

Abstract
We have previously reported that tissue inhibitor of metalloproteinases-2 (TIMP-2), an endogenous inhibitor of matrix metalloproteinase, modulates angiogenic responses through the MMP inhibition-independent activity. In this study, we investigate the molecular mechanisms of TIMP-2-mediated growth inhibition in response to fibroblast growth factor-2 (FGF-2). Pretreatment with a protein tyrosine phosphatase inhibitor orthovanadate or expression of a dominant negative Shp-1 mutant fails to induce TIMP-2 inactivation of FGF-2 signaling pathways in human microvascular endothelial cells. We also show that TIMP-2 inhibition of FGF-2-induced p42/44MAPK activation and cell proliferation is associated with TIMP-2 binding to integrin α3β1 on endothelial cell surfaces, as demonstrated by use of anti-integrin α3 or β1 blocking antibodies, or disruption of integrin α3 experssion by siRNA.
Collectively, our results indicate that TIMP-2 inhibits FGF-2 signaling pathways through association with integrin α3β1 and Shp-1-dependent inhibition of p42/44MAPK signaling, which in turn, results in suppression of FGF-2-stimulated endothelial cell mitogenesis.
Keywords: TIMP-2, angiogenesis, integrin α3β1, Shp-1, FGF-2
Introduction
Angiogenesis, the development of new blood capillaries from the pre-existing vessels, is important for a variety of processes such as embryonic development, wound healing, and organ repair, and also contributes to various pathological processes including primary tumor growth, and initial progression from a pre-malignant tumor into an invasive cancer, as well as a variety of chronic inflammatory diseases (Carmeliet, 2005; Ferrara and Kerbel, 2005; Folkman, 2006). A growing list of growth factors and cytokines are commonly associated with a series of cellular events leading to endothelial cell proliferation during these angiogenic responses. The angiogenic response to these factors is efficiently controlled directly by a variety of endogenous angiogenesis inhibitors such as angiostatin, endostatin, tumstatin or thrombospondin. Alternatively the anti-angiogenic activity of some agents is indirect in that they block the action of mitogen-associated receptor on endothelial cells or production of pro-angiogenic proteins. The balance of matrix metalloproteinases (MMPs) and endogenous tissue inhibitors of metalloproteinases (TIMPs) activities plays a pivotal role in these angiogenic processes by altering biological functions of extracellular matrix (ECM) macromolecules through specific proteolysis and/or release of membrane- or matrix-anchored pro-angiogenic growth factors (Sternlicht and Werb, 2001; Stetler-Stevenson, 1999).
Many investigations demonstrate that TIMPs can inhibit cellular proliferation, invasion, and metastasis through inhibition of MMP activity (Baker et al., 2002; Sternlicht and Werb, 2001). In addition to their MMP-inhibitory activity, several laboratories propose that TIMPs also function to directly modulate cell fate through MMP-independent mechanisms (Corcoran and Stetler-Stevenson, 1995; Fernandez et al., 2003; Guedez et al., 1998; Hoegy et al., 2001; Oh et al., 2004; Perez-Martinez and Jaworski, 2005; Qi et al., 2003; Seo et al., 2003; Seo et al., 2006; Wingfield et al., 1999).
In previous studies, we have reported that TIMP-2 suppresses the effects of mitogenic growth factors such as fibroblast growth factor-2 (FGF-2), vascular endothelial growth factor-A (VEGF-A), platelet derived growth factor (PDGF), or epidermal growth factor (EGF)-induced cell proliferation in vitro as well as angiogenesis in vivo (Hoegy et al., 2001; Seo et al., 2003; Wingfield et al., 1999). TIMP-2 ligation of integrin α3β1 and subsequent induction of SH2-containing protein tyrosine phosphatase-1 (PTP Shp-1) activity mediates TIMP-2 anti-angiogenic activity, and this effect is entirely independent of anti-MMP activity, as demonstrated by the use of Ala+TIMP-2, a form of TIMP-2 that is essentially devoid of MMP-inhibitory activity (Oh et al., 2004; Seo et al., 2003; Seo et al., 2006). However, details of the cellular signaling mechanisms involving TIMP-2 regulation of cell proliferation are incompletely understood.
Intracellular signaling pathways that involve receptor tyrosine kinases (RTKs) play an important role in the control of most cellular processes such as cell proliferation, differentiation, survival, migration, and cell cycle. Binding of a ligand to its cognate RTKs induces dimerization and autophosphorylation of multiple tyrosine residues in the RTK cytoplasmic domain (Schlessinger, 2000; Schlessinger, 2004; Schlessinger and Lemmon, 2003). In the case of the fibroblast growth factor receptor (FGFR) family of RTKs, FGFR activation requires the binding of heparin as well as FGF ligand, which results in the recruitment of FGFR substrate-2 (FRS-2), growth factor receptor-bound protein 2 (Grb2), son of sevenless nucleotide exchange factor (Sos1), Src homology 2 phosphatase-2 (Shp-2). These initial events promote the activation of Ras and mitogen-activated protein kinase (p42/44MAPK) signaling pathways, leading to a broad spectrum of downstream cellular signaling events and responses (Schlessinger and Lemmon, 2003). RTK activation is also controlled by protein tyrosine phosphatases (PTPs). Although some PTPs, including Shp-2, positively regulate the signaling of RTKs, it has widely been appreciated that PTPs can inhibit RTK activation and function as tumor suppressors (Ostman et al., 2006). Understanding the regulatory mechanisms and specific targets of TIMP-2 that inhibits FGF-2-stimulated signaling pathways represents important new insights and potentially novel therapeutic strategies for the treatment of pathophysiologic states such as cancer, cardiovascular diseases, and inflammatory disorders.
Here we investigate the biochemical and molecular mechanisms of TIMP-2 in the regulation of FGF-2-induced mitogenic responses, specifically focusing on the early events in the FGF-2/FGFR-1 signaling cascade in human microvascular endothelial cells and lung carcinoma cells. Using the null-MMP inhibitor form of TIMP-2, referred to as Ala+TIMP-2, dominant negative (dn) Shp-1 mutant, and the integrin α3 siRNA-technology, we demonstrate that the suppressive effect of TIMP-2 on FGF-2-induced signaling pathway and cell proliferation requires induction of Shp-1 activity through TIMP-2 binding to integrin α3β1, and that these effects are independent of MMP-inhibitory activity.
Materials and methods
Reagents
Recombinant human fibroblast growth factor-2 (FGF-2) was obtained from BD biosciences (Bedford, MA). The following antibodies were purchased from commercial sources: monoclonal anti-phosphotyrosine (PY20), anti-Sos1, and anti-integrin β1 (18) (BD transduction laboratories, Lexington, KY); monoclonal anti-FRS-2 (Upstate Biotechnology, Lake Placid, NY); polyclonal anti-phosphor-Raf-1 (S259) and anti-phospho-p44/42MAPK (T202/Y204) (Cell Signaling, Beverly, MA); monoclonal anti-integrin α3 (P1B5), anti-integrin α3 (ASC-1), anti-integrin α5 (P1D6), anti-integrin β1 (P5D2), and anti-integrin β1 (LM534) (Chemicon International Inc., Temecula, CA); monoclonal anti-Raf-1, polyclonal anti-p44/42MAPK, anti-Shp-2, anti-integrin α3 (I-19), and mouse, rabbit and goat IgG-horseradish peroxidase conjugates (Santa Cruz Biotechnology, Santa Cruz, CA). TIMP-2 and Ala+TIMP-2 were prepared and characterized as described previously (Wingfield et al., 1999).
Cell culture conditions
Primary cultures of human microvascular endothelial cells (hMVECs), prepared from lung, were purchased from Cambrex (Walkersville, MD) and used between passages 4 or 5. Cells were cultured in EGM®-2 MV BulletKit media, according to the manufacturer’s instructions. Human lung carcinoma cells (A549, CCL-185) from American Tissue Culture Collection (Manassas, VA) were grown in 10% fetal bovine serum (FBS)-Dulbecco’s Modified Eagle’s Media (DMEM, Invitrogen, Carlsbad, CA).
Immunoprecipitation and Western blot analysis
Subconfluent cells in gelatin-coated 100 mm dishes (BD biosciences) were serum-starved for 24 h in endothelial cell basal medium-2 (EBM-2) and replaced with fresh media, followed by treatments for different time periods, as indicated, at 37 °C. Endothelial cells were rinsed twice with ice-cold phosphate-buffered saline (PBS) and lysed by incubation in 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 10% glycerol, 1% Nonidet P-40, 1 mM EDTA, 100 μg/ml 4-(2-aminoethyl)benzenesulfonyl fluoride, 10 μg/ml aprotinin, 1 μg/ml pepstatin A, 0.5 μg/ml leupeptin, 80 mM β-glycerophosphate, 25 mM NaF and 1 mM sodium orthovanadate for 30 min at 4 °C. Cell lysates were clarified at 13,000 × g for 20 min at 4 °C, and the supernatants were subjected to immunoprecipitation and Western blot as described previously (Seo et al., 2006; Wingfield et al., 1999). All immunoprecipitations and Western blots were performed at least duplicate or triplicate experiments and representative gels are shown.
Dominant negative Shp-1 preparation
The role of Shp-1 on TIMP-2-mediated inhibition of FGF-2-stimulated signaling pathways was investigated using dominant negative mutant Shp-1 as previously described (Seo et al., 2003; Seo et al., 2006). Expression and loss of activity of dominant negative Shp-1 in endothelial cells was verified by Western blot analysis using anti-Shp-1 antibody (BD biosciences) and PTP assay (Upstate Biotechnology) of whole cell lysates, respectively.
Cell growth assays
hMVECs, plated on gelatin-coated 96-well plates (5×103 cells/well, Costar, Corning, NY), were grown in basal medium EBM-2 without serum or growth factors for 24 h to synchronize cells in G1/G0 phase of the cell cycle prior to treatment with TIMP-2 (50 nM) or orthovanadate (1 μM) alone for 24 h. Where indicated, cells were pretreated with Ala+TIMP-2 (50 nM) for 15 min in the presence or absence of anti-integrin antibodies (10-25 μg/ml). Following subsequent FGF-2 (50 ng/ml) stimulation and culture for 24 h, the cell numbers were quantified using CellTiter 96® AQueous One Solution reagent (Promega, Madison, WI)(Seo et al., 2003). Subconfluent A549 lung carcinoma cells, plated on 96-well plates (5×103 cells/well), were serum-starved for 24 h before treatment with Ala+TIMP-2. After subsequent FGF-2 stimulation for 24 h, the cell numbers were determined as described previously (Seo et al., 2006). The results from triplicate determinations (mean ± standard deviation) are presented as the percentage of maximal TIMP-2 inhibition of FGF-2-stimulated cell growth and maximal FGF-2-stimulated cell growth, or the fold-increase of non-treated growth.
Integrin α3 siRNA preparation and transfection
For design of siRNA inserts, a cDNA sequence of integrin α3 AGCAACACAGACTACCTGGAG was selected according to the InvivoGen siRNAWizard program based on a BLAST search. As a control we used the scrambled oligonucleotide sequence AGCATATGTGCGTACCTAGCT, available prepackaged in the psiRNA-hH1zeo vector (InvivoGen, San Diego, CA). The siRNA-targeting integrin α3 gene and scrambled sequences cloned into psiRNA-hH1zeo vector were transfected into A549 cells. After 48 h, the cells were selected with zeocin in 10% FBS-DMEM for 1-2 weeks until positive colonies formed (Seo et al., 2006). For transfection of hMVECs, synthetic integrin α3 and control siRNA duplexes were purchased from Santa Cruz Biotechnology. Subconfluent hMVECs in gelatin-coated 100 mm dishes were transfected with siRNA (integrin α3 siRNA (sc-35684): 50 nM; control siRNA (sc-37007): 50 nM) using Lipofectamine RNAiMAX (Invitrogen) and OPTI-MEM I reduced serum medium (Invitrogen), according to the manufacturer’s instructions. The concentrations of siRNAs were chosen based on dose-response studies.
Results
Down-regulation of FGFR signaling pathway by TIMP-2 is associated with enhanced protein tyrosine phosphatase activity
In previous studies, we have demonstrated that TIMP-2 suppresses FGF-2- or VEGF-A-stimulated human microvascular endothelial cell (hMVEC) proliferation in vitro and angiogenesis in vivo (Fernandez et al., 2003; Oh et al., 2004; Seo et al., 2003; Seo et al., 2006), and these TIMP-2 anti-angiogenic effects are mainly mediated through integrin α3β1 and protein tyrosine phosphatase (PTP) activity-dependent induction of cell cycle inhibitor p27Kip1 levels (Seo et al., 2006). To further examine the mechanism by which TIMP-2 inhibits FGF-2-stimulated endothelial cell proliferation in vitro, we searched for the changes in down-stream events of FGFR-1 in the presence or absence of 50 nM TIMP-2. FGF-2 binding to FGFR enhances intrinsic tyrosine kinase activity and generates docking sites for SH2 or phosphotyrosine binding (PTB) domain-containing proteins, such as FRS-2, phospholipase C-γ (PLCγ) and Shc (Kouhara et al., 1997; Rhee, 2001; Schlessinger, 2000). Phosphorylation of FGF-2-induced FRS-2, a lipid-anchored docking protein, induces recruitment of signaling molecules such as Grb2, Sos1, and Shp-2, which play pivotal roles in linking FGFR activation with the Ras/Raf-1/p42/44MAPK signaling pathway. We first examined the ability of TIMP-2 to suppress FGF-2-induced FRS-2 phosphorylation in hMVECs. The amount of phosphorylated FRS-2 was measured by Western blot analysis of FRS-2 immunoprecipitates prepared from hMVECs. As shown in Fig. 1A, stimulation of quiescent hMVECs with FGF-2 led to tyrosine phosphorylation of FRS-2, however, pre-treatment with TIMP-2 prior to FGF-2 stimulation significantly reduced FRS-2 phosphorylation to the level of un-treated cells. This inhibitory effect of TIMP-2 on FRS-2 phosphorylation was completely abrogated by addition of PTP inhibitor orthovanadate. We next determined the levels of Sos1 and Shp-2 associated with FRS-2 to confirm the effects of TIMP-2 on FRS-2 phosphorylation. TIMP-2 pre-treatment dramatically reduced FGF-2-induced association of Sos1 and Shp-2 with FRS-2 (Fig. 1A). The decrease in amount of Sos1 and Shp-2 associated with FRS-2 correlated with the decreased FRS-2 phosphorylation following TIMP-2 pre-treatment. However, pretreatment with orthovanadate (1 μM), followed by TIMP-2 and then FGF-2, completely ablated the suppressive effect of TIMP-2 on recruitment of Sos1 and Shp-2 to FRS-2. We also examined the effects of TIMP-2 and orthovanadate on the FRS-2 activation in the absence of FGF-2. As shown in Fig. 1B, alone orthovanate treatment did not alter the levels of FRS-2 phosphorylation and association of Sos1 and Shp-2, compared with those in basal hMVECs. Although TIMP-2 treatment resulted in a slight increase in the association of Sos1 and Shp-2 with FRS-2, orthovanate or TIMP-2 treatment of hMVECs without subsequent FGF-2 stimulation did not alter the proliferative response of hMVECs under basal conditions (Fig. 2A) (Oh et al., 2004; Seo et al., 2003). The definitive biological roles and effects of TIMP-2-mediated slight increase on FRS-2/p42/44MAPK activity in the absence of FGF-2 remain to be addressed, however, these signaling pathways may contribute to the inhibition of cell migration, promotion of cell spreading and adhesion, or cell cycle arrest at G1 phase as previously reported (Chang et al., 2006; Oh et al., 2004; Seo et al., 2006).
Fig. 1. TIMP-2 suppression of FGF-2-induced signaling pathway is dependent on protein tyrosine phosphatase activity.
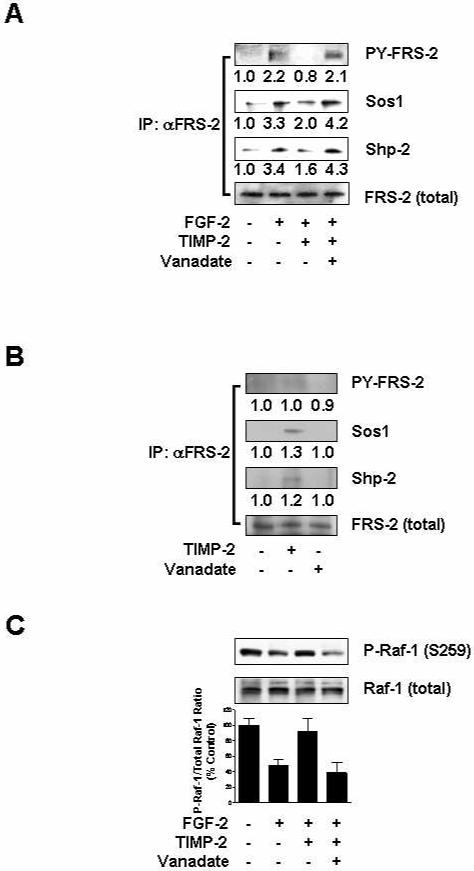
Quiescent hMVECs were pre-treated with or without orthovanadate (1 μM) for 15 min, followed by TIMP-2 (50 nM) for 15 min prior to FGF-2 (50 ng/ml) stimulation for 5 min. Quiescent cells were also treated with TIMP-2 or orthovanadate alone for 15 min. (A) TIMP-2 treatment reduces FRS-2 phosphorylation and recruitment of Sos1 and Shp-2 to FRS-2. (B) TIMP-2 slightly induces the association of Sos1 and Shp-2 with FRS-2. Cell lysates (1 mg) were immunoprecipitated with anti-FRS-2 monoclonal antibodies. Immunoprecipitates were resolved by SDS-PAGE and Western-blotted with anti-phosphotyrosine, anti-Sos1, anti-Shp-2 or anti-FRS-2 antibodies. Integrated density values for the blots were normalized to total FRS-2 levels. The results are representative of two independent experiments. (C) Inhibitory effect of TIMP-2 on FGF-2-stimulated Raf-1 activity. Lysates were Western-blotted with anti-phopho-Raf-1 (S259) or anti-Raf-1 antibodies. Values are normalized to total Raf-1 levels and represent the mean ± S.D. of triplicate determinations.
Fig. 2. Shp-1 mediates TIMP-2 inhibition of p42/44MAPK activity.
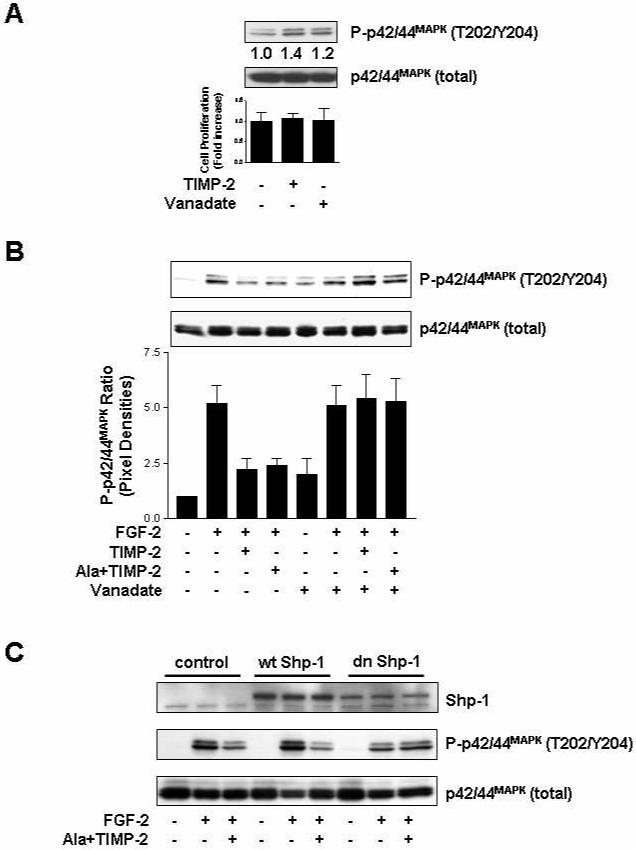
(A) TIMP-2 slightly induces p42/44MAPK activation, but does not alter the proliferative responses of hMVECs in the absence of FGF-2. Quiescent cells were treated with TIMP-2 or orthovanadate alone for 15 min (upper panel) or 24 h (lower panel). Cell proliferation results from triplicate determinations (mean ± S.D.) are presented as the fold-increase of non-treated growth. (B) Orthovanadate reverses TIMP-2 inhibitory effect on FGF-2-stimulated p42/44MAPK activation. Quiescent hMVECs were pre-treated with or without orthovanadate (1 μM) for 15 min, followed by TIMP-2 or Ala+TIMP-2 (50 nM) for 15 min prior to FGF-2 (50 ng/ml) stimulation for 15 min, and were lysed and Western-blotted with anti-phospho-p42/44MAPK (T202/Y204) or anti-p42/44MAPK antibodies. Values are normalized to total p42/44MAPK levels and represent the mean ± S.D. of triplicate determinations. (C) Dominant negative (dn) Shp-1-transduced hMVECs are resistant to TIMP-2 inhibition of FGF-2-stimulated p42/44MAPK activation. hMVECs were transduced with vector control, wild type (wt) Shp-1 or dn Shp-1 mutant as previously reported (Seo et al., 2003; Seo et al., 2006), and were treated and Western-blotted as described in (A).
To further investigate molecular mechanism of TIMP-2 inhibition of FGF-2-induced FGFR activation, we next examined the changes in activation of Raf-1 kinase, a key mediator of FRS-2 to p42/44MAPK signaling pathway. Raf-1 activation involves phosphorylation on serine 338 and tyrosine 341 residues. However, in resting cells Raf-1 is phosphorylated on serine 43, 259, and 621. In particular, phosphorylation of serine 259 residue in Raf-1 is the principal site mediating cAMP-regulated protein kinase (PKA)-induced inhibition of cellular responses (Dhillon et al., 2002). Previously we have reported the growth modulating effects of TIMP-2 on Hs68 fibroblasts, HT1080 fibrosarcoma or A549 lung carcinoma cells through a heterotrimeric G protein-dependent mechanism involves adenylate cyclase and PKA activity (Corcoran and Stetler-Stevenson, 1995; Hoegy et al., 2001). These events are also associated with a decrease in Grb-2 association with the epidermal growth factor receptor. To examine whether TIMP-2-mediated down-regulation of FGFR signaling is associated with the phosphorylation of Raf-1 on serine 259 residue, we first tested the ability of TIMP-2 to increase intracellular cyclic AMP (cAMP) in quiescent hMVECs. TIMP-2 treatment induced a transient increase in cAMP levels that peaked (an approximately 3-4-fold compared to un-treated cells) at 1 min and returned to basal levels by 5 min (data not shown). As shown in Fig. 1B, the levels of phosphorylated Raf-1 on serine 259 site in FGF-2-stimulated hMVECs were significantly reduced, compared with those in un-treated cells. However, TIMP-2 pre-treatment completely reversed the phosphorylation status of Raf-1 at serine 259 to levels observed in the basal state. This effect was completely blocked by addition of orthovanadate. These findings suggest that TIMP-2 inhibitory effect on FGF-2 signaling transduction is exerted at the receptor levels through PTP activity and is PKA-dependent, consistent with previous findings on TIMP-2-induced association of Shp-1 with FGFR-1 (Seo et al., 2003) and inhibition of FRS-2 activation (Fig. 1A). However, a definitive molecular mechanism demonstrating the association of cAMP or PKA with orthovanadate-sensitive PTPases remains to be demonstrated.
We have previously demonstrated that the potent anti-proliferative activity of Ala+TIMP-2, a form of TIMP-2 that lacks MMP-inhibitory activity, and lack of inhibition by other MMP inhibitors in hMVEC cultures are independent of MMP-inhibitory activity (Seo et al., 2003; Seo et al., 2006). To investigate the activation of p42/44MAPK, downstream kinase of Ras/Raf pathway in FGF-2-stimulated hMVECs, we first examined the effect of TIMP-2 and orthovanadate on the phosphorylation/activation of p42/44MAPK. TIMP-2 or orthovanadate treatment of non-stimulated hMVECs resulted in a slight increase in phosphorylation of p42/44MAPK when compared with untreated controls, but did not alter the proliferative responses of hMVECs under basal conditions (Fig. 2A), as we have previously reported (Oh et al., 2004; Seo et al., 2003).
Utilizing Ala+TIMP-2, we next examined the changes of p42/44MAPK phosphorylation in FGF-2-stimulated hMVECs. The experiment presented in Fig. 2B shows that TIMP-2 or Ala+TIMP-2 significantly blocks p42/44MAPK phosphorylation (∼70% decrease), compared to activation induced by FGF-2 in these cells. Pre-treatment with orthovanadate completely reversed the TIMP-2-mediated reduction of p42/44MAPK phosphorylation to levels observed in FGF-2-stimulated cells. Shp-1, a Src homology 2 (SH2)-containing PTP, has been shown to be recruited to the multiple receptor complexes as a negative regulator of signaling pathways in many hematopoietic and non-hematopoietic cells (Ostman et al., 2006). Based on our previous report that TIMP-2 anti-angiogenic activity is Shp-1-dependent (Seo et al., 2003; Seo et al., 2006), we analyzed the phosphorylation status of p42/44MAPK, using the wild type (wt) and dominant negative (dn) Shp-1 mutant-transduced hMVECs, in order to directly assess the requirement of PTP Shp-1 activity in TIMP-2 inhibition of p42/44MAPK phosphorylation. Comparison of the Ala+TIMP-2 effects on FGF-2-stimulated p42/44MAPK phosphorylation demonstrated that in vector control and wt Shp-1 expressing hMVECs p42/44MAPK was dramatically inhibited by Ala+TIMP-2 (Fig. 2C). However, dn Shp-1 cells were resistant to Ala+TIMP-2 suppression of FGF-2-induced p42/44MAPK phosphorylation. These findings demonstrate that PTP Shp-1 activity is specifically required for the TIMP-2-mediated inhibition of FGF-2-induced activation of p42/44MAPK, and are consistent with our previous observations that the suppressive effects of TIMP-2 on the early events in FGFR signaling pathway and cell proliferation are abrogated by the PTP inhibitor orthovanadate (Fig. 1) (Seo et al., 2003). In addition, FGF-2-induced p42/44MAPK activation was sustainedly blocked by pretreatment with Ala+TIMP-2 (Fig. 3).
Fig. 3. TIMP-2 inhibits FGF-2-stimulated p42/44MAPK phosphorylation.
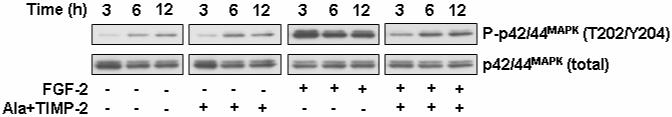
Quiescent hMVECs were pre-treated with Ala+TIMP-2 (50 nM) for 15 min, and further incubated for the indicated time points with or without FGF-2 (50 ng/ml) stimulation. Cell lysates were separated by SDS-PAGE and Western-blotted for phospho-p42/44MAPK and p42/44MAPK.
TIMP-2 interaction with integrin α3β1 is required for TIMP-2 inhibition of FGF-2-induced cell proliferation and p42/44MAPK phosphorylation
Coordinated interactions of integrin and growth factor signaling pathways have been demonstrated to regulate angiogenesis (ffrench-Constant and Colognato, 2004; Stupack et al., 2004). We have previously reported that interaction of TIMP-2 with integrin α3β1 mediates TIMP-2 anti-angiogenic activity (Seo et al., 2003). To investigate whether integrin α3β1 is required for TIMP-2 inhibition of FGF-2-induced mitogenic responses, we examined the effects of TIMP-2 on cell proliferation and p42/44MAPK activation by pretreatment with anti-integrin antibodies or integrin α3 siRNA. Using function-blocking α3- and β1-integrin antibodies, we first confirmed that TIMP-2 inhibition of FGF-2-induced endothelial cell proliferation was mediated by integrin α3β1 (Fig. 4). A function-blocking anti-integrin α5 (P1D6) or non-blocking anti-integrin β1 (LM534), however, did not affect the suppressive effect of TIMP-2 on hMVEC proliferation (Fig. 4). To specifically silence the integrin α3 gene, hMVECs were transfected with siRNA-targeting integrin α3 mRNA. Western blot analysis demonstrated that integrin α3 siRNA successfully reduced expression of α3 subunit (>90% decrease) as compared with the controls (Fig. 5A). As shown in Fig. 5, TIMP-2 pre-treatment dramatically reduced FGF-2-induced association of Sos1/Shp-2 with FRS-2 and p42/44MAPK phosphorylation in control siRNA-transfected hMVECs, identical to our previous observations (Fig. 1 and 2). However, integrin α3 siRNA-transfected cells were unresponsive to the suppressive effect of TIMP-2 on FRS-2/p42/44MAPK activation and cell proliferation in response to FGF-2. Similar results were observed with integrin α3 siRNA transfection of A549 tumor cells. This reduction in integrin α3 expression was sufficient to completely ablate the ability of TIMP-2 to reduce FGF-2-stimulated association of Shp-2 with FRS-2, p42/44MAPK phosphorylation and proliferation of A549 cells (Fig. 6).
Fig. 4. Integrin α3β1 is required for TIMP-2 inhibition of FGF-2-stimulated cell proliferation.
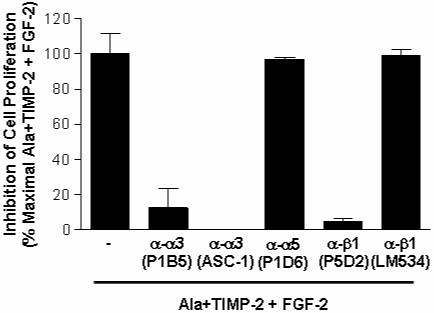
Quiescent hMVECs were pre-treated with or without function-blocking anti-integrin antibodies (except for β1 (LM534), 10-25 μg/ml) for 20 min, followed by Ala+TIMP-2 (50 nM) for 15 min prior to FGF-2 (50 ng/ml) stimulation for 24 h. Results shown are the percentage of maximal inhibition of cell proliferation obtained by Ala+TIMP-2 and FGF-2 treatment. Values represent the mean ± S.D. of triplicate determinations.
Fig. 5. Down-regulation of integrin α3 expression abrogates the suppressive effects of TIMP-2 on FGF-2-induced p42/44MAPK and cell proliferation.
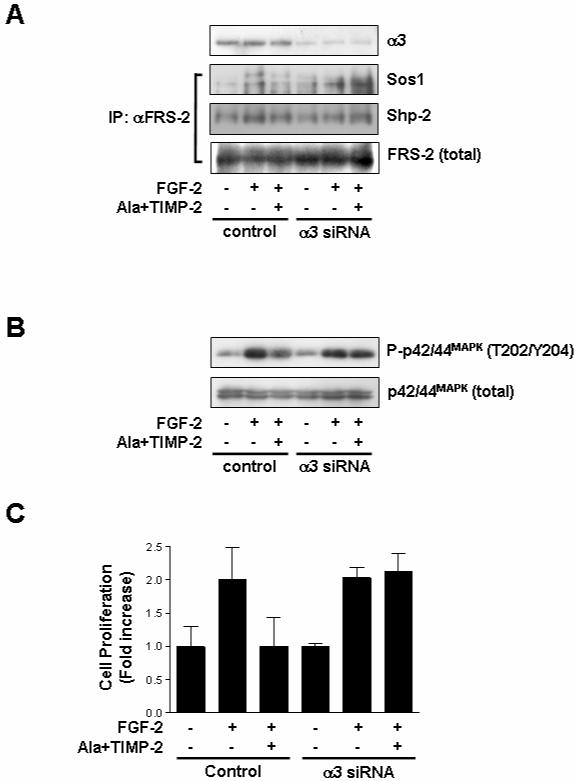
Subconfluent hMVECs in gelatin-coated 100 mm dishes were transfected with integrin α3 siRNA or control siRNA as described in materials and methods. Quiescent cells were pre-treated with Ala+TIMP-2 (50 nM) for 15 min prior to FGF-2 (50 ng/ml) stimulation for 5 min (A), 15 min (B) or 24 h (C). (A) Cell lysates were Western-blotted with anti-integrin α3 (I-19) antibody. Immunoprecipitates (1mg) with anti-FRS-2 monoclonal antibodies were resolved by SDS-PAGE and Western-blotted with anti-Sos1, anti-Shp-2 or anti-FRS-2 antibodies. The results are representative of two independent experiments. (B) Cell lysates were Western-blotted with anti-phospho-p42/44MAPK or anti-p42/44MAPK antibodies. The results are representative of three independent experiments. (C) Cell proliferation results from triplicate determinations (mean ± S.D.) are presented as the fold-increase of non-treated growth.
Fig. 6. Effect of TIMP-2 on FGF-2/FGFR signaling pathways and cell proliferation in integrin α3 siRNA-transfected A549 cancer cells.
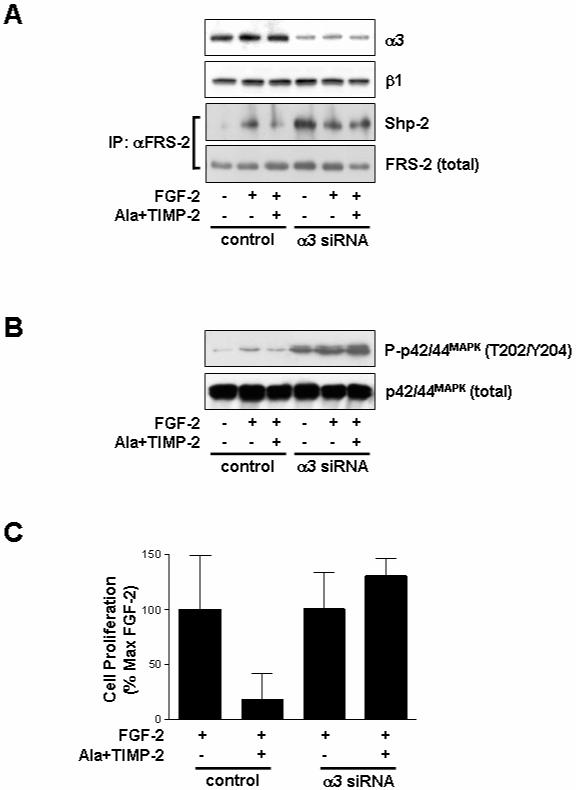
A549 lung carcinoma cells were transfected with integrin α3 siRNA or control (scrambled) siRNA as described in materials and methods. Quiescent cells were pre-treated with Ala+TIMP-2 (50 nM) for 15 min prior to FGF-2 (50 ng/ml) stimulation for 5 min (A), 15 min (B) or 24 h (C). (A) Cell lysates were Western-blotted with anti-integrin α3 or anti-integrin β1 antibodies. Anti-FRS-2 immunoprecipitates (1mg) were resolved by SDS-PAGE and Western-blotted with anti-Shp-2 or anti-FRS-2 antibodies. The results are representative of two independent experiments. (B) Cell lysates were Western-blotted with anti-phospho-p42/44MAPK or anti-p42/44MAPK antibodies. The results are representative of three independent experiments. (C) Integrin α3 siRNA abrogates TIMP-2 inhibition of cell proliferation in response to FGF-2. Values represent the mean ± S.D. of triplicate determinations.
Collectively, these findings from the integrin function-blocking antibodies-treated and integrin α3 siRNA-transfected cells clearly demonstrate that integrin α3β1 is specifically required for TIMP-2-mediated inhibition of FRS-2/Raf-1/p42/44MAPK signaling pathway and cell growth.
Discussion
The degradation of the extracellular matrix (ECM) is likely to release and generate active molecules in the matrix components which influence diverse and pathologic processes, including embryonic development, tissue morphogenesis, wound repair, inflammation diseases, tumor invasion and angiogenesis (Sternlicht and Werb, 2001). Matrix metalloproteinases (MMPs) can degrade all components of the ECM, cell surface molecules, and other pericellular non-matrix proteins, resulting in the regulation of cell behavior, tissue development and remodeling. Tissue inhibitors of metalloproteinases (TIMPs) are physiological inhibitors, which can control MMP functions. It has long been presumed that the regulatory effect of TIMPs on tumorigenic and angiogenic phenotypes in vitro and in vivo models is mediated by MMP-inhibitory activity of TIMPs, which led to the development of synthetic MMP inhibitors for therapeutic application. However, synthetic MMP inhibitors have proved disappointing in clinical trials of cancer patients (Coussens et al., 2002). The lack of a clinically significant effect of these synthetic MMP inhibitors suggests that further understanding of the precise roles and regulation of MMPs is necessary prior to the development of therapeutic agents for diseases associated with dysregulation of ECM degradation. In addition to MMP-inhibitory activity of TIMPs, many investigations have demonstrated that TIMPs have pluripotential effects on cell fates including growth, differentiation, apoptosis, and migration through MMP-independent mechanisms (Baker et al., 2002; Nyberg et al., 2005; Stetler-Stevenson and Seo, 2005). These MMP-independent TIMP functions in modulating cellular responses appears to be mediated via a mechanism involving direct binding to cell surface receptors (i.e. CD63 for TIMP-1; integrin α3β1 for TIMP-2; VEGFR-2 for TIMP-3) (Jung et al., 2006; Qi et al., 2003; Seo et al., 2003).
Cell fate is the result of integration of cellular signaling pathways from multiple signals including soluble factors, cell-to-cell, and/or cell-to-matrix interactions in the tissue microenvironment. Receptor tyrosine kinases (RTKs) on cell surfaces activate the diverse array of signaling pathways in response to multiple extracellular stimuli such as their cognate ligands, cell adhesion via integrin receptors, or agonists of G protein-coupled receptors, resulting in the control of the biochemical processes that regulate cell fate (ffrench-Constant and Colognato, 2004; Hanahan and Weinberg, 2000; Stupack et al., 2004).
We have previously reported that TIMP-2 has suppressive effects on mitogenic growth factors such as FGF-2, VEGF-A, PDGF, or EGF-induced cell proliferation in vitro and angiogenesis in vivo (Hoegy et al., 2001; Seo et al., 2003; Wingfield et al., 1999). In the present study, we report the intracellular signaling mechanisms by which TIMP-2 controls cell growth in response to mitogenic stimuli. Our focus in this report is on the down-stream signaling events after activation of PTP Shp-1 through the binding of TIMP-2 to integrin α3β1. TIMP-2 effectively inhibits the phosphorylation and association of FRS-2 with Sos1/Shp-2, and the activation of Raf-1/p42/44MAPK, the down-stream targets in FGFR-1 signaling pathway in response to FGF-2, however, inclusion of orthovanadate, expression of dn Shp-1, or disruption of integrin α3 completely abrogates the inhibitory activity of TIMP-2 on p42/44MAPK activation and cell proliferation. These findings confirm a model that TIMP-2 ligation to integrin α3β1 suppresses mitogenic growth factors-mediated cell growth through orthovanate-sensitive Shp-1-dependent inactivation of mitogenic signaling pathway, and suggest that regulation of PTP Shp-1 function is an important target for the treatments of pathophysiologic states as well as control of normal cellular homeostasis.
Acknowledgment
This work was supported by intramural research funds from the NCI, Center for Cancer Research Project Z01SC 009179, the Korea Research Foundation Grant from the Korean Government (MOEHRD) (KRF-2006-311-C00547), the Vascular System Research Center Grant from the Korea Science and Engineering Foundation, and 2006 Research Grant from Kangwon National University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baker AH, Edwards DR, Murphy G. Metalloproteinase inhibitors: biological actions and therapeutic opportunities. J. Cell Sci. 2002;115:3719–3727. doi: 10.1242/jcs.00063. [DOI] [PubMed] [Google Scholar]
- Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–936. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- Chang H, Lee J, Poo H, Noda M, Diaz T, Wei B, Stetler-Stevenson WG, Oh J. TIMP-2 promotes cell spreading and adhesion via upregulation of Rap1 signaling. Biochem. Biophys. Res. Commun. 2006;345:1201–1206. doi: 10.1016/j.bbrc.2006.05.044. [DOI] [PubMed] [Google Scholar]
- Corcoran ML, Stetler-Stevenson WG. Tissue inhibitor of metalloproteinase-2 stimulates fibroblast proliferation via a cAMP-dependent mechanism. J. Biol. Chem. 1995;270:13453–13459. doi: 10.1074/jbc.270.22.13453. [DOI] [PubMed] [Google Scholar]
- Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer-trials and tribulations. Science. 2002;295:2387–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- Dhillon AS, Pollock C, Steen H, Shaw PE, Mischak H, Kolch W. Cyclic AMP-dependent kinase regulates Raf-1 kinase mainly by phosphorylation of serine 259. Mol. Cell. Biol. 2002;22:3237–3246. doi: 10.1128/MCB.22.10.3237-3246.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez CA, Butterfield C, Jackson G, Moses MA. Structural and functional uncoupling of the enzymatic and angiogenic inhibitory activities of tissue inhibitor of metalloproteinase-2 (TIMP-2): LOOP 6 is a novel angiogenesis inhibitor. J. Biol. Chem. 2003;278:40989–40995. doi: 10.1074/jbc.M306176200. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438:967–974. doi: 10.1038/nature04483. [DOI] [PubMed] [Google Scholar]
- ffrench-Constant C, Colognato H. Integrins: versatile integrators of extracellular signals. Trends Cell Biol. 2004;14:678–686. doi: 10.1016/j.tcb.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Folkman J. Angiogenesis. Annu. Rev. Med. 2006;57:1–18. doi: 10.1146/annurev.med.57.121304.131306. [DOI] [PubMed] [Google Scholar]
- Guedez L, Stetler-Stevenson WG, Wolff L, Wang J, Fukushima P, Mansoor A, Stetler-Stevenson M. In vitro suppression of programmed cell death of B cells by tissue inhibitor of metalloproteinases-1. J. Clin. Invest. 1998;102:2002–2010. doi: 10.1172/JCI2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hoegy SE, Oh H-R, Corcoran ML, Stetler-Stevenson WG. Tissue inhibitor of metalloproteinases-2 (TIMP-2) suppresses TKR-growth factor signaling independent of metalloproteinase inhibition. J. Biol. Chem. 2001;276:3203–3214. doi: 10.1074/jbc.M008157200. [DOI] [PubMed] [Google Scholar]
- Jung KK, Liu XW, Chirco R, Fridman R, Kim HR. Identification of CD63 as a tissue inhibitor of metalloproteinase-1 interacting cell surface protein. EMBO J. 2006;25:3934–3942. doi: 10.1038/sj.emboj.7601281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouhara H, Hadari YR, Spivak-Kroizman T, Schilling J, Bar-Sagi D, Lax I, Schlessinger J. A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell. 1997;89:693–702. doi: 10.1016/s0092-8674(00)80252-4. [DOI] [PubMed] [Google Scholar]
- Nyberg P, Xie L, Kalluri R. Endogenous inhibitors of angiogenesis. Cancer Res. 2005;65:3967–3979. doi: 10.1158/0008-5472.CAN-04-2427. [DOI] [PubMed] [Google Scholar]
- Oh J, Seo D-W, Diaz T, Wei B, Ward Y, Ray JM, Morioka Y, Shi S, Kitayama H, Takahashi C, Noda M, Stetler-Stevenson WG. Tissue inhibitors of metalloproteinase 2 inhibits endothelial cell migration through increased expression of RECK. Cancer Res. 2004;64:9062–9069. doi: 10.1158/0008-5472.CAN-04-1981. [DOI] [PubMed] [Google Scholar]
- Ostman A, Hellberg C, Bohmer FD. Protein-tyrosine phosphatases and cancer. Nat. Rev. Cancer. 2006;6:307–320. doi: 10.1038/nrc1837. [DOI] [PubMed] [Google Scholar]
- Perez-Martinez L, Jaworski DM. Tissue inhibitor of metalloproteinase-2 promotes neuronal differentiation by acting as an anti-mitogenic signal. J. Neurosci. 2005;25:4917–4929. doi: 10.1523/JNEUROSCI.5066-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi JH, Ebrahem Q, Moore N, Murphy G, Claesson-Welsh L, Bond M, Baker A, Anand-Apte B. A novel function for tissue inhibitor of metalloproteinases-3 (TIMP3): inhibition of angiogenesis by blockage of VEGF binding to VEGF receptor-2. Nat. Med. 2003;9:407–415. doi: 10.1038/nm846. [DOI] [PubMed] [Google Scholar]
- Rhee SG. Regulation of phosphoinositide-specific phospholipase C. Annu. Rev. Biochem. 2001;70:281–312. doi: 10.1146/annurev.biochem.70.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- Schlessinger J. Common and distinct elements in cellular signaling via EGF and FGF receptors. Science. 2004;306:1506–1507. doi: 10.1126/science.1105396. [DOI] [PubMed] [Google Scholar]
- Schlessinger J, Lemmon MA. SH2 and PTB domains in tyrosine kinase signaling. Sci. STKE. 2003;2003:re12. doi: 10.1126/stke.2003.191.re12. [DOI] [PubMed] [Google Scholar]
- Seo D-W, Li H, Guedez L, Wingfield PT, Diaz T, Salloum R, Wei B.-y., Stetler-Stevenson WG. TIMP-2 mediated inhibition of angiogenesis: An MMP-independent mechanism. Cell. 2003;114:171–180. doi: 10.1016/s0092-8674(03)00551-8. [DOI] [PubMed] [Google Scholar]
- Seo D-W, Li H, Qu C-K, Oh J, Kim Y-S, Diaz T, Wei B, Han J-W, Stetler-Stevenson WG. Shp-1 mediates the antiproliferative activity of tissue inhibitor of metalloproteinase-2 in human microvascular endothelial Cells. J. Biol. Chem. 2006;281:3711–3721. doi: 10.1074/jbc.M509932200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell. Dev. Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetler-Stevenson WG. Matrix metalloproteinases in angiogenesis: a moving target for therapeutic intervention. J. Clin. Invest. 1999;103:1237–1241. doi: 10.1172/JCI6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetler-Stevenson WG, Seo D-W. TIMP-2: an endogenous inhibitor of angiogenesis. Trends Mol. Med. 2005;11:97–103. doi: 10.1016/j.molmed.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Stetler-Stevenson WG. The tumor microenvironment: regulation by MMP-independent effects of tissue inhibitor of metalloproteinases-2. Cancer Metastasis Rev. 2008;27:57–66. doi: 10.1007/s10555-007-9105-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupack DG, Cheresh DA, Gerald PS. Integrins and angiogenesis. In: Schatten GP, editor. Current topics in developmental biology. Vol. 64. Academic Press; 2004. pp. 207–238. [DOI] [PubMed] [Google Scholar]
- Wingfield PT, Sax JK, Stahl SJ, Kaufman J, Palmer I, Chung V, Corcoran ML, Kleiner DE, Stetler-Stevenson WG. Biophysical and functional characterization of full-length, recombinant human tissue inhibitor of metalloproteinases-2 (TIMP-2) produced in Escherichia coli. Comparison of wild type and amino-terminal alanine appended variant with implications for the mechanism of TIMP functions. J. Biol. Chem. 1999;274:21362–21368. doi: 10.1074/jbc.274.30.21362. [DOI] [PubMed] [Google Scholar]