

Abstract
Mithramycin is an antitumor drug produced by Streptomyces argillaceus. It consists of a tricyclic aglycone and five deoxyhexoses that form a disaccharide and a trisaccharide chain, which are important for target interaction and therefore for the antitumor activity. Using a combinatorial biosynthesis approach, we have generated nine mithramycin derivatives, seven of which are new compounds, with alterations in the glycosylation pattern. The wild-type S. argillaceus strain and the mutant S. argillaceus M7U1, which has altered d-oliose biosynthesis, were used as hosts to express various “sugar plasmids”, each one directing the biosynthesis of a different deoxyhexose. The newly formed compounds were purified and characterized by MS and NMR. Compared to mithramycin, they contained different sugar substitutions in the second (d-olivose, d-mycarose, or d-boivinose instead of d-oliose) and third (d-digitoxose instead of d-mycarose) sugar units of the trisaccharide as well as in the first (d-amicetose instead of d-olivose) sugar unit of the disaccharide. All compounds showed antitumor activity against different tumor cell lines. Structure–activity relationships are discussed on the basis of the number and type of deoxyhexoses present in these mithramycin derivatives.
Keywords: aureolic acid, deoxysugars, polyketides, Streptomyces
Introduction
Many bioactive compounds are glycosylated by mono- or oligosaccharide chains. Most of these sugars are 6-deoxyhexoses (6-DOHs) derived from glucose-1-phosphate. These sugar moieties are important for the bioactivity of the compounds and they participate in the interaction with the cell target. Consequently changing the sugar profile of these compounds can result in more active compounds or compounds with improved pharmacological properties. Sugar profile modifications can be approached by chemical synthesis, chemoenzymatically, or by combinatorial biosynthesis.[1-4] Mithramycin (1, Scheme 1 A) is an antitumor antibiotic of the aureolic acid family of antitumor compounds,[5] which has been used clinically to treat hypercalcemia in patients with bone metastases from various malignancies, Paget's disease, and several types of cancer, including testicular carcinoma, chronic myeloid leukemia, and acute myeloid leukemia. Recently, it has been found that mithramycin could be used in combination with bevacizumab, a neutralizing antibody against VEGF (vascular endothelial growth factor), as a novel antiangiogenic therapy for pancreatic cancer and other cancers.[6] Furthermore, it has been shown that mithramycin acts as a neuroprotective drug and it has potential application for the alleviation of symptoms underlying β-thalassemia and sickle cell anemia.[7,8] The mechanism of action of mithramycin involves its noncovalently binding to GC-rich nucleotide sequences located in the minor groove of DNA.[9-13] This binding is carried out by complexes of dimers of mithramycin together with bivalent cations such as Mg2+ , with the chromophores parallel to the sugar-phosphate backbone and the saccharide chains partially wrapping the DNA minor groove.[9,10] During these interactions, several H bonds are created among the chromophore hydroxyl groups with the guanine amino protons determining the selectivity for GC-rich sequences.[9,10] As a consequence of this sequence selectivity, mithramycin blocks the binding of proteins to GC-rich sequences in gene promoters, and inhibits transcription of genes regulated by these factors, such as those regulated by the Sp1 family of transcription factors.[14-17] Recent studies carried out with mithramycin–FeII complexes suggest that these complexes promote single-stranded breakage of DNA by producing reactive hydroxyl radicals in the presence of H2O2[18] Structurally, mithramycin consists of a tricyclic chromophore (aglycone) with two aliphatic side chains attached at C3 and C7, and trisaccharide (d-olivose-d-oliose-d-mycarose) and di-saccharide (d-olivose-d-olivose) chains attached at positions two and six of the aglycone, respectively.[19] The aglycone is biosynthesized through the condensation of one acetyl-CoA and nine malonyl-CoA units. The resultant carbon chain is then aromatized, cyclized, oxygenated, and methylated to render the tetracyclic intermediate premithramycinone (compound 2, Scheme 1 A).[20] After this, five consecutive glycosylation steps occur (together with a further C-methylation step). First, the trisaccharide chain is formed by the sequential action of glycosyltransferases MtmGIV and MtmGIII, which are responsible for the transfer of the first and second sugar residues, respectively.[21] No glycosyltransferase has been identified for the attachment of the third sugar. However, indirect evidence suggests that MtmGIV might be responsible for the transfer of the third sugar.[22] Formation of the disaccharide requires the sequential action of glycosyltransferases MtmGI and MtmGII.[23,24] These glycosylation and methylation steps render a fully glycosylated tetracyclic compound, premithramycin B. This compound has low cytotoxic activity and its conversion by the action of oxygenase MtmOIV into a tricyclic compound by the oxidative cleavage of the fourth ring is a key event in mithramycin biosynthesis, as it results in the formation of intermediates with high cytotoxic activities.[25-27]
Scheme 1.

Chemical structures of A) mithramycin (1), premithramycinone (2), premithramycin A1 (3), and mithramycin derivatives with altered glycosylation profiles produced by recombinant strains of B) S. argillaceus wild-type and C) S. argillaceus M7U1. Demycarosyl-mithramycin (4), dideolivosyl-6-β-d-amicetosyl-mithramycin (5), demycarosyl-3D-β-d-digitoxosyl-mithramycin (6), deoliosyl-demycarosyl-3C-β-d-boivinosyl-mithramycin (7), deoliosyl-3C-β-d-mycarosyl-mithramycin (8), deoliosyl-demycarosyl-3C-β-d-olivosyl-3D-β-d-digitoxosyl-mithramycin (9), dideolivosyl-6-β-d-amicetosyl-deoliosyl-3C-β-d-olivosyl-mithramycin (10), dideolivosyl-6-β-d-amicetosyl-deoliosyl-demycarosyl-3C-β-d-olivosyl-3D-β-d-digitoxosyl-mithramycin (11), and dideolivosyl-6-β-d-amicetosyl-deoliosyl-demycarosyl-3C-β-d-boivinosyl-mithramycin (12). Structural differences from mithramycin are highlighted with gray circles.
Sugars in mithramycin seem to play an important role in its activity, participating in the binding process of mithramycin to DNA.[28-29] Consequently, changes in the sugar profile of mithramycin would probably modify the antitumor activities of the compound. In this paper we report the use of combinatorial biosynthesis of sugar biosynthesis genes to generate novel mithramycins with differences in the type and position of sugars in the saccharide chains. Using this approach nine mithramycin derivatives with antitumor activity were produced, seven of which were new compounds.
Results
Modifying the sugar biosynthesis profile of S. argillaceus to produce novel glycosylated mithramycins
In order to generate new derivatives of mithramycin with novel saccharide profiles, the mithramycin producer strain was transformed independently with different plasmids able to direct the biosynthesis of different DOHs. The plasmids and genes used are mentioned in Table 1.[30-33] The rationale behind this experiment was that by introducing these high copy number plasmids containing DOH biosynthesis gene clusters to S. argillaceus, either synthesis of already synthesized DOHs will be facilitated (d-olivose, d-oliose, or d-mycarose) or novel DOHs could be produced in the recombinant strains. Assuming the possible existence of substrate flexibility for the mithramycin glycosyltransferases, one could expect the production of novel mithramycin derivatives with altered glycosylated profiles. As four different glycosyltransferases are involved in the biosynthesis of mithramycin,[21,23] the new mithramycins could contain not only new sugars but also sugars located at different positions of the disaccharide and trisaccharide chains.
Table 1.
Plasmids used for the biosynthesis of deoxysugars.
| Plasmid | Genes | Deoxysugar | Ref. |
|---|---|---|---|
| pLN2Δ | oleU,oleY, oleL,oleS,oleE | dTDP-l-rhamnose | [30] |
| pLN2 | oleV,oleW,oleU,oleY,oleL,oleS,oleE | dTDP-l-olivose | [30] |
| pLNRHO |
oleV,oleW,urdZ3,oleY,oleL,oleS,oleE, urdQ |
dTDP-l-rhodinose | [30] |
| pFL844 |
oleV,oleW,eryBIV,oleY,oleL,oleS,oleE, urdQ |
dTDP-l-amicetose | [32] |
| pFL845 | oleV,oleW,urdR,oleY,oleL,oleS,oleE,urdQ | dTDP-d-amicetose | [32] |
| pFL942 |
mtmE,mtmD,oleV,eryBII,eryBIV,eryBIII, eryBVII |
dTDP-l-mycarose | [31] |
| pFL947 |
mtmE,mtmD,oleV,oleW,eryBIV,mtmC, eryBVII |
dTDP-l-chromose B | [31] |
| pMP1*UI | mtmE,mtmD,oleV,oleW,cmmUI,oleY | dTDP-d-olivose | [33] |
| pMP1*UII | mtmE,mtmD,oleV,oleW,cmmUII,oleY | dTDP-d-oliose | [33] |
| pMP3*BII | mtmE,mtmD,oleV,eryBII,urdR,oleY | dTDP-d-digitoxose | [33] |
| pMP1*BII | mtmE,mtmD,oleV,eryBII,oleU,oleY | dTDP-d-boivinose | [33] |
Two types of compounds were expected to be formed. On one hand, tetracyclic compounds (premithramycin-like) known to have low activity, and on the other hand tricyclic compounds (mithramycin-like), known to be highly active. It was therefore convenient to have a simple way to differentiate between these types of compounds to focus our attention on the highly active mithramycin-like compounds. We took advantage of the absorption spectrum of both types of compounds: although they absorb in similar regions of the UV/Vis spectrum they have slightly different absorption maxima (230, 278, 317, and 411 nm for mithramycin-like compounds versus 232, 282, 329, and 424 nm for premithramycin-like compounds).
After introduction of various “sugar plasmids” in the mithramycin producer, selected transformants were cultivated on R5 A medium in the presence of thiostrepton and cultures were extracted with ethyl acetate. The organic extracts were analyzed by HPLC (Figure 1) and HPLC-MS. From the analysis of the chromatograms we classified the plasmids into three different groups based on the generated compounds: 1) Plasmids not altering the glycosylation profile: expression of some “sugar plasmids” (pLNRHO, pFL947, pMP1*UI, and pMP1*UII) did not result in the formation of new glycosylated mithramycins. A chromatogram corresponding to the strain S. argillaceus (pFL947) is shown in Figure 1 A as a representative example of this group; 2) Plasmids inducing formation of premithramycin-like compounds: expression of some “sugar plasmids” (pLN2Δ and pLN2) resulted in the formation of some new premithramycin-like compounds. S. argillaceus (pLN2Δ) produced a premithramycin-like compound with an m/z value in positive mode of 559 (indicated by an empty asterisk in Figure 1 B). This is in agreement with a premithramycinone aglycone with a mycarose moiety attached. Compounds produced by S. argillaceus (pLN2) (indicated by two empty asterisks in Figure 1 C) showed an m/z value of 675 for both, which is in accordance with premithramycinone aglycones with two units of 2,6-DOHs attached in each. Most probably they will correspond to novel compounds containing one or two l-olivose moieties; 3) Plasmids inducing formation of mithramycin-like compounds: a third group of “sugar plasmids” (pFL844, pFL942, pMP1*BII, pMP3*BII, and pFL845) led to the formation of mithramycin-like compounds (indicated by full asterisks in Figure 1 D–H). Mass analyses of these compounds confirmed that they contain a tricyclic aglycone, most of them harboring four or five DOHs attached.
Figure 1.

HPLC analyses of cultures of S. argillaceus recombinant strains harboring different “sugar plasmids”. ✩ Premithramycin-like compounds. ★ Mithramycin-like compounds. Peaks corresponding to the different compounds are indicated as follows: M: mithramycin; A: premithramycin A1; P: premithramycinone; peaks a–i: mithramycin derivatives purified from the recombinant strains.
Some of these compounds were selected for further purification and characterization. In total we analyzed nine peaks (indicated in Figure 1 by the letters a to i) from different recombinant strains. NMR and MS structure analyses (Supporting Information) showed that those peaks corresponded to the following compounds (Scheme 1B): demycarosyl-mithramycin (peaks a and d; compound 4); dideolivosyl-6-β-d-amicetosyl-mithramycin (peaks b and c; compound 5); demycarosyl-3D-β-d-digitoxosyl-mithramycin (peaks e, g, and i; compound 6); deoliosyl-demycarosyl-3C-β-d-boivinosyl-mithramycin (peaks f and h; compound 7). Three out of these four compounds are new compounds (compounds 4, 5, and 7) whereas compound 6 has been recently described.[34]
The analysis of the structures in relation to the plasmids harbored in the different recombinant strains showed up three different situations. Firstly, some of the compounds contained one DOH whose biosynthesis is encoded by the “sugar plasmid” used. This was the case for compounds 5, 6, and 7, which contain a d-amicetose, a d-digitoxose, or a d-boivinose residue respectively, and those sugars are encoded by pFL845, pMP3*BII, and pMP1*BII, respectively. Secondly, some compounds contain DOHs that were synthesized by the concerted action of enzymes encoded by the “sugar plasmid” and enzymes from the mithramycin sugars biosynthetic machinery. This was the case of compound 5 when produced by S. argillaceus (pFL844). In this case, formation of d-amicetose can be explained by the action of a 4-ketoreductase from the host, either MtmC involved in d-olivose biosynthesis[35] or the MtmTIII involved in d-mycarose biosynthesis,[22] on the intermediate NDP-4-keto-2,3,6-trideoxyglucose (compound a; Scheme 2 A). A similar situation can explain formation of compound 6 isolated from two different strains harboring pMP1*BII or pFL942. In both cases formation of d-digitoxose can be explained by the action of a ketoreductase, either MtmC or MtmTIII on the intermediate NDP-2,6-dideoxy-d-glycero-4-hexulose (compound b; Scheme 2 A). In the case of compound 7, formation of d-boivinose would result from the action of 4-ketoreductase MtmU on the intermediate NDP-2,6-dideoxy-d-glycero-4-hexulose (compound b; Scheme 2 A). MtmU is a 4-ketoreductase involved in the biosynthesis of d-oliose.[35] Thirdly, in one case there is no alteration in the sugar profile but rather one of the usual mithramycin sugar is missing. This is the case of compound 4, which lacks the d-mycarose residue in the trisaccharide chain.
Scheme 2.
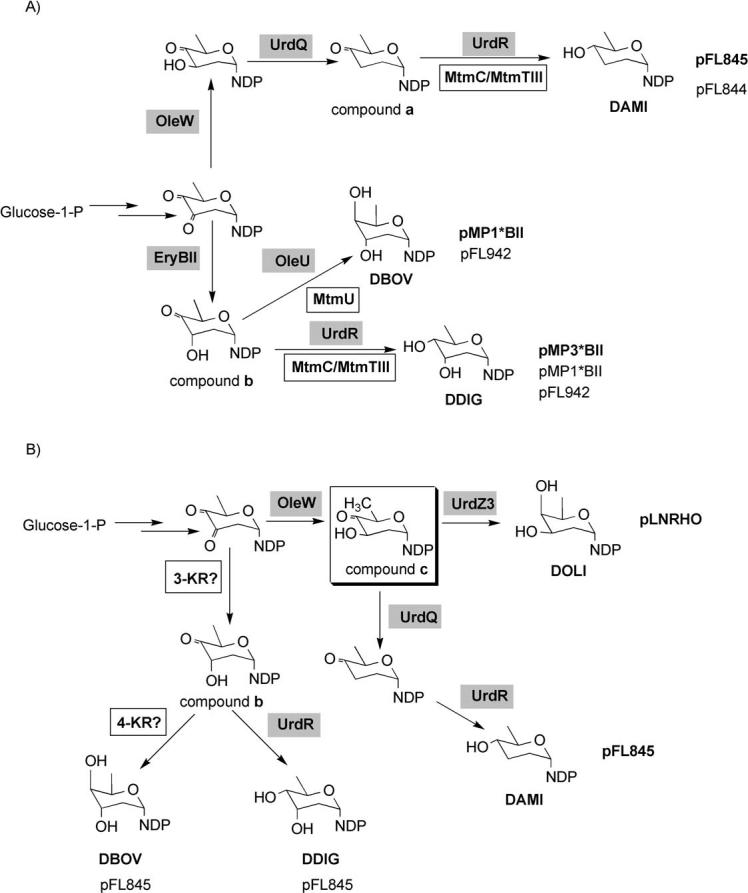
Proposed pathways for the biosynthesis of the new deoxyhexoses produced in recombinant strains of S. argillaceus wild-type (A) and S. argillaceus M7U1 (B). Compound a: NDP-4-keto-2,3,6-trideoxyglucose; compound b: NDP-2,6-dideoxy-d-glycero-4-hexulose; compound c: NDP-4-keto-2,6-dideoxy-d-glucose. Gray boxes indicate enzymes codified by the “sugar plasmids”. White boxes indicate enzymes from the host. Plasmid names in bold or plain indicate that the biosynthesis of the corresponding DOH is codified by the plasmid itself or by the concerted action of plasmid and host-coded enzymes, respectively. Highlighted by a square is the substrate of MtmU 4-ketoreductase: DAMI, d-amicetose; DBOV, d-boivinose; DDIG, d-digitoxose; DOLV, d-olivose; DOLI, d-oliose.
Use of mutant S. argillaceus M7U1 to generate novel mithramycins with substitutions on the d-oliose residue
Once we validated this combinatorial approach to generate mithramycins with altered glycosylation profiles, our next goal was to try to generate mithramycin derivatives with changes affecting a specific sugar position in the molecule. It has been shown that in the mithramycin–DNA binding complex, interactions between the saccharides and DNA are common. Specifically, the d-olivose and d-oliose moieties of the trisaccharide chain interact in an antiparallel alignment with the sugar-phosphate backbone of C-N steps of individual strands in the complex.[9] As these interactions could be important for the specificity and activity of mithramycin, we intended to force the substitution of one of these sugars, in particular d-oliose. To do that, we used the S. argillaceus mutant M7U1 as a host in which the mtmU gene has been inactivated.[35] This mutant accumulates the intermediate premithramycin A1 (compound 3, Scheme 1 A) as it cannot produce NDP-d-oliose. For most of the plasmids, the use of this mutant did not result in the generation of new mithramycin derivatives. Exceptions were strains harboring pLNRHO or pFL844. In the case of pLNRHO, no derivatives were found when this plasmid was introduced in the wild-type strain (Figure 2A); however, its presence in the mutant resulting in the detection of at least four peaks in HPLC (Figure 2B) with the absorption spectrum characteristic of mithramycin-like compounds. In the case of pFL845, the number of peaks containing mithramycin-like compounds was twice as many in the mutant (Figure 2D) as in the wild-type strain (Figure 2C). Two compounds from S. argillaceus M7U1 (pLNRHO) (peaks j and k), and four compounds (peaks l to 0) from cultures of S. argillaceus M7U1 (pFL845) were purified and their structure elucidated by NMR and MS (Supporting Information). Compound k corresponded to the previously identified compound 5 (Scheme 1 B). Compound j has been recently characterized and corresponded to deoliosyl-3C-β-d-mycarosyl-mithramycin (8).[34] Compounds in peaks l, m, n, and o corresponded to the new mithramycin derivatives: deoliosyl-demycarosyl-3C-β-d-olivosyl-3D-β-d-digitoxosyl-mithramycin (9), dideolivosyl-6-β-d-amicetosyl-deoliosyl-3C-β-d-olivosyl-mithramycin (10), dideolivosyl-6-β-d-amicetosyl-deoliosyl-demycarosyl-3C-β-d-olivosyl-3D-β-d-digitoxosyl-mithramycin (11), and dideolivosyl-6-β-d-amicetosyl-deoliosyl-demycarosyl-3C-β-d-boivinosyl-mithramycin (12), respectively (Scheme 1 C).
Figure 2.

HPLC analyses of cultures of S. argillaceus and S. argillaceus M7U1 containing pLNRHO and pFL845. ★ Mithramycin-like compounds. Peaks corresponding to the different compounds are indicated as follows: M: mithramycin; A: premithramycin A1; P: premithramycinone; peaks j–o: mithramycin derivatives purified from the recombinant strains.
As expected, as the biosynthesis of d-oliose was blocked in mutant M7U1, all of these compounds lacked the d-oliose residue and instead they contained another sugar: d-olivose (compounds 9, 10, and 11), d-mycarose (compound 8), or d-boivinose (compound 12). Moreover, some of the compounds contained additional substitutions in the sugar profile: replacement of the first d-olivose of the disaccharide chain by d-amicetose (compounds 10, 11, and 12) or the d-mycarose unit (the terminal sugar) of the trisaccharide chain by d-digitoxose (compounds 9 and 11). The host strain synthesizes NDP-d-olivose and NDP-d-mycarose, whereas NDP-d-amicetose is encoded by plasmid pFL845. However, formation of d-digitoxose and d-boivinose can be explained if the host contains a 3-ketoreductase activity, which will introduce the C3 hydroxyl group in axial configuration, that is, rendering the sugar biosynthesis intermediate NDP-2,6-dideoxy-d-glycero-4-hexulose (compound b; Scheme 2 B). Reduction at the 4-position of this intermediate by the plasmid-encoded 4-ketoreductase UrdR could result in the synthesis of d-digitoxose. Formation of d-boivinose would require the involvement of an additional host 4-ketoreductase (Scheme 2 B). Unexpectedly, compound 5 contains a d-oliose moiety as the second sugar of the trisaccharide chain. Formation of this compound by the strain M7U1 (pLNRHO) could be explained by the presence of 4-ketoreductase UrdZ3 in plasmid pLNRHO, which would act on the sugar biosynthesis intermediate NDP-4-keto-2,6-dideoxy-d-glucose (compound c; Scheme 2B). In accordance with this, it has been reported that 4-ketoreductase UrdZ3 was able to reduce l- and d-sugar biosynthesis intermediates, rendering in this last case a hydroxyl group in axial configuration.[36]
Antitumor activity
The antitumor activity of the different mithramycin derivatives was tested against three tumor cell lines (Table 2). Compilation of the average GI50 (50% growth inhibition) values showed that all the compounds, with the exception of compound 11, were active. Compounds 6, 8, 9, 10, and 12 showed the highest levels of activity. Among these compounds 6 and 9 are fully glycosylated with five DOHs, whereas compounds 8 and 10 lack one DOH, and compound 12 lacks two DOHs. Compounds 4, 7, and 8, all lacking the third DOH of the trisaccharide chain, but varying in the second DOH of the trisaccharide, showed different levels of antitumor activity, compound 8 being more active than compounds 4 and 7. Similarly, compounds 5 and 10, which only differ at the second position of the trisaccharide chain showed different levels of antitumor activity, with compound 10 being about three times more active than compound 5.
Table 2.
Antitumor activity of compounds.
| GI50[a] [μm] | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Tumor cell lines | 1[b] | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
| Breast MDA-MB-231 | 0.13 | 1.28 | 1.38 | 0.23 | 3.72 | 0.37 | 0.69 | 0.47 | >10 | 0.40 |
| NSCL[c] A549 | 0.15 | 1.81 | 1.49 | 0.19 | >10 | 0.48 | 0.91 | 0.44 | >10 | 0.75 |
| Colon HT29 | 0.22 | >10 | 3.19 | 0.27 | >10 | 0.93 | 5.98 | 0.94 | >10 | 2.01 |
GI50, 50% growth inhibition.
For comparison, values for mithramycin (M) are also shown.
NSCL, non-small cell lung.
Discussion
We have used a combinatorial biosynthesis approach to generate derivatives of the antitumor drug mithramycin with glycosylation profiles different from that of the parental compound. To do this, we provided either the mithramycin producer strain or a d-oliose-minus mutant with the capability of synthesizing novel DOHs. This was achieved by introducing different “sugar plasmids” in these strains, each directing the biosynthesis of specific l- and d-DOHs. Nine mithramycin derivatives were produced, differing from the parental compound either in the absence of a sugar moiety, the presence of a mithramycin sugar at an unusual position, and/or the presence of a new DOH normally not found in mithramycin. In fact, the d-boivinose- and d-amicetose-containing compounds described herein are the first aureolic acid derivatives with this type of DOH.
The first sugar attached during the biosynthesis of mithramycin is a d-olivose in position C. Only premithramycin-like compounds were detected, with possible exchange of a sugar in this position. This suggests either that glycosyltransferase MtmGIV (responsible for the incorporation of this first d-olivose) shows limited sugar donor substrate flexibility or that the incorporation of a different DOH in this position C hinders the growth of the saccharide chain. However, several mithramycin derivatives were obtained with substitutions at the second position of the trisaccharide chain. In these mithramycins the d-oliose moiety was replaced either by d-olivose, d-boivinose, or d-mycarose. This indicates that glycosyltransferase MtmGIII, which transfers the second sugar (normally a d-oliose) into the d-position, is quite flexible. In contrast, the third position of the trisaccharide chain was only occupied either by d-mycarose (the original sugar) or by d-digitoxose. This confirms that MtmGIV, which is thought to be responsible to complete the trisaccharide chain,[22] has limited sugar donor flexibility. However, this glycosyltransferase shows broad acceptor substrate flexibility, as it was able to transfer those sugars to intermediates with various sugars occupying this second position (position D) of the trisaccharide chain.
The d-olivose moiety, normally at the first position of the disaccharide chain (position A) could only be substituted by d-amicetose. The incorporation of this DOH was quite efficient as four out of the nine newly-glycosylated mithramycins generated in this work contained this DOH, and the production yields of some of these compounds were quite high. In particular, compound 5 was the main compound produced by strain S. argillaceus (pFL844). Obviously, incorporation of d-amicetose into position A blocks further elongation of the saccharide chain, as this DOH lacks a hydroxyl group at C3, which is essential to establish the 1,3-linkage with the next sugar. These data suggest that the glycosyltransferase MtmGI, normally responsible for the transfer of d-olivose into the A position, shows limited sugar donor substrate flexibility, although it efficiently transfers d-amicetose, and MtmGII, which normally finishes the sugar pattern of mithramycin by adding a d-olivose into position B, is unable to establish a 1,4-linkage. Overall, no mithramycin derivative was ever detected with a sugar substitution at the second position of the disaccharide chain (position B), which suggests that MtmGII is also rather inflexible with respect to its sugar donor substrate.
It is remarkable that some of the sugars incorporated in the new mithramycin derivatives were neither fully synthesized by the host enzymes nor by the “sugar plasmid”-encoded enzymes alone, but rather by a concerted action of both “sugar plasmid”-encoded and host enzymes. This is the case for recombinant strains producing d-amicetose-, d-boivinose-, or d-digitoxose-containing derivatives. Formation of these DOHs suggests substrate flexibility for some of the enzymes involved. For instance, the 4-ketoreductase MtmU can act on its normal substrate NDP-4-keto-2,6-dideoxy-d-glucose and on NDP-2,6-dideoxy-d-glycero-4-hexulose, for the biosynthesis of d-boivinose.
All of the new mithramycin derivatives, with the exception of compound 11, showed antitumor activity at the micromolar level. Some of the new compounds bear five DOHs, but others lack one DOH (either in the trisaccharide or in the disaccharide chain), or even two DOHs (one in each saccharide chain). This indicates that the presence of all five sugars is not absolutely essential for the antitumor activity, and that the existence of a disaccharide chain at C6 or a trisaccharide chain at C2 is not compulsory for the antitumor activity. Four out of the six new mithramycins with modifications at the second position of the trisaccharide chain, position D, showed high antitumor activity. Interestingly, compounds only differing in their sugar moiety at this position show quite different antitumor activities. This confirms the importance of the sugar d-residue as a potential target for the generation of mithramycins with antitumor activity.
In conclusion, we have shown that a combinatorial biosynthesis approach of sugar biosynthesis genes could be a potent strategy to generate new glycosylated mithramycins with sugar substitutions at different positions of the molecule. Many of the new derivatives show antitumor activity, and could be useful as anticancer drugs.
Experimental Section
Strains, culture conditions, and plasmids
Streptomyces argillaceus ATCC 12956 (mithramycin producer) and S. argillaceus M7U1 were used as hosts for gene expression and production.[35] For sporulation they were grown for 7 days at 30 °C on plates containing medium A.[23] For protoplast regeneration, the organisms were grown on R5 solid medium plates.[37] Liquid and solid media for production and isolation of mithramycin derivatives was modified R5 medium (R5A).[23] When plasmid-containing clones were grown, the medium was supplemented with 5 or 50 μgmL−1 thiostrepton for liquid or solid cultures respectively.
DNA manipulation
Plasmid DNA preparations, restriction endonuclease digestions, and other DNA manipulations were according to standard procedures for Streptomyces and for E. coli.[37,38] Preparation of Streptomyces protoplasts, transformation, and selection of transformants were carried out as described.[37]
HPLC analysis and purification of mithramycin derivatives
HPLC-MS analyses were carried out using an Alliance chromatographic system coupled to a ZQ4000 mass spectrometer (Waters-Micromass), using acetonitrile and 0.1% trifluoroacetic acid (TFA) in water as solvents and a reversed phase column (Symmetry C18, 2.1×150 mm, Waters). Samples were eluted with 10% acetonitrile during the first 4 min, followed by a linear gradient from 10 to 88 % acetonitrile over 26 min, at a flow rate of 0.25 mL min−1. Detection and spectral characterization of peaks were performed with a photodiode array detector and Empower software (Waters). MS analyses were done by electrospray ionization in the positive mode, with a capillary voltage of 3 kV and cone voltages of 20 and 100 V.
For purification of mithramycin derivatives, S. argillaceus (pFL844) and S. argillaceus (pMP3*BII) were grown in a two-step culture method, as previously described.[23] The cultures were centrifuged and filtered, and the broth was solid-phase extracted and fractionated as previously described.[39] Fractions containing mithramycin-related compounds (eluting between 40 and 55 min) were pooled and dried in vacuo. These extracts were redissolved in a mixture of methanol/DMSO (50:50, v/v) and chromatographed in a μBondapak C18 radial compression cartridge (PrepPak Cartridge, 25×100 mm, Waters). An isocratic elution with a mixture of acetonitrile and 0.1% TFA in water (42:58, v/v), at 10 mLmin−1, was used. Compound d from strain S. argillaceus (pMP3*BII) was repurified in a semipreparative column (Symmetry C18, 7.8×300 mm, Waters) with isocratic elution with acetonitrile and 0.1% TFA in water (37:63, v/v), at 3 mLmin−1. Compounds c from S. argillaceus (pFL844) and e from S. argillaceus (pMP3*BII) were repurified in a similar way, but using a slightly different mixture (40:60, v/v).
In the case of S. argillaceus (pFL942), S. argillaceus (pFL845), S. argillaceus (pMP1*BII), S. argillaceus M7U1 (pLNRHO), and S. argillaceus M7U1 (pFL845), they were grown on solid R5A medium. For each strain, one hundred agar plates were uniformly inoculated with spores and incubated at 28°C for six days. These cultures were extracted three times with ethyl acetate, as described previously.[40] The extract obtained in each case was chromatographed in a μBondapak C18 radial compression cartridge, using acetonitrile and water (50:50, v/v) at 10 mLmin−1 in isocratic conditions. Subsequent purifications were performed using a SunFire PrepC18 (10×250 mm, Waters) column, with isocratic elution with mixtures of acetonitrile and 0.1% TFA in water as the mobile phase, at 7 mLmin−1. These mixtures, optimized for every compound, ranged from 37:63 to 50:50 (v/v).
For all purifications, whenever TFA was used in the mobile phase, peaks were collected in flasks containing 0.1 m potassium phosphate buffer (pH 7.0) to neutralize TFA in the mobile phase. After every purification step, the collected compounds were diluted four times with water, desalted, and concentrated by solid-phase extraction, being finally lyophilized. Yields obtained for the different compounds were as follows: 12.2 mg and 8.9 mg from peaks a and b of cultures of S. argillaceus (pFL845), respectively; 11.4 mg from peak c of cultures of S. argillaceus (pFL844); 19 mg and 10.8 mg from peaks d and e of cultures of S. argillaceus (pMP3*BII), respectively; 8.2 mg, and 6.4 mg from peaks f and g of cultures of S. argillaceus (pMP1*BII), respectively; 3 mg and 6,9 mg from peaks h and i of cultures of S. argillaceus (pFL942), respectively; 7.3 mg and 3,3 mg from peaks j and k of cultures of S. argillaceus M7U1 (pLNRHO), respectively; 6.7 mg, 17 mg, 12.2 mg and 1.7 mg from peaks l, m, n, and o of cultures of S. argillaceus M7U1 (pFL845), respectively.
Structure elucidation and characterization of new mithramycin derivatives
The structures of the isolated mithramycin derivatives were characterized by NMR spectroscopy and mass spectrometry. Compounds identified as new were fully characterized with physicochemical methods (see the Supporting Information). In general, the following NMR experiments were used: spin systems of each single sugar moiety was identified through H,H-COSY and 1D-TOCSY experiments. Sugar–aglycone connections and intersugar connections were proven through CIGAR-HMBC and/or NOE experiments.[41] The NOE experiments were also useful to identify whether the single sugar moieties were found in 4C1- (typical for d-sugars) or 1C4-conformation (typical for l-sugars). Helpful saccharide fragmentation patterns were observed in the ESI mass spectra confirming the results from the NMR studies, and highly resolved fast atom bombardment (HR-FAB) mass spectra were used to confirm the molecular formulae.
Determination of antitumor activity
The antitumor activity of the mithramycin derivatives was tested against a panel of tumor cell lines. Quantitative measurement of cell growth and viability was carried out by a colorimetric assay, using the sulforhodamine reaction.[42]
Acknowledgements
This work was supported by grants from the Spanish Ministry of Education and Science to C.M. (BMC2002-03599 and BIO2005-04115), a grant of the US National Institutes of Health (CA 91901) to J.R., and from Red Temática de Investigación Cooperativa de Centros de Cáncer (Ministry of Health, Spain; ISCIII-RETIC RD06/0020/0026). M.P. was the recipient of a predoctoral fellowship of the Spanish Ministry of Science and Technology. We thank the University of Kentucky core laboratories for NMR spectroscopy and mass spectrometry for the usage of their instruments, and the mass spectrometry lab from the Department of Chemistry of U. Nebraska–Lincoln for the high-resolution FAB mass spectra. Pharmamar S.A. is acknowledged for helping us with the antitumor assays.
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author: Physicochemical and NMR data.
References
- 1.Fu X, Albermann C, Jiang J, Liao J, Zhang C, Thorson JS. Nat. Biotechnol. 2003;21:1467–1469. doi: 10.1038/nbt909. [DOI] [PubMed] [Google Scholar]
- 2.Losey HC, Jiang J, Biggins JB, Oberthur M, Ye XY, Dong SD, Kahne D, Thorson JS, Walsh CT. Chem. Biol. 2002;9:1305–1314. doi: 10.1016/s1074-5521(02)00270-3. [DOI] [PubMed] [Google Scholar]
- 3.Salas JA, Méndez C. Trends Microbiol. 2007;15:219–232. doi: 10.1016/j.tim.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 4.Yang M, Proctor MR, Bolam DN, Errey JC, Field RA, Gilbert HJ, Davis BG. J. Am. Chem. Soc. 2005;127:9336–9337. doi: 10.1021/ja051482n. [DOI] [PubMed] [Google Scholar]
- 5.Rohr J, Méndez C, Salas JA. Bioorg. Chem. 1999;27:41–54. [Google Scholar]
- 6.Jia Z, Zhang J, Wei D, Wang L, Yuan P, Le X, Li Q, Yao J, Xie K. Cancer Res. 2007;67:4878–4885. doi: 10.1158/0008-5472.CAN-06-3494. [DOI] [PubMed] [Google Scholar]
- 7.Chatterjee S, Zaman K, Ryu H, Conforto A, Ratan RR. Ann. Neurol. 2001;49:345–354. [PubMed] [Google Scholar]
- 8.Fibach E, Bianchi N, Borgatti M, Prus E, Gambari R. Blood. 2003;102:1276–1281. doi: 10.1182/blood-2002-10-3096. [DOI] [PubMed] [Google Scholar]
- 9.Sastry M, Fiala R, Patel DJ. J. Mol. Biol. 1995;251:674–689. doi: 10.1006/jmbi.1995.0464. [DOI] [PubMed] [Google Scholar]
- 10.Sastry M, Patel DJ. Biochemistry. 1993;32:6588–6604. doi: 10.1021/bi00077a012. [DOI] [PubMed] [Google Scholar]
- 11.Remsing LL, Bahadori HR, Carbone GM, McGuffie EM, Catapano CV, Rohr J. Biochemistry. 2003;42:8313–8324. doi: 10.1021/bi034091z. [DOI] [PubMed] [Google Scholar]
- 12.Koutsodontis G, Kardassis D. Oncogene. 2004;23:9190–9200. doi: 10.1038/sj.onc.1208141. [DOI] [PubMed] [Google Scholar]
- 13.Barcelo F, Scotta C, Ortiz-Lombardia M, Mendez C, Salas JA, Portugal J. Nucleic Acids Res. 2007;35:2215–2226. doi: 10.1093/nar/gkm037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller DM, Polansky DA, Thomas SD, Ray R, Campbell VW, Sanchez J, Koller CA. Am. J. Med. Sci. 1987;294:388–394. doi: 10.1097/00000441-198711000-00015. [DOI] [PubMed] [Google Scholar]
- 15.Ray R, Snyder RC, Thomas S, Koller CA, Miller DM. J. Clin. Invest. 1989;83:2003–2007. doi: 10.1172/JCI114110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Snyder RC, Ray R, Blume S, Miller DM. Biochemistry. 1991;30:4290–4297. doi: 10.1021/bi00231a027. [DOI] [PubMed] [Google Scholar]
- 17.Blume SW, Snyder RC, Ray R, Thomas S, Koller CA, Miller DM. J. Clin. Invest. 1991;88:1613–1621. doi: 10.1172/JCI115474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hou MH, Wang AH. Nucleic Acids Res. 2005;33:1352–1361. doi: 10.1093/nar/gki276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wohlert SE, Kunzel E, Machinek R, Méndez C, Salas JA, Rohr J. J. Nat. Prod. 1999;62:119–121. doi: 10.1021/np980355k. [DOI] [PubMed] [Google Scholar]
- 20.Lombó F, Menéndez N, Salas JA, Méndez C. Appl. Microbiol. Biotechnol. 2006;3:1–14. doi: 10.1007/s00253-006-0511-6. [DOI] [PubMed] [Google Scholar]
- 21.Blanco G, Fernández E, Fernández MJ, Braña AF, Weissbach U, Kunzel E, Rohr J, Mendez C, Salas JA. Mol. Gen. Genet. 2000;262:991–1000. doi: 10.1007/pl00008667. [DOI] [PubMed] [Google Scholar]
- 22.Remsing LL, García-Bernardo J, González A, Künzel E, Rix U, Braña AF, Bearden DW, Méndez C, Salas JA, Rohr J. J. Am. Chem. Soc. 2002;124:1606–1614. doi: 10.1021/ja0105156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fernández E, Weißbach U, Reillo CS, Braña AF, Méndez C, Rohr J, Salas JA. J. Bacteriol. 1998;180:4929–4937. doi: 10.1128/jb.180.18.4929-4937.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nur-e-Alam M, Méndez C, Salas JA, Rohr J. ChemBioChem. 2005;6:632–636. doi: 10.1002/cbic.200400309. [DOI] [PubMed] [Google Scholar]
- 25.Prado L, Fernández E, Weissbach U, Blanco G, Quirós LM, Braña AF, Méndez C, Rohr J, Salas JA. Chem. Biol. 1999;6:19–30. doi: 10.1016/s1074-5521(99)80017-9. [DOI] [PubMed] [Google Scholar]
- 26.Remsing LL, Bahadori HR, Carbone GM, McGuffie EM, Catapano CV, Rohr J. Biochemistry. 2003;42:8313–8324. doi: 10.1021/bi034091z. [DOI] [PubMed] [Google Scholar]
- 27.Gibson M, Nur-e-Alam M, Lipata F, Oliveira MA, Rohr J. J. Am. Chem. Soc. 2005;127:17594–17595. doi: 10.1021/ja055750t. [DOI] [PubMed] [Google Scholar]
- 28.Majee S, Sen R, Guha S, Bhattacharyya D, Dasgupta D. Biochemistry. 1997;36:2291–2299. doi: 10.1021/bi9613281. [DOI] [PubMed] [Google Scholar]
- 29.Mir MA, Majee S, Das S, Dasgupta D. Bioorg. Med. Chem. 2003;11:2791–2801. doi: 10.1016/s0968-0896(03)00211-6. [DOI] [PubMed] [Google Scholar]
- 30.Rodríguez L, Aguirrezabalaga I, Allende N, Braña AF, Méndez C, Salas JA. Chem. Biol. 2002;9:721–729. doi: 10.1016/s1074-5521(02)00154-0. [DOI] [PubMed] [Google Scholar]
- 31.Lombó F, Gibson M, Greenwell L, Braña AF, Rohr J, Salas JA, Méndez C. Chem. Biol. 2004;11:1709–1718. doi: 10.1016/j.chembiol.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 32.Pérez M, Lombó F, Zhu L, Gibson M, Braña AF, Rohr J, Salas JA, Méndez C. Chem. Commun. 2005:1604–1606. doi: 10.1039/b417815g. [DOI] [PubMed] [Google Scholar]
- 33.Pérez M, Lombó F, Baig I, Braña AF, Rohr J, Salas JA, Méndez C. Appl. Environ. Microbiol. 2006;72:6644–6652. doi: 10.1128/AEM.01266-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baig I, Perez M, Braña AF, Gomathinayagam R, Damodaran C, Salas JA, Méndez C, Rohr J. J. Nat. Prod. 2008;71:199–207. doi: 10.1021/np0705763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.González A, Remsing LL, Lombó F, Fernández MJ, Prado L, Braña AF, Kunzel E, Rohr J, Méndez C, Salas JA. Mol. Gen. Genet. 2001;264:827–835. doi: 10.1007/s004380000372. [DOI] [PubMed] [Google Scholar]
- 36.Hoffmeister D, Ichinose K, Domann S, Faust B, Trefzer A, Drager G, Kirschning A, Fischer C, Kunzel E, Bearden DW, Rohr J, Bechthold A. Chem. Biol. 2000;7:821–831. doi: 10.1016/s1074-5521(00)00029-6. [DOI] [PubMed] [Google Scholar]
- 37.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. Practical Streptomyces Genetics. The John Innes Foundation; Norwich: 2000. [Google Scholar]
- 38.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd ed. Cold Spring Harbor Laboratory; New York: 1989. [Google Scholar]
- 39.Sánchez C, Butovich IA, Braña AF, Rohr J, Méndez C, Salas JA. Chem. Biol. 2002;9:519–531. doi: 10.1016/s1074-5521(02)00126-6. [DOI] [PubMed] [Google Scholar]
- 40.Menéndez N, Nur-e-Alam M, Braña AF, Rohr J, Salas JA, Méndez C. Chem. Biol. 2004;11:21–32. doi: 10.1016/j.chembiol.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 41.Reynolds WF, Enriquez RG. J. Nat. Prod. 2002;65:221–244. doi: 10.1021/np010444o. [DOI] [PubMed] [Google Scholar]
- 42.Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren J, Bokesch H, Kenney S, Boyd MR. J. Natl. Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]