

Abstract
Adenosine is formed in injured/ischemic tissues where it suppresses the actions of essentially all cells of the immune system. Most of the anti-inflammatory actions of adenosine have been attributed to signaling through the Gs protein-coupled A2A adenosine receptor (AR). Here, we report that the A3AR is highly expressed in murine neutrophils isolated from bone marrow. Selective activation of the A3AR with CP-532,903 potently inhibited mouse bone marrow neutrophil superoxide generation and chemotaxis induced by various activating agents. The selectivity of CP-532,903 was confirmed in assays using neutrophils obtained from A2AAR and A3AR gene “knock-out” mice. In a model of thioglycollate-induced inflammation, treating mice with CP-532,903 inhibited recruitment of leukocytes into the peritoneum by specifically activating the A3AR. Collectively, our findings support the theory that the A3AR contributes to the anti-inflammatory actions of adenosine on neutrophils, and provide a potential mechanistic explanation for the efficacy of A3AR agonists in animal models of inflammation, i.e., inhibition of neutrophil-mediated tissue injury.
INTRODUCTION
The neutrophil is the first cell type recruited to injured tissues where it functions to sterilize the wound of invading bacteria, through phagocytosis and subsequent killing by oxidant mechanisms involving the NADPH oxidase complex (Nathan, 2006). Activated neutrophils also secrete numerous cytokines/chemokines, proteolytic enzymes stored in preformed granules, and pro-inflammatory products of arachidonic acid (PGE2 and leukotrienes), which collectively serve to recruit additional immune cell populations, remove cell debris, and fine-tune the adaptive immune response (Nathan, 2006). While these actions of neutrophils are critical components of normal wound healing, exaggerated or chronic neutrophil activity can contribute to additional tissue injury, particularly when little or no infection is present, for instance, during acute ischemia/reperfusion injury or chronic inflammatory diseases such as rheumatoid arthritis (Nathan, 2006).
Because of their destructive nature, intricate mechanisms have evolved that regulate neutrophil activity at sites of inflammation. As neutrophils invade tissues, the metabolism of arachidonic acid shifts to the formation of anti-inflammatory lipoxins that inhibit additional neutrophil recruitment (Levy et al., 2001). Neutrophil activity is further diminished by TGF β and other anti-inflammatory molecules released from macrophages following the ingestion of expended apoptotic neutrophils (Huynh et al., 2002). In addition to these mechanisms designed to resolve inflammatory reactions, neutrophil activity is also importantly modulated by adenosine. Adenosine is a purine nucleoside generated in inflamed tissues from the catalysis by the ecto-nucleotidases CD39 and CD73 of extracellular ATP and ADP secreted from activated cells (Hasko and Cronstein, 2004; Linden, 2001). Within inflamed tissues, adenosine functions to suppress activity of essentially all cells of the immune system including the neutrophil. Previous studies have shown that adenosine potently inhibits neutrophil superoxide production, adhesion/chemotaxis, and pro-inflammatory mediator production (Cronstein et al., 1990; Cronstein et al., 1983; Cronstein et al., 1992; Flamand et al., 2000; Jordan et al., 1997; McColl et al., 2006; Sullivan et al., 2001; Visser et al., 2000).
All of the actions of adenosine are mediated by four adenosine receptor (AR) subtypes belonging to the super-family of G protein-coupled receptors designated A1, A2A, A2B, and A3. A1 and A3ARs couple to Gi proteins that inhibit adenylyl cyclase while activating multiple other signaling pathways via the release of β γ subunits, whereas A2A and A2BARs couple to Gs proteins that activate adenylyl cyclase resulting in formation of cAMP (Linden, 2001). Most previous work has implicated the A2AAR in mediating the inhibitory effects of adenosine on neutrophil function via the cAMP/PKA signaling axis, based on pharmacological studies using isolated human neutrophils (Bours et al., 2006; Cronstein, 1994; Hasko and Cronstein, 2004; Linden, 2001). Interestingly, mRNA and protein expression of the A2AAR is induced in neutrophils as well as in several other immune cell populations in response to Th1 cytokines or Toll-like receptor agonists, which appears to be an additional feedback mechanism by which adenosine ‘resolves’ inflammatory reactions (Fortin et al., 2006; Khoa et al., 2001; Lappas et al., 2005; Murphree et al., 2005; Nguyen et al., 2003).
Although most previous work has focused on the involvement of the A2AAR in regulating neutrophil activity, a potential role for the A3AR is currently being considered. In this study, we have utilized bone marrow neutrophils isolated from A2A and A3AR gene knock-out mice as well as the newly developed, highly selective mouse A3AR agonist CP-532,903, to explore the possibility that the A3AR mediates some of the suppressive effects of adenosine on neutrophil function. CP-532,903 binds potently to the murine A3AR (Ki = 9.0 nM) with greater than 100- and 1,000-fold selectivity versus murine A1 and A2A/A2BARs, respectively (Wan et al., 2008). Our results demonstrate that, like the A2AAR, activation of the A3AR subtype inhibits neutrophil superoxide production. In addition, our studies reveal that activation of the A3AR inhibits chemotaxis towards various activating agents. Collectively, our findings support the theory that the A3AR contributes to the anti-inflammatory actions of adenosine on neutrophils, and provide a potential mechanistic explanation for the efficacy of A3AR agonists in animal models of inflammation, i.e., inhibition of neutrophil-mediated tissue injury.
MATERIALS AND METHODS
Materials
Cell culture reagents, TRIzol reagent, recombinant human interleukin-8 (IL-8), recombinant mouse tumor necrosis factor a (rm TNF-α) and ThermoScript RT-PCR kits were purchased from Invitrogen (Carlsbad, CA). Calcein-AM, pluronic F-127 and 2 methyl-6-(4-methoxyphenyl)-3,7-dihydroimidazol[1,2-a]pyrazin-3-one,hydrochloride (MCLA) were purchased from Molecular Probes-Invitrogen (Eugene, OR). Percoll was purchased from Amersham Biosciences (Piscataway, NJ). SYBR Green Supermix and Bradford reagent were purchased from Bio-Rad (Hercules, CA). Rat anti-mouse Ly-6G conjugated to Allophycocyanin (Gr-1/APC), rat anti-mouse CD11b conjugated to phycoerythrin (CD11b/PE) and 7AAD were purchased from BD Biosciences Pharmingen (San Jose, CA). Goat anti-rat IgG microbeads were from Miltenyi Biotec Inc, (Auburn, CA). CP-532,903 was a gift from Dr. W. Ross Tracey (Pfizer Global Research and Development, Groton, CT), ZM 241385 was from Tocris Cookson Inc. (Ellisville, MO), adenosine deaminase (ADA) was from Roche Applied Science (Indianapolis, IN), BG 9928 was a gift from Dr. Barry Ticho (Biogen Idec., Cambridge, MA), and all remaining drugs and reagents were purchased from Sigma-Aldrich (St. Louis, MO). N6-(4-Amino-3-[125I]iodobenzyl)adenosine-5′-N-methylcarboxamide ([125I]I-AB-MECA) and 125I-ZM 241385 were synthesized and purified by high-performance liquid chromatography, as described previously (Olah et al., 1994; Palmer et al., 1995).
Mice
C57BL/6 wild-type (WT) mice were purchased from Taconic Farms (Germantown, NY). Congenic C57BL/6 A3KO mice were a kind gift from Dr. Marlene Jacobson (Merck Research Labs, West Point, PA), and congenic A2AKO mice (C57BL/6) created by Dr. Jiang-Fan Chen (Boston University, Boston, MA) were provided by Dr. Joel Linden (University of Virginia, Charlottesville, VA) after crossing to the C57BL/6 genetic background using a speed congenic approach (Sullivan et al., 2004). All animal experiments were conducted with approval of the Institutional Animal Care and Use Committee.
Isolation of Mouse Bone Marrow Neutrophils
Morphologically mature neutrophils were purified from mouse bone marrow by isotonic Percoll gradient centrifugation, as previously described (Lieber et al., 2004). Briefly, mice were euthanized by anoxia with carbon dioxide. Tibias and femurs of mice were flushed with neutrophil isolation buffer (1 × HBSS without Ca2+ and Mg2+, and containing 0.4% sodium citrate) and layered on a 3-step Percoll gradient (72%, 64%, 52%). Following centrifugation at 1,060 × g for 30 min, cells at the 72%:64% interface, were removed and washed once with isolation buffer before use in experiments.
For some studies, neutrophils were further purified by immunomagnetic selection using an antibody directed against mouse Ly-6G (Gr-1). Cells obtained by Percoll gradient centrifugation were incubated with rat anti-mouse Gr-1/APC antibody (1:800 dilution) for 30 min on ice, washed with PBS/2%FBS, and then incubated with goat anti-rat IgG microbeads (1:5 dilution) for 15 min. Cells bound to the magnetic beads were obtained using a MiniMACS magnetic separation column according to the manufacturer’s protocol (Miltenyi Biotec Inc.).
Flow cytometry analysis
Flow cytometry of single cell suspensions was performed using a Bectin Dickinson FACSCaliber flow cyctometer. Cells were incubated with 1:800 dilutions of rat anti-mouse Gr-1/APC antibody and rat anti-mouse CD11b/PE antibody for 30 min on ice. Following washing with PBS/2% FBS to remove unbound antibodies, cells (1 × 106 cells/ml) were re-suspended in PBS/2% FBS containing 7AAD (1:100). Forward scatter and 7AAD fluorescence were used to exclude debris, aggregates, and dead cells, respectively. The remaining fluorescent (PE and APC) and light scatter (side scatter) channels were used to differentiate neutrophils from other cell types.
Quantitative Real-Time RT-PCR
Total RNA was isolated from neutrophils using TRIzol reagent. Subsequently, 1 μg of total neutrophil RNA was reverse transcribed using a mixture of random and poly-T primers according to the manufacturer’s protocol (Invitrogen). Primers were designed for the mouse A1 (FWD, 5′-TGGCTCTGCTTGCTATTG-3′; REV, 5′-GGCTATCCAGGCTTGTTC-3′), A2A (FWD-5′ TCAGCCTCCGCCTCAATG-3′; REV, 5′-CCTTCCTGGTGCTCCTGG-3′), A2B (FWD, 5′-TTGGCATTGGATTGACTC-3′; REV, 5′-TATGAGCAGTGGAGGAAG-3′), and A3AR (FWD, 5′-CGACAACACCACGGAGAC-3′; REV, 5′-GCTTGACCACCCAGATGAC-3′) using Beacon Design software (Bio-Rad). PCR amplification (in SYBR Green Supermix) was performed using an iCycler iQ thermocycler (Bio-Rad) for 40 cycles of 25 s at 95°C followed by 45 s at an optimized annealing temperature for each AR. The cycle threshold, determined as the initial increase in fluorescence above background, was ascertained for each sample. Melt curves were performed upon completion of the cycles to ensure that nonspecific products were absent. For quantification of AR transcripts, a standard curve plotting cycle threshold versus copy number was constructed for each receptor subtype by analyzing 10-fold serial dilutions of plasmids containing the full-length mouse AR clones. AR transcript levels were expressed as copies/50 ng of total RNA.
Radioligand Binding Assays
Binding assays were conducted with membranes prepared from isolated neutrophils. In brief, Percoll gradient purified bone marrow neutrophils were resuspended in homogenization buffer (10 mM Na-HEPES, pH 7.4, 10 mM EDTA, and 0.1 mM benzamidine), homogenized in a glass Dounce homogenizer, and then centrifuged at 20,000g for 30 min. Cell pellets were washed once in binding buffer (10 mM Na-HEPES, pH 7.4, 1 mM EDTA, and 0.1 mM benzamidine), and then resuspended in binding buffer containing 10% (w/v) sucrose. Membranes were stored at −20°C until used for binding assays.
For radioligand binding studies, membrane protein was incubated in a final volume of 100 μl of binding buffer containing 5 mM MgCl2, 1 U/mL ADA, and either ~0.4 nM 125I-ZM 241385 to label A2AARs, or ~0.4 nM [125I]I-AB-MECA to label A1 and A3ARs. In competition experiments, inhibitors were included in the reactions at the concentrations indicated. After incubating at room temperature for 3 h, the incubations were terminated by rapid filtration over glass-fiber filters using a 48-well Brandel cell harvester. Filter discs containing trapped membranes bound with radioligand were quantified using a gamma counter. Nonspecific binding was determined in the presence of 1 μM ZM241385 or 1 μM [125I]I-AB-MECA, respectively.
Superoxide Production
Superoxide production was measured using the chemiluminscent probe MCLA (Nishida et al., 1989). Bone marrow neutrophils (7 × 104 cells/mL) were pre-incubated in HBSS at 37°C for 30 min (unless otherwise specified) with vehicle or AR agonists at the concentrations indicated, in the presence of 1 unit/mL ADA, followed by addition of MCLA (0.5 μM) and stimulation with various agents. In assays involving priming, 100 ng/ml rm TNF-α was included with the AR agonists during the 30-min pre-incubation period. Chemilumunescence was measured using a luminometer (AutoLumat, LB 953) and the cumulative relative light units (RLU) over 3 min (for fMLP, C5a or PAF stimulation) or 30 min (for PMA stimulation) were obtained for each sample. RLUs of unstimulated cells were deducted from the sample RLUs and superoxide produced was calculated as a percentage of that produced with vehicle-treated control samples. A cell free xanthine/xanthine oxidase superoxide generating system was used to determine the chemilumiscence enhancing/quenching properties of each of the agonists. Specificity of the probe for superoxide was verified in all of the assays by adding superoxide dismutase (SOD) in control reactions.
Neutrophil migration
Neutrophil migration was assessed using a standard 48-well micro-chemotaxis chamber (Neuroprobe, Gaithersburg, MD). Neutrophils were labeled with the fluorescent dye Calcein-AM (3 μM) in neutrophil isolation buffer for 30 min at 37° C, followed by pretreatment with vehicle or AR agonists at concentrations indicated in HBSS/0.5% BSA/1 unit/mL ADA for another 30 min at 37° C (unless otherwise specified). Neutrophils (5×104 cells/well) were added to the upper wells of the chemotaxis chamber, which were separated from the lower wells by a polycarbonate membrane (5 μm pores). In chemokinesis studies, various concentrations of the chemoattractant dissolved in HBSS/0.5% BSA were added to both the upper and lower wells, where as the chemoattractants were added only to the lower wells in the chemotaxis studies. The cells were allowed to migrate at 37° C for 35 min after which fluorescence emitted by cells adhered to the underside of the membrane was measured using a fluorimager (Molecular Dynamics Typhoon Imaging System, GE Healthcare Life Sciences, Piscataway, NJ). Following densitometry analysis using Scion Image software (from the National Institutes of Health), the chemotaxis or chemokinesis index was calculated using the following formula: fluorescence density of cells migrated in the presence of chemoattractant/fluorescence density of cells migrated towards medium.
Thioglycollate-induced peritonitis was used as an in vivo model of neutrophil chemotaxis. Briefly, mice were injected intravenously with vehicle or CP-532,903 (100 μg/kg) followed by intraperitoneal injection of 2 ml of 3% thioglycollate. At 4 hours, mice were killed by CO2 asphyxiation and injected intraperitoneally with neutrophil isolation buffer, their abdomens were massaged, and total lavage fluid was collected through a small slit cut into the peritoneal cavity. Collected cells were pelleted by centrifugation, resuspended in neutrophils isolation buffer, diluted with Trypan blue and counted using a standard hemocytometer chamber. Most of the cells found in the peritoneal exudate 4 h after thioglycollate administration are neutrophils (data not shown; (Lagasse and Weissman, 1996)).
Data Analysis
Data are reported as means ± S.E.M. Chemotaxis data were analyzed by two-way ANOVA (chemotactic factor and AR agonist treatment) to determine if there was a main effect of the chemoattractant, a main effect of pretreating cells with AR agonists, or a chemoattractant-AR agonist treatment interaction. All other data were compared by Student’s t test or one-way ANOVA followed by post-hoc contrasts using Dunnett’s t test, as appropriate. A p value <0.05 was considered statistically significant.
RESULTS
Isolation of neutrophils from mouse bone marrow
We determined the purity of neutrophil isolates from mouse bone marrow by immuno-flow cytometry. Neutrophils are found in the subset that show high GR-1 and high CD11b expression (Fig. 1), whereas monocytes and lymphocytes are found in the Gr-1 low/CD11b low and Gr-1 negative/CD11b negative populations, respectively (Lagasse and Weissman, 1996). The results indicate that ~85% of gated mouse bone marrow cells in Percoll density gradient purified fractions are neutrophils, which agree with the results of previous studies (Lieber et al., 2004). The majority of the remaining cells were B-lymphocytes. As shown in Fig. 1, cytospin analysis not only confirmed the purity of the Percoll density gradient purified neutrophil preparations, but also revealed that >95% of the neutrophils exhibited the characteristic size, nuclear morphology, and staining pattern of mature cells. Further purification of the Percoll density gradient separated neutrophil population using the anti-Gr-1 antibody yielded >99% mature neutrophils (Fig. 1). Although immunomagnetic selection provided a purer cell population, we could not use this isolation technique for most subsequent assays because of relatively low yield and because antibody treatment tended to mildly activate the cells.
Figure 1.
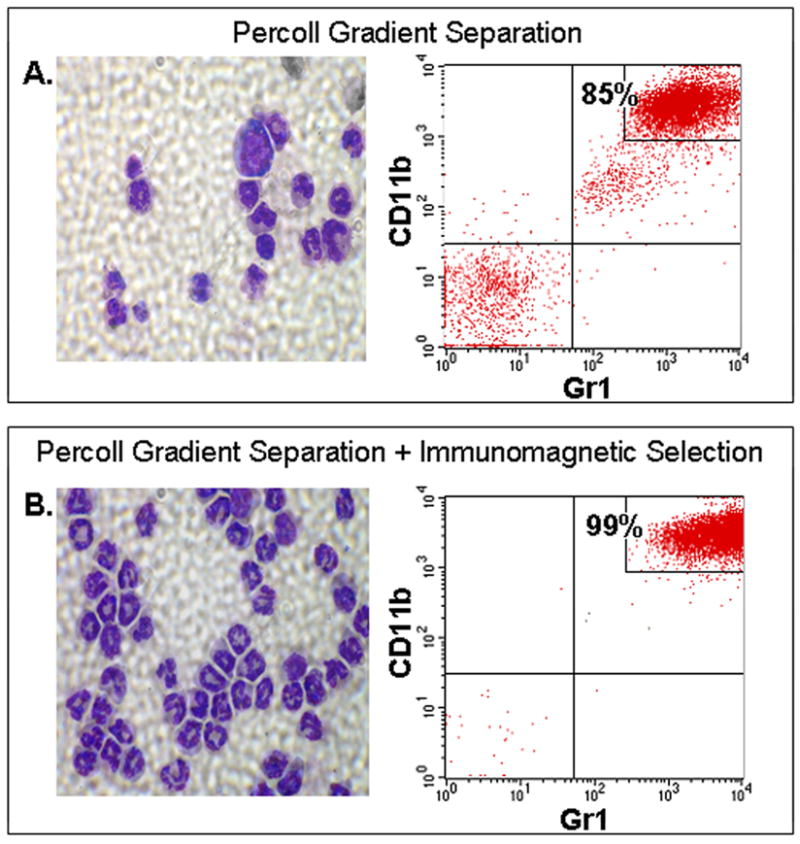
Purity of mouse bone marrow neutrophil preparations obtained by Percoll gradient separation (A) coupled with immunomagnetic selection using the anti-Gr-1 antibody (B). Shown are representative cytospin images and results of flow cytometry analysis using Gr-1 and CD11b as markers. The percentage of neutrophils in the entire cell population is indicated. n=3.
Both A2A and A3ARs are abundantly expressed in mouse bone marrow neutrophils
We used quantitative real-time RT-PCR to measure mRNA expression of ARs in mouse neutrophils. As illustrated in Fig. 2A, we detected transcripts for A2A, A2B, and A3ARs in mouse bone marrow neutrophils isolated by Percoll gradient centrifugation. Based on calculations obtained from standard curves, mRNA expression of the A3AR was the highest (2693 ± 542 copies of mRNA/50 ng of total RNA), followed by the A2AAR (1240 ± 169 copies of mRNA/50 ng of total RNA) and the A2BAR (74 ± 42 copies of mRNA/50 ng of total RNA). We did not detect expression of A1AR mRNA above background levels obtained with control reactions using water in place of cDNA. mRNA expression studies using neutrophil isolates obtained by sequential Percoll gradient purification and immunomagnetic selection with the anti-GR-1 antibody in which ~99% of the cells were neutrophils gave similar results (Fig. 2A).
Figure 2.
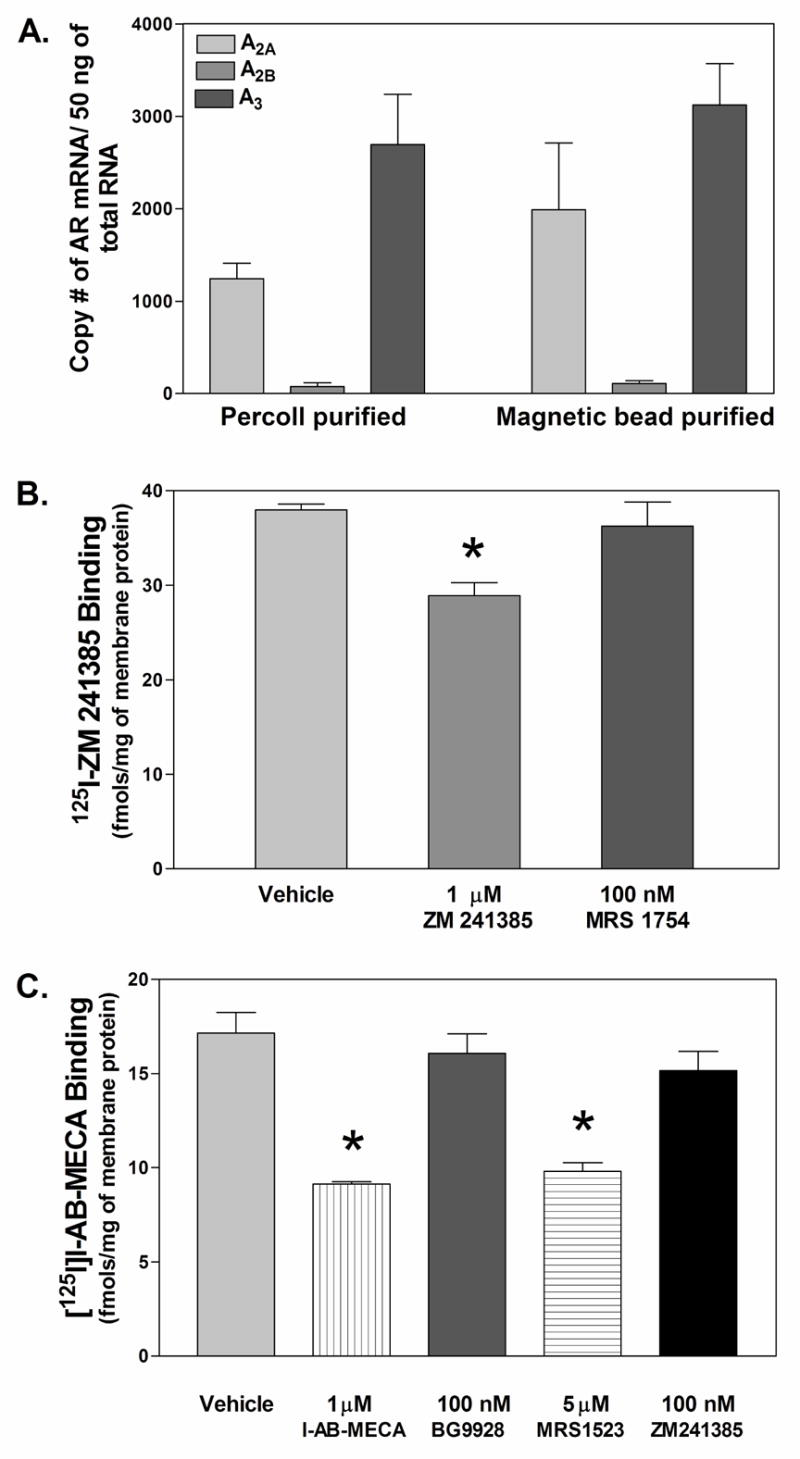
Expression of AR subtypes in mouse bone marrow neutrophils. (A) mRNA levels of AR subtypes in mouse bone marrow neutrophils quantified by real time RT-PCR, n = 5–18. (B and C) Protein levels of AR receptor subtypes in Percoll gradient purified mouse bone marrow neutrophils quantified using radioligand binding analysis. Total binding (fmols/mg of total membrane protein) of the A2AAR antagonist radioligand 125I-ZM241385 ( 0.4 nM; B) or the A1/A3AR agonist radioligand [125I]I-AB-MECA ( 0.4 nM; C) to neutrophil membranes in the presence of vehicle or various competitors at the concentrations indicated. Results are presented as the mean ± SEM. *, p < 0.05 versus the vehicle-treated group by one-way ANOVA and Dunnett’s t test, n = 3.
We subsequently conducted radioligand binding assays with crude membrane proteins prepared from Percoll gradient separated bone marrow neutrophils to assess expression of ARs at the protein level. Membranes were incubated with 0.4 nM 125I-ZM 241385 (A2AAR antagonist) or ~0.4 nM [125I]I-AB-MECA (A1/A3AR agonist). As shown in Fig. 2B, we detected specific binding of 125I-ZM 241385 to neutrophil membranes, defined by inclusion of 1 μM non-radiolabeled ZM 241385. Specific binding of 125I-ZM 241385 was not displaced by the A2BAR antagonist MRS 1754 (100 nM) indicating that 125I-ZM 241385 was specifically labeling A2AARs. We also detected specific binding of [125I]I-AB-MECA to membranes prepared from Percoll density gradient purified mouse bone marrow neutrophils (Fig. 2C), defined by inclusion of 1 M non-radiolabeled I-AB-MECA. Since [125I]I-AB-MECA binds with relatively high affinity to both A1 and A3ARs (Olah et al., 1994), it could have labeled either of these AR subtypes in our assays. However, specific binding of [125I]I-AB-MECA was displaced solely by the A3AR antagonist MRS 1523 (5 μM) and not by BG 9928 (A1AR antagonist; 100 nM) or ZM 241385 (A2AAR antagonist; 100 nM), indicating that [125I]I-AB-MECA was binding to the A3AR. Given that both radioligands were included in the binding assays at a concentration near their Kd values for A2A and A3ARs (Olah et al., 1994; Palmer et al., 1995), the Bmax values for 125I-ZM 241385 and [125I]I-AB-MECA were estimated to be 18.17 ± 3.68 and 16.05 ± 2.30 fmol/mg protein, respectively. Collectively, our quantitative real-time RT-PCR and radioligand binding data indicate that A2A and A3ARs are expressed in mouse bone marrow neutrophils at similar levels. Our PCR data also suggest that mouse bone marrow neutrophils express the A2BAR..
Expression of A2A and A2BARs, but not the A3AR, is induced in murine bone marrow neutrophils during inflammation
Previous studies have shown that A2AARs, and in some reports A2B and A3ARs, are induced by pro-inflammatory Th1 cytokines and Toll like receptor agonists in various immune cell populations, providing an additional feedback mechanism whereby adenosine ‘resolves’ inflammatory responses (Fortin et al., 2006; Gessi et al., 2004; Khoa et al., 2001; Lappas et al., 2005; Murphree et al., 2005; Nguyen et al., 2003). Accordingly, we examined whether inflammation induces the expression of ARs in mouse bone marrow neutrophils. For these studies, neutrophils were isolated from bone marrow by Percoll gradient separation from mice pre-treated 4 h earlier with a large dose of lipopolysaccharide (LPS; 10 mg/kg i.p.). As shown in Fig. 3A, mRNA expression of both A2A and A2BARs was increased ~11- and 15-fold, respectively, in neutrophils isolated from LPS-treated mice compared to neutrophils isolated from vehicle-treated mice, whereas mRNA expression of the A3AR was not significantly changed. Specific binding of 125I-ZM 241385 was also increased ~3-fold in neutrophils isolated from LPS-treated mice, indicating that LPS induced expression of the A2AAR at the protein level (Fig. 3B).
Figure 3.
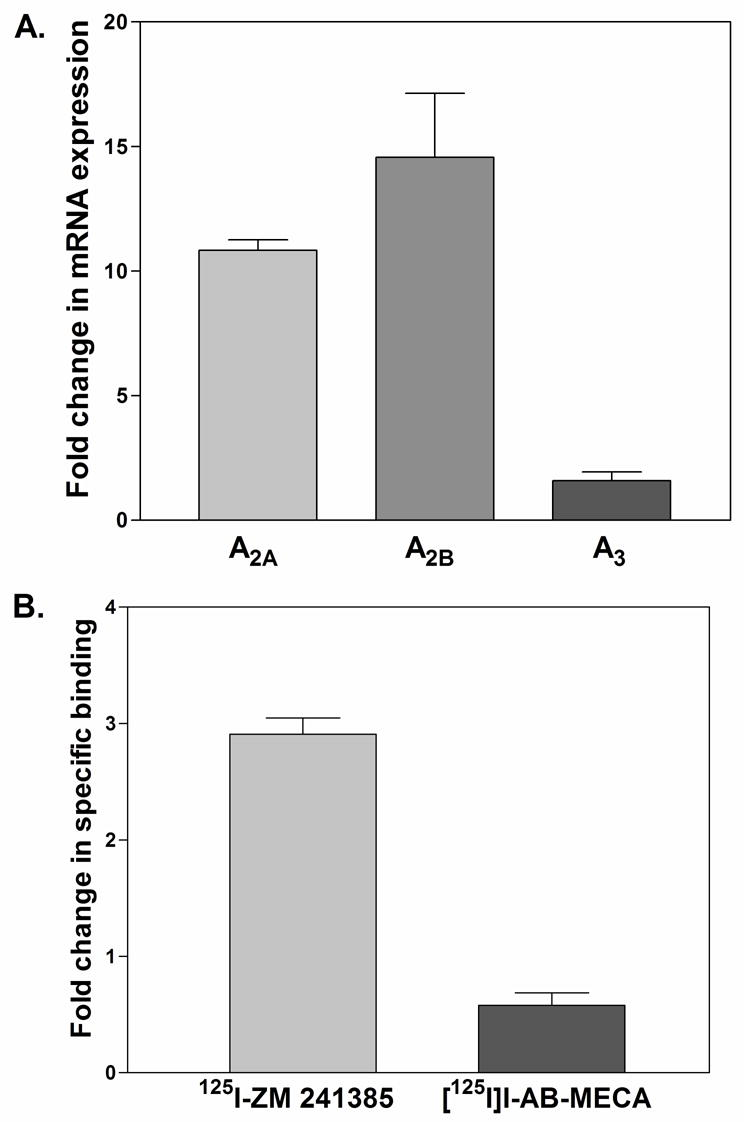
Changes in AR mRNA (A) and protein (B) expression in bone marrow neutrophils from LPS-treated mice. Mice were injected ip with either vehicle or 10 mg/kg LPS. Four hours later, bone marrow neutrophils were obtained by Percoll gradient separation. AR mRNA levels were determined by real time RT-PCR normalized to 18S RNA. A2A and A3AR protein levels were determined by radioligand binding analysis with crude membrane preparations using 125I-ZM 241385 (~0.4 nM) or [125I]I-AB-MECA (~0.4 nM). Specific binding was defined by including 1 μM of the respective non-radiolabeled ligand in the assays. The data (mean ± SEM; n = 3) are presented as the fold increase in mRNA or protein expression in neutrophils from LPS-treated mice compared to vehicle-treated mice.
Both A2A and A3ARs inhibit superoxide production by stimulated neutrophils
In preliminary studies, we examined the ability of a panel of agents to stimulate Percoll gradient purified bone marrow neutrophils to generate superoxide anions, and confirmed that the bacterial tripeptide fMLP, complement component 5a (C5a), platelet-activating factor (PAF), and phorbol myristate acetate (PMA) were all effective stimulants. The time course and magnitude of superoxide production assessed by MCLA chemiluminescence in response to stimulation with 1 μM fMLP is depicted in Fig. 4. Priming the cells with 100 ng/ml of rm TNF-α increased superoxide production 3–4-fold compared to unprimed cells.
Figure 4.
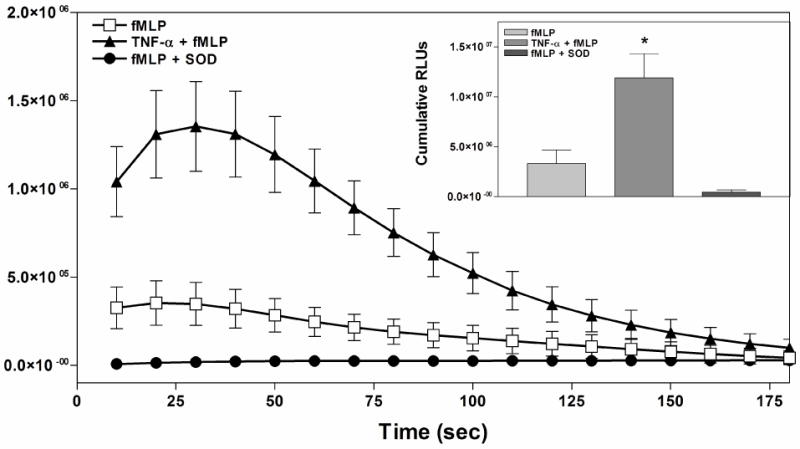
Superoxide production by Percoll gradient purified mouse bone marrow neutrophils in response to fMLP (1 μM). Studies were conducted with naïve neutrophils, neutrophils primed with TNF-α (30 min incubation with 100 ng/mL rm TNF-α), or treated with superoxide dismutase (SOD). Superoxide production was measured using the chemiluminescent probe MCLA. Relative light units (RLUs) were obtained for 3 minutes at 10 second intervals. The inset depicts the cumulative relative light units generated over 3 minutes after subtracting baseline values. Results are presented as the mean ± SEM. *, p < 0.05 versus the unprimed fMLP-induced group by one-way ANOVA followed by a Student’s t test, n = 3.
We subsequently assessed the ability of AR stimulation to inhibit superoxide production by Percoll gradient purified mouse bone marrow neutrophils. Similar to previous reports, pre-treatment (30 min) with the non-selective AR agonist NECA (300 nM) or the A2AAR agonist CGS 21680 (100 nM) effectively attenuated fMLP (1 μM)-stimulated superoxide production by ~45% in assays using unprimed cells and by ~25% using TNF-α-primed cells (Fig. 5A). Interestingly, however, pre-treating neutrophils with the selective A3AR agonist CP-532,903 (100 nM; (Tracey et al., 2003; Wan et al., 2008)) also effectively inhibited fMLP-induced neutrophil superoxide generation regardless of priming with TNF-α (Fig. 5A). These results imply that the A3AR, in addition to the A2AAR, might function to inhibit neutrophil superoxide production. Pre-treatment with NECA (300 nM), CGS 21680 (100 nM), or CP-532,903 (100 nM) also inhibited superoxide production by ~50% from neutrophils stimulated with C5a (3 nM) or PAF (100 nM), but not by PMA (800 nM; Fig. 5B, C and D).
Figure 5.

Effect of pretreating with AR agonists on superoxide production by Percoll gradient purified mouse bone marrow neutrophils in response to fMLP (A), C5a (B), PAF (C), or PMA (D). Neutrophils were pretreated with either vehicle or the AR agonists for 30 min in the presence of ADA (1 unit/mL), and then stimulated with the activating agents while measuring superoxide production using the chemiluminescent probe MCLA. In studies with fMLP, the effect of the agonists on superoxide production was tested with both unprimed and TNF-α-primed (100 ng/mL) neutrophils. The data (mean ± SEM) are presented as the percentage of superoxide produced compared to the vehicle-treated group. *, p < 0.05 versus the vehicle-treated group by one-way ANOVA and Dunnett’s t test, n = 4 - 6.
To conclusively determine whether the A3AR regulates neutrophil superoxide production, we compared concentration-response curves generated with CGS 21680 or CP-532,903 using Percoll gradient purified neutrophils obtained from WT, A2AKO, or A3KO mice. As shown in Fig. 6, pre-treating neutrophils with either CGS 21680 or CP-532,903 produced a concentration-dependent reduction in fMLP-induced superoxide generation by neutrophils obtained from WT mice with equal potencies and efficacies (EC50 and maximum inhibition were 25.3 ± 10.8 nM and 48.6 ± 5.7% for CGS 21680 and 49.3 ± 26.3 nM and 61.2 ± 5.1% for CP-532,903). The ability of CGS 21680 to inhibit superoxide production was lost completely using neutrophils obtained from A2AKO mice, but was unaffected using neutrophils obtained from A3KO mice (Fig. 6A). These results indicate that CGS 21680 reduced superoxide production by selectively activating the A2AAR. On the other hand, the concentration-response relationship obtained with CP-532,903 to inhibit superoxide production was unaffected using A2AKO neutrophils, but was right-shifted ~100-fold using neutrophils obtained from A3KO mice, suggesting that CP-532,903 inhibited superoxide production primarily via the A3AR at low concentrations. These results confirm that activating either the A2AAR or the A3AR effectively inhibits neutrophil superoxide production.
Figure 6.

Inhibition of fMLP-induced (1 μM) superoxide production by Percoll gradient purified bone marrow neutrophils from WT, A2AKO, or A3KO mice by increasing concentrations of CGS 21680 (A) or CP-532,903 (B). Results (mean ± SEM; n = 3) are displayed as percentage of superoxide produced by the vehicle-treated group.
We also compared concentration-response curves for CGS 21680 and CP-532,903 to inhibit superoxide production using Percoll gradient purified neutrophils obtained from mice pretreated 4 h earlier with LPS (10 mg/kg) in which A2AAR message and protein were induced. Both agonists continued to concentration-dependently inhibit fMLP-induced superoxide production. EC50 values for CGS 21680 (30.4 ± 17.6 nM) and CP-532,903 (23.8 ± 11.8 nM) were similar to those obtained in assays using naive neutrophils, although the maximal inhibition (27.2 ± 5.2 % and 36.0 ± 4.0 %, respectively) was less compared to those obtained with naïve cells. The efficacies of the agonists to inhibit superoxide production were similar to values obtained with naïve cells treated with TNF-α (Fig. 5A), suggesting that cells obtained from LPS-treated mice had likely undergone priming.
Prolonged A3AR activation is required to effectively inhibit neutrophil superoxide production
In all previous superoxide assays in this study, cells were pre-treated with the AR agonists for 30 min before the addition of activating agents. We therefore examined the duration of pretreatment that is necessary to produce maximal inhibition of fMLP-induced superoxide production. For these studies, Percoll gradient purified mouse bone marrow neutrophils were pretreated with CGS 21680 (100 nM) or CP-532,903 (100 nM) for 6, 18, or 30 min before stimulating with fMLP (1 μM). As shown in Fig. 7, pretreating unprimed cells with CGS 21680 for as little as 6 min resulted in maximal inhibition (51.1 ± 8.7% of vehicle-treated cells) of fMLP-stimulated superoxide production, whereas maximal inhibition with CP-532,903 (49.0 ± 5.3% of vehicle-treated cells) was only achieved when the cells were pretreated for at least 18 min. Pretreatment with CP-532,903 for 6 min only inhibited superoxide production by 17.8 ± 2.1%.
Figure 7.

Effect of duration of pretreatment with CGS 21680 or CP-532,903 on superoxide production by Percoll gradient purified bone marrow neutrophils in response to fMLP. Neutrophils were incubated (37° C) with vehicle, CGS 21680 (100 nM), or CP-532,903 (100 nM) in the presence of ADA (1 unit/mL) for the times indicated prior to stimulation with fMLP (1 μM). Superoxide produced was measured using the chemiluminescent probe MCLA (0.5 μM). Results (mean ± SEM, n = 4) are presented as the percentage of superoxide produced compared to the corresponding vehicle-treated group.
Activation of the A3AR inhibits chemotaxis of mouse bone marrow neutrophils
We initially examined whether activation of the A3AR affects chemotaxis in a trans-well assay system with Percoll gradient-purified neutrophils in the upper wells and increasing concentrations of CP-532,903 in the lower wells, separated by a polycarbonate membrane. Neutrophils were suspended in a buffer containing 1 unit/ml of adenosine deaminase to remove endogenous sources of adenosine. As shown in Fig. 8A, inclusion of up to 10 μM CP-532,903 in the lower wells did not promote chemotaxis using naïve neutrophils. Inclusion of increasing concentrations of CP-532,903 in the lower wells in combination with 1 μM fMLP also neither promoted nor inhibited chemotaxis of unprimed or TNF-αprimed cells (Fig. 8B).
Figure 8.

Effect of CP-532,903 on neutrophil chemotaxis in trans-well migration assays using Percoll gradient purified mouse bone marrow neutrophils. Fluorescently labeled (Calcein-AM) neutrophils were added into upper wells, with fMLP and/or CP-532,903 in the lower wells in the presence of ADA (1 unit/mL), separated by a polycarbonate membrane. After incubating at 37° C for 35 min, migrated cells were quantified and chemotaxis was calculated as described in Methods. (A) Effect of adding increasing concentrations of CP-532,903 into lower wells. (B) Effect of adding increasing concentration of CP-532,903 into bottom wells on fMLP-induced (1 μM) chemotaxis of unprimed and TNF-αprimed neutrophils. Results are presented as the mean ± SEM. n = 3–5.
Based on the results of our superoxide studies, we subsequently examined whether pre-treating neutrophils with CP-532,903 influences chemotaxis. For this assay, neutrophils were pre-treated with either vehicle or AR agonists for 30 minutes in the presence of adenosine deaminase prior to their addition to the chemotaxis apparatus containing increasing concentrations of fMLP in the lower wells. Using vehicle-treated cells, fMLP produced a typical bell-shaped concentration-response curve observed with chemoattractants with a maximal chemotaxis index of ~4 occurring at a concentration of 1 μM (Fig. 9). Pre-treating the cells with 100 nM CP-532,903 had no effect on the chemotactic response of unprimed cells to fMLP (Fig. 9A). However, pre-treatment with CP-532,903 inhibited migration of TNF-α-primed cells towards higher concentrations of fMLP. In contrast, treatment with the A2AAR agonist CGS 21680 had no significant effect on fMLP-induced chemotaxis of WT neutrophils regardless of whether the cells were primed or not (Fig. 9A and B). To confirm that the effect of CP-532,903 on chemotaxis was mediated via the A3AR, the assays were repeated using neutrophils obtained from either A3KO or A2AKO mice. As shown in Fig. 9, CP-532,903 continued to inhibit fMLP-induced chemotaxis in assays using A2AKO but not A3KO neutrophils, indicating that the effect of CP-532,903 on chemotaxis was mediated by the A3AR. Note that the chemotactic responses of both A2AKO and A3KO neutrophils were similar to WT neutrophils.
Figure 9.

Effect of pretreating with CP-532,903 or CGS 21680 on fMLP-induced chemotaxis of unprimed WT (A), TNF-αprimed WT (B), TNF-α primed A2AKO (C) or TNF-αprimed A3KO (D) Percoll gradient purified mouse bone marrow neutrophils. Neutrophils were incubated (37° C) with vehicle, CP-532,903 (100 nM), or CGS 21680 (100 nM; A and B) for 30 min in the presence of ADA (1 unit/mL) with (B, C and D) or without rm TNF-α (100 ng/mL; A), prior to addition to the upper wells of trans-well assays containing increasing concentrations of fMLP in lower wells. The chemotaxis index was calculated as described in Methods after allowing the cells to migrate for 35 min. Results are presented as the mean ± SEM. *, p < 0.05 versus the vehicle-treated group by two-way ANOVA, n = 5–16.
We further examined whether pretreatment with CP-532,903 influences chemotaxis of neutrophils towards other stimuli including C5a, PAF, and IL-8. We found that pretreatment with CP-532,903 (100 nM) inhibited migration of neutrophils towards C5a regardless of whether they were primed or not, whereas it only inhibited chemotaxis of unprimed neutrophils towards PAF and IL-8 (Fig. 10).
Figure 10.

Effect of pretreating with CP-532,903 on chemotaxis of Percoll gradient purified mouse bone marrow neutrophils towards various chemoattractants. Neutrophils were incubated (37° C) with vehicle or CP-532,903 (100 nM) for 30 min in the presence of ADA (1 unit/mL) with or without rm TNF-α(100 ng/mL), prior to addition to the upper wells of trans-well assays containing increasing concentrations of C5a (A and B), IL-8 (C and D), or PAF (E and F) in lower wells. The chemotaxis index was calculated as described in Methods after allowing the cells to migrate for 35 min. Results are presented as the mean ± SEM. p < 0.05 versus the vehicle-treated group by two-way ANOVA, n = 3–6.
We next sought to determine whether pretreating neutrophils with CP-532,903 influenced neutrophil chemokinesis. Cells were pretreated with 100 nM CP-532,903 for 30 min in the presence of adenosine deaminase and then placed into the upper wells of the trans-well apparatus containing increasing amounts of fMLP in both the upper and lower wells. As depicted in Fig. 11, pre-treatment with CP-532,903 increased chemokinesis of unprimed neutrophils compared to vehicle-treated cells becoming significant at fMLP concentrations ≥1 μM, but had no effect on fMLP-induced chemokinesis of TNF-αprimed cells).
Figure 11.

Effect of pretreating with CP-532,903 on fMLP-induced chemokinesis of Percoll gradient purified mouse bone marrow neutrophils. Neutrophils were incubated (37° C) with vehicle or CP-532,903 (100 nM) for 30 min in the presence of ADA (1 unit/mL) with or without rm TNF-α(100 ng/mL), prior to addition to the upper wells of trans-well assays containing increasing concentrations of fMLP in both the upper and lower wells. The chemokinesis index was calculated as described in Methods after allowing the cells to migrate for 35 min. Results are presented as the mean ± SEM. *, p < 0.05 versus the vehicle-treated group by two-way ANOVA, n = 5.
Given the results of our in vitro chemotaxis assays, we further examined whether activation of the A3AR inhibits leukocyte accumulation in thioglycollate-induced peritonitis. For these studies, WT or A3KO mice were pre-treated with either vehicle or CP-532,903 (100 μg/kg i.v.) prior to intraperitoneal injection of thioglycollate (2 ml of a 3% solution) and the number of cells found within the peritoneal cavity was quantified 4 h later. As shown in Fig 12, treatment with CP-532,903 significantly reduced thioglycollate-induced leukocyte accumulation in WT mice by ~25% compared to vehicle-treated mice, but not in A3KO mice. However, leukocyte recruitment was not different between vehicle-treated WT and A3KO mice. Like the in vitro chemotaxis results, these data suggest that activation of the A3AR inhibits leukocyte chemotaxis, but does not play and endogenous role to regulate migration in this model of acute inflammation.
Figure 12.

Effect of CP-532,903 on leukocyte accumulation during thioglycollate-induced peritonitis. Mice were injected i.v. with either vehicle or CP-532,903 (100 μg/kg) immediately prior to a single intraperitoneal injection of thioglycollate (2 ml of a 3% solution). Four hours later, the number of leukocytes within peritoneal exudates was quantitated, as described in Methods. Results are presented as the mean ± SEM. *, p < 0.05 versus the vehicle-treated group by Student’s t test, n = 6.
DISCUSSION
Previous work has implicated the A2AAR in mediating the anti-inflammatory actions of adenosine on neutrophils. In the present investigation, we have demonstrated using AR gene knock-out mice that the A3AR, in addition to the A2AAR, suppresses the superoxide burst. Moreover, we have demonstrated that activation of the A3AR inhibits neutrophil chemotaxis towards a variety of activating agents.
We found that the A3AR is abundantly expressed in mouse bone marrow neutrophils. Indeed, our data indicate that it is expressed at equal or even higher levels than the A2AAR. Compared to recent reports using RT-PCR, the pattern of AR receptor expression in mouse bone marrow neutrophils appears to be similar to that of circulating human neutrophils, where A2A and A3AR expression is predominant, followed by low levels of expression of the A2BAR subtype, and little or no detectable expression of A1AR message (Chen et al., 2006; Fortin et al., 2006; Zhang et al., 2006). Unlike the A2AAR, expression of the A3AR was not increased in neutrophils obtained from mice treated with LPS, indicating that the A3AR may not be transcriptionally regulated by pro-inflammatory cytokines in neutrophils. It is notable that mRNA expression of the Gs protein-coupled A2BAR was induced nearly 15-fold in neutrophils obtained from LPS-treated mice.
According to our results, the A3AR functions in murine neutrophils to inhibit stimulated superoxide production along with the A2AAR. This conclusion is based on our observation that pretreatment with either the A2AAR agonist CGS 21680 or the A3AR agonist CP-532,903 potently inhibited superoxide production. The selectivity of each of the agonists for their respective AR subtype was confirmed in assays using cells obtained from AR KO mice. Most importantly, we showed that CP-532,903 continued to potently inhibit stimulated superoxide production from neutrophils obtained from A2AAR KO mice (but not from A3AR KO mice), excluding the concern that CP-532,903 was acting non-specifically via the A2AAR. Interestingly, we found that it was necessary to pretreat with CP-532,903 for a much longer period of time (18 min) to achieve maximal inhibition of superoxide production compared to CGS 21680. Pretreatment with CP-532,903 for 6 min only slightly inhibited superoxide production (~18% inhibition), which could be one of the reasons why a role for the A3AR in regulating neutrophil superoxide production has not been detected previously (Jordan et al., 1999). From a functional perspective, our data suggest that the A2AAR serves to provide immediate suppression of superoxide generation as neutrophils migrate into inflamed tissues, whereas the A3AR may assist to sustain inhibition. Although our results show that activating either the A2AAR or the A3AR inhibited superoxide production, activating both receptors simultaneously using the non-selective agonist NECA (Fig. 5) or a combination of CGS 21680 and CP-532,903 (data not shown) did not provide further inhibition. These results suggest that signaling pathways propagated by the Gs protein-coupled A2AAR and the Gi protein-coupled A3AR may cross talk or converge upon a common inhibitory mechanism, such as interference with assembly of the NADPH oxidase complex, inhibition of intracellular calcium mobilization, or cross-desensitization of chemokine receptors.
Our results also suggest that the A3AR functions to inhibit neutrophil chemotaxis. In a standard trans-well chemotaxis assay, we found that prior activation of the A3AR inhibits neutrophil migration towards a variety of chemotactic agents, predominantly in response to high concentrations of chemoattractants with variability depending on the state of cell priming. These results are consistent with the idea that neutrophils exposed to adenosine within hypoxic/ischemic tissues would have reduced ability to migrate towards chemotactic stimuli. Moreover, our results suggest that systemic administration of an A3AR agonist could inhibit neutrophil chemotaxis into inflamed tissues. Indeed, we found that pre-treating mice intravenously with CP-532,903 inhibited migration of neutrophils into the peritoneum following intraperitoneal injection of thioglycollate. Previous studies have shown that adenosine weakly promotes neutrophil chemotaxis at low concentrations, which has previously been attributed to activation of the A1AR (Cronstein et al., 1990; Rose et al., 1988). Considering that murine neutrophils do not express the A1AR and since activation of the A3AR increased fMLP-induced random movement (Fig. 12), we speculate that actions previously attributed to the A1AR in neutrophils may actually be mediated by the A3AR.
Chen and colleagues (Chen et al., 2006) have recently suggested that migrating neutrophils secrete ATP at the leading edge, which signals via P2Y2 receptors to amplify chemoattractant signals. Following hydrolysis to adenosine by ecto-nucleotidases, these investigators (Chen et al., 2006) also provide evidence that the A3AR is recruited from a cytosolic location to the leading edge and further promotes chemotaxis by increasing the speed of migration. This hypothesis is supported by results of trans-well migration studies as well as video-tracking studies of migrating neutrophils showing that activation of the A3AR using N6-(3-iodobenzyl)adenosine-5′-N-methylcarboxamide (IB-MECA) promotes chemotaxis (Chen et al., 2006). In the present investigation, we were not able to demonstrate that the A3AR normally functions to facilitate neutrophil migration. In trans-well migration assays, we failed to observe that including CP-532,903 in bottom wells alone or with fMLP acutely increases neutrophil chemotaxis (Fig. 8). Unlike Chen and colleagues (Chen et al., 2006), we also failed to observe that chemotaxis of neutrophils obtained from A3KO mice is impaired (Fig. 9C). Although several factors including the specific pharmacological agents used to stimulate the A3AR (i.e., CP-532,903 versus IB-MECA), methods to isolate/culture murine neutrophils, stimulation protocols including the time and duration of pretreatment of cells with agonists, and the state of cell priming could have contributed, a definite explanation for the differences in results obtained in our studies and those of Chen and colleagues (Chen et al., 2006) remain unclear. Nevertheless, the results of our studies are more consistent with the theory that the primary function of the A3AR is to inhibit neutrophil chemotaxis rather than to promote it. In agreement with our work, three previous studies have also concluded that activation of the A3AR inhibits chemotaxis of human and murine eosinophils (Knight et al., 1997; Walker et al., 1997; Young et al., 2004).
In summary, we have shown that activation of the A3AR inhibits two pro-inflammatory actions of murine neutrophils, i.e., stimulated superoxide production and chemotaxis. A precise role for the A3AR in regulating these responses was confirmed using A3AR gene knock-out mice and the newly characterized selective A3AR agonist CP-532,903 (Wan et al., 2008). Collectively, our results suggest that the anti-inflammatory actions of adenosine are not only mediated through the A2AAR, but also via the Gi protein-coupled A3AR. Considering that differences in the functional role of the A3AR have been observed between species (Linden, 1994), it will be important to confirm our findings in humans and other species. Several previous studies have demonstrated that A3AR agonists provide benefit in experimental animal models of inflammation. For example, A3AR agonists have been shown to increase survival during sepsis (Lee et al., 2006), to reduce the severity of inflammation in adjunct-induced arthritis (Montesinos et al., 2000), and to reduce ischemia/reperfusion injury (Ge et al., 2006; Jordan et al., 1999; Wan et al., 2008). However, the precise mechanism of action of these agents remains uncertain. The results of this investigation provide evidence that these agents may be effective by inhibiting the pro-inflammatory actions of neutrophils.
Acknowledgments
We acknowledge the assistance of the Cardiovascular Center HPLC Core and the Clinical Immunodiagnostic and Research Laboratory for purification of radioligands and assistance with cytospin analysis, respectively. We also thank Dr. Phillip Pratt (Department of Anesthesiology, Medical College of Wisconsin), Dr. Zhi-Dong Ge (Department of Anesthesiology, Medical College of Wisconsin), and Dr. Jeffrey Woodliff (Department of Pediatrics, Medical College of Wisconsin) for their technical assistance.
This research was supported in part by National Institutes of Health Grants R01 HL 60051 and R01 HL 077707 (to J.A.A.) and by American Heart Association Grant 0615533Z (to D.V.).
Nonstandard Abbreviations
- ADA
adenosine deaminase
- AR
adenosine receptor
- BG 9928
1,3-dipropyl-8-[1-(4-propionate)-bicyclo-[2,2,2]octyl]xanthine
- C5a
complement component 5a
- CGS 21680
2-[p-(2-carboxyethyl)phenethylamino]-5′-N-ethylcarboxamidoadenosine
- Cl-IB-MECA
2-chloro derivative of N6-(3-iodobenzyl)adenosine-5′-N-methyluronamide
- CP-532
903, (2S,3S,4R,5R)-3-amino-5-[6-(2,5-dichlorobenzylamino)purin-9-yl]-4-hydroxytetrahydrofuran-2-carboxylic acid methylamide
- fMLP
formylated-Methionine-Leucine-Phenylalanine
- FWD
forward
- IB-MECA
N6-(3-iodobenzyl)adenosine-5′-N-methyluronamide
- [125I]I-AB-MECA
N6-(4-amino-3-[125I]iodobenzyl)adenosine-5′-N-methylcarboxamide
- IL-8
interleukin-8
- KO
knockout
- MCLA
2 methyl-6-(4-methoxyphenyl)-3,7-dihydroimidazol[1,2-a]pyrazin-3-one,hydrochloride
- MPO
myeloperoxidase
- MRS 1754
1,3-dipropyl-8-[4-[((4-cyanophenyl)carbamoylmethyl)oxy]phenyl]xanthine
- MRS 1523
3-propyl-6-ethyl-5[(ethylthio)carbonyl]-2-phenyl-4-propyl-3-pyridine-carboxylate
- NECA
adenosine-5′-N-ethylcarboxamide
- PAF
platelet activating factor
- PMA
phorbol 12-myristate 13-acetate
- REV
reverse
- RT-PCR
reverse transcription-polymerase chain reaction
- SOD
superoxide dismutase
- rm TNF-α
recombinant mouse tumor necrosis factor- α
- WT
wild-type
- ZM241385
4-{2-[7-amino-2-(2-furyl)[1,2,4]triazolo-[2,3-a][1,3,5]triazin-5-ylamino]ethyl}phenol
Footnotes
This PDF receipt will only be used as the basis for generating PubMed Central (PMC) documents. PMC documents will be made available for review after conversion (approx. 2–3 weeks time). Any corrections that need to be made will be done at that time. No materials will be released to PMC without the approval of an author. Only the PMC documents will appear on PubMed Central -- this PDF Receipt will not appear on PubMed Central.
References
- Bours MJ, Swennen EL, Di Virgilio F, Cronstein BN, Dagnelie PC. Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther. 2006;112:358–404. doi: 10.1016/j.pharmthera.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Chen Y, Corriden R, Inoue Y, Yip L, Hashiguchi N, Zinkernagel A, Nizet V, Insel PA, Junger WG. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science. 2006;314:1792–1795. doi: 10.1126/science.1132559. [DOI] [PubMed] [Google Scholar]
- Cronstein BN. Adenosine, an endogenous anti-inflammatory agent. J Appl Physiol. 1994;76:5–13. doi: 10.1152/jappl.1994.76.1.5. [DOI] [PubMed] [Google Scholar]
- Cronstein BN, Daguma L, Nichols D, Hutchison AJ, Williams M. The adenosine/neutrophil paradox resolved: human neutrophils possess both A1 and A2 receptors that promote chemotaxis and inhibit O2 generation, respectively. J Clin Invest. 1990;85:1150–1157. doi: 10.1172/JCI114547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronstein BN, Kramer SB, Weissmann G, Hirschhorn R. Adenosine: a physiological modulator of superoxide anion generation by human neutrophils. J Exp Med. 1983;158:1160–1177. doi: 10.1084/jem.158.4.1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronstein BN, Levin RI, Philips M, Hirschhorn R, Abramson SB, Weissmann G. Neutrophil adherence to endothelium is enhanced via adenosine A1 receptors and inhibited via adenosine A2 receptors. J Immunol. 1992;148:2201–2206. [PubMed] [Google Scholar]
- Flamand N, Boudreault S, Picard S, Austin M, Surette ME, Plante H, Krump E, Vallee MJ, Gilbert C, Naccache P, Laviolette M, Borgeat P. Adenosine, a potent natural suppressor of arachidonic acid release and leukotriene biosynthesis in human neutrophils. Am J Respir Crit Care Med. 2000;161:S88–94. doi: 10.1164/ajrccm.161.supplement_1.ltta-18. [DOI] [PubMed] [Google Scholar]
- Fortin A, Harbour D, Fernandes M, Borgeat P, Bourgoin S. Differential expression of adenosine receptors in human neutrophils: up-regulation by specific Th1 cytokines and lipopolysaccharide. J Leukoc Biol. 2006;79:574–585. doi: 10.1189/jlb.0505249. [DOI] [PubMed] [Google Scholar]
- Ge ZD, Peart JN, Kreckler LM, Wan TC, Jacobson MA, Gross GJ, Auchampach JA. Cl-IB-MECA [2-chloro-N6-(3-iodobenzyl)adenosine-5′-N-methylcarboxamide] reduces ischemia/reperfusion injury in mice by activating the A3 adenosine receptor. J Pharmacol Exp Ther. 2006;319:1200–1210. doi: 10.1124/jpet.106.111351. [DOI] [PubMed] [Google Scholar]
- Gessi S, Varani K, Merighi S, Cattabriga E, Avitabile A, Gavioli R, Fortini C, Leung E, Mac Lennan S, Borea PA. Expression of A3 adenosine receptors in human lymphocytes: up-regulation in T cell activation. Mol Pharmacol. 2004;65:711–719. doi: 10.1124/mol.65.3.711. [DOI] [PubMed] [Google Scholar]
- Hasko G, Cronstein BN. Adenosine: an endogenous regulator of innate immunity. Trends Immunol. 2004;25:33–39. doi: 10.1016/j.it.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-β1 secretion and the resolution of inflammation. J Clin Invest. 2002;109:41–50. doi: 10.1172/JCI11638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan JE, Thourani VH, Auchampach JA, Robinson JA, Wang NP, Vinten-Johansen J. A3 adenosine receptor activation attenuates neutrophil function and neutrophil-mediated reperfusion injury. Am J Physiol. 1999;277:H1895–1905. doi: 10.1152/ajpheart.1999.277.5.H1895. [DOI] [PubMed] [Google Scholar]
- Jordan JE, Zhao ZQ, Sato H, Taft S, Vinten-Johansen J. Adenosine A2 receptor activation attenuates reperfusion injury by inhibiting neutrophil accumulation, superoxide generation and coronary endothelial adherence. J Pharmacol Exp Ther. 1997;280:301–309. [PubMed] [Google Scholar]
- Khoa ND, Montesinos MC, Reiss AB, Delano D, Awadallah N, Cronstein BN. Inflammatory cytokines regulate function and expression of adenosine A2A receptors in human monocytic THP-1 cells. J Immunol. 2001;167:4026–4032. doi: 10.4049/jimmunol.167.7.4026. [DOI] [PubMed] [Google Scholar]
- Knight D, Zheng X, Rocchini C, Jacobson M, Bai T, Walker B. Adenosine A3 receptor stimulation inhibits migration of human eosinophils. J Leukoc Biol. 1997;62:465–468. doi: 10.1002/jlb.62.4.465. [DOI] [PubMed] [Google Scholar]
- Lagasse E, Weissman IL. Flow cytometric identification of murine neutrophils and monocytes. J Immunol Methods. 1996;197:139–150. doi: 10.1016/0022-1759(96)00138-x. [DOI] [PubMed] [Google Scholar]
- Lappas CM, Rieger JM, Linden J. A2A adenosine receptor induction inhibits IFN- production in murine CD4+ T cells. J Immunol. 2005;174:1073–1080. doi: 10.4049/jimmunol.174.2.1073. [DOI] [PubMed] [Google Scholar]
- Lee HT, Kim M, Joo JD, Gallos G, Chen JF, Emala CW. A3 adenosine receptor activation decreases mortality and renal and hepatic injury in murine septic peritonitis. Am J Physiol Regul Integr Comp Physiol. 2006;291:R959–969. doi: 10.1152/ajpregu.00034.2006. [DOI] [PubMed] [Google Scholar]
- Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol. 2001;2:612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- Lieber JG, Webb S, Suratt BT, Young SK, Johnson GL, Keller GM, Worthen GS. The in vitro production and characterization of neutrophils from embryonic stem cells. Blood. 2004;103:852–859. doi: 10.1182/blood-2003-04-1030. [DOI] [PubMed] [Google Scholar]
- Linden J. Cloned adenosine A3 receptors: pharmacological properties, species differences and receptor functions. Trends Pharmacol Sci. 1994;15:298–306. doi: 10.1016/0165-6147(94)90011-6. [DOI] [PubMed] [Google Scholar]
- Linden J. Molecular approach to adenosine receptors: receptor-mediated mechanisms of tissue protection. Annu Rev Pharmacol Toxicol. 2001;41:775–787. doi: 10.1146/annurev.pharmtox.41.1.775. [DOI] [PubMed] [Google Scholar]
- McColl SR, St-Onge M, Dussault AA, Laflamme C, Bouchard L, Boulanger J, Pouliot M. Immunomodulatory impact of the A2A adenosine receptor on the profile of chemokines produced by neutrophils. Faseb J. 2006;20:187–189. doi: 10.1096/fj.05-4804fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montesinos MC, Yap JS, Desai A, Posadas I, McCrary CT, Cronstein BN. Reversal of the antiinflammatory effects of methotrexate by the nonselective adenosine receptor antagonists theophylline and caffeine: evidence that the antiinflammatory effects of methotrexate are mediated via multiple adenosine receptors in rat adjuvant arthritis. Arthritis Rheum. 2000;43:656–663. doi: 10.1002/1529-0131(200003)43:3<656::AID-ANR23>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Murphree LJ, Sullivan GW, Marshall MA, Linden J. Lipopolysaccharide rapidly modifies adenosine receptor transcripts in murine and human macrophages: role of NF- B in A2A adenosine receptor induction. Biochem J. 2005;391:575–580. doi: 10.1042/BJ20050888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. 2006;6:173–182. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- Nguyen DK, Montesinos MC, Williams AJ, Kelly M, Cronstein BN. Th1 cytokines regulate adenosine receptors and their downstream signaling elements in human microvascular endothelial cells. J Immunol. 2003;171:3991–3998. doi: 10.4049/jimmunol.171.8.3991. [DOI] [PubMed] [Google Scholar]
- Nishida A, Kimura H, Nakano M, Goto T. A sensitive and specific chemiluminescence method for estimating the ability of human granulocytes and monocytes to generate O2. Clin Chim Acta. 1989;179:177–181. doi: 10.1016/0009-8981(89)90164-2. [DOI] [PubMed] [Google Scholar]
- Olah ME, Gallo-Rodriguez C, Jacobson KA, Stiles GL. 125I-4-aminobenzyl-5′-N-methylcarboxamidoadenosine, a high affinity radioligand for the rat A3 adenosine receptor. Mol Pharmacol. 1994;45:978–982. [PMC free article] [PubMed] [Google Scholar]
- Palmer TM, Poucher SM, Jacobson KA, Stiles GL. 125I-4-(2-[7-amino-2-[2-furyl][1,2,4]triazolo[2,3-a][1,3,5] triazin-5-yl-amino]ethyl)phenol, a high affinity antagonist radioligand selective for the A2A adenosine receptor. Mol Pharmacol. 1995;48:970–974. [PMC free article] [PubMed] [Google Scholar]
- Rose FR, Hirschhorn R, Weissmann G, Cronstein BN. Adenosine promotes neutrophil chemotaxis. J Exp Med. 1988;167:1186–1194. doi: 10.1084/jem.167.3.1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan GW, Fang G, Linden J, Scheld WM. A2A adenosine receptor activation improves survival in mouse models of endotoxemia and sepsis. J Infect Dis. 2004;189:1897–1904. doi: 10.1086/386311. [DOI] [PubMed] [Google Scholar]
- Sullivan GW, Rieger JM, Scheld WM, Macdonald TL, Linden J. Cyclic AMP-dependent inhibition of human neutrophil oxidative activity by substituted 2-propynylcyclohexyl adenosine A2A receptor agonists. Br J Pharmacol. 2001;132:1017–1026. doi: 10.1038/sj.bjp.0703893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracey WR, Magee WP, Oleynek JJ, Hill RJ, Smith AH, Flynn DM, Knight DR. Novel N6-substituted adenosine 5′-N-methyluronamides with high selectivity for human adenosine A3 receptors reduce ischemic myocardial injury. Am J Physiol Heart Circ Physiol. 2003;285:H2780–2787. doi: 10.1152/ajpheart.00411.2003. [DOI] [PubMed] [Google Scholar]
- Visser SS, Theron AJ, Ramafi G, Ker JA, Anderson R. Apparent involvement of the A2A subtype adenosine receptor in the anti-inflammatory interactions of CGS 21680, cyclopentyladenosine, and IB-MECA with human neutrophils. Biochem Pharmacol. 2000;60:993–999. doi: 10.1016/s0006-2952(00)00414-7. [DOI] [PubMed] [Google Scholar]
- Walker BA, Jacobson MA, Knight DA, Salvatore CA, Weir T, Zhou D, Bai TR. Adenosine A3 receptor expression and function in eosinophils. Am J Respir Cell Mol Biol. 1997;16:531–537. doi: 10.1165/ajrcmb.16.5.9160835. [DOI] [PubMed] [Google Scholar]
- Wan TC, Ge ZD, Tampo A, Mio Y, Bienengraeber MW, Tracey WR, Gross GJ, Kwok WM, Auchampach JA. The A3 adenosine receptor agonist CP-532,903 [N6-(2,5-dichlorobenzyl)-3′-aminoadenosine-5′-N-methylcarboxamide] protects against myocardial ischemia/reperfusion injury via the sarcolemmal ATP-sensitive potassium channel. J Pharmacol Exp Ther. 2008;324:234–243. doi: 10.1124/jpet.107.127480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young HW, Molina JG, Dimina D, Zhong H, Jacobson M, Chan LN, Chan TS, Lee JJ, Blackburn MR. A3 adenosine receptor signaling contributes to airway inflammation and mucus production in adenosine deaminase-deficient mice. J Immunol. 2004;173:1380–1389. doi: 10.4049/jimmunol.173.2.1380. [DOI] [PubMed] [Google Scholar]
- Zhang N, Yang D, Dong H, Chen Q, Dimitrova DI, Rogers TJ, Sitkovsky M, Oppenheim JJ. Adenosine A2A receptors induce heterologous desensitization of chemokine receptors. Blood. 2006;108:38–44. doi: 10.1182/blood-2005-06-2599. [DOI] [PMC free article] [PubMed] [Google Scholar]