

Abstract
Nitric oxide (NO) has been invoked in nearly every normal and pathological condition associated with human physiology. In tumor biology, nitrogen oxides have both positive and negative affects as they have been implicated in both promoting and preventing cancer. Our work has focused on NO chemistry and how it correlates with cytotoxicity and cancer. Toward this end, we have studied both concentration- and time-dependent NO regulation of specific signaling pathways in response to defined nitrosative stress levels that may occur within the tumor microenvironment. Threshold levels of NO required for activation and stabilization of key proteins involved in carcinogenesis including p53, ERK, Akt, and HIF have been identified. Importantly, threshold NO levels are further influenced by reactive oxygen species (ROS) including superoxide, which can shift or attenuate NO-mediated signaling as observed in both tumor and endothelial cells. Our studies have been extended to determine levels of NO that are critical during angiogenic response through regulation of the anti-angiogenic agent thrombospondin-1 (TSP-1) and pro-angiogenic agent matrix metalloproteinase-9 (MMP-9). The quantification of redox events at the cellular level has revealed potential mechanisms that may either limit or potentiate tumor growth, and helped define the positive and negative function of nitric oxide in cancer.
Keywords: nitric oxide, cancer, nitrosative stress
Introduction
Does nitric oxide (NO) promote or inhibit carcinogenesis? The role of NO in cancer has been studied for more than forty years, yet this fundamental question remains unanswered. Earlier studies found that NO was a critical component of the immune response of macrophages [1, 2]. Subsequent studies have shown that iNOS deletions can lead to the development of several kinds of cancer [3]. In contrast, other studies have shown a role for iNOS in the promotion of tumorigenesis. Recent studies have shown a positive correlation between iNOS and poor prognosis for breast cancer and melanoma patients [4-6]. Together, these observations suggest that NO generated by iNOS elicits multiple physiologic and pathologic effects.
Several important aspects of NO must be considered when examining biological outcome, which is dictated by the chemistry of this diatomic radical. The mechanisms involved in cancer and cancer treatment are diverse and the effect of NO depends on the tumor microenvironment as well as its concentration, spatial, and temporal constraints [7]. Over the last several years, a new picture of NO has emerged where the concentration of this radical can dictate the phenotypic response [8]. Here we discuss the importance of concentration and temporal dependence as they pertain to the role of NO in different aspects of tumor biology.
NO Signaling and Concentration Dependence
Characterizing steady state NO is of particular importance when assessing its effects at the cellular level [9, 10]. MCF-7 breast cancer cells were found to activate specific signaling pathways in response to distinct fluxes of NO. Levels below 50 nM NO were associated with increased cGMP-mediated ERK phosphorylation, intermediate levels (>100 nM) lead to HIF-1α stabilization, and high NO (>300 nM) was associated with p53-P(ser-15), which persisted even after dissipation of NO [10]. These phenotypic responses favor a pro-growth and anti-apoptotic paradigm at steady state NO levels at or below 100nM. However, the pro-survival effects of NO are lost at concentrations above 400nM, which is signified by increases in phosphorylation and acetylation of p53 and the induction of p53 tumor suppressor activity [10-12]. This signaling profile was mimicked by activated macrophages co-cultured with MCF-7 cells at varying ratios [10]. These data indicate that NO released by tumor-associated macrophages can regulate tumor cell responses in vivo. Indeed, tumor phagocyte density and aberrant p53 expression correlated significantly with the phosphorylation of Akt at ser-473 in human breast cancer tissue [4]. Similarly, a strong correlation between p53-P-(ser-15) and iNOS protein expression in human samples of ulcerative colitis provided further evidence that NO causes a p53 pathway activation in vivo [11].
Other proteins including MKP-1, a phosphatase that regulates pERK, also increases at or above 400nM steady state NO [13, 14]). Higher NO levels (>1 _M) are associated with increased nitrosation and nitrosative stress [8]. Moreover, inhibition of proteins including DNA repair enzymes and zinc finger complexes occur at these stress levels [15].
Another important signaling aspect involves the temporal properties of NO. Though NO is short lived, the sustained NO flux generated by NOS can vary in duration from seconds to days. NO mediated HIF-1α stabilization correlates directly with concentration and time, and requires the presence of NO [10]. While pERK increases immediately in response to NO, it also transiently decreases despite the maintenance of steady state NO levels. Moreover, NO-induced p53 phosphorylation remains stably elevated even after the dissipation of steady state NO. Thus, signaling responses to NO are temporally and spatially defined [10].
Several sources of nitric oxide exist in the immune system. Macrophages perform a wide variety of functions from fighting bacteria and tumors to coordinating wound healing and tissue remodeling. Macrophages can generate a number of different levels of NO that mediate a wide range of functions. For instance, low steady state NO released by eNOS activates guanylyl cyclase in the resting macrophage, which is critical during cytokine activation [16]. Moreover, macrophage activating agents demonstrate significant variation in NO output. For example, IFN-γ pretreated macrophages that are stimulated with TNF-α or IL-1β release nearly 10 times less NO than they do after stimulation of Toll-like receptors (e.g. LPS, PIPC and Listeria) [17]. Taken together, macrophages provide an example of a cell type with large variations in NO output upon stimulation by the environment.
MMP and TIMP regulation is an additional example of the regulatory capacity of threshold NO produced by macrophages. MMPs are important mediators of wound healing, while both MMPs and TIMPs have been linked to cancer progression. A recent report demonstrated biphasic NO regulation of MMPs expressed in macrophages via biological (TIMP-1 suppression) and chemical (RNS-mediated activation/inactivation) mechanisms [18]. NO levels ranging from 30-100 nM were associated with increased MMP activity secreted from murine macrophages through the cGMP-mediated suppression of the endogenous TIMP-1 inhibitor. Higher NO levels were associated with inhibition of MMP activity that most likely occurred through the chemical reaction of RNS. This chemical inhibition occurred in IFN-γ/LPS stimulated macrophages as well as by high NO donor concentrations. Similarly, in an epithelial wound model low steady state NO (17.5 nM) up-regulated MMP-9 expression, which declined as NO levels increased [19]. This biphasic regulatory trend may at least in part account for the complexities of how macrophages differentially respond to the high NO microenvironment at the inflammatory site versus the NO levels present during resolution of inflammation and the initiation of wound healing [20].
Inflammation and Wound Healing in Cancer Therapy
While it is clear that NO elicits direct radiosensitizing effects on DNA within a tumor, modulation of wound responses by NO is an additional mechanism for the potentiation of chemo and radiation therapies. Although molecular mechanisms regulating growth differ among cancer types, exposure to radiation, surgery, and/or chemotherapy enhances cell death followed by the onset of inflammation and wound healing. These processes are similar to that of a wound response in normal tissue where inflammatory cells infiltrate the wound followed by chemotactic attraction of fibroblasts, lymphocytes, and endothelial cells. These cell types participate in matrix remodeling and neovascularization processes for the restoration of tissue architecture and perfusion capabilities [21]. Indeed, it has been postulated that tumor associated macrophages (TAMs) recognize tumors as persistent, nonhealing wounds [22]. Moreover, tumor growth is analogous to wound repair as both involve the formation of new tissue and angiogenesis in response to similar local signals including cytokines and proangiogenic factors [22]. Because NOS knockout animals demonstrate impaired wound response, NO modulation via NOS inhibition coupled with radiation and/or drugs aimed at specific targets may be therapeutically beneficial. Modulation of these basic properties, particularly antigenic response has been proposed [8].
Several studies have recently been published which show that patient 5 year survival decreases with enhanced iNOS expression in the tumor [4, 6]. These and other studies also suggest a link between inflammation, COX-2 and iNOS expression in the tumor microenvironment. As stated above the wound healing process is considerably impaired in the iNOS knockout when compared to wild type [23]. It has been suggested that NO levels, which increase HIF-1α ultimately increase VEGF following exposure to ionizing radiation [24]. This leads to increased angiogenesis and an enhanced regrowth of the tumor. Thus application of NOS inhibitors could be beneficial because of the inhibition of macrophages and factors released by them that stimulate tumor growth.
Three cell types (tumor, endothelial, and macrophage) are affected by NO. As discussed in the previous section, tumor cells have quite different NO concentration requirements to cause either a pro-growth or an anti-growth phenotype. Akt and ERK phosphorylation events provide a pro-growth and anti-apoptotic phenotype. Prolonged exposure of human breast cancer cells to NO using the long acting donor DETA/NO (30-60μM), led to increased pAkt and pERK [25]. Moreover, the interruption of ERK and Akt phosphorylation inhibited proliferation of these cells. However, prolonged exposure of breast cancer cells to 1mM DETA/NO for an extended period of time lead to MKP1-mediated pERK and pAkt dephosphorylation with subsequent induction of apoptosis. However, it may not be the induction of tumor cell apoptosis that is most significant in NO-induced tumor regression; instead, the induction of apoptosis in the surrounding stromal tissue may be at least equally as important.
Macrophages coordinate many processes during inflammation and wound response. In a Th1 environment, increased production of Th1 cytokines increase tumor inflammatory processes that sterilize the region by removal of debris. During this process both iNOS and COX-2 expression is enhanced. While iNOS upregulation increases HIF-1α and VEGF [26], COX2 upregulation has been found to increase VEGF via PGE2 and cAMP in some tumor models [27]. Also, NO activation of p53 increases IL- 10 production from Treg cells and activates latent TGF-β all of which is necessary to down regulate Th1 for tissue restoration [28, 29].
An important area of NO regulation involves formation of blood vessels via angiogenic processes. Enhanced angiogenesis prior to radiation or drug treatment can increase the therapeutic efficacy by enhanced oxygen effect or enhanced drug delivery. In contrast, inhibition of angiogenesis post treatment can delay tumor regrowth by suppression of oxygen and nutrient delivery. Several reports have shown eNOS phosphorylation by different pro-angiogenic factors thereby generating a prolonged flux of NO, which leads to increased cGMP mediated angiogenic response [30]. Some antiangiogenic factors inhibit eNOS phosphorylation by increasing PP2A the phosphatase for eNOS serine 1179 [30]. Interestingly, the endogenous angiogenesis inhibitor TSP-1 inhibits eNOS activation via its receptor CD36, inhibits cGMP production by soluble guanylyl cyclase via its receptor CD47, and inhibits its target cGMP-dependent protein kinase [31-33]. Conversely, TSP-1 expression is suppressed by NO at concentrations that are at or below our ability to detect [14]. NO-mediated TSP-1 suppression requires ERK phosphorylation and is inhibited by the MEK1/2 inhibitor. Thus NO/cGMP suppression of TSP-1 involves the pro-growth ERK signaling pathway. However, as steady state NO levels continue to increase, TSP-1 begins to re-accumulate in the media. This may be associated, at least in part, with increased MKP-1 mediated dephosphorylation of ERK and is consistent with the demonstration of growth inhibition or cytostasis by higher steady state levels of NO.
Summary
When NO effects in mammalian cells are grouped according to the concentration of NO, a pro- and anti-tumorigenic response emerges (Figure 1). Increased cGMP promotes angiogenesis and proliferation of endothelial cells and occurs between 1-30 nM [14]. At this level, ERK phosphorylation stimulates proliferation of endothelial cells while suppressing the anti-angiogenic factor TSP-1 [14]. At NO concentrations of 30-100 nM, a marked increase in Akt and ERK signals a proliferative and anti-apoptotic response in tumor cells [4, 10, 13]. In addition, in macrophages, TIMP-1 is suppressed while MMP-9 activity increases [34]. This concentration range of NO appears to protect the tumor cell from apoptosis while increasing angiogenic effects. At 100 nM, HIF-1α is stabilized thereby increasing VEGF [10, 35]. These three levels of NO are protumorigenic and many of the molecules that are activated by NO at these concentrations (i.e. Akt-P, HIF-1 α and MMP-9) have been identified as poor prognostic indicators. Above 300 nM, increased phosphorylation of p53 and expression of MKP1 occurs and cellular respiration is inhibited [10, 13, 14]. These higher levels of NO promote apoptosis and are likely responsible for the anti-tumor activity first observed in macrophages. Sustained fluxes of NO > 0.5 _M can result in nitrosation of thiols, tyrosines, and also amines, which can be genotoxic.
Figure 1.
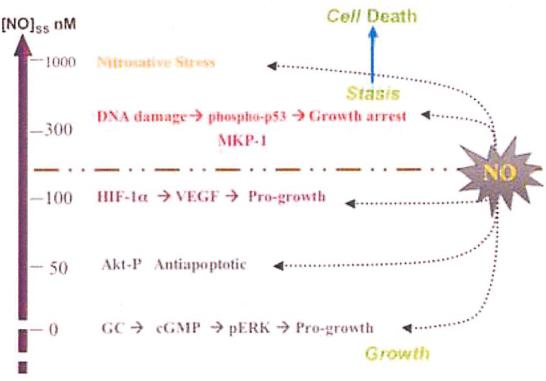
Correlation between NO concentration and molecular mechanisms of cell proliferation and death.
An interesting observation is that superoxide and ROS through the scavenging of NO can convert an anti-tumor concentration to a pro-growth paradigm [36]. Tumors generally exhibit increased superoxide and ROS via NADPH oxidase (NOX) and decreased MnSOD. Within this tumor microenvironment, NO scavenging by ROS and superoxide may provide an important mechanism to abate macrophage mediated tumor cell killing. While mechanisms in cancer are complex, undoubtedly the role(s) of NO will continue to expand. In the process, numerous scenarios are beginning to emerge, which indicate that NO concentration may indeed dictate phenotypic responses and a group of molecular mechanisms that together define a pro- and/or anti-tumorigenic tumor response.
Acknowledgements
This work is supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, and Center for Cancer Research.
References
- 1.Hibbs JBJ. Synthesis of nitric oxide from L-arginine: a recently discovered pathway induced by cytokines with antitumour and antimicrobial activity. Res Immunol. 1991;142:565–569. doi: 10.1016/0923-2494(91)90103-p. [DOI] [PubMed] [Google Scholar]
- 2.Stuehr DJ, Nathan CF. A macrophage product responsible for cytostasis and respiratory inhibition in tumor target cells. J. Exp. Med. 1989;169:1543–1555. doi: 10.1084/jem.169.5.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hussain SP, Trivers GE, Hofseth LJ, He P, Shaikh I, Mechanic LE, Doja S, Jiang W, Subleski J, Shorts L, Haines D, Laubach VE, Wiltrout RH, Djurickovic D, Harris CC. Nitric oxide, a mediator of inflammation, suppresses tumorigenesis. Cancer Res. 2004;64:6849–6853. doi: 10.1158/0008-5472.CAN-04-2201. [DOI] [PubMed] [Google Scholar]
- 4.Prueitt RL, Boersma BJ, Howe TM, Goodman JE, Thomas DD, Ying L, Pfiester CM, Yfantis HG, Cottrell JR, Lee DH, Remaley AT, Hofseth LJ, Wink DA, Ambs S. Inflammation and IGF-I activate the Akt pathway in breast cancer. Int J Cancer. 2007;120:796–805. doi: 10.1002/ijc.22336. [DOI] [PubMed] [Google Scholar]
- 5.Ekmekcioglu S, Ellerhorst J, Smid CM, Prieto VG, Munsell M, Buzaid AC, Grimm EA. Inducible nitric oxide synthase and nitrotyrosine in human metastatic melanoma tumors correlate with poor survival. Clin Cancer Res. 2000;6:4768–4775. [PubMed] [Google Scholar]
- 6.Ekmekcioglu S, Ellerhorst JA, Prieto VG, Johnson MM, Broemeling LD, Grimm EA. Tumor iNOS predicts poor survival for stage III melanoma patients. Int J Cancer. 2006;119:861–866. doi: 10.1002/ijc.21767. [DOI] [PubMed] [Google Scholar]
- 7.Wink DA, Mitchell JB. Chemical biology of nitric oxide: Insights into regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radic Biol Med. 1998;25:434–456. doi: 10.1016/s0891-5849(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 8.Ridnour LA, Thomas DD, Donzelli S, Espey MG, Roberts DD, Wink DA, Isenberg JS. The biphasic nature of nitric oxide responses in tumor biology. Antioxidant Redox Signal. 2006;8:1329–1337. doi: 10.1089/ars.2006.8.1329. [DOI] [PubMed] [Google Scholar]
- 9.Zhou J, Schmid T, Brune B. HIF-1alpha and p53 as targets of NO in affecting cell proliferation, death and adaptation. Curr Mol Med. 2004;4:741–751. doi: 10.2174/1566524043359926. [DOI] [PubMed] [Google Scholar]
- 10.Thomas DD, Espey MG, Ridnour LA, Hofseth LJ, Mancardi D, Harris CC, Wink DA. Hypoxic inducible factor 1alpha, extracellular signal-regulated kinase, and p53 are regulated by distinct threshold concentrations of nitric oxide. Proc Natl Acad Sci U S A. 2004;101:8894–8899. doi: 10.1073/pnas.0400453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hofseth LJ, Saito S, Hussain SP, Espey MG, Miranda KM, Araki Y, Jhappan C, Higashimoto Y, He P, Linke SP, Quezado MM, Zurer I, Rotter V, Wink DA, Appella E, Harris CC. Nitric oxide-induced cellular stress and p53 activation in chronic inflammation. Proc Natl Acad Sci U S A. 2003;100:143–148. doi: 10.1073/pnas.0237083100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang C, Trudel LJ, Wogan GN, Deen WM. Thresholds of nitric oxide-mediated toxicity in human lymphoblastoid cells. Chem Res Toxicol. 2003;16:1004–1013. doi: 10.1021/tx0340448. [DOI] [PubMed] [Google Scholar]
- 13.Pervin S, Singh R, Freije WA, Chaudhuri G. MKP-1-induced dephosphorylation of extracellular signal-regulated kinase is essential for triggering nitric oxide-induced apoptosis in human breast cancer cell lines: implications in breast cancer. Cancer Res. 2003;63:8853–8860. [PubMed] [Google Scholar]
- 14.Ridnour LA, Isenberg JS, Espey MG, Thomas DD, Roberts DD, Wink DA. Nitric oxide regulates angiogenesis through a functional switch involving thrombospondin-1. Proc Natl Acad Sci U S A. 2005;102:13147–13152. doi: 10.1073/pnas.0502979102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wink DA, Mitchell JB. The Chemical Biology of Nitric Oxide: Insights into Regulatory, Cytotoxic and Cytoprotective Mechanisms of Nitric Oxide. Free Rad. Biol. Med. 1998;25:434–456. doi: 10.1016/s0891-5849(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 16.Connelly L, Jacobs AT, Palacios-Callender M, Moncada S, Hobbs AJ. Macrophage endothelial nitric-oxide synthase autoregulates cellular activation and pro-inflammatory protein expression. J Biol Chem. 2003;278:26480–26487. doi: 10.1074/jbc.M302238200. [DOI] [PubMed] [Google Scholar]
- 17.Espey MG, Miranda KM, Pluta RM, Wink DA. Nitrosative capacity of macrophages is dependent on nitric-oxide synthase induction signals. J. Biol. Chem. 2000;275:11341–11347. doi: 10.1074/jbc.275.15.11341. [DOI] [PubMed] [Google Scholar]
- 18.Ridnour LA, Windhausen AN, Isenberg JS, Yeung N, Thomas DD, Vitek MP, Roberts DD, Wink DA. Nitric oxide regulates matrix metalloproteinase-9 activity by guanylyl-cyclase-dependent and -independent pathways. Proc Natl Acad Sci U S A. 2007:16898–903. doi: 10.1073/pnas.0702761104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bove PF, Wesley UV, Greul AK, Hristova M, Dostmann WR, van der Vliet A. Nitric oxide promotes airway epithelial wound repair through enhanced activation of MMP-9. Am J Respir Cell Mol Biol. 2007;36:138–146. doi: 10.1165/rcmb.2006-0253SM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duffield JS. The inflammatory macrophage: a story of Jekyll and Hyde. Clin Sci (Lond) 2003:27–38. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- 21.Crowther M, Brown NJ, Bishop ET, Lewis CE. Microenvironmental influence on macrophage regulation of angiogenesis in wounds and malignant tumors. J Leukoc Biol. 2001;70:478–490. [PubMed] [Google Scholar]
- 22.Whalen GF. Solid tumours and wounds: transformed cells misunderstood as injured tissue? Lancet. 1990;336:1489–1492. doi: 10.1016/0140-6736(90)93188-u. [DOI] [PubMed] [Google Scholar]
- 23.Schwentker A, Vodovotz Y, Weller R, Billiar TR. Nitric oxide and wound repair: role of cytokines? Nitric Oxide. 2002;7:1–10. doi: 10.1016/s1089-8603(02)00002-2. [DOI] [PubMed] [Google Scholar]
- 24.Li F, Sonveaux P, Rabbani ZN, Liu S, Yan B, Huang Q, Vujaskovic Z, Dewhirst MW, Li CY. Regulation of HIF-1alpha stability through S-nitrosylation. Mol Cell. 2007;26:63–74. doi: 10.1016/j.molcel.2007.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pervin S, Singh R, Hernandez E, Wu G, Chaudhuri G. Nitric oxide in physiologic concentrations targets the translational machinery to increase the proliferation of human breast cancer cells: involvement of mammalian target of rapamycin/eIF4E pathway. Cancer Res. 2007;67:289–299. doi: 10.1158/0008-5472.CAN-05-4623. [DOI] [PubMed] [Google Scholar]
- 26.Griffiths EA, Pritchard SA, Welch IM, Price PM, West CM. Is the hypoxia-inducible factor pathway important in gastric cancer. Eur J Cancer. 2005;41:2792–2805. doi: 10.1016/j.ejca.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 27.Sonoshita M, Takaku K, Sasaki N, Sugimoto Y, Ushikubi F, Narumiya S, Oshima M, Taketo MM. Acceleration of intestinal polyposis through prostaglandin receptor EP2 in Apc(Delta 716) knockout mice. Nat Med. 2001;7:1048–1051. doi: 10.1038/nm0901-1048. [DOI] [PubMed] [Google Scholar]
- 28.Niedbala W, Cai B, Liu H, Pitman N, Chang L, Liew FY. Nitric oxide induces CD4+CD25+ Foxp3 regulatory T cells from CD4+CD25 T cells via p53, IL-2, and OX40. Proc Natl Acad Sci U S A. 2007;104:15478–15483. doi: 10.1073/pnas.0703725104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vodovotz Y, Chesler L, Chong H, Kim SJ, Simpson JT, DeGraff W, Cox GW, Roberts AB, Wink DA, Barcellos-Hoff MH. Regulation of transforming growth factor beta1 by nitric oxide. Cancer Res. 1999;59:2142–2149. [PubMed] [Google Scholar]
- 30.Roberts DD, Isenberg JS, Ridnour LA, Wink DA. Nitric oxide and its gatekeeper thrombospondin-1 in tumor angiogenesis. Clin Cancer Res. 2007;13:795–798. doi: 10.1158/1078-0432.CCR-06-1758. [DOI] [PubMed] [Google Scholar]
- 31.Isenberg JS, Ridnour LA, Perruccio EM, Espey MG, Wink DA, Roberts DD. Thrombospondin-1 inhibits endothelial cell responses to nitric oxide in a cGMP-dependent manner. Proc Natl Acad Sci U S A. 2005;102:13141–13146. doi: 10.1073/pnas.0502977102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Isenberg JS, Ridnour LA, Dimitry J, Frazier WA, Wink DA, Roberts DD. CD47 is necessary for inhibition of nitric oxide-stimulated vascular cell responses by thrombospondin-1. J Biol Chem. 2006;281:26069–26080. doi: 10.1074/jbc.M605040200. [DOI] [PubMed] [Google Scholar]
- 33.Isenberg JS, Romeo MJ, Yu C, Yu CK, Nghiem K, Monsale J, Rick ME, Wink DA, Frazier WA, Roberts DD. Thrombospondin-1 stimulates platelet aggregation by blocking the antithrombotic activity of nitric oxide/cGMP signaling. Blood. 2008;111:613–623. doi: 10.1182/blood-2007-06-098392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ridnour LA, Windhausen AN, Isenberg JS, Yeung N, Thomas DD, Vitek MP, Roberts DD, Wink DA. Nitric oxide regulates matrix metalloproteinase-9 activity by guanylyl-cyclase-dependent and -independent pathways. Proc Natl Acad Sci U S A. 2007;104:16898–16903. doi: 10.1073/pnas.0702761104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Metzen E, Zhou J, Jelkmann W, Fandrey J, Brune B. Nitric Oxide Impairs Normoxic Degradation of HIF-1{alpha} by Inhibition of Prolyl Hydroxylases. Mol Biol Cell. 2003;14:3470–3481. doi: 10.1091/mbc.E02-12-0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thomas DD, Ridnour LA, Espey MG, Donzelli S, Ambs S, Hussain SP, Harris CC, DeGraff W, Roberts DD, Mitchell JB, Wink DA. Superoxide fluxes limit nitric oxide-induced signaling. J Biol Chem. 2006;281:25984–25993. doi: 10.1074/jbc.M602242200. [DOI] [PubMed] [Google Scholar]