

Summary
The rapid development of our understanding of the diverse biological roles fulfilled by non-coding RNA has motivated interest in the basic macromolecular behavior, structure, and function of RNA. We focus on two areas in the behavior of complex RNAs. First, we present advances in the understanding of how RNA folding is accomplished in vivo by presenting a mechanism for the action of DEAD-box proteins. Members of this family are intimately associated with almost all cellular processes involving RNA, mediating RNA structural rearrangements and chaperoning their folding. Next, we focus on advances in understanding and characterizing the basic biophysical forces that govern the folding of complex RNAs. Ultimately we expect that a confluence and synergy between these approaches will lead to profound understanding of RNA and its biology.
The explosion in our knowledge about noncoding RNA (ncRNA) – RNA that is not translated into protein – has expanded interest in RNA beyond the traditional RNA community. Diverse ncRNAs include the recently-discovered riboswitches, whose novel gene-regulation function is induced by the binding of small metabolites [1]; most of the so-called “Human Accelerated Regions” (HAR) of the human genome, whose recent evolution may be linked to the emergence of the human brain [2]; and many ncRNAs of unknown function [3]. These discoveries underscore the need to understand the fundamental macromolecular behavior of RNA, its structure and dynamics, and how these RNAs are handled and exploited in biology.
Study of catalytic RNAs, which allow detection of correct folding via activity measurements, has established basic thermodynamic and kinetic properties of RNA folding. Large catalytic RNAs such as the group I intron from Tetrahymena thermophila and the RNase P RNA from Bacillus subtilis fold at vastly different rates under different conditions upon addition of Mg2+ and have been shown to fold via multiple parallel pathways, in which different molecules follow different routes with different rates and intermediates, to a common final folded structure [4,5]. These observations support the common view that RNAs traverse rugged energy landscapes as they fold [6,7]. However, little is known about the true shape of these landscapes, and our understanding of the underlying physical behavior of RNA is severely limited.
The rugged folding landscape predisposes RNA to forming long-lived misfolded states. Indeed, essentially all functional RNAs studied in vitro have been found to form such alternative states, from alternative tRNA folds discovered in the 1960’s [8–10] to a recently-suggested topological isomer of a group I intron that takes hours or days to refold [7,11].
RNA’s propensity to misfold has been attributed to its intrinsic “character flaws”: the low information content of its four side chains (relative to the 20 for proteins), the sequestration of these side chains in helices (relative to the outward-facing side chains in peptide beta sheets and alpha helices), the promiscuous ability of any base to form multiple hydrogen bonds with any other base, and the high stability of stacking interactions of rigidly oriented groups upon the cyclic base (relative to the more flexible protein side chains) [12,13].
It has been suggested that the problems encountered by RNA provided an inroad for peptides and proteins in the presumed early RNA world [13–15]. RNA chaperones, proteins that can reorient RNA structures, presumably evolved to address this misfolding, and considerable evidence for the existence and roles of RNA chaperones has accumulated over the last decade [16–19].
In this review, we first focus on recent advances in the field of chaperone-assisted RNA folding – one of the more complex processes in RNA biology. We then turn to recent conceptual and experimental advances in understanding the basic physical forces that underlie complex RNAs and RNA/protein complexes. We end with a brief discussion of recent technical advances that will be critical for developing a deep energetic, dynamic, and structural understanding of RNA and, ultimately, for drawing profound connections between these physical properties and behaviors and the biology of RNAs and RNA/protein complexes.
By necessity, more has been omitted than included in this short review. Fortunately, there are insightful reviews in these omitted areas (e.g., [20–28]). It is a remarkable testament to those in the rather small world of RNA folding and function that so much progress has been made in the relatively short time of modern studies of RNA folding and dynamics.
How ATP-dependent RNA chaperones assist folding
The predisposition of RNA for misfolding or slow folding under physiological conditions poses serious challenges for RNA in vivo [7,13]. Paradoxically, some RNA structural elements seem to exacerbate misfolding; studies of the group I intron from Tetrahymena have shown that its P5abc domain stabilizes misfolded intermediates and dramatically slows refolding of these intermediates into the native state [11,29,30]. P5abc’s detrimental effect on folding kinetics is balanced by its ability to confer thermodynamic stability to the native state and enhance its catalytic activity [31,32]. RNA chaperones that rescue misfolded structures allow an escape from these opposing demands for catalytic and folding efficiency by relieving selective pressure against slow-folding or misfolding RNAs.
Members of the DExD/H-box protein family, principally from the major subfamily DEAD-box (the eponymous DEAD motif is one-letter code for the amino-acid sequence Asp-Glu-Ala-Asp), are involved in virtually every cellular process involving RNA, ranging from splicing, transport, localization, translation, and decay [23,33]. Although DEAD-box proteins have been casually referred to as “RNA helicases” on the basis of their structural similarity to DNA helicases, they are unlikely to processively unwind helices, as RNA rarely contains the long helical stretches found in genomic DNA. Furthermore, functional RNAs often require conformational transitions other than simple helical rearrangements. Since DEAD-box proteins may function differently from canonical DNA helicases, they have been less-judgmentally referred to as RNA-dependent ATPases or RNA “unwindases” [34–36].
The discovery of DEAD-box proteins that promote splicing in the well-characterized group I and II introns opened the door for recent mechanistic breakthroughs [18,37–39]. Interestingly, these DEAD-box proteins were shown to be interchangeable to a substantial extent, demonstrating that they function rather promiscuously. Deletion of Mss116p, a mitochondrial DEAD-box protein from Saccharomyces cerevisiae, adversely affected the splicing of group I and II introns in vivo; these deleterious effects could be largely rescued by the expression of a related DEAD-box protein, CYT-19 from Neurospora crassa [38]. Further, Mss116p and CYT-19 were shown to promote group I and group II intron splicing in vitro, as was the cytosolic DEAD-box protein, Ded1p [37–39]. Each of these proteins was also shown to unwind RNA helices non-specifically in an ATP-dependent manner [36,37,39–41].
The ability of these DEAD-box proteins to promote proper folding of structurally diverse RNAs and their common structural features suggest that mechanistic details obtained for one particular DEAD-box protein and RNA substrate may yield general insights for a wide variety of DEAD-box proteins. Recently, the mechanism of CYT-19 has been probed in the folding and function of a ribozyme derived from the Tetrahymena group I intron. CYT-19 was shown to act non-specifically, disrupting both misfolded and native forms of the ribozyme. However, the efficiency of unfolding activity was dependent on the stability of the RNA species, with CYT-19 unfolding the misfolded RNA at least 50-fold faster than the native ribozyme [42,43].
Unexpectedly, CYT-19 was also able to unwind the base paired P1 duplex, formed between the ribozyme and its oligonucleotide substrate, much more efficiently than the same duplex free in solution, demonstrating that local unwinding activity was enhanced by the presence of the larger intron structure. Further, unwinding activity was abolished if tertiary contacts were formed between P1 and the intron. This curious result immediately suggested a physical model for the chaperone activity of CYT-19 and other DEAD-box proteins. Chaperones function by first binding nonspecifically to structured RNA, then rearranging RNA structure by unwinding double-stranded regions and perhaps other accessible structures [36]. This general model was also supported by a clever experiment in which the unwinding activity of Ded1p was shown to be enhanced by a ssRNA tethered through a streptavidin linker to a model RNA duplex [44].
These and related results lead to a general model for RNA chaperone function (Figure 1) [40,45]. The proteins do not recognize any specific structural features of their target RNAs; unfolding efficiency is dependent on the accessibility of the target. If a structural element is loosely associated with the rest of the RNA, it is easy to “grab” and unwind. This model elegantly explains the enhanced activity of CYT-19 on misfolded relative to native RNA, as properly folded structure is more compact and will have fewer conformational excursions of its structural elements.
Figure 1.
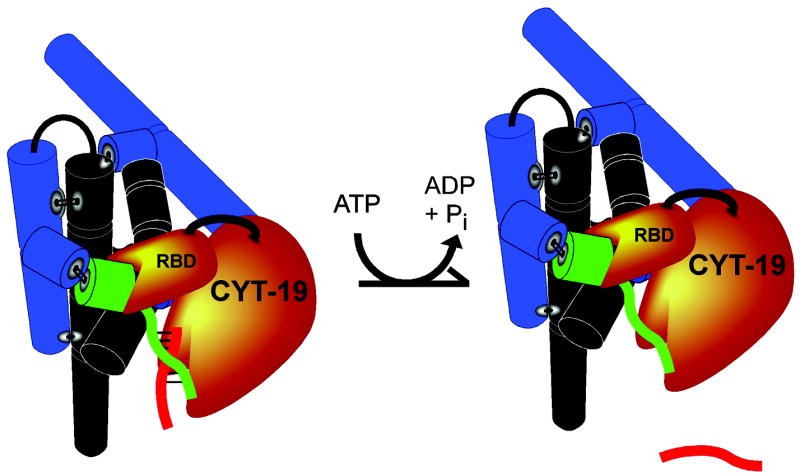
Model for general ATP-driven chaperone activity by CYT-19 and other DEAD-box proteins. CYT-19 first binds to structured RNAs using a binding site (RBD). Binding of CYT-19 then allows for structural rearrangement through local, nonprocessive unwinding of helical elements such as the P1 duplex (green and red strands). In general, the accessibility of the targeted structural element determines CYT-19’s unwinding efficiency. Reprinted from [36]. Copyright © 1993–2008 by The National Academy of Sciences of the United States of America, all rights reserved.
A consequence of non-specific unfolding activity is that DExD/H-box proteins can drive folding away from thermodynamic equilibrium, and such effects have recently been demonstrated for Ded1p and CYT-19 [42,43,46]. These results suggest that, subsequent to each ATP-driven unfolding event, the RNA repartitions between folding forms based on the kinetics of this partitioning rather than the stability of the ultimate folded states. If the stability and accessibility differences between alternative folded states of the RNA are not too large, the chaperone can unfold both species and allow accumulation of a less stable but kinetically preferred state.
The ability of DExD/H-box proteins to perturb RNAs away from thermodynamic equilibrium may also have profound biological implications beyond its implications for the chaperone mechanism. Structured RNAs are involved in complex, multi-step processes, such as pre-mRNA splicing and translation. Although the details of these processes remain elusive, it is plausible that DExD/H-box proteins capture the energy of ATP hydrolysis to drive the transient formation of thermodynamically unstable states that are part of the normal reaction cycle. Although non-specific DEAD-box chaperones could be important for these processes, presumably most of the required structural changes are mediated by dedicated DExD/H-box proteins, which use specific RNA or protein interactions to position themselves within complexes for rearrangement of the desired structural element [47–49].
A future challenge in this area is dissecting the individual reaction steps in complex RNA-mediated processes and unraveling the physical roles played by DExD/H-box proteins. The recently-attained ability to follow pre-mRNA splicing by FRET at the single molecule level represents a technical breakthrough toward this important goal [50].
Simple Forces Underlying the Complex Behavior of RNA
As noted in the Introduction, one of the primary lessons from prior work is that RNA folding is highly complex. However, work from several groups, aimed at understanding the underlying physical properties of RNA and the behavior of RNA components, suggests the feasibility and potential of a “bottom up” approach to RNA folding.
Figure 2 presents a simple hierarchy of forces and features that underlie the folding of all RNAs. Understanding the underlying behavior of each component, from theory or empirical observation, would provide the foundation for describing and understanding of the behavior of complex RNAs. In this section, we explain this hierarchy and emphasize some recent foundational advances in our understanding.
Figure 2.
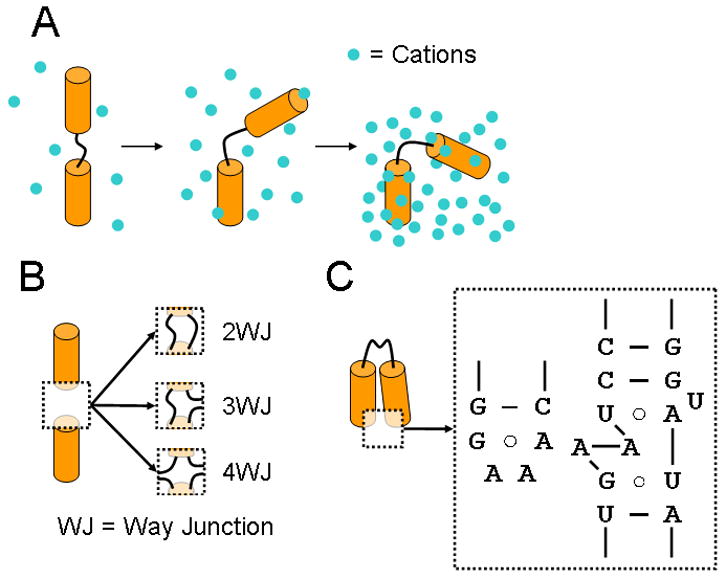
A schematic of the hierarchy of forces that determine RNA folding. Electrostatic repulsion between helices (depicted as cylinders) dictates the energetic cost of forming a compact structure. Electrostatic screening from an associated ion atmosphere (blue dots) mitigates this cost (A). Junctions that join helical regions bias the relative positions of the helices that they join, bringing different regions of secondary structure in close proximity (B). Recurring tertiary motifs bind secondary structure elements together and stabilize the final folded state. Some motifs, such as the tetraloop/tetraloop receptor (whose secondary structure is pictured) are highly conserved and ubiquitous throughout RNA structure (C).
The formation of secondary structure, encoded in RNA sequence, is typically an early step in folding. The stability of RNA secondary structure and the fact that formation of secondary and tertiary structure is often temporally separated simplifies consideration of the folding process [51] (see also [28]). However, RNA secondary structure topology is more complex than the topology of proteins, and understanding this complexity and the folding constraints introduced by secondary structure topology will require advanced modeling and experimental approaches [52,53].
In the absence of other forces, the predominant force on secondary structure helices (depicted in Figure 2 as cylinders) is the enormous inter-helical electrostatic repulsion due to the polyelectrolyte nature of the RNA. Draper has written extensively on the importance of the “ion atmosphere” that surrounds any polyelectrolyte and has noted that the ions in this atmosphere are likely to provide the dominant electrostatic contribution to the folding energetics, far greater than any ions bound to specific sites within the RNA [54,55].
Nevertheless, the ion atmosphere has been a difficult concept for experimentalists to grasp, both because of limited familiarity with the underlying theory and because of our inability to directly visualize this dynamic sheath of ions via x-ray crystallography or other direct approaches. Thus, the recent ability to decipher the shape of this atmosphere by anomalous small angle x-ray scattering provided an unusually direct experimental window into properties of the atmosphere [56,57]. Further, the contents of the atmosphere have recently been quantitatively dissected, by essentially counting the ions associated with a nucleic acid under a variety of ionic conditions. This technique allows RNA systems to be characterized stoichiometrically in terms of the number of each cation and anion species present in the ion atmosphere [58].
The next challenge is the calculation of electrostatic energies, as these energies determine the energetic penalty of assembling helices in densely packed arrangements. Most simply, helices at low ionic concentrations repel; at higher ionic concentration, this repulsion is reduced due to the enhanced screening, allowing helices to approach one another (Figure 2A). However, some have proposed the existence of attractive forces in polyelectrolyte systems under certain ionic conditions that may be responsible for the coalescence of helices in the folded state [59,60].
While simulation experiments and the aggregation of DNA and RNA in ethanol during standard precipitation procedures and other conditions demonstrate the existence of such attractive forces under some conditions, their exact magnitude and significance for folding in vitro was unknown [61,62]. The magnitude of these attractive forces was tested with a simple model: two DNA helices tethered together by a neutral polyethylene glycol tether. Small angle x-ray scattering (SAXS) revealed no significant attractive forces under standard in vitro folding conditions [63]. Thus, the observed “electrostatic collapse” of large RNAs upon addition of ions appears to arise from a screening of repulsive force rather than the action of an attractive one (e.g., [64–66]).
There have also been recent advances in understanding and quantitating electrostatic forces. The predominant electrostatic theory used by biochemists has been Poisson-Boltzmann (PB) theory, a computationally convenient method for calculation of approximate electrostatic forces [67]. PB theory neglects potentially important excluded-volume effects from ion-size and positional correlation effects from ion-ion electrostatic interactions. In particular, correlation effects are predicted to be more important for multivalent ions.
Simulation has suggested that PB theory is inadequate for describing electrostatic energies for nucleic acids in the presence of divalent ions such as Mg2+, a conclusion supported by experiments assessing the effects of ions on DNA duplex stability ([68,69] and references therein). However, the opposite conclusion has been drawn from fitting RNA folding transitions to PB predictions [70–74]. Ion-counting and SAXS experiments have provided a simple test for these effects and have revealed effects of ion size and valence that are not accounted for by PB theory, with deviations up to an order of magnitude in the systems investigated [58,75]. Advances in theory and simulation are required to account for these discrepancies and obtain accurate electrostatic energies for use in dissecting RNA folding and RNA/protein energetics [76,77].
The junctions in secondary structure bias the conformations of the helices they join (Figure 2B). Groundbreaking work by Lilley on Holliday junctions – four-way DNA junctions involved in recombination – and junctions found in structured RNAs has shown that these junctions position their attached helices into preferred conformations [78–80]. Such preferred geometries are integral to the adoption of stable, active structure by functional RNAs. Ha and Lilley have used single molecule FRET to probe the underlying kinetics and thermodynamics of hairpin ribozyme folding, showing that the conformational dynamics of its junction accelerate folding and promote formation of its active site [81]. An important challenge for understanding and predicting RNA structure and dynamics remains to interrogate the behaviors and structures of isolated RNA junctions to reveal their intrinsic behaviors and then how these behaviors determine and are modulated by the rest of the RNA molecule in which they are found. The development of a database of RNA junctions extracted from structured RNAs will undoubtedly help advance this goal [82].
RNAs often utilize motifs for “gluing” RNA secondary structure together to fold to precise tertiary structures (Figure 2C). Long range interactions in RNA are often found by phylogenetic covariation, beginning with tRNA whose tertiary structure was first modeled in 1969 [83]. The tetraloop/tetraloop receptor motif is perhaps the most famous RNA motif. It was first identified by phylogeny in group I and II introns by Michel, Westhof, and colleagues and observed for the first time in the seminal crystal structure of the P4-P6 domain of the Tetrahymena group I intron, in which this motif reinforces the side-by-side packing architecture of RNA helices [84–86]. This motif and others are ubiquitous in the ribosome and other structured RNAs [87–91]. We wish to emphasize that information on motif partners and secondary structure – determined from phylogeny – provides a powerful tool in modeling RNA structure. Westhof has developed and exploited this tool to create impressive first generation three dimensional models for many structured RNAs [92].
Early studies emphasized special roles for Mg2+ in RNA folding and function. However, some RNAs have been shown to fold without Mg2+ [93–95]. Some, such as the hairpin and hammerhead ribozymes, can even function in its absence [96,97]. A recent dissection of folding the P4–P6 domain from the Tetrahymena group I intron has helped clarify the roles of metal ions in RNA folding and, more generally, the underlying energetics of folding [94]. This RNA can adopt a compact shape with its tetraloop motif formed in the presence of monovalent ions alone.
Instead of a special role for Mg2+, folding of P4–P6 and of RNAs in general can be accounted for by a simple balance of forces and interactions outlined in Figure 2. Unfavorable electrostatic repulsion and loss of conformational entropy in the folded state resist folding, whereas junction preferences and tertiary motif formation favor it. Electrostatic forces are modulated by ion concentration and valence; thus, even monovalent cations can mitigate electrostatic repulsion to allow folding to be favorable if present at sufficient concentrations. Fewer divalent ions are needed since they are much more effective at charge screening (localizing a single divalent ion around RNA is entropically less-costly than two monovalents). Specific divalent ion binding sites – if present in the folded RNA –contribute additional stability. The contributions from specifically-bound divalent cations have been isolated by saturating the ion atmosphere with monovalent cations and following the binding of two specific divalent metal ions to the core of the P4-P6 RNA [98,99] (see also [100]).
RNA: Present and Future
We have highlighted some of the conceptual and experimental advances over the past few years and some of the future challenges. In this section, we briefly note some additional methodological advances in the context of challenges that remain in unraveling the structure, energetics and dynamics of RNA.
Single molecule force experiments have far exceeded simple proof-of-principle experiments and have been used to elucidate the folding thermodynamics and kinetics for several RNAs [101–106]. This approach has answered one of the most fundamental questions in nucleic acid structure formation, providing direct evidence for the long-held nucleation model for duplex involving formation of 2–3 base pairs prior to rapid formation of the remainder of the duplex [107]. Further, it has been shown that force unfolding data can be deconstructed into a complete energy landscape [108]. It will be fascinating to see if this approach can be applied to increasingly complex RNAs.
There have been important advances in studying the kinetics of the folding of medium and large RNAs. Time-resolved hydroxyl radical footprinting experiments at millisecond resolution are now routinely conducted using standard rapid-quench devices, without the need for synchrotron radiation to generate radicals [109]. Thus, a large body of kinetic data on RNA folding at the residue-level is readily obtainable. This large body of data can be analyzed in minutes instead of in hours using freely available semi-automated software (https://simtk.org/home/safa) [110]. Still higher-throughput data acquisition is now possible using capillary electrophoresis and a software analysis package called CAFA [111]. Parallel advances have been made with a new and powerful footprinting approach called “SHAPE”, which shows protection data for residues in secondary and tertiary structures [112–114]. New kinetic modeling approaches have greatly simplified and systematized the development of kinetic models from large sets of footprinting data; such an approach has been recently applied to characterize the folding pathways of wild type and mutant Tetrahymena group I intron [115,116].
Early single molecule FRET studies on RNA folding kinetics revealed a wealth of information inaccessible to traditional pre-steady state kinetic methods, demonstrating the utility of these studies [117–119]. Since then, there has been an explosion of single molecule fluorescence studies on RNA [120]. One of the most puzzling observations in these studies is of molecular “memory” – the fact that different molecules tend to display similar kinetic behavior over long time scales that differ from that of other molecules of the same sequence [81,121]. Future work will undoubtedly address the basis for such quixotic behavior by single molecules.
RNA structures remain difficult to obtain relative to protein structures despite the solution of the ribosome structure other RNA structures. Piccirilli and coworkers, following on observations that proteins may help RNA crystallization, have pioneered a potentially high-throughput approach for crystallization of RNA/antibody complexes [122]. Nevertheless, many important RNA structures may be partially or intrinsically unstructured and thus resistant to crystallization, necessitating the development of new approaches to RNA structure. Medium resolution structural information is now obtainable from a method that allows pairwise distance constraints to be read from a single polyacrylamide gel. This method has been used to obtain structural information about a molten globule-like state of the P4–P6 RNA [123]. Also, additional modeling approaches are needed for RNA, and a first pass fragment assembly method, following the highly successful approach of David Baker for protein structure prediction, has been pioneered for RNA [124].
The intrinsically dynamic behavior of RNA that complicates structure determination has also been highly resistant to study. However, recent advances in NMR techniques over the past two years are now providing an unprecedented view into the structural dynamics of RNA [125–127]. Particularly powerful is the ability to determine the amplitude and relative directionality of the internal motion of helical domains through residual dipolar coupling measurements [128,129].
In this review we have emphasized advances in mechanistic and physical understanding using model RNAs of varying complexity. In parallel, the biology community is increasingly recognizing the diverse roles of RNA in basic gene expression and regulation. We suspect that biological and physical approaches to understanding RNA will converge in the coming decade as biological systems are reconstituted and characterized and the tools for understanding RNA are developed and perfected. While there is much work ahead, we eagerly anticipate that union.
Reference Annotations
Of Special Interest (•)
[42] – The authors demonstrate the ability of CYT-19, a DEAD-box protein from Neurospora crassa, to unwind the native form of the Tetrahymena ribozyme, albeit 50-fold less efficiently than the misfolded form. Furthermore, the ATP-driven unfolding activity actively redistributes the conformational ensemble of structured RNAs, populating intermediates that otherwise would be rare at thermodynamic equilibrium
[46] – The authors investigate the activity of Ded1p, a DEAD-box protein, in a model system and observe its ability to populate less stable structures by converting more stable structures to misfolded ones using the energy from ATP hydrolysis.
[82] – The authors present a database of RNA junctions extracted from structural RNA data, offering researchers a centralized collection of data to perform structural analyses and structure design.
[87] – A detailed review about the vastly increased amount of structural data from the ribosome crystal structure and its implications for our understanding of RNA folding, structure, and function
[105] – Using optical tweezers, the authors probe the energetics of the formation and unfolding of a kissing-loop motif. They find that the stability of the kissing-loop interaction is unusually strong
[109] – The authors demonstrate the feasibility of rapid acquisition of structural footprinting data at the millisecond time scale using hydroxyl radicals generated using a chemical source and standard quench-flow mixers. Such a technique makes hydroxyl radical footprinting techniques accessible to the wide scientific community without the use of a synchrotron source.
[115] – The authors present a model for the folding landscape of a group I intron, including folding intermediates and pathways, using the computational analysis of a large body of footprinting data
[122] – RNA structures are more difficult to obtain versus proteins. The authors present a new method for RNA crystallization, based on the idea that protein binding may help chaperone RNA crystal formation. These ideas are demonstrated by the crystallization of the P4-P6 domain of Tetrahymena in complex with an antibody.
Of Outstanding Interest (••)
[36] – The authors elucidate the mechanism for CYT-19, a DEAD-box RNA chaperone, finding that its RNA duplex unwinding action is driven by ATP and enhanced by the presence of surrounding RNA structure. These results led to the proposal of a model for CYT-19’s activity: non-specific binding of the chaperone, followed by structural disruption via local duplex unwinding
[38] – The authors demonstrate the promiscuity of DEAD-box proteins CYT-19 (from Neurospora crassa) and Mss116p (from Saccharomyces cerevisiae) by showing that splicing reactions in Mss116p-deletion mutants can be rescued by expression of CYT-19 in vivo.
[45] – The authors demonstrate that DEAD-box RNA chaperones are qualitatively different from canonical DNA helicases by examining the activity Ded1p and Mss116p. They find that these two DEAD-box proteins unwind RNA helices nonspecifically, acting as local “strand separators”. Their lack of processive or directional translocation along helices makes their unwinding activity distinct from the mechanism of typical DNA helicases
[81] – The authors probe the influence of a naturally-occurring four-way junction in the folding hairpin ribozyme using single molecule spectroscopy demonstrating that its junction accelerates the folding of the molecule and the organization of the active site.
[106] – Using optical tweezers, the authors follow the entire folding trajectory of a simple adenine riboswitch aptamer. With extremely high accuracy, the authors observe the formation of secondary and tertiary structure, as well as the ligand-induced stabilization of the folded aptamer state, resolving the hierarchical folding of this important regulatory ncRNA.
Acknowledgments
The authors thank and apologize to the many investigators in the RNA world whose work has contributed greatly to our overall understanding of RNA but could not be cited due to space limitations. It is truly a testament to this vigorous community that so much has been accomplished of note in the recent years. We would like to thank Yu Bai, Max Greenfeld, Jan Lipfert, Jungwon Park, Adelene Sim, and Sergey Solomatin for critical discussion and insightful comments and all of our collaborators. Our work on RNA folding is supported by NIH grant PO1 GM066275. VBC is supported by a Pfizer Bio-X Graduate fellowship. We have adopted an agnostic view in highlighting references to our own work to avoid the moral dilemma of assigning “significance stars” to our own work.
Footnotes
Conflicts of Interest
The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Daniel Herschlag, Department of Biochemistry, Stanford University, B400, Beckman Center, Stanford, CA 94305, herschla@stanford.edu.
Vincent B. Chu, Department of Applied Physics, Stanford University, GLAM, McCullough 318, 476 Lomita Mall, Stanford, CA 94305, vincentc@stanford.edu
References
- 1.Mandal M, Breaker RR. Gene regulation by riboswitches. Nat Rev Mol Cell Biol. 2004;5:451–463. doi: 10.1038/nrm1403. [DOI] [PubMed] [Google Scholar]
- 2.Pollard KS, Salama SR, Lambert N, Lambot MA, Coppens S, Pedersen JS, Katzman S, King B, Onodera C, Siepel A, et al. An RNA gene expressed during cortical development evolved rapidly in humans. Nature. 2006;443:167–172. doi: 10.1038/nature05113. [DOI] [PubMed] [Google Scholar]
- 3.Eddy SR. Non-coding RNA genes and the modern RNA world. Nat Rev Genet. 2001;2:919–929. doi: 10.1038/35103511. [DOI] [PubMed] [Google Scholar]
- 4.Pan T, Fang X, Sosnick T. Pathway modulation, circular permutation and rapid RNA folding under kinetic control. J Mol Biol. 1999;286:721–731. doi: 10.1006/jmbi.1998.2516. [DOI] [PubMed] [Google Scholar]
- 5.Russell R, Zhuang X, Babcock HP, Millett IS, Doniach S, Chu S, Herschlag D. Exploring the folding landscape of a structured RNA. Proc Natl Acad Sci U S A. 2002;99:155–160. doi: 10.1073/pnas.221593598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thirumalai D, Lee N, Woodson SA, Klimov D. Early events in RNA folding. Annu Rev Phys Chem. 2001;52:751–762. doi: 10.1146/annurev.physchem.52.1.751. [DOI] [PubMed] [Google Scholar]
- 7.Treiber DK, Williamson JR. Exposing the kinetic traps in RNA folding. Curr Opin Struct Biol. 1999;9:339–345. doi: 10.1016/S0959-440X(99)80045-1. [DOI] [PubMed] [Google Scholar]
- 8.Gartland WJ, Sueoka N. Two interconvertible forms of tryptophanyl sRNA in E. coli. Proc Natl Acad Sci U S A. 1966;55:948–956. doi: 10.1073/pnas.55.4.948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adams A, Lindahl T, Fresco JR. Conformational differences between the biologically active and inactive forms of a transfer ribonucleic acid. Proc Natl Acad Sci U S A. 1967;57:1684–1691. doi: 10.1073/pnas.57.6.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ishida T, Sueoka N. Effect of ambient conditions on conformations of tryptophan transfer ribonucleic acid of Escherichia coli. J Biol Chem. 1968;243:5329–5336. [PubMed] [Google Scholar]
- 11.Russell R, Das R, Suh H, Travers KJ, Laederach A, Engelhardt MA, Herschlag D. The paradoxical behavior of a highly structured misfolded intermediate in RNA folding. J Mol Biol. 2006;363:531–544. doi: 10.1016/j.jmb.2006.08.024. [DOI] [PubMed] [Google Scholar]
- 12.Sigler PB. An analysis of the structure of tRNA. Annu Rev Biophys Bioeng. 1975;4:477–527. doi: 10.1146/annurev.bb.04.060175.002401. [DOI] [PubMed] [Google Scholar]
- 13.Herschlag D. RNA chaperones and the RNA folding problem. J Biol Chem. 1995;270:20871–20874. doi: 10.1074/jbc.270.36.20871. [DOI] [PubMed] [Google Scholar]
- 14.Karpel RL, Swistel DG, Miller NS, Geroch ME, Lu C, Fresco JR. Acceleration of RNA renaturation by nucleic acid unwinding proteins. Brookhaven Symp Biol. 1975:165–174. [PubMed] [Google Scholar]
- 15.Karpel RL, Miller NS, Fresco JR. Mechanistic studies of ribonucleic acid renaturation by a helix-destabilizing protein. Biochemistry. 1982;21:2102–2108. doi: 10.1021/bi00538a019. [DOI] [PubMed] [Google Scholar]
- 16.Rein A, Henderson LE, Levin JG. Nucleic-acid-chaperone activity of retroviral nucleocapsid proteins: significance for viral replication. Trends Biochem Sci. 1998;23:297–301. doi: 10.1016/s0968-0004(98)01256-0. [DOI] [PubMed] [Google Scholar]
- 17.Lorsch JR. RNA chaperones exist and DEAD box proteins get a life. Cell. 2002;109:797–800. doi: 10.1016/s0092-8674(02)00804-8. [DOI] [PubMed] [Google Scholar]
- 18.Mohr S, Stryker JM, Lambowitz AM. A DEAD-box protein functions as an ATP-dependent RNA chaperone in group I intron splicing. Cell. 2002;109:769–779. doi: 10.1016/s0092-8674(02)00771-7. [DOI] [PubMed] [Google Scholar]
- 19.Schroeder R, Barta A, Semrad K. Strategies for RNA folding and assembly. Nat Rev Mol Cell Biol. 2004;5:908–919. doi: 10.1038/nrm1497. [DOI] [PubMed] [Google Scholar]
- 20.Collins K. The biogenesis and regulation of telomerase holoenzymes. Nat Rev Mol Cell Biol. 2006;7:484–494. doi: 10.1038/nrm1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williamson JR. Assembly of the 30S ribosomal subunit. Q Rev Biophys. 2005;38:397–403. doi: 10.1017/S0033583506004264. [DOI] [PubMed] [Google Scholar]
- 22.Lipfert J, Doniach S. Small-angle X-ray scattering from RNA, proteins, and protein complexes. Annu Rev Biophys Biomol Struct. 2007;36:307–327. doi: 10.1146/annurev.biophys.36.040306.132655. [DOI] [PubMed] [Google Scholar]
- 23.Jankowsky E, Fairman ME. RNA helicases--one fold for many functions. Curr Opin Struct Biol. 2007;17:316–324. doi: 10.1016/j.sbi.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 24.Hammann C, Westhof E. Searching genomes for ribozymes and riboswitches. Genome Biol. 2007;8:210. doi: 10.1186/gb-2007-8-4-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoogstraten CG, Sumita M. Structure-function relationships in RNA and RNP enzymes: recent advances. Biopolymers. 2007;87:317–328. doi: 10.1002/bip.20836. [DOI] [PubMed] [Google Scholar]
- 26.Cristofari G, Darlix JL. The ubiquitous nature of RNA chaperone proteins. Prog Nucleic Acid Res Mol Biol. 2002;72:223–268. doi: 10.1016/s0079-6603(02)72071-0. [DOI] [PubMed] [Google Scholar]
- 27.Wolin SL, Cedervall T. The La protein. Annu Rev Biochem. 2002;71:375–403. doi: 10.1146/annurev.biochem.71.090501.150003. [DOI] [PubMed] [Google Scholar]
- 28.Pan T, Sosnick T. RNA folding during transcription. Annu Rev Biophys Biomol Struct. 2006;35:161–175. doi: 10.1146/annurev.biophys.35.040405.102053. [DOI] [PubMed] [Google Scholar]
- 29.Russell R, Tijerina P, Chadee AB, Bhaskaran H. Deletion of the P5abc peripheral element accelerates early and late folding steps of the Tetrahymena group I ribozyme. Biochemistry. 2007;46:4951–4961. doi: 10.1021/bi0620149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Treiber DK, Rook MS, Zarrinkar PP, Williamson JR. Kinetic intermediates trapped by native interactions in RNA folding. Science. 1998;279:1943–1946. doi: 10.1126/science.279.5358.1943. [DOI] [PubMed] [Google Scholar]
- 31.Doherty EA, Herschlag D, Doudna JA. Assembly of an exceptionally stable RNA tertiary interface in a group I ribozyme. Biochemistry. 1999;38:2982–2990. doi: 10.1021/bi982113p. [DOI] [PubMed] [Google Scholar]
- 32.Engelhardt MA, Doherty EA, Knitt DS, Doudna JA, Herschlag D. The P5abc peripheral element facilitates preorganization of the tetrahymena group I ribozyme for catalysis. Biochemistry. 2000;39:2639–2651. doi: 10.1021/bi992313g. [DOI] [PubMed] [Google Scholar]
- 33.Tanner NK, Linder P. DExD/H box RNA helicases: from generic motors to specific dissociation functions. Mol Cell. 2001;8:251–262. doi: 10.1016/s1097-2765(01)00329-x. [DOI] [PubMed] [Google Scholar]
- 34.Staley JP, Guthrie C. Mechanical devices of the spliceosome: motors, clocks, springs, and things. Cell. 1998;92:315–326. doi: 10.1016/s0092-8674(00)80925-3. [DOI] [PubMed] [Google Scholar]
- 35.Mayas RM, Staley JP. DEAD on. Nat Struct Mol Biol. 2006;13:954–955. doi: 10.1038/nsmb1106-954. [DOI] [PubMed] [Google Scholar]
- 36.Tijerina P, Bhaskaran H, Russell R. Nonspecific binding to structured RNA and preferential unwinding of an exposed helix by the CYT-19 protein, a DEAD-box RNA chaperone. Proc Natl Acad Sci U S A. 2006;103:16698–16703. doi: 10.1073/pnas.0603127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mohr S, Matsuura M, Perlman PS, Lambowitz AM. A DEAD-box protein alone promotes group II intron splicing and reverse splicing by acting as an RNA chaperone. Proc Natl Acad Sci U S A. 2006;103:3569–3574. doi: 10.1073/pnas.0600332103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang HR, Rowe CE, Mohr S, Jiang Y, Lambowitz AM, Perlman PS. The splicing of yeast mitochondrial group I and group II introns requires a DEAD-box protein with RNA chaperone function. Proc Natl Acad Sci U S A. 2005;102:163–168. doi: 10.1073/pnas.0407896101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Halls C, Mohr S, Del Campo M, Yang Q, Jankowsky E, Lambowitz AM. Involvement of DEAD-box proteins in group I and group II intron splicing. Biochemical characterization of Mss116p, ATP hydrolysis-dependent and -independent mechanisms, and general RNA chaperone activity. J Mol Biol. 2007;365:835–855. doi: 10.1016/j.jmb.2006.09.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grohman JK, Del Campo M, Bhaskaran H, Tijerina P, Lambowitz AM, Russell R. Probing the mechanisms of DEAD-box proteins as general RNA chaperones: the C-terminal domain of CYT-19 mediates general recognition of RNA. Biochemistry. 2007;46:3013–3022. doi: 10.1021/bi0619472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang Q, Jankowsky E. ATP- and ADP-dependent modulation of RNA unwinding and strand annealing activities by the DEAD-box protein DED1. Biochemistry. 2005;44:13591–13601. doi: 10.1021/bi0508946. [DOI] [PubMed] [Google Scholar]
- 42.Bhaskaran H, Russell R. Kinetic redistribution of native and misfolded RNAs by a DEAD-box chaperone. Nature. 2007;449:1014–1018. doi: 10.1038/nature06235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jankowsky E. Biochemistry: indifferent chaperones. Nature. 2007;449:999–1000. doi: 10.1038/449999a. [DOI] [PubMed] [Google Scholar]
- 44.Yang Q, Jankowsky E. The DEAD-box protein Ded1 unwinds RNA duplexes by a mode distinct from translocating helicases. Nat Struct Mol Biol. 2006;13:981–986. doi: 10.1038/nsmb1165. [DOI] [PubMed] [Google Scholar]
- 45.Yang Q, Del Campo M, Lambowitz AM, Jankowsky E. DEAD-box proteins unwind duplexes by local strand separation. Mol Cell. 2007;28:253–263. doi: 10.1016/j.molcel.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 46.Yang Q, Fairman ME, Jankowsky E. DEAD-box-protein-assisted RNA structure conversion towards and against thermodynamic equilibrium values. J Mol Biol. 2007;368:1087–1100. doi: 10.1016/j.jmb.2007.02.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Karginov FV, Caruthers JM, Hu Y, McKay DB, Uhlenbeck OC. YxiN is a modular protein combining a DEx(D/H) core and a specific RNA-binding domain. J Biol Chem. 2005;280:35499–35505. doi: 10.1074/jbc.M506815200. [DOI] [PubMed] [Google Scholar]
- 48.Silverman E, Edwalds-Gilbert G, Lin RJ. DExD/H-box proteins and their partners: helping RNA helicases unwind. Gene. 2003;312:1–16. doi: 10.1016/s0378-1119(03)00626-7. [DOI] [PubMed] [Google Scholar]
- 49.Wang S, Hu Y, Overgaard MT, Karginov FV, Uhlenbeck OC, McKay DB. The domain of the Bacillus subtilis DEAD-box helicase YxiN that is responsible for specific binding of 23S rRNA has an RNA recognition motif fold. RNA. 2006;12:959–967. doi: 10.1261/rna.5906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crawford DJ, Hoskins AA, Friedman LJ, Gelles J, Moore MJ. Visualizing the splicing of single pre-mRNA molecules in whole cell extract. RNA. 2008;14:170–179. doi: 10.1261/rna.794808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brion P, Westhof E. Hierarchy and dynamics of RNA folding. Annu Rev Biophys Biomol Struct. 1997;26:113–137. doi: 10.1146/annurev.biophys.26.1.113. [DOI] [PubMed] [Google Scholar]
- 52.Cao S, Chen SJ. Predicting RNA folding thermodynamics with a reduced chain representation model. RNA. 2005;11:1884–1897. doi: 10.1261/rna.2109105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cao S, Chen SJ. Biphasic folding kinetics of RNA pseudoknots and telomerase RNA activity. J Mol Biol. 2007;367:909–924. doi: 10.1016/j.jmb.2007.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Draper DE, Grilley D, Soto AM. Ions and RNA folding. Annu Rev Biophys Biomol Struct. 2005;34:221–243. doi: 10.1146/annurev.biophys.34.040204.144511. [DOI] [PubMed] [Google Scholar]
- 55.Draper DE. A guide to ions and RNA structure. RNA. 2004;10:335–343. doi: 10.1261/rna.5205404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Andresen K, Das R, Park HY, Smith H, Kwok LW, Lamb JS, Kirkland EJ, Herschlag D, Finkelstein KD, Pollack L. Spatial distribution of competing ions around DNA in solution. Phys Rev Lett. 2004;93:248103. doi: 10.1103/PhysRevLett.93.248103. [DOI] [PubMed] [Google Scholar]
- 57.Das R, Mills TT, Kwok LW, Maskel GS, Millett IS, Doniach S, Finkelstein KD, Herschlag D, Pollack L. Counterion distribution around DNA probed by solution X-ray scattering. Phys Rev Lett. 2003;90:188103. doi: 10.1103/PhysRevLett.90.188103. [DOI] [PubMed] [Google Scholar]
- 58.Bai Y, Greenfeld M, Travers KJ, Chu VB, Lipfert J, Doniach S, Herschlag D. Quantitative and comprehensive decomposition of the ion atmosphere around nucleic acids. J Am Chem Soc. 2007;129:14981–14988. doi: 10.1021/ja075020g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Heilman-Miller SL, Thirumalai D, Woodson SA. Role of counterion condensation in folding of the Tetrahymena ribozyme. I Equilibrium stabilization by cations. J Mol Biol. 2001;306:1157–1166. doi: 10.1006/jmbi.2001.4437. [DOI] [PubMed] [Google Scholar]
- 60.Murthy VL, Rose GD. Is counterion delocalization responsible for collapse in RNA folding? Biochemistry. 2000;39:14365–14370. doi: 10.1021/bi001820r. [DOI] [PubMed] [Google Scholar]
- 61.Grosberg AY, Nguyen TT, Shklovskii BI. Colloquium: The physics of charge inversion in chemical and biological systems. Rev Mod Phys. 2002;74:329–345. [Google Scholar]
- 62.Bloomfield VA. DNA condensation by multivalent cations. Biopolymers. 1997;44:269–282. doi: 10.1002/(SICI)1097-0282(1997)44:3<269::AID-BIP6>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 63.Bai Y, Das R, Millett IS, Herschlag D, Doniach S. Probing counterion modulated repulsion and attraction between nucleic acid duplexes in solution. Proc Natl Acad Sci U S A. 2005;102:1035–1040. doi: 10.1073/pnas.0404448102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Buchmueller KL, Webb AE, Richardson DA, Weeks KM. A collapsed non-native RNA folding state. Nat Struct Biol. 2000;7:362–366. doi: 10.1038/75125. [DOI] [PubMed] [Google Scholar]
- 65.Das R, Kwok LW, Millett IS, Bai Y, Mills TT, Jacob J, Maskel GS, Seifert S, Mochrie SG, Thiyagarajan P, et al. The fastest global events in RNA folding: electrostatic relaxation and tertiary collapse of the Tetrahymena ribozyme. J Mol Biol. 2003;332:311–319. doi: 10.1016/s0022-2836(03)00854-4. [DOI] [PubMed] [Google Scholar]
- 66.Russell R, Millett IS, Tate MW, Kwok LW, Nakatani B, Gruner SM, Mochrie SG, Pande V, Doniach S, Herschlag D, et al. Rapid compaction during RNA folding. Proc Natl Acad Sci U S A. 2002;99:4266–4271. doi: 10.1073/pnas.072589599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Honig B, Nicholls A. Classical electrostatics in biology and chemistry. Science. 1995;268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- 68.Tan ZJ, Chen SJ. Nucleic acid helix stability: effects of salt concentration, cation valence and size, and chain length. Biophys J. 2006;90:1175–1190. doi: 10.1529/biophysj.105.070904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tan ZJ, Chen SJ. RNA helix stability in mixed Na+/Mg2+ solution. Biophys J. 2007;92:3615–3632. doi: 10.1529/biophysj.106.100388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Misra VK, Draper DE. The linkage between magnesium binding and RNA folding. J Mol Biol. 2002;317:507–521. doi: 10.1006/jmbi.2002.5422. [DOI] [PubMed] [Google Scholar]
- 71.Misra VK, Shiman R, Draper DE. A thermodynamic framework for the magnesium-dependent folding of RNA. Biopolymers. 2003;69:118–136. doi: 10.1002/bip.10353. [DOI] [PubMed] [Google Scholar]
- 72.Soto AM, Misra V, Draper DE. Tertiary structure of an RNA pseudoknot is stabilized by "diffuse" Mg2+ ions. Biochemistry. 2007;46:2973–2983. doi: 10.1021/bi0616753. [DOI] [PubMed] [Google Scholar]
- 73.Grilley D, Soto AM, Draper DE. Mg2+-RNA interaction free energies and their relationship to the folding of RNA tertiary structures. Proc Natl Acad Sci U S A. 2006;103:14003–14008. doi: 10.1073/pnas.0606409103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Grilley D, Misra V, Caliskan G, Draper DE. Importance of partially unfolded conformations for Mg(2+)-induced folding of RNA tertiary structure: structural models and free energies of Mg2+ interactions. Biochemistry. 2007;46:10266–10278. doi: 10.1021/bi062284r. [DOI] [PubMed] [Google Scholar]
- 75.Bai Y, Chu VB, Lipfert J, Pande VS, Herschlag D, Doniach S. Experimental and computational reconstruction of the unfolded state ensemble in nucleic acid folding. 2008 doi: 10.1021/ja800854u. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chu VB, Bai Y, Lipfert J, Herschlag D, Doniach S. Evaluation of ion binding to DNA duplexes using a size-modified Poisson-Boltzmann theory. Biophys J. 2007;93:3202–3209. doi: 10.1529/biophysj.106.099168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tan ZJ, Chen SJ. Electrostatic correlations and fluctuations for ion binding to a finite length polyelectrolyte. J Chem Phys. 2005;122:44903. doi: 10.1063/1.1842059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lilley DM. Structures of helical junctions in nucleic acids. Q Rev Biophys. 2000;33:109–159. doi: 10.1017/s0033583500003590. [DOI] [PubMed] [Google Scholar]
- 79.Hohng S, Wilson TJ, Tan E, Clegg RM, Lilley DM, Ha T. Conformational flexibility of four-way junctions in RNA. J Mol Biol. 2004;336:69–79. doi: 10.1016/j.jmb.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 80.Walter F, Murchie AI, Lilley DM. Folding of the four-way RNA junction of the hairpin ribozyme. Biochemistry. 1998;37:17629–17636. doi: 10.1021/bi9821115. [DOI] [PubMed] [Google Scholar]
- 81.Tan E, Wilson TJ, Nahas MK, Clegg RM, Lilley DM, Ha T. A four-way junction accelerates hairpin ribozyme folding via a discrete intermediate. Proc Natl Acad Sci U S A. 2003;100:9308–9313. doi: 10.1073/pnas.1233536100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bindewald E, Hayes R, Yingling YG, Kasprzak W, Shapiro BA. RNAJunction: a database of RNA junctions and kissing loops for three-dimensional structural analysis and nanodesign. Nucleic Acids Res. 2008;36:D392–397. doi: 10.1093/nar/gkm842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Levitt M. Detailed molecular model for transfer ribonucleic acid. Nature. 1969;224:759–763. doi: 10.1038/224759a0. [DOI] [PubMed] [Google Scholar]
- 84.Costa M, Michel F. Frequent use of the same tertiary motif by self-folding RNAs. EMBO J. 1995;14:1276–1285. doi: 10.1002/j.1460-2075.1995.tb07111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Michel F, Westhof E. Modelling of the three-dimensional architecture of group I catalytic introns based on comparative sequence analysis. J Mol Biol. 1990;216:585–610. doi: 10.1016/0022-2836(90)90386-Z. [DOI] [PubMed] [Google Scholar]
- 86.Cate JH, Gooding AR, Podell E, Zhou K, Golden BL, Kundrot CE, Cech TR, Doudna JA. Crystal structure of a group I ribozyme domain: principles of RNA packing. Science. 1996;273:1678–1685. doi: 10.1126/science.273.5282.1678. [DOI] [PubMed] [Google Scholar]
- 87.Noller HF. RNA structure: reading the ribosome. Science. 2005;309:1508–1514. doi: 10.1126/science.1111771. [DOI] [PubMed] [Google Scholar]
- 88.Golden BL, Kim H, Chase E. Crystal structure of a phage Twort group I ribozyme-product complex. Nat Struct Mol Biol. 2005;12:82–89. doi: 10.1038/nsmb868. [DOI] [PubMed] [Google Scholar]
- 89.Torres-Larios A, Swinger KK, Krasilnikov AS, Pan T, Mondragon A. Crystal structure of the RNA component of bacterial ribonuclease P. Nature. 2005;437:584–587. doi: 10.1038/nature04074. [DOI] [PubMed] [Google Scholar]
- 90.Doherty EA, Batey RT, Masquida B, Doudna JA. A universal mode of helix packing in RNA. Nat Struct Biol. 2001;8:339–343. doi: 10.1038/86221. [DOI] [PubMed] [Google Scholar]
- 91.Nissen P, Ippolito JA, Ban N, Moore PB, Steitz TA. RNA tertiary interactions in the large ribosomal subunit: the A-minor motif. Proc Natl Acad Sci U S A. 2001;98:4899–4903. doi: 10.1073/pnas.081082398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lehnert V, Jaeger L, Michel F, Westhof E. New loop-loop tertiary interactions in self-splicing introns of subgroup IC and ID: a complete 3D model of the Tetrahymena thermophila ribozyme. Chem Biol. 1996;3:993–1009. doi: 10.1016/s1074-5521(96)90166-0. [DOI] [PubMed] [Google Scholar]
- 93.Rangan P, Woodson SA. Structural requirement for Mg2+ binding in the group I intron core. J Mol Biol. 2003;329:229–238. doi: 10.1016/s0022-2836(03)00430-3. [DOI] [PubMed] [Google Scholar]
- 94.Takamoto K, Das R, He Q, Doniach S, Brenowitz M, Herschlag D, Chance MR. Principles of RNA compaction: insights from the equilibrium folding pathway of the P4-P6 RNA domain in monovalent cations. J Mol Biol. 2004;343:1195–1206. doi: 10.1016/j.jmb.2004.08.080. [DOI] [PubMed] [Google Scholar]
- 95.Takamoto K, He Q, Morris S, Chance MR, Brenowitz M. Monovalent cations mediate formation of native tertiary structure of the Tetrahymena thermophila ribozyme. Nat Struct Biol. 2002;9:928–933. doi: 10.1038/nsb871. [DOI] [PubMed] [Google Scholar]
- 96.Nesbitt S, Hegg LA, Fedor MJ. An unusual pH-independent and metal-ion-independent mechanism for hairpin ribozyme catalysis. Chem Biol. 1997;4:619–630. doi: 10.1016/s1074-5521(97)90247-7. [DOI] [PubMed] [Google Scholar]
- 97.Murray JB, Seyhan AA, Walter NG, Burke JM, Scott WG. The hammerhead, hairpin and VS ribozymes are catalytically proficient in monovalent cations alone. Chem Biol. 1998;5:587–595. doi: 10.1016/s1074-5521(98)90116-8. [DOI] [PubMed] [Google Scholar]
- 98.Travers KJ, Boyd N, Herschlag D. Low specificity of metal ion binding in the metal ion core of a folded RNA. RNA. 2007;13:1205–1213. doi: 10.1261/rna.566007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Das R, Travers KJ, Bai Y, Herschlag D. Determining the Mg2+ stoichiometry for folding an RNA metal ion core. J Am Chem Soc. 2005;127:8272–8273. doi: 10.1021/ja051422h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bukhman YV, Draper DE. Affinities and selectivities of divalent cation binding sites within an RNA tertiary structure. J Mol Biol. 1997;273:1020–1031. doi: 10.1006/jmbi.1997.1383. [DOI] [PubMed] [Google Scholar]
- 101.Vieregg J, Cheng W, Bustamante C, Tinoco I., Jr Measurement of the effect of monovalent cations on RNA hairpin stability. J Am Chem Soc. 2007;129:14966–14973. doi: 10.1021/ja074809o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tinoco I, Jr, Li PT, Bustamante C. Determination of thermodynamics and kinetics of RNA reactions by force. Q Rev Biophys. 2006;39:325–360. doi: 10.1017/S0033583506004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li PT, Collin D, Smith SB, Bustamante C, Tinoco I., Jr Probing the mechanical folding kinetics of TAR RNA by hopping, force-jump, and force-ramp methods. Biophys J. 2006;90:250–260. doi: 10.1529/biophysj.105.068049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Li PT, Bustamante C, Tinoco I., Jr Real-time control of the energy landscape by force directs the folding of RNA molecules. Proc Natl Acad Sci U S A. 2007;104:7039–7044. doi: 10.1073/pnas.0702137104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Li PT, Bustamante C, Tinoco I., Jr Unusual mechanical stability of a minimal RNA kissing complex. Proc Natl Acad Sci U S A. 2006;103:15847–15852. doi: 10.1073/pnas.0607202103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Greenleaf WJ, Frieda KL, Foster DA, Woodside MT, Block SM. Direct observation of hierarchical folding in single riboswitch aptamers. Science. 2008;319:630–633. doi: 10.1126/science.1151298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Woodside MT, Behnke-Parks WM, Larizadeh K, Travers K, Herschlag D, Block SM. Nanomechanical measurements of the sequence-dependent folding landscapes of single nucleic acid hairpins. Proc Natl Acad Sci U S A. 2006;103:6190–6195. doi: 10.1073/pnas.0511048103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Woodside MT, Anthony PC, Behnke-Parks WM, Larizadeh K, Herschlag D, Block SM. Direct measurement of the full, sequence-dependent folding landscape of a nucleic acid. Science. 2006;314:1001–1004. doi: 10.1126/science.1133601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shcherbakova I, Mitra S, Beer RH, Brenowitz M. Fast Fenton footprinting: a laboratory-based method for the time-resolved analysis of DNA, RNA and proteins. Nucleic Acids Res. 2006;34:e48. doi: 10.1093/nar/gkl055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Das R, Laederach A, Pearlman SM, Herschlag D, Altman RB. SAFA: semiautomated footprinting analysis software for high-throughput quantification of nucleic acid footprinting experiments. RNA. 2005;11:344–354. doi: 10.1261/rna.7214405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mitra S, Shcherbakova I, Altman RB, Brenowitz M, Laederach A. High-throughput single-nucleotide structural map-ping by CAFA (Capillary Automated Footprinting Analysis) In Preparation. 2008 doi: 10.1093/nar/gkn267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wilkinson KA, Merino EJ, Weeks KM. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution. Nat Protoc. 2006;1:1610–1616. doi: 10.1038/nprot.2006.249. [DOI] [PubMed] [Google Scholar]
- 113.Wilkinson KA, Merino EJ, Weeks KM. RNA SHAPE chemistry reveals nonhierarchical interactions dominate equilibrium structural transitions in tRNA(Asp) transcripts. J Am Chem Soc. 2005;127:4659–4667. doi: 10.1021/ja0436749. [DOI] [PubMed] [Google Scholar]
- 114.Merino EJ, Wilkinson KA, Coughlan JL, Weeks KM. RNA structure analysis at single nucleotide resolution by selective 2′-hydroxyl acylation and primer extension (SHAPE) J Am Chem Soc. 2005;127:4223–4231. doi: 10.1021/ja043822v. [DOI] [PubMed] [Google Scholar]
- 115.Laederach A, Shcherbakova I, Jonikas MA, Altman RB, Brenowitz M. Distinct contribution of electrostatics, initial conformational ensemble, and macromolecular stability in RNA folding. Proc Natl Acad Sci U S A. 2007;104:7045–7050. doi: 10.1073/pnas.0608765104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Laederach A, Shcherbakova I, Liang MP, Brenowitz M, Altman RB. Local kinetic measures of macromolecular structure reveal partitioning among multiple parallel pathways from the earliest steps in the folding of a large RNA molecule. J Mol Biol. 2006;358:1179–1190. doi: 10.1016/j.jmb.2006.02.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ha T, Zhuang X, Kim HD, Orr JW, Williamson JR, Chu S. Ligand-induced conformational changes observed in single RNA molecules. Proc Natl Acad Sci U S A. 1999;96:9077–9082. doi: 10.1073/pnas.96.16.9077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kim HD, Nienhaus GU, Ha T, Orr JW, Williamson JR, Chu S. Mg2+-dependent conformational change of RNA studied by fluorescence correlation and FRET on immobilized single molecules. Proc Natl Acad Sci U S A. 2002;99:4284–4289. doi: 10.1073/pnas.032077799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhuang X, Bartley LE, Babcock HP, Russell R, Ha T, Herschlag D, Chu S. A single-molecule study of RNA catalysis and folding. Science. 2000;288:2048–2051. doi: 10.1126/science.288.5473.2048. [DOI] [PubMed] [Google Scholar]
- 120.Zhuang X. Single-molecule RNA science. Annu Rev Biophys Biomol Struct. 2005;34:399–414. doi: 10.1146/annurev.biophys.34.040204.144641. [DOI] [PubMed] [Google Scholar]
- 121.Zhuang X, Kim H, Pereira MJ, Babcock HP, Walter NG, Chu S. Correlating structural dynamics and function in single ribozyme molecules. Science. 2002;296:1473–1476. doi: 10.1126/science.1069013. [DOI] [PubMed] [Google Scholar]
- 122.Ye JD, Tereshko V, Frederiksen JK, Koide A, Fellouse FA, Sidhu SS, Koide S, Kossiakoff AA, Piccirilli JA. Synthetic antibodies for specific recognition and crystallization of structured RNA. Proc Natl Acad Sci U S A. 2008;105:82–87. doi: 10.1073/pnas.0709082105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Das R, Kudaravalli M, Jonikas MA, Laederach A, Fong R, Schwans JP, Baker D, Piccirilli JA, Altman RB, Herschlag D. Visualizing Multiple Conformational States of a Large RNA with High-Throughput Contact Mapping. Proc Natl Acad Sci U S A. 2008 doi: 10.1073/pnas.0709032105. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Das R, Baker D. Automated de novo prediction of native-like RNA tertiary structures. Proc Natl Acad Sci U S A. 2007;104:14664–14669. doi: 10.1073/pnas.0703836104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Getz M, Sun X, Casiano-Negroni A, Zhang Q, Al-Hashimi HM. NMR studies of RNA dynamics and structural plasticity using NMR residual dipolar couplings. Biopolymers. 2007;86:384–402. doi: 10.1002/bip.20765. [DOI] [PubMed] [Google Scholar]
- 126.Shajani Z, Varani G. NMR studies of dynamics in RNA and DNA by 13C relaxation. Biopolymers. 2007;86:348–359. doi: 10.1002/bip.20650. [DOI] [PubMed] [Google Scholar]
- 127.Zhang Q, Sun X, Watt ED, Al-Hashimi HM. Resolving the motional modes that code for RNA adaptation. Science. 2006;311:653–656. doi: 10.1126/science.1119488. [DOI] [PubMed] [Google Scholar]
- 128.Getz MM, Andrews AJ, Fierke CA, Al-Hashimi HM. Structural plasticity and Mg2+ binding properties of RNase P P4 from combined analysis of NMR residual dipolar couplings and motionally decoupled spin relaxation. Rna. 2007;13:251–266. doi: 10.1261/rna.264207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sun X, Zhang Q, Al-Hashimi HM. Resolving fast and slow motions in the internal loop containing stem-loop 1 of HIV-1 that are modulated by Mg2+ binding: role in the kissing-duplex structural transition. Nucleic Acids Res. 2007;35:1698–1713. doi: 10.1093/nar/gkm020. [DOI] [PMC free article] [PubMed] [Google Scholar]