

Abstract
Enantioselective syntheses of the alkaloids (−)-aurantioclavine, (+)-amurensinine, (−)-lobeline, and (−)- and (+)-sedamine are described. The syntheses demonstrate the effectiveness of the Pd-catalyzed asymmetric oxidation of secondary alcohols in diverse contexts and the ability of this methodology to set the absolute configuration of multiple stereocenters in a single operation. The utility of an aryne C–C insertion reaction in accessing complex polycyclic frameworks is also described.
Introduction
Chiral amines are a key functional group in numerous biologically active natural products and synthetic drugs. The enantioselective synthesis of amines has been extensively investigated and continues to be a field of intense interest.1 Chiral amines can be accessed from the corresponding alcohols using several possible approaches. This transformation could, in principle, be achieved via functional group interconversion of a hydroxyl to an amine functionality at a stereogenic carbon (Scheme 1a, path A).2 Alternatively, desymmetrization of a meso-substrate bearing multiple pro-stereogenic centers with alcohol and amine functional groups could deliver the corresponding enantioenriched product (Scheme 1a, path B).3 In addition, kinetic resolution of a racemic, diastereomerically pure substrate with stereocenters bearing hydroxyl and amine functionalities provides a route to chiral amino alcohols (Scheme 1a, path C). Finally, diastereoselective installation of a stereocenter bearing an amine group can be accomplished from a substrate that already features a hydroxyl-bearing stereocenter (Scheme 1a, path D). In this article, we present syntheses of natural products bearing chiral amines or chiral amino alcohols employing each of the above strategies.
Scheme 1a.
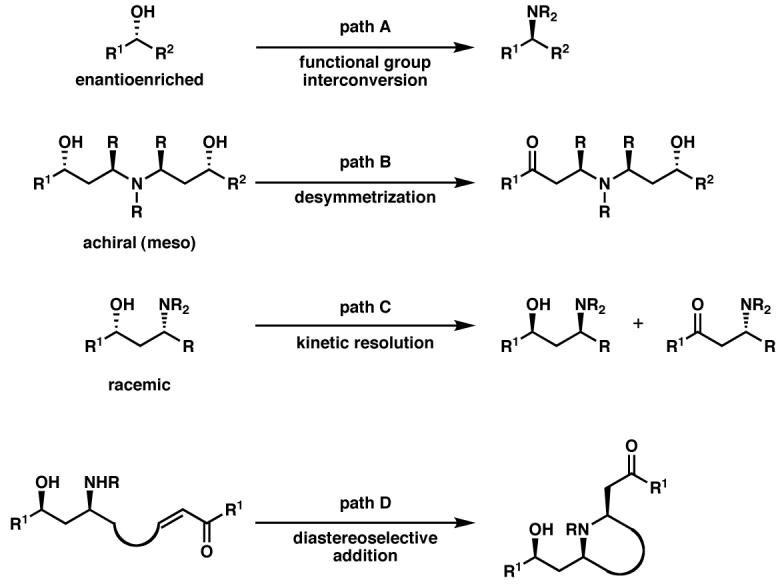
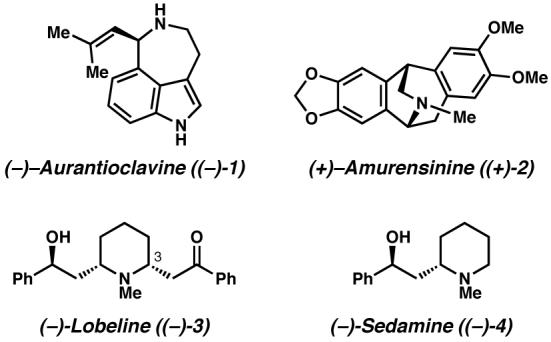
We sought to apply the Pd-catalyzed aerobic oxidative kinetic resolution4 (Scheme 1b) developed in our laboratory toward the synthesis of alkaloids. This transformation enables the catalytic enantioselective synthesis of chiral alcohols, with benzylic alcohols being particularly excellent substrates.5 We present here syntheses of the natural products (−)-aurantioclavine ((−)-1), (+)-amurensinine ((+)-2), (−)-lobeline ((−)-3), (−)-sedamine ((−)-4), and (+)-sedamine (Figure 1) by kinetic resolution or desymmetrization of benzylic alcohols as the key steps.
Scheme 1b.
Figure 1.
Alkaloids with nitrogen-bearing stereocenters.
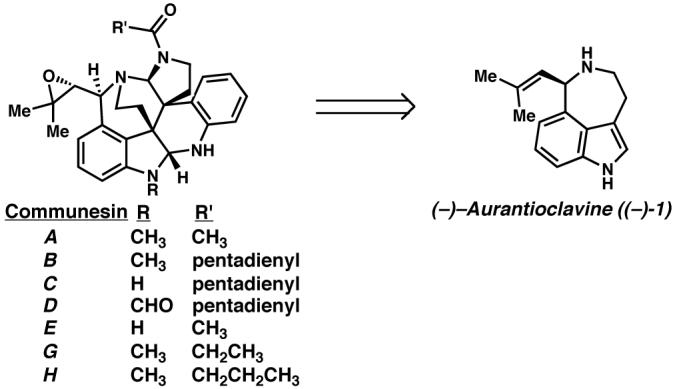
(−)-Aurantioclavine ((−)-1) is an ergot alkaloid that was first isolated from Penicillium aurantiovirens.6 The alkaloid possesses a 3,4,5,6-tetrahydro-6-(2-methyl-1-propenyl)azepino[5,4,3-cd]indole tricyclic ring system bearing a single stereocenter. We were particularly interested in aurantioclavine as an intermediate en route to the complex polycyclic alkaloids of the communesin family7 (Scheme 2). While syntheses of racemic aurantioclavine8 have been reported in the literature, an asymmetric synthesis of aurantioclavine has, to the best of our knowledge, not been reported. Our objective was to develop an enantioselective synthesis of (−)-aurantioclavine ((−)-1) as a means toward an asymmetric synthesis of members of the communesin family.9
Scheme 2.
(−)-Amurensinine ((−)-2) belongs to a family of alkaloids, the isopavines (Figure 2), originally isolated from Papaveraceae plants.10 The isopavines display biological activity relevant to neurological disorders such as Parkinson's disease, Down's syndrome, Alzheimer's disease, amyotropic lateral sclerosis and Huntington's chorea. Despite the potential medicinal applications of the isopavines and their analogues,11 relatively few total syntheses of these natural products have been reported.12 The majority of these syntheses involve intramolecular acid-promoted cyclization to form the azabicyclo[3.2.2]nonane core of the isopavines. We envisioned a catalytic enantioselective approach to rapidly access the isopavine core in a modular fashion.13
Figure 2.
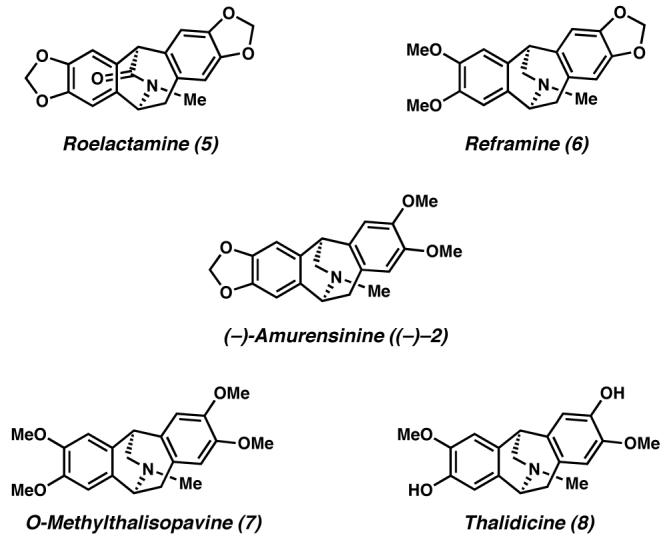
The isopavine natural products.
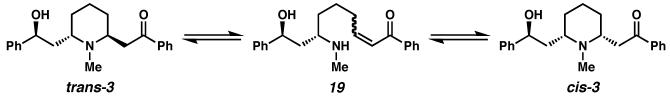
(−)-Lobeline ((−)-3) is a primary alkaloid constituent of Lobelia inflata, a plant commonly known as “Indian tobacco” because it was previously used by native North Americans as a tobacco substitute.14, 15 A respiratory stimulant, the plant's crude extracts have been widely used for the treatment of respiratory illnesses, including asthma, bronchitis, pneumonia, and whooping cough. (−)-Lobeline mildly mimics the effect of nicotine, is an antagonist of nicotine acetylcholine receptors, and has thus been applied as a smoking cessation agent.16 (−)-Lobeline also inhibits the neurochemical and behavioral effects of methamphetamine and is an inhibitor of dopamine and vesicular monoamine transporter function. (−)-Lobeline is thus a promising lead in the development of treatments for methamphetamine abuse.17 While it may be isolated from its natural source and crystallized as a single isomer in salt form, in the solution state the free base of (−)-lobeline ((−)-3) is known to exist in equilibrium with its epimer at C(3).18 Due to this known equilibrium, early enantioselective syntheses19 by Marazano and Lebreton report the synthesis of (−)-lobeline as a mixture with its C(3) epimer. Our goal was to access the alkaloid 3 in diastereomerically pure form, and we believed that the application of the Pd-catalyzed enantioselective oxidation would allow efficient access to (−)-lobeline ((−)-3) from a symmetric meso-intermediate. We also believed that access to either enantiomer of sedamine (4),20, 21 a piperidine derivative found in various Sedum species, would be possible through the application of a Pd-catalyzed oxidative kinetic resolution.
Results and Discussion
Retrosynthesis of (−)-aurantioclavine
Our retrosynthetic analysis for (−)-aurantioclavine is depicted in Scheme 3. We believed that (−)-aurantioclavine ((−)-1) could be accessed from amino alcohol derivative 9 via dehydration of the tertiary alcohol to form the trisubstituted olefin and subsequent removal of the sulfonamide protecting groups. Sulfonamide 9 could in turn be derived from amino-diol derivative 10 via cyclization to form the 3,4,5,6-tetrahydroazepino[5,4,3-cd]indole ring system of the natural product. Diol 10 could then be derived from enantioenriched diol 12 via amino alcohol derivative 11. Diol 12 could then be accessed from the known aldehyde 13.
Scheme 3.

Retrosynthesis of (+)-amurensinine
Our approach to the isopavines, and specifically to amurensinine, is depicted in Scheme 4. Disconnection of the bridging amine in the natural product reveals azidoester 14, which can be derived from hydroxyester (−)-15. We envisioned the enantioselective synthesis of this benzylic alcohol by Pd-catalyzed oxidative kinetic resolution. Hydroxyester (±)-15 could then arise from ketoester (±)-16, which we believed could be derived from arylsilyl triflate 17 and β-ketoester 18 via aryne C–C insertion22 methodology recently developed in our laboratory.
Scheme 4.

Retrosynthesis of (−)-lobeline
We believed that the late stage Pd-catalyzed oxidative desymmetrization of meso-diol 20, a natural product known as lobelanidine, could deliver (−)-lobeline ((−)-3, Scheme 5). We also envisioned that the diastereoselective formation of the desired cis-2,6-disubstituted piperidine moiety of amino alcohol (−)-3 could be achieved under equilibrating conditions via intermediate enone 19. Diol 20 could then be derived via disconnection of a C–C bond to arrive at amino alcohol (±)-21. Enantioselective oxidation of amino alcohol derivative (±)-21 in turn would allow us to access either (−)-sedamine ((−)-4) or its enantiomer. We believed that amino alcohol derivative (±)-21 could be readily accessed from aldehyde 22.
Scheme 5.

Synthesis of (−)-aurantioclavine
Our synthesis of the ergot alkaloid (−)-aurantioclavine commenced with aldehyde 13 (Scheme 6).23 Addition of the dianion derived from isobutylene oxide24 to the aldehyde furnished racemic diol (±)-12. At this stage, application of a Pd-catalyzed oxidative kinetic resolution delivered enantioenriched diol, (−)-12 in 96% ee and 37% yield (91% of the theoretical maximum, selectivity factor25 s = 18.2). Product ketone 23 could be readily recycled by reduction with lithium aluminum hydride, affording diol (±)-12 in 95% yield.
Scheme 6.

The enantioenriched diol (−)-12 was then transformed to tricyclic alcohol 9 (Scheme 7). Diol (−)-12 was treated with hydrazoic acid under Mitsunobu conditions26 to furnish azidoalcohol 24. This transformation was conducted at low temperature to minimize racemization at the sensitive benzylic stereocenter. Hydrogenation of the azide and protection of the amine as a 2-nitrobenzenesulfonamide27 delivered sulfonamide 11. Bromination of the indole nucleus was followed by vinylation of bromoindole 25 under Stille conditions28 to furnish vinyl indole 26. Hydroboration-oxidation29 of the olefin of 26 to amino diol derivative 10 was followed by formation of the azepine under Mitsunobu conditions to deliver tricyclic alcohol 9 in excellent yield. We then investigated the dehydration of the tertiary alcohol 9 to (−)- aurantioclavine (Scheme 8).
Scheme 7.

Scheme 8.

Unfortunately, under various reaction conditions30 dehydration of the tertiary alcohol delivered a mixture of inseparable olefin isomers, with the undesired isomer 28 predominating in all cases. In an attempt to access the desired trisubstituted olefin isomer, we then turned to dehydration of intermediate 26 prior to formation of the azepine ring (Scheme 8).
We found that employing phosphorus oxychloride as the dehydrating agent in pyridine as solvent delivered the desired trisubstituted olefin isomer 29 as the major product. The olefin isomers 29 and 30 were separable by employing silica gel impregnated with silver nitrate.31
The completion of the synthesis of (−)-aurantioclavine is depicted in Scheme 9. Selective hydroboration-oxidation of the terminal olefin in vinyl indole 29 delivered the protected amino alcohol 31, which was then transformed to the tricyclic alcohol 27 under Mitsunobu conditions in excellent yield. Removal of the o-nitrobenzenesulfonyl protecting group delivered amine 32. Subsequent removal of the tosyl group using tetrabutylammonium fluoride (TBAF)32 then delivered (−)-aurantioclavine ((−)-1).
Scheme 9.

Synthesis of (+)-amurensinine
The synthesis of amurensinine began from diazoketone 33, readily available in 5 steps and 95% overall yield from homoveratric acid (Scheme 10). Selective intramolecular C-H insertion provided β-ketoester 18 in excellent yield.33 Treatment of β-ketoester 18 with arylsilyl triflate 17, available in 5 steps from sesamol,34 in the presence of cesium fluoride22a afforded ketoester (±)-16, thereby generating the polycyclic carbon framework of amurensinine in rapid fashion.
Scheme 10.
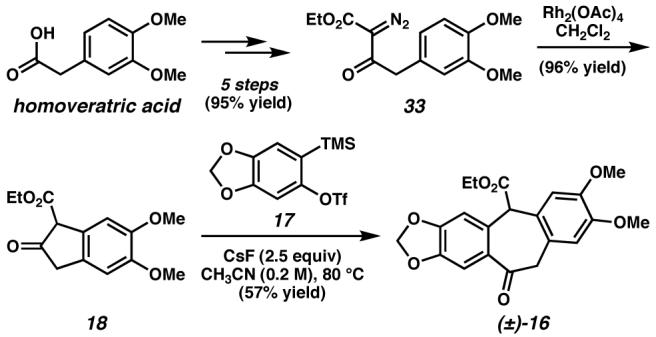
Chemo- and diastereoselective reduction of the ketone of (±)-16 with L-Selectride generated hydroxyester (±)-15 as a single diastereomer, presumably resulting from equatorial delivery of hydride to the ketone (Scheme 11).
Scheme 11.
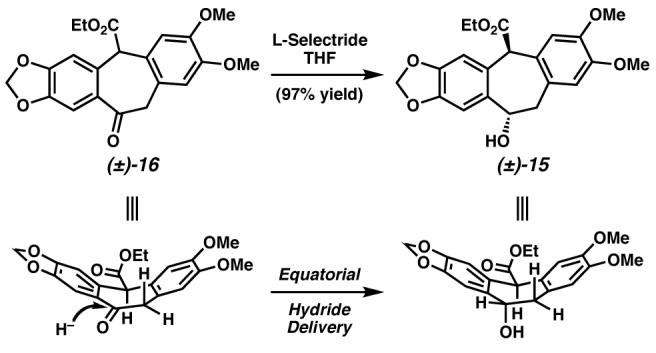
We then examined the Pd-catalyzed oxidative kinetic resolution of hydroxyester (±)-15. Under optimized reaction conditions, the racemic alcohol could be resolved to highly enantioenriched hydroxyester (−)-15 with good selectivity (Scheme 12). Though the overall mass recovery of (−)-15 and (+)-16 was not ideal, significant quantities of enantioenriched alcohol (−)-15 could be accessed from this pathway.
Scheme 12.

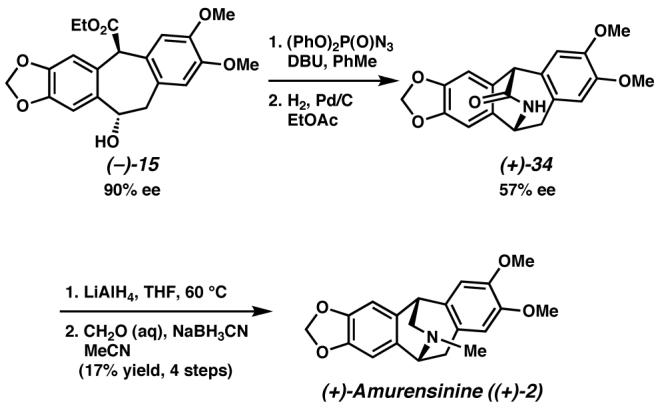
We next investigated introduction of the amine required for amurensinine from alcohol (−)-15. We found that alcohol (−)-15 was unreactive to many common nitrogen-bearing nucleophiles under Mitsunobu conditions, or it reacted to form a stilbene system by elimination. However, we were able to install an azide by employing diphenylphosphoryl azide in a procedure developed specifically for electron-rich benzylic alcohols by Thompson and co-workers (Scheme 13).35 Reduction of the resulting azide afforded bridged lactam (+)-34. Lactam reduction and reductive methylation afforded (+)-amurensinine ((+)-2).
Scheme 13.
While (+)-amurensinine could be accessed by this route, the sequence to (+)-2 from (±)-15 was complicated by the formation of several side products. Treatment of hydroxyester (−)-15 with DBU in the absence of diphenylphosphoryl azide produced lactone 35, demonstrating the sensitivity of the bis-benzylic stereocenter to epimerization (Scheme 14). Of further concern was the formation of lactam (+)-34 in only 57% ee. This partial racemization required two stereocenters to be inverted in the azide installation reaction, presumably via an intermediate such as o-quinonedimethide 36.
Scheme 14.
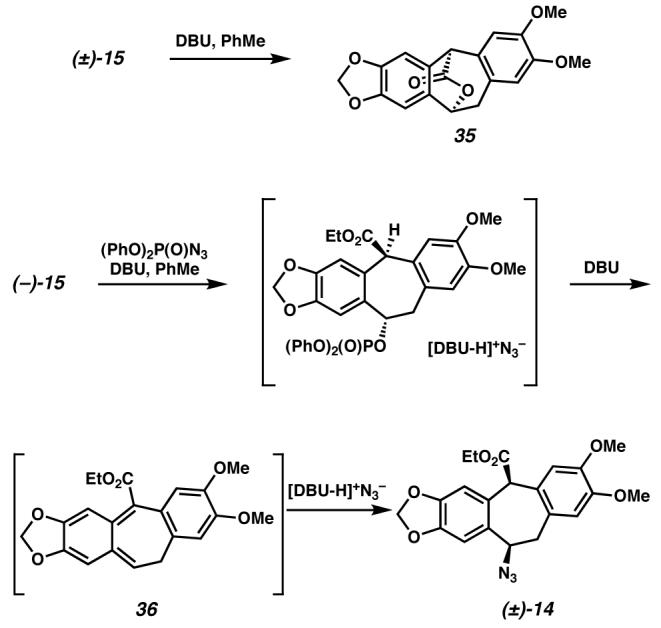
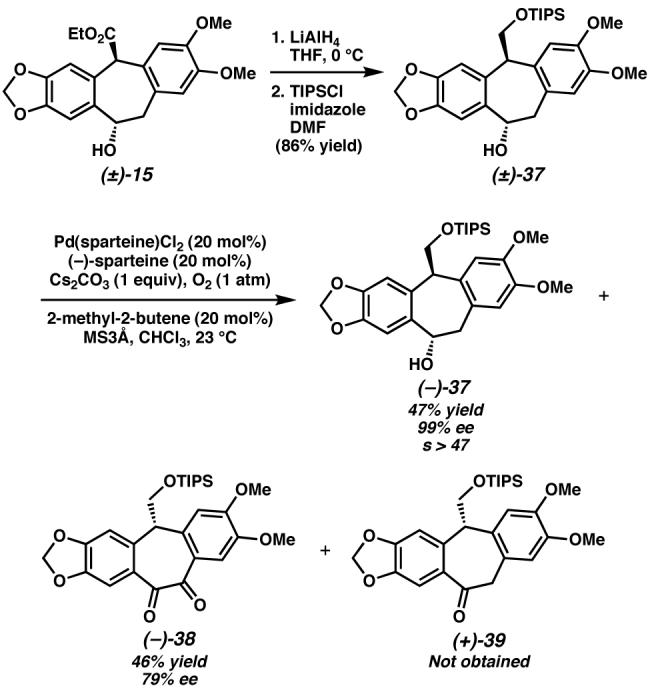
Our difficulty in suppressing epimerization at the bis-benzylic stereocenter bearing the ester in the azide displacement led us to pursue an alternate route to amurensinine via intermediates with attenuated acidity at the bis-benzylic carbon (Scheme 15). Also, we hoped an alternate benzylic alcohol substrate would lead to improved mass recovery in the oxidative kinetic resolution. To this end, reduction of hydroxyester (±)-15 was followed by selective silylation of the primary alcohol to afford alcohol (±)-37. Oxidative kinetic resolution of this alcohol proved highly selective (selectivity factor s>47), providing highly enantioenriched alcohol (−)-37. Interestingly, resolutions that were allowed to proceed to high ee of (−)-37 did not afford any of the expected ketone (+)-39. Instead, diketone (−)-38 was formed in good yield and high ee. Monitoring of the reaction demonstrated that the ketone (+)-39 was being generated, but it slowly underwent further oxidation to the diketone (−)-38. In fact, isolated samples of ketone 39 were slowly oxidized to the diketone in C6D6. Handling of this ketone under argon delayed this oxidative decomposition. We hypothesized that ketone (+)-39 was reacting with molecular oxygen via a radical pathway to afford the diketone (−)-38. This hypothesis is supported by our observations on non-enantioselective oxidations performed on alcohol (±)-37. Thus, oxidation of (±)-37 with Dess-Martin periodinane36 cleanly provided ketone (±)-39, while diketone (±)-38 was obtained when either MnO2 or Pd(OAc)2/O237 was employed as the oxidant. We therefore screened various radical inhibitors as additives in kinetic resolutions of alcohol (±)-37. Tetracyanoethylene led to little alcohol oxidation. Neither BHT nor 2-methyl-2-butene suppressed ketone overoxidation; however, incorporation of even catalytic quantities of 2-methyl-2-butene enhanced mass recovery and improved selectivity in the kinetic resolution. While the role of 2-methyl-2-butene is not yet clear, it was included in our preparative experiments because of its beneficial effect. Thus, under our optimized conditions, (±)-37 was resolved with Pd(sparteine)Cl2 as a catalyst in the presence of O2 to 99% ee and was isolated in 47% yield (94% of the theoretical maximum).
Scheme 15.
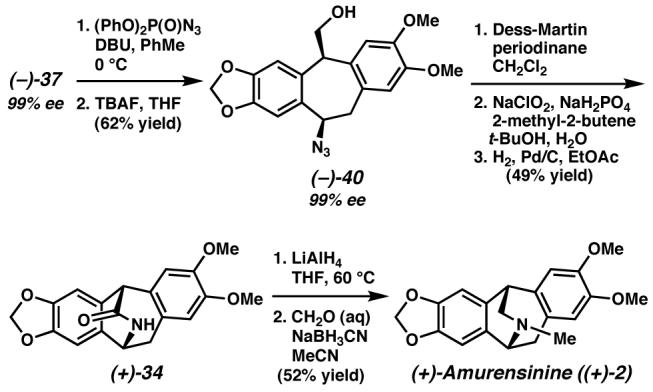
Having accessed substantial quantities of highly enantioenriched alcohol (−)-37, we next proceeded with the installation of the nitrogen required in the natural product. Azide displacement under Thompson's conditions was followed by desilylation to afford azidoalcohol (−)-40 (Scheme 16). Gratifyingly, this azide was obtained with clean inversion in 99% ee, indicating that decreasing the acidity of the bis-benzylic center did indeed minimize SN1 pathways. Oxidation of alcohol (−)-40 in two steps to the corresponding carboxylic acid and subsequent azide reduction delivered lactam (+)-34. Reduction and methylation as before afforded (+)-amurensinine ((+)-2) in 99% ee.38
Scheme 16.
Synthesis of (−)-lobeline and (−)- and (+)-sedamine
Our synthesis commenced from known aldehyde 22.39 Addition of the anion derived from Horner-Emmons reagent 41 followed by hydrolysis of the resulting methyl enol ether provided ketone (±)-42 in good overall yield (Scheme 17). DIBAL emerged as the preferred reductant from a screen of conditions for the subsequent reduction of aryl ketone (±)-42, providing the amino alcohol derivative (±)-21 with 12:1 diastereoselectivity.40 The diastereomers were separable by chromatography, and the benzylic alcohol of the major diastereomer (±)-21 was protected as a TBS ether. Formylation with DMF under anionic conditions furnished aldehyde 43 as a single diastereomer. Aldehyde 43 was then treated with the anion derived from Horner-Emmons reagent 41. Hydrolysis of the intermediate enol ether afforded ketone 44. As before, diastereoselective ketone reduction was accomplished with DIBAL, providing a single alcohol diastereomer. Subsequent desilylation with TBAF, followed by exhaustive reduction of the carbamate delivered the meso-diol lobelanidine (20). The relative stereochemistry of lobelanidine was confirmed by x-ray crystallography.
Scheme 17.

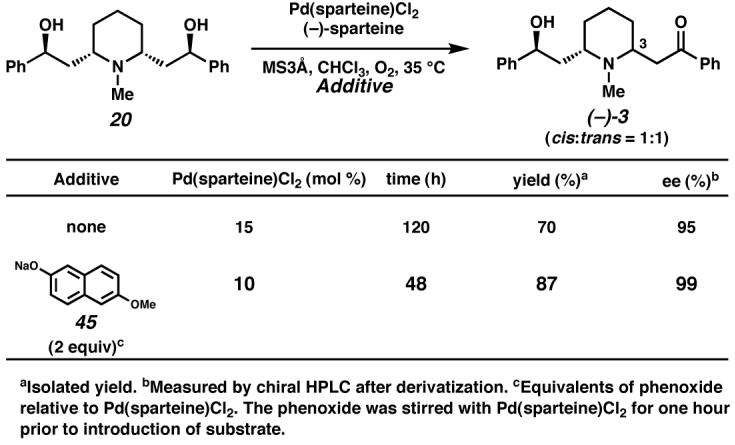
At this stage, we investigated the key desymmetrization of 20 to access (−)-lobeline ((−)-3). Our previously optimized reaction conditions,41 involving catalytic Pd(sparteine)Cl2 in chloroform under an oxygen atmosphere, furnished lobeline (3) as a 1:1 mixture of diastereomers at C(3) in 70% yield and 95% ee (Scheme 18).42 Synthetic alkaloid 3 was found to be spectroscopically identical to the equilibrated natural isolate and possessed identical optical rotation. Interestingly, a survey of additives revealed that incubation of Pd(sparteine)Cl2 with the sodium salt of 6-methoxy-2-naphthol (45) for one hour prior to exposure to meso-diol 20 had a dramatic impact on the reaction. Inclusion of the phenoxide salt permitted smooth conversion to amino alcohol (−)-3 with a lowered catalyst loading (10 mol%) and reaction time (48h) and furnished amino alcohol (−)-3 in enhanced isolated yield (87%) and ee (99%). Although the role of the phenoxide is presently unclear, one possibility is that the observed improvement in overall reaction profile may result from the formation of a more reactive, catalytically competent Pd(sparteine)phenoxide salt.
Scheme 18.
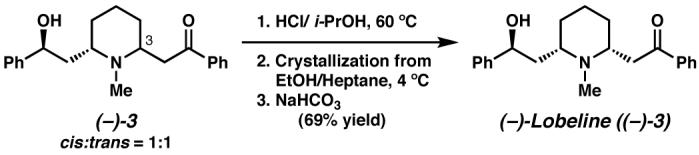
After implementing a late stage Pd-catalyzed oxidative desymmetrization to access (−)-lobeline ((−)-3) as a 1:1 mixture of equilibrating diastereomers, we began efforts to control the epimerization at C(3) in order to access (−)-lobeline as a single diastereomer. The rate of epimerization of natural (−)-lobeline was found to increase with increasing basicity of the reaction medium and by protic solvents.43 Furthermore, we found that the hydrochloride salt 3•HCl (cis:trans = 1:1) could be equilibrated to a 3:1 mixture of the cis:trans isomers in i-PrOH. Prompted by these observations, we developed a dynamic crystallization method allowing for selective precipitation of the cis-isomer of aminoketone 3 (Scheme 19). Heating a 1:1 mixture of cis:trans isomers of the hydrochloride of aminoketone 3 in i-PrOH followed by a slow crystallization permitted a 69% recovery of (−)-lobeline (cis-(−)-3) after liberation of the free base.
Scheme 19.
Having demonstrated a concise total synthesis of isomerically pure (−)-lobeline ((−)-3), we explored the utility of protected amino alcohol (±)-21 for the synthesis of related Sedum alkaloids, namely, both enantiomers of sedamine (i.e., (−)- and (+)-4, Scheme 20). Exposure of protected amino alcohol (±)-21 to our optimized Pd(II)-catalyzed aerobic oxidative kinetic resolution conditions furnished protected amino alcohol (−)-21 in 94% ee and the oxidation product, ketone (−)-42, in 81% ee with excellent yield and selectivity.
Scheme 20.

Direct reduction of amino alcohol derivative (−)-21 with lithium aluminum hydride provided natural (−)-sedamine ((−)-4). Reiteration of the highly diastereoselective DIBAL reduction of ketone (−)-42 generated enantiomeric alcohol (+)-21, and further reduction of the carbamate delivered the non-natural (+)-sedamine ((+)-4).
Conclusion
Herein, we have demonstrated the power of palladium-catalyzed enantioselective aerobic oxidation toward the synthesis of alkaloid natural products. We have developed an enantioselective synthesis of the ergot alkaloid (−)-aurantioclavine ((−)-1), a promising intermediate en route to the communesin family of alkaloids. We have also demonstrated that the strategic combination of an aryne insertion followed by enantioselective oxidation allows efficient access to the isopavine alkaloids exemplified by (+)-amurensinine ((+)-2). Finally, our concise enantioselective synthesis of (−)-lobeline ((−)-3) in diastereomerically pure form, as well as the synthesis of either enantiomer of sedamine ((−)- or (+)-4), demonstrates the extraordinarily facile generation of multiple stereogenic centers in a catalytic enantioselective manner in a single operation.
Supplemental Material
ACKNOWLEDGMENT
The authors thank the NIH-NIGMS (R01 GM65961-01), California TRDRP (postdoctoral fellowships to YKR, and SK, and predoctoral fellowship to JTB), NDSEG (predoctoral fellowships to DCE and UKT), NSF (predoctoral fellowship to DCE), Caltech, AstraZeneca, Boehringer Ingelheim, Johnson & Johnson, Pfizer, Merck, Amgen, Abbott, Research Corporation, Roche, and GlaxoSmithKline for generous funding.
Footnotes
SUPPORTING INFORMATION. Supporting information for this article including full experimental details and characterization data for all new compounds is available on the World Wide Web at http://pubs.acs.org.
REFERENCES
- 1.For recent reviews on the enantioselective synthesis of amines, see: Moody CJ. Angew. Chem., Int. Ed. 2007;46:9148. doi: 10.1002/anie.200703016.Ouellet SG, Walji AM, MacMillan DWC. Acc. Chem. Res. 2007;40:1327. doi: 10.1021/ar7001864.Skucas E, Ngai M-Y, Komanduri V, Krische MJ. Acc. Chem. Res. 2007;40:1394. doi: 10.1021/ar7001123.Masson G, Housseman C, Zhu J. Angew. Chem., Int. Ed. 2007;46:4614. doi: 10.1002/anie.200604366.Li G, Saibau K, Timmons C. Eur. J. Org. Chem. 2007;17:2745.Bräse S, Baumann T, Dahmen S, Vogt H. Chem. Commun. 2007:1881. doi: 10.1039/b611619a.Maruoka K, Ooi T, Kano T. Chem. Commun. 2007:1487. doi: 10.1039/b613049f.Ellman JA. Pure Appl. Chem. 2003;75:39.Roesky PW, Mueller TE. Angew. Chem., Int. Ed. 2003;42:2708. doi: 10.1002/anie.200301637.Enders D, Reinhold U. Tetrahedron: Asymmetry. 1997;8:1895.
- 2.For selected examples of synthesis of amines directly from alcohols, their corresponding esters, carbonates, sulfonate esters or alkyl halides, see: Kan T, Fukuyama T. Chem. Commun. 2004:353. doi: 10.1039/b311203a.Fujita K-i., Fujii T, Yamaguchi R. Org. Lett. 2004;6:3525. doi: 10.1021/ol048619j.Evans PA, Robinson JE, Nelson JD. J. Am. Chem. Soc. 1999;121:6761.Scriven EFV, Turnbull K. Chem. Rev. 1988;88:297.Kolasa T, Miller MJ. J. Org. Chem. 1987;52:4978.Bestmann HJ, Wölfel G. Chem. Ber. 1984;117:1250.Trost BM, Keinan E. J. Org. Chem. 1979;44:3451.Nordlander JE, Catalane DB, Eberlein TH, Farkas LV, Howe RS, Stevens RM, Tripoulas NA, Stansfield RE, Cox JL. Tetrahedron Lett. 1978;50:4987.Jonczyk A, Ochal Z, Makosza M. Synthesis. 1978:882.Zwierzak A, Podstawczynska I. Angew. Chem., Int. Ed. Engl. 1977;16:702.Hendrickson JB, Joffee I. J. Am. Chem. Soc. 1973;95:4083.Mitsunobu O, Wada M, Sano T. J. Am. Chem. Soc. 1972;94:679.Gibson MS, Bradshaw RW. Angew. Chem., Int. Ed. Engl. 1968;7:919.
- 3.For recent reviews of asymmetric desymmetrization, see: Rendler S, Ostreich M. Angew. Chem., Int. Ed. 2008;47:248. doi: 10.1002/anie.200704210.Rovis T. Recent Advances in Catalytic Asymmetric Desymmetrization Reactions. In: Mikami K, Lautens M, editors. New Frontiers in Asymmetric Catalysis. John Wiley & Sons, Inc.; Hoboken, NJ: 2007. pp. 275–311.Atodiresei I, Schiffers I, Bolm C. Chem. Rev. 2007;107:5683. doi: 10.1021/cr068369f.Schneider C. Synthesis. 2006;23:3919.Garcia-Urdiales E, Alfonso I, Gotor V. Chem. Rev. 2005;105:313. doi: 10.1021/cr040640a.
- 4.(a) Ferreira EM, Stoltz BM. J. Am. Chem. Soc. 2001;123:7725. doi: 10.1021/ja015791z. [DOI] [PubMed] [Google Scholar]; (b) Jensen DR, Pugsley JS, Sigman MS. J. Am. Chem. Soc. 2001;123:7475. doi: 10.1021/ja015827n. [DOI] [PubMed] [Google Scholar]
- 5.For an experimentally derived model for stereoselectivity in the oxidative kinetic resolution of benzylic alcohols catalyzed by Pd(sparteine)Cl2, see Trend RM, Stoltz BM. J. Am. Chem. Soc. 2004;126:4482. doi: 10.1021/ja039551q.
- 6.Kozlovskii AG, Solov'eva TF, Sahkarovskii VG, Adanin VM. Dokl. Akad. Nauk SSSR. 1981;260:230. [PubMed] [Google Scholar]
- 7.(a) Numata A, Takahashi C, Ito Y, Takada T, Kawai K, Usami Y, Matsumura E, Imachi M, Ito T, Hasegawa T. Tetrahedron Lett. 1993;34:2355. [Google Scholar]; (b) Jadulco R, Edrada RA, Ebel R, Berg A, Schaumann K, Wray V, Steube K, Proksch P. J. Nat. Prod. 2004;67:78. doi: 10.1021/np030271y. [DOI] [PubMed] [Google Scholar]; (c) Hayashi H, Matsumoto H, Akiyama K. Biosci. Biotechnol. Biochem. 2004;68:753. doi: 10.1271/bbb.68.753. [DOI] [PubMed] [Google Scholar]; (d) Dalsgaard PW, Blunt JW, Munro MHG, Frisvad JC, Christophersen C. J. Nat. Prod. 2005;68:258. doi: 10.1021/np049646l. [DOI] [PubMed] [Google Scholar]
- 8.For total syntheses of aurantioclavine, see: Somei M, Yamada F. Heterocycles. 2007;74:943.Yamada F, Makita Y, Suzuki T, Somei M. Chem. Pharm. Bull. 1985;33:2162.Hegedus LS, Toro JL, Miles WH, Harrington PJ. J. Org. Chem. 1987;52:3319.
- 9.(a) May JA, Stoltz BM. Tetrahedron. 2006;62:5262. [Google Scholar]; (b) May JA, Zeidan RK, Stoltz BM. Tetrahedron Lett. 2003;44:1203. [Google Scholar]
- 10.(a) Boit HG, Flentje H. Naturwiss. 1960;47:180. [Google Scholar]; (b) Gözler B, Lantz MS, Shamma M. J. Nat. Prod. 1983;46:293. [Google Scholar]
- 11.For discussions of the biological activity of the isopavine alkaloids, see: Weber E, Keana J, Barmettler P. PCT Int. Appl. WO 9012575, 1990. Chem. Abstr. 1991;115:106019w.Childers WE, Jr., Abou-Gharbia MA. U.S. Patent 4,940,789, 1990 Chem. Abstr. 1990;113:191190w.
- 12.For selected syntheses of isopavine alkaloids, see: Gözler B. Pavine and Isopavine alkaloids. In: Brossi A, editor. The Alkaloids. Vol. 31. Academic Press; New York: 1987. pp. 343–356.Meyers AI, Dickman DA, Boes M. Tetrahedron. 1987;43:5095.Gottlieb L, Meyers AI. J. Org. Chem. 1990;55:5659.Carillo L, Badía D, Domínguez E, Vicario JL, Tellitu IJ. J. Org. Chem. 1997;62:6716. For a synthesis of (−)-amurensinine, see:Shinohara T, Takeda A, Toda J, Sano T. Heterocycles. 1998;48:981.Hanessian S, Mauduit M. Angew. Chem., Int. Ed. 2001;40:3810.Dragoli DR, Burdett MT, Ellman JA. J. Am. Chem. Soc. 2001;123:10127. doi: 10.1021/ja016349j.
- 13.For our preliminary communication of the enantioselective synthesis of (+)-amurensinine, see: Tambar UK, Ebner DC, Stoltz BM. J. Am. Chem. Soc. 2006;128:11752. doi: 10.1021/ja0651815.
- 14.Felpin F-X, Lebreton J. Tetrahedron. 2004;60:10127. For a review of chemistry and biology of the Lobelia alkaloids, see: [Google Scholar]
- 15.For syntheses of racemic (±)-lobeline, see: Wieland H, Drishaus I. Annalen. 1929;473:102.Wieland H, Koschara W, Dane E. Annalen. 1929;473:118.Scheuing G, Winterhalder L. Justus Liebigs Ann. Chem. 1929;473:126.Wieland H, Koshara W, Dane E, Renz J, Schwarze W, Linde W. Justus Liebigs Ann. Chem. 1939;540:103.Parker W, Raphael RA, Wilkinson DI. J. Chem. Soc. 1959:2433.Glaser R, Hug P, Drouin M, Michael A. J. Chem. Soc., Perkin Trans. 2. 1992:1071. For an X-ray crystallography study of (−)-lobeline salts, see:Keogh MF, O'Donovan DG. J. Chem. Soc. 1970:2470. For a study of the biosynthesis of (−)-lobeline, see:
- 16.(a) Millspaugh CF. American medicinal plants: an illustrative and descriptive guide to plants indigenous to and naturalized in the United States which are used in medicine. Dover; New York: 1974. Lobelia inflata; pp. 385–388. [Google Scholar]; (b) Dwoskin LA, Crooks PA. Biochem. Pharmacol. 2002;63:89. doi: 10.1016/s0006-2952(01)00899-1. [DOI] [PubMed] [Google Scholar]; (c) Thayer AM. Chem. Eng. News. 2006;84:21. [Google Scholar]
- 17.Zheng G, Dwoskin LP, Deacuic AG, Norrholm SD, Crooks PA. J. Med. Chem. 2005;48:5551. doi: 10.1021/jm0501228. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
18.The epimerization at C(3) is believed to be a base-catalyzed equilibration via transient retro-conjugate addition intermediate 19. See also ref 18a, 18b and 18d.
- 19.For enantioselective syntheses of (−)-lobeline, see: Compere D, Marazano C, Das BC. J. Org. Chem. 1999;64:4528.Felpin F-X, Lebreton J. J. Org. Chem. 2002;67:9192. doi: 10.1021/jo020501y.Klingler F-D, Sobotta R. (Boehringer Ingelheim) US 2006014791 Birman VB, Jiang H, Li X. Org. Lett. 2007;9:3237. doi: 10.1021/ol071064i.
- 20.(a) Marion L, Lavigne R, Lemay L. Can. J. Chem. 1951;29:347. doi: 10.1139/v51-040. [DOI] [PubMed] [Google Scholar]; (b) Franck B. Chem. Ber. 1958;91:2803. [Google Scholar]; (c) Logar C, Mesicek M, Perpar M, Seles E. Farm. Vestn. 1974;21 [Google Scholar]; Chem. Abstr. 1975;82:82916h. [Google Scholar]; (d) Krasnov EA, Petrova LV, Bekker EF. Khim. Prir. Soedin. 1977:585. [Google Scholar]; Chem. Abstr. 1977;87:164249k. [Google Scholar]
- 21.For a review of syntheses of the Sedum alkaloids, see: Bates RW, Sa-Ei K. Tetrahedron. 2002;58:5957.For recent syntheses of either enantiomer of sedamine, see: Bates RW, Nemeth JA, Snell RH. Synthesis. 2008:1033.Fustero S, Jiménez D, Moscardó J, Catalán S, del Pozo C. Org. Lett. 2007;9:5283. doi: 10.1021/ol702447y.Yadav JS, Reddy MS, Rao PP, Prasad AR. Synthesis. 2006:4005.Yadav JS, Reddy MS, Rao PP, Prasad AR. Tetrahedron Lett. 2006;47:4397.Bates RW, Boonsoombat J. Org. Biomol. Chem. 2005;3:520. doi: 10.1039/b415297b.Josephson NS, Snapper ML, Hoveyda AH. J. Am. Chem. Soc. 2004;126:3734. doi: 10.1021/ja049388e.Zheng G, Dwoskin LP, Crooks PA. J. Org. Chem. 2004;69:8514. doi: 10.1021/jo048848j.Angoli M, Barilli A, Lesma G, Passarella D, Riva S, Silvani A, Danieli B. J. Org. Chem. 2003;68:9525. doi: 10.1021/jo035215g.Cossy J, Willis C, Bellosta V, BouzBouz S. J. Org. Chem. 2002;67:1982. doi: 10.1021/jo010653d.
- 22.Tambar UK, Stoltz BM. J. Am. Chem. Soc. 2005;127:5340. doi: 10.1021/ja050859m.Others have disclosed similar aryne insertions after our initial report, see: Yoshida H, Watanabe M, Ohshita J, Kunai A. Chem. Commun. 2005:3292. doi: 10.1039/b505392g.Yoshida H, Watanabe M, Ohshita J, Kunai A. Tetrahedron Lett. 2005;46:6729.
- 23.Kozikowski AP, Ishida H, Chen Y-Y. J. Org. Chem. 1980;45:3350. [Google Scholar]
- 24.Bachki A, Foubelo F, Yus M. Tetrahedron: Asymmetry. 1996;7:2997. [Google Scholar]
- 25.The selectivity factor, ‘s’ for a kinetic resolution is defined as the ratio of the rates of reaction of the fast reacting enantiomer of the racemic substrate, kfast to the rate of reaction of the slow reacting enantiomer, kslow in the same transformation, i.e. s = (kfast/kslow). The selectivity factor can be expressed in terms of enantiomeric excess (ee) of the remaining alcohol and the conversion (c) of the alcohol to the corresponding ketone, i.e. s = ln[(1−c)(1−ee)]/ln[(1−c)(1+ee)] . For s > 10, synthetically useful quantities of enantioenriched alcohol can be accessed. For example, an oxidative kinetic resolution with s = 10 at 62% conversion would afford recovery of alcohol of 90% ee. See also Kagan HB, Fiaud JC. In: Topics in Stereochemistry. Eliel EL, editor. Vol. 18. Wiley & Sons; New York: 1988. pp. 249–330.
- 26.Mitsunobu O. Synthesis. 1981:1. [Google Scholar]
- 27.Fukuyama T, Jow C-K, Cheung M. Tetrahedron Lett. 1995;36:6373. [Google Scholar]
- 28.For reviews of the Stille cross-coupling reaction, see: Stille JK. Angew. Chem., Int. Ed. Engl. 1986;25:508.Farina V, Krishnamurthy V, Scott WJ. Org. React. 1997;50:1.Littke AF, Fu GC. Angew. Chem., Int. Ed. 2002;41:4176. doi: 10.1002/1521-3773(20021115)41:22<4176::AID-ANIE4176>3.0.CO;2-U.Espinet P, Echavarren AM. Angew. Chem., Int. Ed. 2004;43:4704. doi: 10.1002/anie.200300638.
- 29.(a) Knights EF, Brown HC. J. Am. Chem. Soc. 1968;90:5281. [Google Scholar]; (b) Brown HC, Knights EF, Scouten CG. J. Am. Chem. Soc. 1974;96:7765. [Google Scholar]
- 30.See supporting information for details.
- 31.Morris LJ. J. Lipid. Res. 1966;7:717. [PubMed] [Google Scholar]
- 32.Yasuhara A, Sakamoto T. Tetrahedron Lett. 1998;39:595. [Google Scholar]
- 33.(a) Wenkert E, Davis LL, Mylari BL, Solomon MF, Da Silva RR, Shulman S, Warnet RJ, Ceccherelli D, Curini M, Pellicciari RJ. J. Org. Chem. 1982;47:3242. [Google Scholar]; (b) Taber DF, Petty EH. J. Org. Chem. 1982;47:4808. [Google Scholar]
- 34.Himeshima Y, Sonoda T, Kobayashi H. Chem. Lett. 1983:1211. [Google Scholar]
- 35.Thompson AS, Humphrey GR, DeMarco AM, Mathre DJ, Grabowski EJJ. J. Org. Chem. 1982;47:4808. [Google Scholar]
- 36.Dess DB, Martin JC. J. Org. Chem. 1983;48:4155. [Google Scholar]
- 37.Nishimura T, Onoue T, Ohe K, Uemura S. J. Org. Chem. 1999;64:6750. doi: 10.1021/jo9906734. [DOI] [PubMed] [Google Scholar]
- 38.We have recently accomplished a formal synthesis of the naturally occurring enantiomer of amurensinine using a readily accessible diamine ligand that serves as a mimic for (+)-sparteine in the oxidative kinetic resolution. See Ebner DC, Trend RM, Genet C, McGrath MJ, O'Brien P, Stoltz BM. Angew. Chem. Int. Ed. 2008 doi: 10.1002/anie.200801865. Early View.
- 39.Molander GA, Romero JAC. Tetrahedron. 2005;61:2631. [Google Scholar]
- 40.A rationale for the observed selectivity follows from the calculated conformational equilibria of cis,cis-1,4-cyclooctadiene. See: Anet FAL, Yavari I. J. Am. Chem. Soc. 1977;99:6986. Also see supporting information for a stereochemical model.
- 41.Bagdanoff JT, Stoltz BM. Angew. Chem., Int. Ed. 2004;43:353. doi: 10.1002/anie.200352444. [DOI] [PubMed] [Google Scholar]
- 42.The enantiomeric excess was determined after derivatization of lobeline by the method described in reference 21(h). See supporting information for details.
- 43.Crooks, P. A., and co-workers (reference 21(h)) noted that the rate of epimerization of diastereomerically pure cis-lobeline to 46:54 cis:trans-lobeline in various deuterated solvents by 1H NMR (CD3OD > CD3CN > CD3COCD3 > CDCl3), see ref. 21(h)]. In our hands, the epimerization of diastereomerically pure cis-lobeline to 1:1 cis:trans-lobeline was much slower in C6D6 (5 days) as opposed to CDCl3 (2 days) and CD3OD (2 hours).

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.