

Abstract
Immunocytokines (IC), consisting of tumor-specific monoclonal antibodies fused to the immunostimulatory cytokine interleukin 2 (IL2), exert significant antitumor effects in several murine tumor models. We investigated whether intratumoral (IT) administration of IC provided enhanced antitumor effects against subcutaneous tumors. Three unique ICs (huKS-IL2, hu14.18-IL2, and GcT84.66-IL2) were administered systemically or IT to evaluate their antitumor effects against tumors expressing the appropriate IC-targeted tumor antigens. The effect of IT injection of the primary tumor on a distant tumor was also evaluated. Here, we show that IT injection of IC resulted in enhanced antitumor effects against B16-KSA melanoma, NXS2 neuroblastoma, and human M21 melanoma xenografts when compared to intravenous (IV) IC injection. Resolution of both primary and distant subcutaneous tumors and a tumor-specific memory response were demonstrated following IT treatment in immunocompetent mice bearing NXS2 tumors. The IT effect of huKS-IL2 IC was antigen-specific, enhanced compared to IL2 alone, and dose-dependent. Hu14.18-IL2 also showed greater IT effects than IL2 alone. The antitumor effect of IT IC did not always require T cells since IT IC induced antitumor effects against tumors in both SCID and nude mice. Localization studies using radiolabeled 111In-GcT84.66-IL2 IC confirmed that IT injection resulted in a higher concentration of IC at the tumor site than IV administration. In conclusion, we suggest that IT IC is more effective than IV administration against palpable tumors. Further testing is required to determine how to potentially incorporate IT administration of IC into an antitumor regimen that optimizes local and systemic anticancer therapy.
Keywords: Intratumoral, Immunocytokine, Hu14.18-IL2, HuKS-IL2, Melanoma, Neuroblastoma
Introduction
Immune-based cancer therapy involves a combination of two separate modalities to eradicate tumor cells. Therapies must be targeted to the tumor and stimulate the immune system to kill cancer cells selectively [38]. Immunocytokines (IC) are synthetic proteins that combine both of these strategies via a tumor antigen-specific mAb fused to an immune-stimulating cytokine. Members of this novel class of immunotherapy have been shown to have significant antitumor and anti metastatic effects in several murine tumor models and are currently being investigated for clinical safety and anticancer potential in human patients with neuroblastoma (NB), melanoma, ovarian and prostate cancers [8, 21, 22, 32]. Our laboratories have been involved in the preclinical and clinical development of two ICs [39]: huKS-IL2, which consists of the humanized mAb huKS1/4 that recognizes epithelial cell adhesion molecule (EpCAM) and is linked to IL2; and hu14.18-IL2, which is an anti-disialoganglioside GD2 mAb also linked to IL2. These ICs selectively bind tumor cells, activate IL2-responsive immune cells, and facilitate antibody dependent cell-mediated cytotoxicity (ADCC) by FcR+ effector cells [15]. Both ICs were shown to activate a strong antitumor innate immune response involving NK cells that induced antimetastatic effects [18, 24]. These antimetastatic effects occurred when ICs were administered systemically via tail vein injection. However, IV hu14.18-IL2 IC at the doses used resulted in only a transient antitumor response against well-established primary subcutaneous (s.c.) murine NXS2 NB tumors, followed by tumor recurrence [29]. In this setting, addition of s.c. continuous infusion IL2 to systemic hu14.18-IL2 IC enabled durable resolution of established NXS2 tumors [30]. Clinically, administration of higher doses of IC has been limited by the IL2-related toxicities of the IC [21, 33]. Thus, administration of additional constant infusion IL2 would be expected to cause unacceptable IL2 toxicity if given to patients already receiving hu14.18-IL2 IC at the maximum tolerated dose. Therefore, in this study we hypothesized that IT administration of IC to established, primary tumors would result in greater local tumor destruction compared to IV administration. Since IT delivery of IL2 alone has been shown to mediate tumor regression via an enhanced tumor-specific cytotoxic T lymphocyte (CTL) response [19], IT IC therapy may induce a T cell memory response due to enhanced sequestration of the IL2-containing IC in the tumor microenvironment and subsequent improved local tumor-specific T cell sensitization.
Our studies focus on the antitumor activity of IT huKS-IL2 and hu14.18-IL2 ICs in s.c. murine tumor models. HuKS-IL2 IC recognizes B16-KSA melanoma, a subclone of the parental B16 cell line transfected with human EpCAM. Hu14.18-IL2 IC targets the GD2-expressing cells, including the murine NXS2 NB and human M21 melanoma [15, 23]. The antitumor activity of IT hu14.18-IL2 was tested in a xenograft model consisting of human M21 melanoma tumors grown in nude mice. To determine the in vivo distribution of IT IC, we used the anti-carcinoembryonic antigen (CEA) 111In-GcT84.66-IL2 IC because it is radiolabeled and tumor localization studies following systemic administration have previously been done [42]. T84.66-IL2 has been shown to target CEA-positive MC-38.CEA murine tumors and inhibit tumor growth [7]. The ICs and tumor models used in this study are summarized in Table 1.
Table 1.
Tumor models, specific antigen expression and corresponding ICs
| Tumor model | Tumor type | Mouse strain | Tumor antigen | Immunocytokine |
|---|---|---|---|---|
| B16-KSA | Melanoma | C57BL/6 | EpCAM | HuKS-IL2a |
| B16-KSA | Melanoma | SCID | EpCAM | HuKS -IL2 |
| NXS2 | Neuroblastoma | A/J | GD2 | Hu14.18-IL2 |
| M21 (human) | Melanoma | NCr nude | GD2 | Hu14.18-IL2 |
| MC-38.CEA | Colon carcinoma | C57BL/6.CEA | CEA | 111In-GcT84.66-IL2 |
aDesignated huKS1/4-IL2 in some studies [9]
In the in vivo studies reported here, we demonstrate a greater antitumor response with IT IC compared to systemic IV IC injection in the treatment of localized palpable s.c. tumors that was antigen-specific, dose-dependent, and greater than IL2 alone. In addition, IT IC delivery resulted in resolution of both the directly treated primary tumor, as well as the non-locally treated distant tumor. A tumor-specific memory response was also seen. Localization studies using a radiolabeled IC confirmed that IT injection resulted in a higher concentration of IC at the tumor site compared to IV administration. Therefore, we suggest that IT IC administration may be combined with other immune or cytotoxic therapies to enhance local and systemic antitumor effects.
Materials and methods
Mice
Female A/J and C57BL/6 mice, 7–8 weeks old, were obtained from Harlan Sprague Dawley (Madison, WI, USA); B6.CB17 scid/scid mice, 5–6 weeks old, were obtained from Jackson (Bar Harbor, ME, USA); and male NCr nude mice, 6–7 weeks old, were obtained from Taconic (Germantown, NY, USA). All animals were housed in university-approved facilities and were handled according to National Institutes of Health and University of Wisconsin-Madison Research Animal Resource Center guidelines. The C57BL/6.CEA transgenic mouse was developed at City of Hope by Clarke et al. [7] and used in IC localization studies as previously described.
Cell lines
NXS2 is a poorly immunogenic, highly metastatic, murine NB hybrid cell line that was created as previously described [23]. This GD2 + cell line is sensitive to NK cell-mediated therapies [25]. The murine NXS2 cell line was grown in DMEM medium (Mediatech, Herndon, VA, USA) supplemented with penicillin (100 U/ml), streptomycin (100 μg/ml), l-glutamine (2 mM) (all from Life Technologies, Inc., Grand Island, NY, USA) and 10% heat-inactivated fetal calf serum (FCS, Sigma Chemicals, St. Louis, MO, USA). Cells were maintained at 37°C in a humidified 5% CO2 atmosphere. The B16-KSA cell line was generated by transfecting the murine melanoma cell line B16 with the gene encoding human EpCAM. Constitutive expression of EpCAM on a subclone designated B16-KSA was maintained by growing the cells as monolayers in the presence of 1 mg/ml G418. This cell line was cultured in RPMI-1640 medium (Mediatech, Herndon, VA, USA) with the same additives as described for NXS2 cells above. The GD2 + M21 human melanoma cell line was also cultured in RPMI medium with the same additives as described above [15]. The MC-38.CEA cell line was generated by transfection of CEA into MC-38 cells as previously described, and cultured in RPMI media in the absence of antibiotics [7].
Flow cytometry
Expression of antigens on tumor cell lines was evaluated by flow cytometry. Briefly, tumor cells were collected and resuspended in PBS with 2% FCS (flow buffer) at a concentration of 3 × 106 cells/ml, and 3 × 105 NXS2 or B16-KSA cells were incubated with hu14.18-IL2 IC or huKS-IL2 IC, respectively, 10 μg per 3 × 105 cells at 4°C for 40 min. Cells were washed and stained with a secondary Ab, anti-human IL2-PE (BD Biosciences, San Diego, CA, USA), 2 μg per 3 × 105 cells, for an additional 40 min. at 4°C. Staining of cells with secondary Ab only, without IC, was used as a negative control. Cells were washed and resuspended in 0.3 ml flow buffer and analyzed using a FACScan cytofluorometer (Becton Dickinson, San Jose, CA, USA). Analysis of data collected for 10,000 events/sample was performed using the CellQuest software (Becton Dickinson, San Jose, CA).
ICs and immunotherapy
The humanized hu14.18-IL2 and huKS-IL2 ICs were generated and provided by EMD-Lexigen Research Center (Billerica, MA, USA). HuKS-IL2 was originally designated huKS1/4-IL2, and shown to be effective against human prostate carcinoma metastases in SCID mice [9]. For the B16-KSA melanoma tumor model, C57BL/6 or SCID mice were injected s.c. with 3 × 105 cells in 100 μl PBS on the abdomen. For the NXS2 NB single tumor and tumor re-challenge models, A/J mice were injected s.c. with 2 × 106 NXS2 NB cells in 100 μl PBS on the abdomen. For the primary and distant tumor model, 1 × 106 NXS2 NB cells were implanted on one flank on day 0 and again on day 4 on the opposite flank. Smaller tumor cell inoculates were used in this model to allow the implantation of two adjacent tumors. The specific IC, doses and treatment schedule are indicated for each experiment. Mice were given IV IC by tail vein injection in 200 μl PBS. IT injections consisted of IC in 50 μl PBS, placed into the center of the s.c. tumor with the needle perpendicular to the plane of the skin. Whenever possible, the same site of injection was used for each injection; however, in rare cases, the 30 gauge needle tract had not completely healed on subsequent injections and a new injection entry site was selected. Control treatments consisted of an equivalent volume of PBS administered by IV or IT injection. Human recombinant IL2 (Tecin®, 5 million IU/vial) was purchased from Hoffmann-La Roche Inc. (Nutley, NJ, USA), reconstituted and stored at 4°C according to manufacturer’s instructions. Appropriate amounts of IL2 were delivered based on 1 μg IC containing approximately 3,000 IU of human recombinant IL2, as determined by molar components and in vitro functional studies [30]. For the human M21 melanoma model, 3 × 106 cells were implanted s.c. into nude mice. Since nude mice do not have T-cells and do not develop a mouse anti-human antibody (MAHA) response following IC treatment (our unpublished data), two courses of the humanized IC were administered. Tumor size was measured every third day with a digital caliper, and volume was calculated by applying the formula 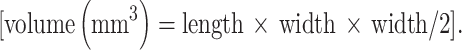 The end point of these studies was death of the animal or excessive tumor burden as determined by both tumor size and the condition and behavior of the animal. The decision to euthanize an animal was made by an independent observer without regard for treatment group.
The end point of these studies was death of the animal or excessive tumor burden as determined by both tumor size and the condition and behavior of the animal. The decision to euthanize an animal was made by an independent observer without regard for treatment group.
IC localization studies
These studies, performed at City of Hope, involved Indium labeling of the anti-CEA IC, designated 111In-GcT84.66-IL2. CEA transgenic C57BL/6 mice bearing day 10 MC38.CEA s.c. tumors were treated IV (25 μg of IC with 4 μCi of In-111 DOTA conjugated radiolabeled IC). Following IV IC injection, mice were sacrificed (five per group) at a variety of time points and radioactivity was measured in a gamma-counter. Parallel studies were performed after IT injections (2.3 μg of IC with 6.3 μCi of In-111 DOTA conjugated radiolabeled IC). Two mice were imaged on a dedicated small animal gamma camera (BioSpace γ-Imager) and regions of interest drawn around the tumor to measure radioactivity using GammaVision + software. For comparison purposes, all data was converted to percent injected dose (ID) and normalized to the initial scan after correcting for radionuclide decay.
Statistical analysis
A two-tailed Student’s t-test was used to determine significance of differences between experimental and relevant control values. Survival curves were generated and statistically compared using the method described by Kaplan and Meier. We determined whether the difference between the proportion of tumor-free animals in particular treatment groups (Table 2) was significant by using a standard comparison of two proportions and calculating 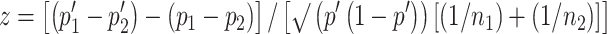 , where
, where 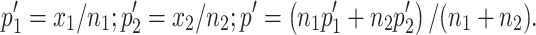 Variables include: z = Z score (estimate of the area under the curve of the standard normal distribution),
Variables include: z = Z score (estimate of the area under the curve of the standard normal distribution), 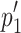 and
and 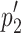 are proportions 1 and 2 given by the number of tumor free animals (x) divided by the total animals in that group (n), and n
1 and n
2 are the total number of animals in each group. Data are presented as mean ± SE and considered significant for p values less than 0.05.
are proportions 1 and 2 given by the number of tumor free animals (x) divided by the total animals in that group (n), and n
1 and n
2 are the total number of animals in each group. Data are presented as mean ± SE and considered significant for p values less than 0.05.
Table 2.
IT treatment induced tumor regression of NXS2 tumors with hu14.18-IL2 IC
| Treatment | Number of tumor-free A/J mice | |||
|---|---|---|---|---|
| Exp 1 | Exp 2 | Exp 3 | Total | |
| IC dose | 15 μg | 15 μg | 5 μg | |
| IV PBS | 0 of 3 | 0 of 6 | 0 of 6 | 0/15 |
| IV hu14.18-IL2 | 0 of 5 | 3 of 6 | 0 of 6 | 3/17 |
| IT hu14.18-IL2 | 5 of 5 | 5 of 6 | 2 of 6 | 12/17 |
A/J mice were implanted with s.c. NXS2 tumors and treated with hu14.18-IL2 IC on days 7–11, as described in “Materials and methods”
The numbers of animals showing complete tumor regressions without tumor recurrence for at least 3 weeks are noted for each treatment group from three separate experiments
The increased proportion of tumor-free mice in groups treated IT vs. IV with hu14.18-IL2 IC treatment for the combination of all 3 experiments was significant (p = 0.002)
Results
IT IC resulted in greater antitumor effects than IV IC
First, we confirmed that the tumor cell lines used in our antitumor studies expressed the antigens recognized by the hu14.18-IL2 and huKS-IL2 ICs. The specific expression of tumor-associated antigens, EpCAM on B16-KSA cells and disialoganglioside GD2 on NXS2 cells, is shown in Fig. 1A. Next, the antitumor effect of IC was tested via IT or IV administration. The dose of IC utilized corresponded to IV doses previously determined to be sub-optimal against s.c. tumors, but which resulted in less systemic toxicity [29]. In our experiments, IC was given via IV or IT administration to animals bearing established, palpable day 7 tumors. Figure 1b shows the effect of IT huKS-IL2 IC against s.c. B16-KSA melanoma tumors. Administration of IT huKS-IL2 IC resulted in greater suppression of tumor growth compared to both the equivalent IV administered IC dose and IT PBS (p = 0.02 day 16, p = 0.03 day 19 for IT IC vs. IV IC; p = 0.01 day 16, p < 0.001 day 19 for IT IC vs. PBS). In a second model (Fig. 1c), treatment of s.c. NXS2 tumors with hu14.18-IL2 IC administered IT also resulted in significant antitumor effects compared to PBS-treated tumors (p < 0.0015, beginning on day 11), and enhanced antitumor effects compared to the same dose of hu14.18-IL2 IC given IV (p < 0.02, days 11–18). In addition to significant differences in the mean calculated tumor volume based on caliper measurements of length and width (see “Materials and methods” section), the IT IC-treated tumors also appeared visually flatter, with less elevation above the abdominal wall, compared to either PBS or IV IC-treated tumors. Thus, because tumor height is not serially measurable with calipers, the calculated values for tumor volumes were overestimated for the flatter tumors resulting from IT IC treatment. The specificity of the antitumor effects of IT IC are addressed in Fig. 3. Thus, IT delivery of IC resulted in enhanced antitumor effects compared to systemic delivery in these two murine tumor models.
Fig. 1.

Tumor-associated antigens expression and enhanced antitumor effect of IT ICs compared to IV IC administration. a Flow cytometric expression of tumor-associated antigens, EpCAM on B16-KSA cells (left panel) and GD2 on NXS2 cells (right panel), is shown by binding of huKS-IL2 (filled histogram, left panel) and hu14.18-IL2 (filled histogram, right panel). Gray lines represent control staining with anti-human IL2-PE alone. The y-axis shows the relative cell counts and the x-axis shows the fluorescence intensity units. b Groups of C57BL/6 mice (eight per group) were implanted with 3 × 105 B16-KSA tumor cells on day 0 and treated with 15 μg IV huKS-IL2 (in 200 μl) or IT IC (in 50 μl) on days 7–11. Control mice were treated with IT PBS (50 μl). (p = 0.02 day 16, p = 0.03 day 19 for IT IC vs. IV IC; p = 0.01 day 16, p < 0.001 day 19 for IT IC vs. IT PBS) Results are representative of 2 similar experiments. c Groups of A/J mice (five per group) were implanted with 2 × 106 NXS2 cells (day 0) and treated with IT PBS, 15 μg IV hu14.18-IL2 (in 200 μl) or IT IC (in 50 μl) on days 7–11. (p < 0.0015 for IT hu14.18-IL2 vs. IT PBS, days 11–18; p < 0.02 for IT hu14.18-IL2 vs. IV hu14.18-IL2 days 11–18.) Results are representative of three similar experiments
Fig. 3.

Specificity and dose response of IT IC. a C57BL/6 mice (eight per group) were implanted with 3 × 105 B16-KSA tumor cells on the abdomen on day 0. Mice were treated with 15 μg IT huKS-IL2, hu14.18-IL2, or PBS (in 50 μl) on days 7–11. Data are representative of two experiments. (p = 0.02 day 16 for huKS-IL2 vs. hu14.18-IL2, p = 0.06 day 19 for huKS-IL2 vs. hu14.18-IL2; p = 0.003 day 16 hu14.18-IL2 vs. PBS, p = 0.04 day 19 hu14.18-IL2 vs. PBS). b C57BL/6 mice (eight per group) were implanted with 3 × 105 B16-KSA cells on day 0. Mice were treated with 15 μg IT huKS-IL2, 45,000 IU IL2, or PBS (in 50 μl) on days 7–11. (p = 0.01 for IT PBS vs. IT huKS-IL2, p = 0.002 for IT huKS-IL2 vs. IT IL2, p = NS for IT PBS vs. IT IL2 days 16–19) Data are representative of two experiments. c Groups of A/J mice (5–6 per group) were implanted with 2 × 106 NXS2 cells on day 0. Mice were treated with 15 μg IT hu14.18-IL2, 45,000 IU IL2, or PBS (in 50 μl) on days 7–11. (p < 0.001 for IT PBS vs. IT hu14.18-IL2 days 10–17, p < 0.01 for IT PBS vs. IT IL2 days 14–17, p = 0.03 for IT hu14.18-IL2 vs. IT IL2 days 10–17). d C57BL/6 mice (six per group) were implanted with 3 × 105 B16-KSA cells on day 0. Mice were treated IT with 15, 20, or 30 μg of huKS-IL2 (in 50 μl) on days 7–11. Control mice received 50 μl IT PBS on days 7–11. (p = 0.05 for 15 ug huKS-IL2 and 20 μg huKS-IL2 vs. 30 μg huKS-IL2 day 16)
NXS2 tumor regressions were seen in a majority of animals (12 of 17) that received IT treatment (Table 2, p = 0.002 for the proportion of tumor-free animals following IT vs. IV IC treatment). In Experiment 1 (Table 2), mice with palpable tumors on day 7 received IT IC on days 7–11, and five of five mice became tumor free by day 14 compared to none of five in the IV IC group. All five mice continued to be tumor-free through day 28, though two of these mice showed delayed tumor re-growth at the primary tumor site on days 30 and 32. On day 35, the remaining three tumor-free mice were re-challenged with 2 × 106 NXS2 cells to determine if anti-NXS2 memory had developed. Four naive A/J mice received s.c. injection of 2 × 106 NXS2 cells as a control, and developed palpable tumors by day 5. Of the three tumor-free mice that were re-challenged, two mice remained tumor-free for greater than 35 days after re-challenge, while one showed tumor development beginning on day 13. The remaining tumor-free mice also rejected a second NXS2 cell re-challenge on day 49 after the first re-challenge (84 days after the original NXS2 tumor implantation). In Experiment 2 (Table 2), all mice remained tumor-free for at least 3 weeks after initial NXS2 tumor resolution. These animals also showed a specific antitumor immune response where NXS2 tumor cells, but not the non-specific YAC-1 tumor cells, were rejected following tumor challenge (data not shown). IT hu14.18-IL2 IC even resulted in two of six tumor resolutions at very low doses (Experiment 3, Table 2). Taken together, these experiments show that IT IC administration resulted in enhanced antitumor effects against well-established primary NXS2 NB compared to IV IC, as well as resolution of NXS2 tumors and induction of a tumor-specific memory response in some mice after IT IC treatment.
IT IC resulted in antitumor effects against both primary and distant tumors
Previous studies have demonstrated enhanced survival after IV administration of IC and a therapeutic effect on systemic metastases [18, 23]. We sought to determine the effect of IT hu14.18-IL2 IC on primary and distant NXS2 tumors in A/J mice. NXS2 tumor cells were implanted s.c. on one side of the abdomen on day 0 and a second tumor was initiated on the opposite flank on day 4, as described in the “Material and methods” section. The primary abdominal tumor was treated with either IT IC or IT PBS and both tumors were serially measured. Two additional groups, each bearing only one abdominal tumor were included as controls: one group was treated with IT IC and the other group received s.c. injection of IC at a tumor-free site on the flank. As expected, Fig. 2a demonstrates that IT IC had a significant antitumor effect on the IT-treated primary abdominal tumor compared to IT PBS (p = 0.001 for IC vs. PBS treatment, day 16 of primary tumor growth). Fig. 2b shows that treatment of the primary tumor starting on day 7 (corresponding to day 3 after implantation for the distant tumor) also resulted in a significant antitumor effect on the growth of the distant flank tumor (p = 0.007 for IC vs. PBS treatment, measured on day 16 of flank tumor growth). In this experiment, two of five primary abdominal tumors showed complete resolution after IT IC, and three of five animals showed complete resolution of the flank tumor after IT IC treatment of the primary tumor. All five primary and distant tumors progressed in animals that received IT PBS injection of the primary abdominal tumor (p = 0.02 for proportion of distant, flank tumor-free animals for IC vs. PBS; p = 0.057 for proportion of primary, abdominal tumor-free animals for IC vs. PBS). This result was confirmed in a second experiment using five animals per group. In the second experiment (not shown), three of five animals resolved the primary tumor and four of five resolved the distant tumor. Overall, the antitumor effect seen in Fig. 2b suggests that IT IC at the primary tumor site resulted in an immune effect seen at the distant flank tumor. In contrast, Fig. 2c shows that s.c. IC injected at a site on the flank away from the tumor had little antitumor effect on the tumor. Here, one of three animals receiving IT IC became tumor-free, while none of the three mice receiving s.c. IC in the flank showed tumor resolution (p = 0.04 for IT IC vs. s.c. IC, days 13–16). This result suggests that the antitumor effect seen in the non-injected distant flank tumors following IT IC treatment of the primary tumor (Fig. 2b) is related to the local effects of IC at the primary tumor site. IT IC may induce a systemic immunologic response that provides a greater antitumor effect compared to the effect of systemic IC after s.c. injection at the doses tested. Further studies are required to determine the mechanisms underlying the effect of IT IC on the distant tumor site.
Fig. 2.

IT IC effect on primary and distant tumor growth. Groups of A/J mice (five per group) were injected with 1 ×106 NXS2 cells on day 0 on the abdomen (primary tumor). A second injection of 1 × 106 NXS2 cells was placed on the opposite flank on day 4 (distant tumor). The abdominal tumor was treated IT with either 50 μl PBS or 15 μg hu14.18-IL2 on days 7–11, while the flank tumor was not treated. a Tumor volume of the IT hu14.18-IL2- or IT PBS-treated primary tumors (p = 0.001 for IT IC vs. IT PBS, day 16). Day 0 corresponds to implantation of the primary tumor. b Tumor volume of the non-treated distant tumor in animals where the primary tumor was treated with 15 μg IT hu14.18-IL2 or IT PBS (p = 0.007 for IT IC vs. IT PBS, day 16). Day 0 corresponds to implantation of the distant tumor (day 4 of primary tumor growth), where treatment of the primary tumor occurred on days 3–7 of distant tumor growth (days 7–11 of primary tumor growth). c Groups of A/J mice (three per group) were injected with 1 × 106 NXS2 cells on day 0 on the abdomen. Mice were treated with 15 μg IT hu14.18-IL2 or an equivalent dose of s.c. IC into the flank at a site away from the tumor. Tumor volume of the single abdominal tumor is shown (p = 0.04 for IT IC vs. s.c. IC, days 13–16)
We also note that the distant flank tumors in PBS treated mice grew somewhat slower compared to the primary abdominal tumors (compare PBS treated groups in Fig. 2a, b). This may reflect different rates of tumor growth at these 2 anatomic sites, as well as innate or adaptive mechanisms induced by prior implantation of the primary abdominal tumor. The mechanisms underlying this difference in flank and abdominal tumor sizes are not the focus of this study.
IT IC antitumor efficacy has Ab-specific and non-specific components.
To determine whether the antitumor effect of IT IC is tumor specific, we compared huKS-IL2 IC with the non-specific hu14.18-IL2 IC in the treatment of B16-KSA tumors. The data presented in Fig. 3a show that huKS-IL2 IC, which specifically targets huEpCAM expressed on B16-KSA cells, resulted in greater antitumor effects compared to the effects of the non-specific hu14.18-IL2 IC (p = 0.02 day 16; p = 0.06 day 19 for specific, huKS-IL2 IC vs. non-specific, hu14.18-IL2 IC). Notably, the non-specific hu14.18-IL2 IC had significant antitumor activity against B16-KSA tumors (p = 0.003, day 16; p = 0.04, day 19 for IT hu14.18-IL2 vs. IT PBS). This may be due to the effect of sustained, localized IL2 within the tumor microenvironment related to the large molecular size of the IC, as well as to non-specific interactions that occur through the Fc-component of the IC. When compared to IT treatment with an equivalent amount of soluble recombinant IL2 as contained in the dose of IC, the tumor-specific IC had greater antitumor effects in both tumor models (p = 0.002, days 16 and 19 for huKS-IL2 vs. IL2 in Fig. 3b; p = 0.03, day 17 for hu14.18 IL2 vs. IL2 in Fig. 3c). Suppression of B16-KSA tumor growth was also shown to be huKS-IL2 IC dose-dependent in Fig. 3d (p = 0.05, day16 for 15 μg and 20 μg vs. 30 μg huKS-IL2). These data show that IT IC resulted in a specific antitumor response that was dose-dependent and due to the unique properties of the IC, which combine tumor specificity with the antitumor effects of localized IL2.
Effective IT response in the absence of T cells
Systemic huKS-IL2 and hu14.18-IL2 ICs have been shown to mediate antitumor effects that can be T cell-independent and involve NK cells [18, 23]. To determine whether T cells were required in the antitumor effects of IT huKS-IL2 IC, SCID mice bearing established B16-KSA tumors were treated with IT huKS-IL2 or PBS as a control. Fig. 4a shows that significant antitumor effects due to IT huKS-IL2 persisted in the absence of T cells (p = 0.006, day 16). In a separate model, GD2 + human M21 melanoma tumors were xenografted into nude mice and treated with IT hu14.18-IL2 (Fig. 4b). Again, even in the absence of mature T cells, IT hu14.18-IL2 resulted in significant antitumor effects (p = 0.04 for IT IC vs. IT PBS, days 22–28). IC treatment also resulted in a significant survival advantage where three of five IC-treated mice were still alive on day 79, while PBS-treated mice required euthanasia between days 22–40 due to large tumor burden (data not shown, p < 0.0018, day 79). Together, these studies indicate that the antitumor immunotherapeutic effects of IT IC against B16-KSA and M21 melanoma can occur in SCID and nude mice, suggesting a T-cell independent activity. Since SCID and nude mice have functional NK cells, the role of NK cells in this model requires further study.
Fig. 4.

T-cell independent antitumor effect of IT IC. a Groups of SCID mice were implanted with 3 × 105 B16-KSA cells on day 0. Mice were treated with 15 μg IT huKS-IL2 or PBS (50 μl) on days 7–11. (p = 0.006 for IT IC vs. IT PBS, day 16). b Groups of nude mice (five per group) were implanted with 5 × 107 M21 cells on day 0. Mice were treated with 5 μg IT hu14.18-IL2 or or IT PBS (50 μl) on days 9–13 and 16–20. (p = 0.04 for IT IC vs. IT PBS, days 22–28)
IT administration of radiolabeled IC induced greater sustained IC tumor localization
As previously introduced, T84.66-IL2 is an anti-CEA IC with antitumor activity against the CEA+ murine MC-38.CEA colon carcinoma [42]. The availability of a radiolabeled form of this IC, 111In-GcT84.66-IL2, enabled the determination of tumor localization following systemic or IT administration. For this tumor model, transgenic C57BL/6.CEA mice were implanted with s.c.MC-38.CEA tumors and treated with a single dose of IV (25 μg) or IT (2.3 μg) 111In-GcT84.66-IL2. Whole blood and tumor samples were obtained from IV-treated mice for gamma counter analysis, and IT-treated mice were imaged with a small animal gamma camera, as described in “Materials and methods”. Figure 5 demonstrates that the relative amount of total radioactivity that accumulated at the tumor site was significantly greater after IT IC administration compared to IV IC. The detectable radioactivity after IT IC had a higher level and longer persistence at the tumor site, with over 50% of the injected dose (ID) present at 8 h and still detectable after 72 h. We hypothesize that most of this radioactivity is intact IC that was bound to the tumor cell surface or has been internalized by the tumor cells, thereby representing delivered drug. There was a rapid decline in detectable radiolabeled IC in circulating blood after IV injection (data not shown) that corresponded to previously determined pharmacokinetics of ICs in mice [20], which may have led to less accumulation of IC at the tumor site over time. The IV administered IC should be metabolized in the same manner, suggesting that IT administration results in more than an order of magnitude difference in drug delivery at the earliest time points. Although IV IC does localize to the tumor as shown previously [23, 42], our data show that IT injection increased the amount of IC delivered and retained at the tumor site.
Fig. 5.

IC-localization after systemic or IT administration. C57BL/6.CEA mice bearing day 10 s.c. CEA-positive MC-38.CEA tumors were treated with 111In-GcT84.66-IL2. Animals received a single dose of 25 μgs IV (five per group) or 2.3 μg IT (two per group). At various time points, tumors were harvested from the IV-treated mice, and the percent injected dose (ID) was determined by a gamma counter. Intact IT-treated mice were imaged with a small animal gamma camera, acquiring percent ID by gating on the s.c. tumor. Data represent the percent of the original ID at various time points
Discussion
While ICs enable targeting of immunomodulatory cytokines to the tumor microenvironment, systemic administration is still limited by dose-limiting toxicities [21, 33]. In this study, we show enhanced antitumor effects and increased tumor localization of IC following IT administration. Using the B16-KSA melanoma and NXS2 NB models, we demonstrated a greater antitumor response with IT IC compared to an equivalent systemic IC dose in the treatment of localized palpable s.c. tumors. Interestingly, IT hu14.18-IL2 IC delivery resulted in resolution of both the directly treated primary NXS2 tumor, as well as the non-locally treated distant NXS2 tumor in some animals. Evidence of a tumor-specific memory response was seen after primary NXS2 tumor regression. In the B16-KSA model, we show that the antitumor effect of IT IC was antigen-specific, dose-dependent, and greater than IL2 alone. The enhanced antitumor effect of IT IC compared to recombinant IL2 was also shown in the NXS2 model. These preclinical data suggest that the antitumor activities of huKS-IL2 and hu14.18-IL2, two ICs already undergoing clinical testing via systemic delivery, may result in enhanced tumor control when administered directly into the tumor microenvironment.
The advantage of local administration of IC compared to IV IC has been described previously using SCID mice bearing human tumor xenografts and infused with HLA identical human peripheral blood lymphocytes as effector cells [6]. In that study, ICs consisting of anti-EGFR mAb linked to IL2 or TNF showed greater in vivo antitumor effects when administered IT compared to IV. Furthermore, IT injection of intact IC resulted in a survival advantage compared to IT injection of the mAb and IL2 as separate IT injections. In another model, IT injection of huKS-IL12/IL2, where huKS1/4 mAb was linked to the synergizing cytokine combination of IL2 and IL12, resulted in complete resolution of s.c. Lewis lung carcinomas transfected with EpCAM [12]. Our studies confirm and extend this previous work by evaluating IT administration of IC in several unique systems, including immunodeficient mice (Table 1). HuKS-IL2 IC delivered IT resulted in significant antitumor effects in both immunocompetent and T-cell deficient SCID mice bearing syngeneic B16-KSA tumors. Hu14.18-IL2 IC was also effective against human M21 melanoma xenograft tumors in nude mice. These data provide further insight into the mechanism of IC-mediated tumor cell destruction, suggesting that T cells are not the only potential effectors of antitumor activity in IT IC immunotherapy. Indeed, our studies show that IT IC induced antitumor effects in a T-cell independent manner. Based on previous studies that have shown a role for NK cells in the antitumor effects of IC, these innate immune cells are likely to be playing a significant role in the antitumor effects of IT IC [18]. Since SCID and nude mice have expanded NK cell populations, the specific contribution of NK cells needs to be further evaluated through the use of depletion studies in immunocompetent animals to specifically characterize the immune effector cells involved in IT IC therapy.
The preclinical data presented in this report demonstrate several unique antitumor effects of IT IC compared to equivalent doses of systemic IC. In the NXS2 model, IC administration resulted in significantly more tumor resolutions when given IT. Complete tumor regressions are not typical at these doses when given systemically [30]. Interestingly, IT IC also induced a tumor-specific memory response in tumor-free mice as evidenced by multiple rejections of NXS2 tumor rechallenges. These data indicate the likely development of a T cell memory response following initial tumor eradication, similar to what we have previously shown in an NXS2 model where T cells were involved in the resolution of tumors due to IV IC supplemented with systemic IL2 [30]. The exact mechanism of this specific immune response in our IT IC model remains to be determined. While we observed complete resolution of some NXS2 tumors with IT IC therapy, we have not been able to demonstrate this curative effect against B16-KSA melanoma at the doses tested. This may be reflective of the quickly replicating, aggressive nature of the B16 cell line compared to the much slower growth pattern of the NXS2 NB cell line, as well as to differences in effector cell functions between the A/J and C57BL/6 mouse strains. In the same way that combination with chemotherapy seems to augment the efficacy of IV IC [16], we hypothesize that combination therapy using IT IC will be more effective than IT IC alone against aggressive or more established tumors. On-going murine studies are attempting to augment the antitumor effect of IT IC with chemotherapeutic agents or antitumor immune stimulants such as αCD40 or CpG [2, 3, 26]. In addition, since IV IC has been shown to have significant antimetastatic effects, combinatorial IT and IV IC therapy is a clinical possibility.
Also in the NXS2 model, we showed IT hu14.18-IL2 IC had significant antitumor effects on both the primary abdominal tumor treated directly with IT IC as well as a distant s.c. NXS2 tumor. Although the distant tumor was smaller at the initiation of treatment to the primary tumor, our data support a significant antitumor effect due to IT IC that is evident soon after IT IC administration. Here again, IT IC therapy induced tumor resolution in some animals. Additionally, we found that the effects at the distant tumor required IC delivery into the tumor microenvironment. Only IT IC, but not s.c. IC injected at a tumor-free site, resulted in the antitumor effect seen in the non-injected distant NXS2 flank tumors. These data suggest that the local effects of IC at the primary tumor site may induce systemic immunologic activity that provides a significant antitumor effect at the doses tested. Another model of intralesional immunomodulatory therapy, involving the chemotactic CC chemokine CCL16, also showed both local antitumor effects as well as reduced mortality due to distant metastatic disease [13]. In this model, IT immunotherapy caused attraction and activation of immune effector cells in the tumor environment that led to significant antitumor effects. Local administration has been tested with other anticancer therapeutics as well. Altenschmidt, et al. used the antibody toxin scFV(FRP6)-ETA, consisting of a mAb against erbB2-receptor tyrosine kinase fused to Pseudomonas endotoxin A, against established schwannomas in a preclinical murine model. They similarly demonstrated greater antitumor effect with IT administration compared to IV injection [1]. Administration of the non-specific immune stimulant CpG directly into s.c. CT-26 colon adenocarcinoma and B16 melanoma has also been shown to suppress tumor growth and increase survival in mice via CD8+ T effector cells [35]. Finally, IT injection of the chemotherapeutic agent doxorubicin in liposomes into Meth-A tumors in mice showed greater suppression of tumor growth when compared to the same agent administered IV [17]. In the future, these individual agents may be combined with IC to enhance the therapeutic efficacy of each agent, with the ultimate goal of both local and systemic tumor eradication.
Localization studies using radiolabeled 111In-GcT84.66-IL2 IC demonstrated that IT delivery resulted in more than an order of magnitude greater concentration of IC in the tumor microenvironment compared to systemic IC administration (Fig. 5). Our results are consistent with a previous report that showed approximately 5% of the ID of systemically injected IC was detectable in s.c. tumors 8 h later [23]. We found that over 50% of the ID was detectable at the earliest time point after IT injection, and that radiolabeled-IC remained detectable at later time points than after IV IC, representing persistence of the drug in the tumor microenvironment. These analyses suggest that the enhanced efficacy of IT IC compared to IV IC may be due to higher and sustained concentrations of IC at the tumor site. We hypothesize that most of this radioactivity is retained at the tumor by cells which have bound the IC through specific Fab-mediated recognition of tumor antigen by the antibody component of the IC, thereby representing delivered drug. As non-specific IC (which is unable to recognize tumor antigen with its Fab component) also provides some antitumor effect when given IT (Fig. 3a), further studies are needed to determine how each component of the IC molecule (mAb, FcR binding capability, IL2, or the larger complete protein) may be additionally contributing to the overall retention of the IC in the tumor microenvironment and the antitumor efficacy after IT delivery. Finally, it should be noted that IL2-containing IC are only one method of providing enhanced IL2-related effects. IT injection of liposomal IL2 or polyethylene glycol-modified recombinant IL2 have both been shown to induce significant antitumor effects against both primary and distant non-injected tumors in animal models [27, 31]. Comparison of IL2-containing IC to other methods of increasing the delivery of IL2 are indicated to understand the relative efficacy and toxicities related to these modalities. Additionally, comparisons of the dose-response curves of IT soluble IL2, formulations of IL2 with longer half-lives, and IL2-containing IC (with specific vs. non-specific antibody components) are necessary to further characterize the antitumor effects related to the IL2 component of the IC.
In our studies, the antitumor effect of IT huKS-IL2 was found to be significantly greater than that of IT administration of either IL2 alone or the non-specific IC hu14.18-IL2 against B16-KSA melanoma. Similarly, specific IC hu14.18-IL2 was also found to be more effective than IL2 alone in the treatment of NXS2 tumors. The mechanisms of IC-mediated antitumor activity are hypothesized to involve IC binding to specific antigens expressed on the tumor cell surface, followed by ADCC or complement mediated cytotoxicity (CMC). The studies performed in this study confirm that IC can localize to tumors after IV and IT administration. Tumor cell killing after antibody binding involves activation of FcR+ cells such as macrophages, granulocytes, monocytes, and NK cells, as well as CMC [37]. Since the non-specific IC resulted in a limited but significant antitumor effect, it is likely that the localization of IL2 and/or non-specific activation through the Fc-component of the IC contributed to this effect. In this regard, a previous study involving genetically-modified ICs such that FcR-binding capability was eliminated showed that IC-mediated antitumor effects could be obtained without FcR-binding [11]. MAb therapy has been used effectively in clinical trials as single agents, including the anti-GD2 Ab 3F8 and precursors to the hu14.18-IL2 IC including the 14.G2a murine mAb and ch14.18 chimeric mAb [4, 5, 14, 34]. The IL2 component of the IC augments the effects of mAb, and has been shown to increase the number and activation state of NK cells, as well as to stimulate tumor cell killing by antigen-specific T-cells [36]. In addition, the IL2 component can stimulate both NK and T-cells via the IL2 receptor, independent of Fc or T-cell receptor binding, respectively [10, 28, 40, 41]. Taken together, our studies suggest that the proposed mechanisms of action for systemic IC also occur when IC therapy is administered directly to the tumor microenvironment. Currently, we are attempting to further characterize the immune effector cells within the tumor microenvironment and their activation following IT IC through immunohistochemistry and flow cytometry studies.
In summary, this study demonstrates that IT administration of IC is more effective than IV injection. Furthermore, the effect is greater than IT injection of IL2 alone or nonspecific IC. Complete tumor eradication of treated and untreated tumors, with development of a memory response, was demonstrated in immunocompetent mice. Furthermore, in immunocompetent mice, IT administration of IC induced an antitumor effect at a distant site that far exceeded the antitumor effect of s.c. injection of IC into a distant site of normal skin. Further characterization of the effector cell populations involved in the local antitumor effect at the IT site and at the distant, non-injected tumor site following this immunotherapeutic strategy may help to determine the local and systemic mechanisms involved in immunocompetent mice, and enable development of novel strategies to simultaneously activate multiple arms of the host antitumor immune response. Further testing is required to determine how to potentially incorporate IT administration of IC into a regimen with IV IC in order to optimize local and systemic antitumor effects, and in combination with other antitumor therapies.
Acknowledgments
The authors thank Dr. J Gan, for helpful discussions. Support for this work comes from: NIH Grants CA032685, CA87025, CA14520, GM067386, T32-CA090217, The Midwest Athletes for Childhood Cancer Fund, The Crawdaddy Foundation, The UW- Cure Kids Cancer Coalition, and The Super Jake Foundation.
References
- 1.Altenschmidt U, Schmidt M, Groner B, Wels W. Targeted therapy of schwannoma cells in immunocompetent rats with an erbB2-specific antibody-toxin. Int J Cancer. 1997;73(1):117–124. doi: 10.1002/(SICI)1097-0215(19970926)73:1<117::AID-IJC18>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 2.Buhtoiarov IN, Lum H, Berke G, Paulnock DM, Sondel PM, Rakhmilevich AL. CD40 ligation activates murine macrophages via an IFN-gamma-dependent mechanism resulting in tumor cell destruction in vitro. J Immunol. 2005;174(10):6013–6022. doi: 10.4049/jimmunol.174.10.6013. [DOI] [PubMed] [Google Scholar]
- 3.Buhtoiarov IN, Lum HD, Berke G, Sondel PM, Rakhmilevich AL. Synergistic activation of macrophages via CD40 and TLR9 results in T cell independent antitumor effects. J Immunol. 2006;176(1):309–318. doi: 10.4049/jimmunol.176.1.309. [DOI] [PubMed] [Google Scholar]
- 4.Cheung NK, Lazarus H, Miraldi FD, Abramowsky CR, Kallick S, Saarinen UM, Spitzer T, Strandjord SE, Coccia PF, Berger NA. Ganglioside GD2 specific monoclonal antibody 3F8: a phase I study in patients with neuroblastoma and malignant melanoma. J Clin Oncol. 1987;5(9):1430–1440. doi: 10.1200/JCO.1987.5.9.1430. [DOI] [PubMed] [Google Scholar]
- 5.Cheung NK, Kushner BH, Yeh SJ, Larson SM. 3F8 monoclonal antibody treatment of patients with stage IV neuroblastoma: a phase II study. Prog Clin Biol Res. 1994;385:319–328. [PubMed] [Google Scholar]
- 6.Christ O, Seiter S, Matzku S, Burger C, Zoller M. Efficacy of local versus systemic application of antibody-cytokine fusion proteins in tumor therapy. Clin Cancer Res. 2001;7(4):985–998. [PubMed] [Google Scholar]
- 7.Clarke P, Mann J, Simpson JF, Rickard-Dickson K, Primus FJ. Mice transgenic for human carcinoembryonic antigen as a model for immunotherapy. Cancer Res. 1998;58(7):1469–1477. [PubMed] [Google Scholar]
- 8.Connor JP, Felder M, Hank J, Harter J, Gan J, Gillies SD, Sondel P. Ex vivo evaluation of anti-EpCAM immunocytokine huKS-IL2 in ovarian cancer. J Immunother. 2004;27(3):211–9. doi: 10.1097/00002371-200405000-00005. [DOI] [PubMed] [Google Scholar]
- 9.Dolman CS, Mueller BM, Lode HN, Xiang R, Gillies SD, Reisfeld RA. Suppression of human prostate carcinoma metastases in severe combined immunodeficient mice by interleukin 2 immunocytokine therapy. Clin Cancer Res. 1998;4(10):2551–2557. [PubMed] [Google Scholar]
- 10.Gillies SD, Reilly EB, Lo KM, Reisfeld RA. Antibody-targeted interleukin 2 stimulates T-cell killing of autologous tumor cells. Proc Natl Acad Sci USA. 1992;89(4):1428–1432. doi: 10.1073/pnas.89.4.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gillies SD, Lan Y, Lo KM, Super M, Wesolowski J. Improving the efficacy of antibody-interleukin 2 fusion proteins by reducing their interaction with Fc receptors. Cancer Res. 1999;59(9):2159–2166. [PubMed] [Google Scholar]
- 12.Gillies SD, Lan Y, Brunkhorst B, Wong WK, Li Y, Lo KM. Bi-functional cytokine fusion proteins for gene therapy and antibody-targeted treatment of cancer. Cancer Immunol Immunother. 2002;51(8):449–460. doi: 10.1007/s00262-002-0302-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guiducci C, Di CE, Parenza M, Hitt M, Giovarelli M, Musiani P, Colombo MP. Intralesional injection of adenovirus encoding CC chemokine ligand 16 inhibits mammary tumor growth and prevents metastatic-induced death after surgical removal of the treated primary tumor. J Immunol. 2004;172(7):4026–4036. doi: 10.4049/jimmunol.172.7.4026. [DOI] [PubMed] [Google Scholar]
- 14.Handgretinger R, Baader P, Dopfer R, Klingebiel T, Reuland P, Treuner J, Reisfeld RA, Niethammer D. A phase I study of neuroblastoma with the anti-ganglioside GD2 antibody 14.G2a. Cancer Immunol Immunother. 1992;35(3):199–204. doi: 10.1007/BF01756188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hank JA, Surfus JE, Gan JC, Jaeger P, Gillies SD, Reisfeld RA, Sondel PM. Activation of human effector cells by a tumor reactive recombinant anti-ganglioside GD (2) interleukin-2 fusion protein (ch14.18-IL2) Clin Cancer Res. 1996;2(12):1951–1959. [PubMed] [Google Scholar]
- 16.Holden SA, Lan Y, Pardo AM, Wesolowski JS, Gillies SD. Augmentation of antitumor activity of an antibody-interleukin 2 immunocytokine with chemotherapeutic agents. Clin Cancer Res. 2001;7(9):2862–2869. [PubMed] [Google Scholar]
- 17.Idani H, Matsuoka J, Yasuda T, Kobayashi K, Tanaka N. Intra-tumoral injection of doxorubicin (adriamycin) encapsulated in liposome inhibits tumor growth, prolongs survival time and is not associated with local or systemic side effects. Int J Cancer. 2000;88(4):645–651. doi: 10.1002/1097-0215(20001115)88:4<645::AID-IJC20>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 18.Imboden M, Murphy KR, Rakhmilevich AL, Neal ZC, Xiang R, Reisfeld RA, Gillies SD, Sondel PM. The level of MHC class I expression on murine adenocarcinoma can change the antitumor effector mechanism of immunocytokine therapy. Cancer Res. 2001;61(4):1500–1507. [PubMed] [Google Scholar]
- 19.Jackaman C, Bundell CS, Kinnear BF, Smith AM, Filion P, Van HD, Robinson BW, Nelson DJ. IL–2 intratumoral immunotherapy enhances CD8+T cells that mediate destruction of tumor cells and tumor-associated vasculature: a novel mechanism for IL-2. J Immunol. 2003;171(10):5051–5063. doi: 10.4049/jimmunol.171.10.5051. [DOI] [PubMed] [Google Scholar]
- 20.Kendra K, Gan J, Ricci M, Surfus J, Shaker A, Super M, Frost JD, Rakhmilevich A, Hank JA, Gillies SD, Sondel PM. Pharmacokinetics and stability of the ch14.18-interleukin-2 fusion protein in mice. Cancer Immunol Immunother. 1999;48(5):219–229. doi: 10.1007/s002620050569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.King DM, Albertini MR, Schalch H, Hank JA, Gan J, Surfus J, Mahvi D, Schiller JH, Warner T, Kim K, Eickhoff J, Kendra K, Reisfeld R, Gillies SD, Sondel P. Phase I clinical trial of the immunocytokine EMD 273063 in melanoma patients. J Clin Oncol. 2004;22(22):4463–4473. doi: 10.1200/JCO.2004.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ko YJ, Bubley GJ, Weber R, Redfern C, Gold DP, Finke L, Kovar A, Dahl T, Gillies SD. Safety, pharmacokinetics, and biological pharmacodynamics of the immunocytokine EMD 273066 (huKS-IL2): results of a phase I trial in patients with prostate cancer. J Immunother. 2004;27(3):232–239. doi: 10.1097/00002371-200405000-00008. [DOI] [PubMed] [Google Scholar]
- 23.Lode HN, Xiang R, Varki NM, Dolman CS, Gillies SD, Reisfeld RA. Targeted interleukin-2 therapy for spontaneous neuroblastoma metastases to bone marrow. J Natl Cancer Inst. 1997;89(21):1586–1594. doi: 10.1093/jnci/89.21.1586. [DOI] [PubMed] [Google Scholar]
- 24.Lode HN, Xiang R, Becker JC, Gillies SD, Reisfeld RA. Immunocytokines: a promising approach to cancer immunotherapy. Pharmacol Ther. 1998;80(3):277–292. doi: 10.1016/S0163-7258(98)00033-3. [DOI] [PubMed] [Google Scholar]
- 25.Lode HN, Xiang R, Dreier T, Varki NM, Gillies SD, Reisfeld RA. Natural killer cell-mediated eradication of neuroblastoma metastases to bone marrow by targeted interleukin-2 therapy. Blood. 1998;91(5):1706–1715. [PubMed] [Google Scholar]
- 26.Lum HD, Buhtoiarov IN, Schmidt BE, Berke G, Paulnock DM, Sondel PM, Rakhmilevich AL. In vivo CD40 ligation can induce T-cell-independent antitumor effects that involve macrophages. J Leukoc Biol. 2006;79(6):1181–1192. doi: 10.1189/jlb.0405191. [DOI] [PubMed] [Google Scholar]
- 27.Mattijssen V, Balemans LT, Steerenberg PA, De Mulder PH. Polyethylene-glycol-modified interleukin-2 is superior to interleukin-2 in locoregional immunotherapy of established guinea-pig tumors. Int J Cancer. 1992;51(5):812–817. doi: 10.1002/ijc.2910510524. [DOI] [PubMed] [Google Scholar]
- 28.Mule JJ, Yang JC, Afreniere RL, Shu SY, Rosenberg SA. Identification of cellular mechanisms operational in vivo during the regression of established pulmonary metastases by the systemic administration of high-dose recombinant interleukin 2. J Immunol. 1987;139(1):285–294. [PubMed] [Google Scholar]
- 29.Neal ZC, Imboden M, Rakhmilevich AL, Kim KM, Hank JA, Surfus J, Dixon JR, Lode HN, Reisfeld RA, Gillies SD, Sondel PM. NXS2 murine neuroblastomas express increased levels of MHC class I antigens upon recurrence following NK-dependent immunotherapy. Cancer Immunol Immunother. 2004;53(1):41–52. doi: 10.1007/s00262-003-0435-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neal ZC, Yang JC, Rakhmilevich AL, Buhtoiarov IN, Lum HE, Imboden M, Hank JA, Lode HN, Reisfeld RA, Gillies SD, Sondel PM. Enhanced activity of hu14.18-IL2 immunocytokine against murine NXS2 neuroblastoma when combined with interleukin 2 therapy. Clin Cancer Res. 2004;10(14):4839–4847. doi: 10.1158/1078-0432.CCR-03-0799. [DOI] [PubMed] [Google Scholar]
- 31.Neville ME, Robb RJ, Popescu MC. In situ vaccination against a non-immunogenic tumour using intratumoural injections of liposomal interleukin 2. Cytokine. 2001;16(6):239–250. doi: 10.1006/cyto.2001.0963. [DOI] [PubMed] [Google Scholar]
- 32.Osenga K, Hank J, Albertini M, Gan J, Sternberg A, Seeger R, Matthay K, Reynolds P, Krailo M, Adamson P, Reisfeld R, Gillies S, Sondel P. A phase I trial of immunocytokine HU14.18-IL2 in children with recurrent or refractory neuroblastoma and other GD2 positive malignancies: a study of the children’s oncology group. J Immunother. 2004;27(6):S55–S56. doi: 10.1097/00002371-200411000-00198. [DOI] [Google Scholar]
- 33.Osenga KL, Hank JA, Albertini MR, Gan J, Sternberg AG, Eickhoff J, Seeger RC, Matthay KK, Reynolds CP, Twist C, Krailo M, Adamson PC, Reisfeld RA, Gillies SD, Sondel PM. A phase I clinical trial of the hu14.18-IL2 (EMD 273063) as a treatment for children with refractory or recurrent neuroblastoma and melanoma: a study of the children’s oncology group. Clin Cancer Res. 2006;12(6):1750–1759. doi: 10.1158/1078-0432.CCR-05-2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saleh MN, Khazaeli MB, Wheeler RH, Dropcho E, Liu T, Urist M, Miller DM, Lawson S, Dixon P, Russell CH. Phase I trial of the murine monoclonal anti-GD2 antibody 14G2a in metastatic melanoma. Cancer Res. 1992;52(16):4342–4347. [PubMed] [Google Scholar]
- 35.Sharma S, Karakousis CP, Takita H, Shin K, Brooks SP. Intra-tumoral injection of CpG results in the inhibition of tumor growth in murine Colon-26 and B-16 tumors. Biotechnol Lett. 2003;25(2):149–153. doi: 10.1023/A:1021927621813. [DOI] [PubMed] [Google Scholar]
- 36.Sondel PM, Hank JA. Combination therapy with interleukin-2 and antitumor monoclonal antibodies. Cancer J Sci Am. 1997;3(Suppl 1):S121–S127. [PubMed] [Google Scholar]
- 37.Sondel PM, Hank JA. Antibody-directed, effector cell-mediated tumor destruction. Hematol Oncol Clin North Am. 2001;15(4):703–721. doi: 10.1016/S0889-8588(05)70243-4. [DOI] [PubMed] [Google Scholar]
- 38.Sondel PM, Gillies SD (2004) Immunocytokines for Cancer Immunotherapy. In: Morse MA, Blay TM, Lyerly HK, (eds). Handbook of Cancer Vaccines. Humana Press, Totowa pp 341–358
- 39.Sondel PM, Hank JA, Albertini MR, Gillies SD (2007). Novel strategies for cytokine administration via targeting. In: Caligiuri MA, Lotze MT, (eds). Cancer Drug Discovery and Development, Cytokines in the Genesis and Treatment of Cancer. Humana Press, Totowa, pp 399–422
- 40.Voss SD, Robb RJ, Weil-Hillman G, Hank JA, Sugamura K, Tsudo M, Sondel PM. Increased expression of the interleukin 2 (IL-2) receptor beta chain (p70) on CD56 + natural killer cells after in vivo IL-2 therapy: p70 expression does not alone predict the level of intermediate affinity IL-2 binding. J Exp Med. 1990;172(4):1101–1114. doi: 10.1084/jem.172.4.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weil-Hillman G, Fisch P, Prieve AF, Sosman JA, Hank JA, Sondel PM. Lymphokine-activated killer activity induced by in vivo interleukin 2 therapy: predominant role for lymphocytes with increased expression of CD2 and leu19 antigens but negative expression of CD16 antigens. Cancer Res. 1989;49(13):3680–3688. [PubMed] [Google Scholar]
- 42.Xu X, Clarke P, Szalai G, Shively JE, Williams LE, Shyr Y, Shi E, Primus FJ. Targeting and therapy of carcinoembryonic antigen-expressing tumors in transgenic mice with an antibody-interleukin 2 fusion protein. Cancer Res. 2000;60(16):4475–4484. [PubMed] [Google Scholar]