, using the Cochran-Armitage test for trend and stratified analysis. We also assessed the time from ADT to AI, ADT to PSA nadir and PSA nadir to AI using the Kaplan-Meier method for patients in the D0 and D2 groups, and then combined the analysis by using the standard and stratified forms of the exact log-rank test by stage at ADT. The study was approved by the institutional review board.
RESULTS
We compared the Gleason scores of patients in the stage groups at the time of ADT and with genotype (Table 1). There was no significant difference in the proportion of Gleason 8–10 tumours in patients carrying one or two copies of the T allele vs those with two copies of the G allele (D0; P = 0.53; D2, P = 0.49; combined group with stratified analysis, P = 0.34).
TABLE 1.
The distribution of Gleason scores in each patient group by stage at the time of ADT, and SLCO1B3 genotype
| Gleason score |
n (%) per genotype and stage at ADT
|
|||
|---|---|---|---|---|
| D0 (T/T + T/G) | D0 (G/G) | D2 (T/T + T/G) | D2 (G/G) | |
| 2–5 | 0 | 1 (4) | 0 | 2 (9) |
| 6–7 | 3 | 9 (32) | 2 | 8 (36) |
| 8–10 | 9 | 18 (64) | 4 | 11 (50) |
| Not known | 0 | 0 | 0 | 1 (5) |
| Total | 12 | 28 | 6 | 22 |
The data for all the median times from ADT to AI, ADT to PSA nadir and PSA nadir to AI by stage and genotype are summarized in Table 2. Notably, when the time from ADT to AI for the D0 and D2 groups were combined and stratified by stage at ADT, there was an earlier onset of AI that was statistically significant for patients who had at least one copy of the T allele compared with those who had two copies of the G allele (P = 0.048). A comparison of the time from ADT to AI for patients treated with ADT at stage D0 (Fig. 1A) shows that patients with one or two copies of the wild-type allele (T/T or T/G genotype) had a clear trend to a shorter time to AI than patients with two copies of the variant (G/G) allele, although the difference was not statistically significant (1.55 vs 1.87 years; P = 0.11). In patients who were treated with ADT when metastatic disease was evident (D2), those with the T/T or T/G genotype also had a shorter time to AI (Fig. 1B) than those carrying the G/G genotype (0.96 vs 1.41 years; P = 0.18). Figure 1C shows the combined data for the D0 and D2 groups; although this combines the power of the groups in the individual stages, the caveat is that the natural history of time to AI in those with D0 and D2 disease is inherently different [6,8,9].
TABLE 2.
A summary of the median time from ADT to AI, ADT to PSA nadir, and PSA nadir to AI for patients in each stage and genotype. The first row indicates these times combined for patients of stage D0 and D2 and stratified by stage
| Median time, years, from
|
|||||
|---|---|---|---|---|---|
| Stage (n) | Genotype | N patients | ADT to AI | ADT to PSA nadir | PSA nadir to AI |
| D0 and D2 (68) | T/T, T/G | 18 | 1.20 | 0.38 | 0.57 |
| G/G | 50 | 1.57 | 0.32 | 0.67 | |
| P | 0.048 | 0.540 | 0.240 | ||
| D0 (40) | T/T, T/G | 12 | 1.55 | 0.50 | 0.57 |
| G/G | 28 | 1.87 | 0.28 | 0.86 | |
| P | 0.110 | 0.920 | 0.250 | ||
| D2 (28) | T/T, T/G | 6 | 0.96 | 0.30 | 0.57 |
| G/G | 22 | 1.41 | 0.35 | 0.58 | |
| P | 0.180 | 0.230 | 0.610 | ||
FIG. 1.

Kaplan-Meier curves of the probabilities of freedom from AIPC from the time of ADT for patients who were treated with ADT at stage D0 (A) or at stage D2 (B) and C, both groups combined in one plot.
A shorter time from ADT to AIPC associated with the SLCO1B3 334T polymorphism that increases testosterone import into prostate cancer cells could be caused by either (i) a shorter time from ADT to PSA nadir, by effectively increasing the intracellular concentration of testosterone with castrate levels of serum testosterone and minimizing the decline to PSA nadir, or (ii) a shorter time from PSA nadir to AI by ‘priming’ a subset of prostate cancer cells for growth and facilitating the induction of other gain-of-function changes in the androgen pathway. To investigate these two possibilities, we analysed the time from ADT to PSA nadir and PSA nadir to AI by genotype and stage at ADT.
In the D0 group, patients with the T/T or T/G genotype had a median time from ADT to PSA nadir of 0.50 years, compared with 0.28 years with the G/G genotype (P = 0.92). The slightly longer time with the T/T or T/G genotype, which was not statistically significant, is in the opposite direction to what would be expected. Although the median time for the T/T or T/G genotype was slightly longer, the curves on the Kaplan-Meier plots cross (Fig. 2A) and the tail on the G/G curve is longer than that of the T/T or T/G curve. In the D2 group the median time from ADT to PSA nadir with the T/T or T/G genotype, vs the G/G genotype (Fig. 2B), was 0.30 vs 0.35 years (P = 0.23). The tail of the G/G curve is also longer for the D2 group. The stratified test for differences in the genotypes for the combination of the D0 and D2 groups gave P = 0.54.
FIG. 2.

Kaplan-Meier curves of the probabilities of not reaching PSA nadir from the time of ADT for patients who were treated with ADT at stage D0 (A) or stage D2 (B).
We then examined the time from PSA nadir to AI by stage and genotype. In the D0 group the median time from PSA nadir to AI (Fig. 3A) in the T/T or T/G genotype was 0.57 years, vs 0.86 years for the G/G genotype (P = 0.25). In the D2 group the values for the T/T or T/G genotype vs the G/G genotype (Fig. 3B) were 0.57 and 0.58 years, respectively (P = 0.61). Combining and stratifying the values for the D0 and D2 groups, the differences in the median time from PSA nadir to AI by genotype gave P = 0.24.
FIG. 3.
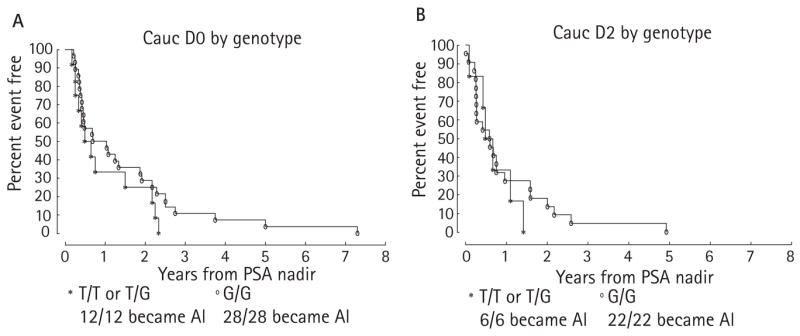
Kaplan-Meier curves of the probabilities of freedom from AIPC from the time of PSA nadir for patients who were treated with ADT at stage D0 (A) or stage D2 (B).
DISCUSSION
Patients with prostate cancer treated with ADT who have at least one copy of the T allele at the 334 T > G polymorphism in the SLCO1B3 gene have a shorter time from ADT to AI than those with two copies of the G allele. We previously showed that SLCO1B3 is overexpressed in prostate cancer, that this transporter has testosterone uptake activity, and that the protein expressed from the T allele has higher testosterone transport activity than the protein expressed from the G allele [5]. AIPC still depends on the AR [10,11], but in addition to mechanisms that lead to a net gain-of-function in AR, other mechanisms also exist that increase the net concentration of androgen ligands in the tumour environment [12], despite castrate levels of serum testosterone, by the local synthesis of androgens or the conversion of adrenal androgens to testosterone [13]. Whether the mechanisms of AI rely on an increase in testosterone concentration in the tumour, or other mechanisms that directly affect AR, the previously described mechanisms occur via somatic mutations in prostate cancer cells. Here, we describe a germline polymorphism in a gene that appears to predispose to AIPC. Although the difference between the T/T or T/G vs G/G genotypes in time from ADT to AI was not statistically significant when patients were examined by stage at the time when ADT was used, there were relatively few patients, and the difference between genotypes is significant when the these two groups were combined using a stratified analysis.
All of the 68 patients analysed in this dataset were Caucasian; the frequency of each respective polymorphism in SLCO1B3 varies with ethnicity and we previously showed that the T/T and T/G haplotypes are more frequent in African-American than Caucasian patients [5,7]. Although somewhat controversial, it is widely thought that African-American patients with prostate cancer have a more aggressive clinical course than Caucasian patients. Therefore, a combined analysis of Caucasian and African-American patients together confound the finding of a shorter time to AI with the T/T and T/G haplotypes. The effect of SLCO1B3 polymorphisms on AI in African-American patients and other ethnic groups will require larger studies.
If the mechanism of a shorter time from ADT to AI conferred by the T/T or T/G haplotype is indeed due to an increase in testosterone uptake as we postulate, then this would occur through (i) a shorter time from ADT to PSA nadir or (ii) a shorter time from PSA nadir to AI. In (i) the PSA nadir might be reached more quickly as an increase in testosterone uptake would in part counteract the depleted serum levels of testosterone by increasing intracellular testosterone concentrations. In (ii), the SLCO1B3 T/T or T/G haplotype would affect a subset of residual prostate cancer cells after the initiation of ADT, rendering them capable of growth and the ability to acquire somatic mutations to further the process of AI. Although neither the median times from ADT to PSA nadir nor the times from PSA nadir to AI were statistically significant, the tails on the curves that correspond to the less active SLCO1B3 G/G haplotype are longer in all of the comparisons. Therefore, the effect of the SLCO1B3 haplotype might have an effect on both possibilities (i) and (ii).
Importantly, this was a retrospective study and it requires confirmation with other patient groups. Ideally, this would be tested in a prospective clinical trial of ADT. Examination of the SLCO1B3 haplotype with the responsiveness and duration of response to ADT in more patients should also resolve the issue of whether the shorter time from ADT to AI occurs via a shorter time from ADT to PSA nadir, shorter time from PSA nadir to AI, or both. Prognostication of a patient’s response to ADT might help to guide the management and treatment of prostate cancer, by anticipating how an individual patient will respond. In addition, it would aid in designing clinical trials that would test more intensive therapies, e.g. chemotherapy or a combination of chemotherapy and hormonal therapy, in selected patients who are destined to have an inadequate or transient response to ADT alone.
In conclusion, a polymorphism in a gene that encodes for a testosterone transporter, SLCO1B3, is associated with the response duration to ADT in patients with prostate cancer. These findings potentially have important clinical implications in prognosticating the response to hormonal therapy and in the design of clinical trials for hormonal therapy in prostate cancer. However, our findings require validation in an independent group of patients with prostate cancer treated with ADT.
Acknowledgments
This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
Abbreviations
- AIPC
androgen-independent prostate cancer
- ADT
androgen-deprivation therapy
- AR
androgen receptor
- OATP
organic anion transporter polypeptide
Footnotes
CONFLICT OF INTEREST None declared.
References
- 1.Sharifi N, Gulley JL, Dahut WL. Androgen deprivation therapy for prostate cancer. JAMA. 2005;294:238–44. doi: 10.1001/jama.294.2.238. [DOI] [PubMed] [Google Scholar]
- 2.Mostaghel EA, Montgomery RB, Lin DW. The basic biochemistry and molecular events of hormone therapy. Current Urol Reports. 2007;8:224–32. doi: 10.1007/s11934-007-0010-z. [DOI] [PubMed] [Google Scholar]
- 3.Sharifi N, Farrar WL. Androgen receptor as a therapeutic target for androgen independent prostate cancer. Am J Ther. 2006;13:166–70. doi: 10.1097/00045391-200603000-00013. [DOI] [PubMed] [Google Scholar]
- 4.Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocrine Rev. 2004;25:276–308. doi: 10.1210/er.2002-0032. [DOI] [PubMed] [Google Scholar]
- 5.Hamada A, Sissung T, Price P, et al. Effect of SLCO1B3 haplotype on testosterone transport and clinical outcome in Caucasian patients with androgen-independent prostatic cancer. Clin Cancer Res. 2008 doi: 10.1158/1078-0432.CCR-07-4118. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharifi N, Dahut WL, Steinberg SM, et al. A retrospective study of the time to clinical endpoints for advanced prostate cancer. BJU Int. 2005;96:985–9. doi: 10.1111/j.1464-410X.2005.05798.x. [DOI] [PubMed] [Google Scholar]
- 7.Smith NF, Marsh S, Scott-Horton TJ, et al. Variants in the SLCO1B3 gene: interethnic distribution and association with paclitaxel pharmacokinetics. Clin Pharm Ther. 2007;81:76–82. doi: 10.1038/sj.clpt.6100011. [DOI] [PubMed] [Google Scholar]
- 8.Smith MR, Kabbinavar F, Saad F, et al. Natural history of rising serum prostate-specific antigen in men with castrate nonmetastatic prostate cancer. J Clin Oncol. 2005;23:2918–25. doi: 10.1200/JCO.2005.01.529. [DOI] [PubMed] [Google Scholar]
- 9.Eisenberger MA, Blumenstein BA, Crawford ED, et al. Bilateral orchiectomy with or without flutamide for metastatic prostate cancer. N Engl J Med. 1998;339:1036–42. doi: 10.1056/NEJM199810083391504. [DOI] [PubMed] [Google Scholar]
- 10.Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005;23:8253–61. doi: 10.1200/JCO.2005.03.4777. [DOI] [PubMed] [Google Scholar]
- 11.Gelmann EP. Molecular biology of the androgen receptor. J Clin Oncol. 2002;20:3001–15. doi: 10.1200/JCO.2002.10.018. [DOI] [PubMed] [Google Scholar]
- 12.Titus MA, Schell MJ, Lih FB, Tomer KB, Mohler JL. Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clin Cancer Res. 2005;11:4653–7. doi: 10.1158/1078-0432.CCR-05-0525. [DOI] [PubMed] [Google Scholar]
- 13.Stanbrough M, Bubley GJ, Ross K, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815–25. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]