

Summary
In E. coli, the chemotaxis receptor protein Tsr localizes abundantly to cell poles. The current study, utilizing a Tsr-GFP fusion protein and time lapse fluorescence microscopy of individual cell lineages, demonstrates that Tsr accumulates approximately linearly with time at the cell poles and that, in consequence, more Tsr is present at the old pole of each cell than at its newborn pole. The rate of pole-localized Tsr accumulation is large enough that old and new poles can always be reliably distinguished, even for cells whose old poles have had only one generation to accumulate signal. Correspondingly, Tsr-GFP can be reliably used to assign new and old poles to any cell without use of information regarding pole heritage, thus providing a useful tool to analyze cells whose prior history is not available. The absolute level of Tsr-GFP at the old pole of a cell also provides a rough estimate of pole (and thus cell) age.
Keywords: Tsr, chemosensory receptor, E. coli, cell pole, cell lineage, aging
Introduction
The cell poles of rod-shaped bacteria are functionally specialized regions. This specialization is particularly prominent with regard to transport of molecules to the cell exterior. In E.coli, polysaccharide biosynthesis and export across the outer membrane occur specifically in these regions (McNulty et al., 2006). So, too, does secretion of autotransporters, a family of large virulence-associated proteins also present in several other bacterial species (Jain et al., 2006, Brandon et al., 2003). Similarly, the type II secretion system of Vibrio cholerae, devoted to transport across the outer membrane and the type IV system of Legionella pneumophilia and Agrobacterium tumefaciens, which moves proteins across both membranes, are pole-localized (for references, see Jain et al., 2006). Streptococcus F protein is also secreted specifically at cells poles, though it finally distributes throughout the cell surface (Carlsson et al., 2006). Chemotaxis proteins also localize abundantly to poles via membrane anchoring of chemoreceptors, resulting in highly ordered clusters that are thought to be required for normal signaling responses (for recent discussions, see Sourjik and Berg, 2004, Shiomi et al., 2005). Cluster assembly appears to be a stochastic process involving competition between nucleation of new clusters and growth of existing clusters (Thiem et al., 2007, Thiem and Sourjik, 2008). Similarly, secreted proteins such as alkaline phosphatase or maltose binding protein collect differentially in the periplasmic spaces of the poles, a phenomenon known as formation of “polar caps” (Dietzel et al., 1978, Wetzel et al., 1970) Additional complexities in pole function arise because the two poles of each cell are not identical. Each cell division yields two sister cells. In each sister, one pole has just arisen via septum formation at the time of birth (the “new pole”) while the other was already present in the parent cell, having arisen during some prior division (the “old pole”). In several cases, functional and/or morphological differences mirror this asymmetry in lineage. For example, In Shigella and Listeria, the old pole is used specifically to direct formation of an actin tail. In Caulobacter, old/new pole asymmetry is used to create a swimming “swarmer cell”, in which flagella and chemotaxis proteins are localized at the old pole; when a suitable environment is identified, the flagellum is lost and a stalk grows in its place which fixes the cell to its food source (reviewed in Shapiro et al., 2002). Also, in some strains of Rhizobium leguminosarum, an extracellular calcium-binding protein and a high molecular weight surface polysaccharide were both detected exclusively on one pole of the cell which, in the case of the polysaccharide is used for polar adhesion of the bacterium to the legume root hair during colonization (Ausmees et al., 2001, Laus et al., 2006).
In E.coli, analysis of individual cell lineages has revealed that cells harboring old and new poles are not functionally identical with respect to growth and division kinetics (Stewart et al., 2005). The older the “old” pole of a cell, the slower its rate of growth, a phenomenon that has been interpreted to represent cell “aging”; in contrast, cells whose old poles are in their first generation exhibit increased growth rates, a phenomenon interpreted to represent “rejuvenation”. The basis for the observed correlation between cell age and slower growth is unknown but it has been suggested that the accumulation of mis-folded proteins at the poles could play a role (Stewart et al., 2005, Woldringh, 2005).
Preferential occurrence of certain processes at cell poles may be a function of their unique structure. For example, at E.coli poles, neither the external peptidoglycan nor multiple components of the underlying outer membrane exhibit turnover or dilution by new synthesis (for discussion, see Jain et al., 2006). Moreover, as this stability is more prominent at old poles than at new poles (de Pedro et al., 1997, Mileykovskaya and Dowhan, 2000), these structural features may also relate to functional differences between the two poles.
Old/new cell polarity is also of interest with regard to chromosome dynamics. In slowly growing E.coli where, analogously to the eukaryotic cell cycle, one round of replication is completed before another begins, a cell undergoing division carries its origin of replication closer to its old pole while the terminus region is close to the newly-arising new pole (e.g. Bates and Kleckner, 2005). Dynamic changes in the locations of these and other regions then ensue which might, or might not, be influenced by old/new heritage of the two cell poles (Bates and Kleckner, 2005, Wang et al., 2006).
In the present study we have analyzed in further detail the polar abundance of the serine chemotaxis receptor protein Tsr (Fig. 1A). Tsr is the sensory element of a downstream signalling pathway, all protein components of which interact with the cytoplasmic domain of Tsr. Two of these components, histidine autokinase CheA and the adapter protein CheW, as well as Tsr are essential for polar clustering of chemotaxis components (Zhang et al., 2007). Other proteins such as CheR (Banno et al., 2004) and CheB (Sourjik and Berg, 2004) are involved in the methylation and demethylation, respectively, of four specific glutamate residues on the Tsr molecule (green balls with arrows in Fig. 1A). Interestingly, however, polar clustering of Tsr is largely independent of chemotactic activity (Liberman et al., 2004).
Figure 1.
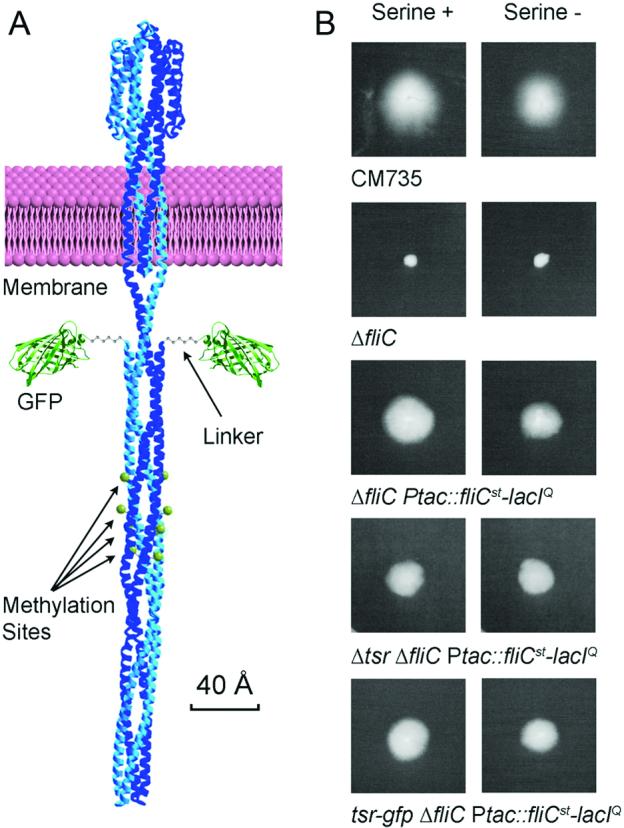
Construction of Tsr-GFP strain. (A) The predicted structure of the Tsr-GFP fusion protein. All components are proportional to their actual size. The Tsr protein picture is adapted from (Kim et al., 1999). Green balls with arrows indicate the sites of CheR/CheB-medited methylation/demehylation. (B) Swarm assay verifying the functionality of the fusion protein. On the left are results of colonies grown in the presence of 1 mM serine. The right column is growth in the absence of serine. Strain CM735 is the original wild type. The genotype of each strain is shown beneath each comparison. Based on 4 replications, the size of the occupation area of the tsr-GFP strain (NK9456, bottom panel) on serine plus plates is 1.367 ± 0.248 times larger than the same strain on serine minus plates; while that of NK9386 (middle panel) on serine plus plates is 1.380 ± 0.254 times larger than the same strain on serine minus plates. The serine blind strain (second panel from bottom) showed no difference (0.999 ± 0.031).
We were interested not only in exactly how differential localization of Tsr to the two poles arises but also whether the relative intensities of the two poles could be a reliable indicator for specifically identifying the “old” and the “new” poles of an individual cell in the absence of any information as to the ancestry of that cell. If so, Tsr-GFP could be used to ask whether other processes, e.g. relating to chromosome dynamics, are asymmetric with regard to a cell’s old and new poles. To this end, we have analyzed Tsr-GFP distributions in individual cell lineages and pairs of just-divided agar-immobilized sister cells. We find that Tsr-GFP fluorescence accumulates progressively at both the old and new poles of each cell and fails to localize to mid-cell until around the time of division. As a result, Tsr-GFP is more abundant at the old pole of the cell than at the new pole. The rate of increase in fluorescence is such that old and new poles are reliably identified and distinguished in virtually every cell. Analysis of synchronous populations of newborn cells and asynchronous populations in several media suggest that the described approach is widely applicable to old/new pole identification in diverse growth conditions.
Results and Discussion
Creation of a fusion construct expressing Tsr-GFP
We created a tsr allele that expressed a Tsr protein with GFP fused to its C-terminus (Tsr-GFP; Fig. 1A). In order to avoid interference between the C-terminal GFP tag and access of Tsr-interacting proteins (Introduction), sequences encoding a six-glycine linker, ∼2.5 nm predicted length, was placed between the GFP and Tsr sequences in the fusion gene (Fig. 1A). This tsr-GFP fusion gene was substituted for the wildtype tsr gene in an E.coli strain that contains additional markers making it convenient for use in obtaining synchronous cell populations (Bates et al., 2005). The resulting strain was assayed for Tsr activity. E.coli exhibits a chemotactic response to serine, mediated by Tsr, which can be detected by enhanced movement of cells outwards from a focal inoculation point on 0.35% agar plates containing 1 mM serine, as compared to identical plates lacking serine. This difference can be seen for a fully wild-type strain (Fig. 1B, first line) and for the multiply marked derivative into which the fusion gene was integrated (Fig. 1B, third line). Correspondingly, strains deleted either for the tsr gene or for a gene involved in making the flagella needed for chemotactic movement (fliC) exhibit the same response in the presence and absence of serine (Fig.1B, second and fourth lines). The tsr-GFP fusion strain exhibits the same chemotactic response as its isogenic tsr counterpart (Fig. 1B, fifth versus third lines, respectively) with no detectable defect in activity.
Visualization of Tsr-GFP
Fluorescence visualization of Tsr-GFP in cells of asynchronous populations confirms the earlier finding (Introduction) that, in every cell one pole is brighter than the other (e.g. Figs. 2AB). These images further show that fluorescence is rarely detectable at mid-cell except in the few especially long cells where the presence of invaginations suggests that septation is underway (Fig. 2B, arrowheads). Small aggregates are occasionally also seen associated with the edge of a cell at a non-polar position (in 5-8% of cells; e.g. Fig. 2AC, also see Fig. 5B, lineage 4) consistent with other studies. Lateral aggregates have been observed in wild-type cells by immuno-electron microscopy using gold-particle labeled anti-Tsr antibody (Lybarger and Maddock, 2000). In addition lateral clusters were observed of CheR by fluorescence microscopy (Thiem et al., 2007). Interestingly, these authors suggested that chemosensory receptors occur not only at the poles but also at positions corresponding to future division sites
Figure 2.

Epifluorescence microscopy of bacteria expressing Tsr-GFP showing that Tsr-GFP occurs preferentially at one of the two cell poles. (A,C), phase contrast image and corresponding fluorescent image were superimposed. (A) A typical field with living cells from an exponential culture mounted on agar slab. One representative cell with two lateral Tsr-GFP foci is marked by a square, and shown enlarged in (C). Arrows indicate lateral foci. (B) Representative cells from a dilute asynchronous population show the difference between the two poles more clearly than in (A) due to reduced background level. Arrowheads indicate appearance of Tsr-GFP at cell midpoints in two about-to - divide cells, also enlarged and shown to the right. (D) A diagram depicts the distribution of detectable GFP signal on the surface of a representative cell. (E) The 3D reconstruction of Tsr-GFP signal used to construct the image in (D). The cell was placed at a ca. 45 degree angle towards the horizontal plane, and rotated around the vertical axis, which passes the cell center. Diagrams below show the corresponding projections on the horizontal plane. Broken arrows indicate the original position. Solid arrows show the directions in the above image.
Figure 5.

Pole brightness increases in growing cells. (A) Diagram of the pole numbering system used in lineage analysis, with each pole represented by a different color. Brightness was quantified for only the first 8 poles of a given lineage. The generations after the start of observation are listed to the right side. (B) Brightness increases of individual poles over time. Four lineages representative of the different types observed were chosen (Figs. S1-4) and analyzed. Poles identified as in (A). Broken lines demark the beginning of each generation. Images acquired for Lineage 1 are those shown in Fig. 4B. Founder cell in Lineage 4 contained a large internal aggregate as shown in inset. Asterisks (*) indicate cases in which poles 3 and 4 (red and green, respectively) exhibit reciprocally higher and lower rates of brightness increase as compared to the average (text). (C) Distribution of average rates of increase in pole brightness, in arbitrary brightness units (AUs) (Thiem et al., 2007). For each pole of each lineage, the straight line that best fits the curve of brightness versus time was determined. (i) The distributions of the slopes of all of these lines. The mean of brightness units per 120min generation is shown inside the plot. (ii) Average rates of brightness increase for each type of pole as defined in (A). (D) Old pole minus new pole brightness in each kind of cell from all four analyzed movies. (E) Old pole minus new pole brightness on each time point in (B). Generation numbers are shown on top, followed by enumeration of cells by their pole contents.
Three-dimensional imaging of individual fixed cells, with z-sections separated by 200 nm, reveals that, at relatively bright poles, the Tsr-GFP forms a symmetrical “cap” that hugs the inner surface of the cell while, at less bright poles, the signal occurs as an irregular aggregate, rather than being smoothly distributed. These features are seen in 3D reconstructions (Fig. 2DE). These patterns also correspond to findings from cryoelectron tomography showing that Tsr form arrays composed of about 6,500 receptor molecules, often on one side of the pole dome (Zhang et al., 2007).
Quantification of Tsr-GFP fluorescence at cell poles
To permit detailed analysis of Tsr-GFP abundance, pole fluorescence was quantitated from 0.2μm mid-cell focal-plane images (Experimental Procedures). For each cell of interest, the two poles and a nearby blank background area, were each surrounded with a semicircle (Fig. 3A). Fluorescence intensities were then recorded for every pixel within each shape (∼500 total) and the numbers obtained sorted from highest to lowest (Fig. 3B inset). Such plots make it apparent that the level of external background is quite low and very constant; moreover, even at the dimmest poles, the brightest ∼50 pixels are always all above background. The fluorescence intensity at each pole was therefore determined by subtracting the average background pixel intensity from each of the pixel intensities for that pole (Fig. 3B main plot) and then averaging the background-subtracted intensities for the 50 brightest pixels (cut-off indicated by dotted vertical line).
Figure 3.


Method for analyzing brightness of poles. (A). Top panel; Overlayed phase and fluorescence images of a single cell. To quantify the fluorescent signal on the cell poles, only the fluorescence image (bottom panel) was employed. The phase overlay was used to define the cell boundary, especially when one of the cell poles is very dim. To measure the gray level, a semicircle was drawn big enough to include the whole cell pole within it. The same semicircle was moved and rotated so that the other cell pole was caught completely. The semicircle was further moved and rotated to an area free of cells to measure the background gray level. B) Rank order plots of the signals acquired from A. Inset is the plot of raw data from the bright pole (blue), dim pole (red) and background (black). The outside plot shows the results after background subtraction. The blue line is the net bright pole intensity and the red line the net dim pole intensity. The vertical broken line indicates the brightest 50 pixels for each pole. The average of these was used in subsequent analysis. For the dim pole the intensities of pixels lower than the 50 brightest is strongly dependent on exactly the amount of background subtracted, By using only the 50 brightest pixels for comparison, the contribution of fluctuations in background levels is minimized.
Analysis of Tsr-GFP pole fluorescence at poles in single cell lineages
To determine precisely the dynamics of Tsr-GFP accumulation, single cell lineages were analyzed at 30°C in AB medium (Fig. 4, Fig. S1-4; Experimental Procedures). Under these conditions, as characterized in previous studies from this laboratory (Bates and Kleckner, 2005), E. coli undergoes a “linear” cell cycle involving stages analogous to G1-S-G2-M of the eukaryotic cycle. Lineages were recorded for cells constrained within agarose microfluidic channels (Fig. 4A). Periodic infusion of fresh growth medium ensured that cells in a favorable region (Fig. 4A, legend) exhibited a constant growth rate, with a doubling time of ∼120min, as previously observed for asynchronous batch populations growing under the same conditions (Bates and Kleckner, 2005). For each analysis, a single cell was identified and photographed for phase contrast and fluorescence imaging every 20 minutes for two to three generations (e.g. Fig. 4B; Figs. S1-4).
Figure 4.

Cell lineage pedigree analysis. (A) Schematic of time lapse methodology. An agarose slab containing microfluidic channels of 1um in both width and depth, formed in a PDMS mold, was set on a microscopic slide (central square with lines). Bacteria were pipetted into the channels then a cover glass applied and its edges covered by grease (shown in grey) leaving three openings. The sample was maintained at constant temperature by use of a suitably-positioned hair dryer (Experimental Procedures), which produces an air current as depicted by the big gray arrows. The vacuum generated by this air flow causes air to exit from under the coverslip as shown by the small hollow arrows. The nutrient solution was supplemented every 60 minutes during imaging by pipetting in through the bottom opening (open arrowhead). The oval shape indicates where the cells were photographed. Cells in this area exhibit a constant rate of division, comparable to that observed under the same growth conditions in batch culture, throughout the period of data collection. Regions closer to the air flow, and along the left and right edges of the slab, tend to dry out: cells closer to the point of nutrient addition tend to float away during the infusion process. (B) One cell pedigree (Lineage 1 in the following analysis). On the left are the phase contrast images. On the right are the fluorescence images of the same cells. Arrowheads point to invaginations indicating that cells have divided and to the fluorescent signals at mid-cell which appear at the same time. The time points are listed to the left of the pictures.
Tsr-GFP intensities were quantified as described above at all cell poles of individual cell lineages over three generations. Results obtained by this method are not significantly affected by photo-bleaching of the GFP signal. As a control experiment, single cell lineages were traced by phase contrast images taken every 20 min as in Figs. 4 and 5 but subjected to UV excitation for fluorescence images only occasionally, just before and just after each cell division. Levels and rates of increase of pole fluorescence observed by this protocol were not different from those observed when fluorescence images were collected in concert with phase images every 20 min over three generations (data not shown).
Dynamics of Tsr-GFP accumulation
Qualitative inspection of cell lineages reveals the basis for unequal Tsr abundance at the two poles of individual cells. During a given cell cycle, Tsr-GFP accumulates at both poles but does not accumulate detectably at mid-cell until the time of division. Times of cell division during any particular lineage were defined as the points at which mid-cell invaginations become prominent between the two cells (e.g. in Fig. 4B, arrowheads at t = 160min and 280min). For each of the four analyzed lineages, visible fluorescence signal appears at the midcell division site either in the same frame denoted as “division” or the immediately subsequent frame (e.g. Fig. 4B, 160 min and Figs. 1-4). As a result, cell division produces two sister progeny cells with abundant fluorescence at their old poles and little or no detectable fluorescence at their new poles.
Progressive accumulation at old and new poles
Additional information emerges from quantitative analysis of pole fluorescence of four lineages, including that in Fig. 4B (= Lineage 1). Collages of all corresponding movie frames are presented in Figs. S1-4. For ease of reference, poles of each lineage are numbered 1-8 in order of appearance (Fig. 5A).
Tsr-GFP intensities increase progressively with time at all poles, with relatively similar accumulation rates in most cases, albeit with significant variations. These effects can be seen by simple visual inspection of accumulation curves (Fig. 5B). For more quantitative treatment, we considered the slopes of best-fit lines for the curves for each pole which, despite relatively poor fits in some cases (r2 values; mean=0.91, median=0.95) provide a general picture. Rates of brightness increase vary among all analyzed poles by a factor of ∼six, with a mean of ∼68±32 brightness units per 120 min generation; a majority (53%) of all values falling in the range of 45-75; and no obvious variation by pole type (Fig. 5Ci, ii). Tsr-GFP will accumulate linearly at the poles as along as the rate of synthesis exceeds the rate of turnover. It might be expected, however, that accumulation might saturate and/or trigger some special response and/or a catastrophe (death). The kinetics of accumulation at Pole 1 of Lineage 3 might suggest such saturation as reported elsewhere (Thiem and Sourjik, 2008)
Other irregularities also occur. (a) In Lineages 1, 3 and 4, sister poles 3 and 4 exhibit significantly different accumulation rates (Fig. 5B, asterisks) suggesting that an asymmetry may have been set up at the time of division. (b) In Lineage 3, the oldest pole (Pole 1) is particularly bright in the first cell of the lineage and increases in brightness at a lower rate than the other three “oldest” poles. This may reflect a tendency for saturation of pole brightness (Thiem and Sourjik, 2008). (c) In Lineage 4, a lateral aggregate is present in the initial cell (Fig. 5B; Fig.S4). This aggregate, which may evolve into an “ectopic pole”, appears to influence accumulation of Tsr-GFP at certain poles. In the initial cell, i.e. during the first generation of observation (“G1”), the rate of Tsr-GFP accumulation at the pole nearest the aggregate (Pole 2, blue) is much lower than normal while accumulation at the distal pole is more normal (Pole 1, black). Severely reduced accumulation at Pole 2 continues into the next generation. Mysteriously, however, the rate of increase returns to normal in the third generation of observation (“G3”), despite the fact that the aggregate remains prominent. These phenomena are presumably related to competition between nucleation of new chemosensory clusters and growth of existing clusters observed by Thiem and Sourjik (2008).
The old pole is always brighter than the new pole
Within all of the examined cells, the old pole is always brighter than the new pole. This relationship can be inferred from the primary plots of pole brightness: when new poles appear, the fluorescence levels of those poles is lower than that of all pre-existing poles, implying that, necessarily, every new pole is dimmer than its partner old pole (Fig. 5B; compare Poles 3 and 4 versus Poles 1 and 2 at the onset of G2 and Poles 5-8 versus Poles 1-4 at the onset of G3). This relationship can be seen directly in plots of [old-new] pole differences for every cell in each lineage (Figs. 5D,E). It is particularly notable that this relationship holds even for cells whose old poles are in their first generation as old poles, and thus are the dimmest old poles in the population (i.e. cells 2,4; 3,6 and 4,7; Figs. 5A, 5D). Such plots also show that the [old-new] pole difference of any given cell usually remains approximately constant over the time of a single cell cycle (Fig. 5E), a predicted consequence of linear accumulation at both poles.
Robustness of the [old-new] pole brightness differences
An important implication of the findings presented above is that the old and new poles of any individual cell should be identifiable simply from their relative intensities, irrespective of any lineage information. For this approach to be of practical value, relative pole intensities must correctly identify new and old poles in essentially every cell, with few if any exceptions. While this is true for every analyzed frame of each of the four lineages analyzed by time lapse analysis (above; Fig. 6Bi), we sought to confirm and extend this conclusion to larger, more representative populations of cells.
Figure 6.

Comparisons of pole brightnesses among different cell populations. (A) Depiction of cells obtained from baby cell column. Note particularly: “newborn” and “just-divided” cells. Also: new poles (N), first-generation old pole (O1) and another old pole (O). Finally: note that any pole in a newborn or just-divided cell is at the start of the cell cycle (“start cc”; C below). (B) Top row: [old-new] or [bright-dim] differences in pole brightness for the indicated populations. (i) Lineage cell population was obtained by considering every frame of each of the four presented lineages as a single cell (and thus is not fully representative of a random cell population). (ii, iii) Just-divided and newborn cells are defined above; note that the corresponding poles are all at the beginnings of their cell cycles. (iv-vi) Asynchronous populations growing exponentially in liquid cultures of the indicated media. Middle row: expansion of area in small boxes in lower left corner of top row plots. Bottom row; rank plots of pole brightnesses for each type of pole in the indicated populations. (C) Average brightnesses of various categories of poles in the populations defined in (B) and the types of poles as defined for (A) (see legend above). (D) Left; relative frequencies of pole brightnesses predicted for an asynchronous population assuming average rate of brightness increase observed in Fig. 5. Right; observed distribution of pole brightnesses for all poles in an asynchronous AB population (black line) compared with that predicted (grey line, which connects the midpoints of the bars in (D;left).
We therefore carried out a second, entirely different type of analysis in which a population of nearly-synchronous cells was allowed to undergo division on an agar surface, where the identities of old and new poles are given by the relative positions of daughter cell pairs. This situation was achieved as follows. Using our recently-described “baby cell column” (Bates et al., 2005), we collected an aliquot of synchronous newborn cells growing under the same conditions as in experiments described above (Fig. 6A, “newborn”). We then further incubated those cells for ∼110 minutes in liquid medium to allow near-completion of the ongoing division cycle and transferred the cells to agar for an additional ten minutes to permit completion of the division. In such populations, the majority of cells undergo division during the ten minutes after immobilization, thus providing a large population of “just-divided” cells for which old-versus-new pole fluorescence can be compared (Fig. 6A, “just divided cell”).
Among such “just-divided cells”, both sister cells have “just-emerged” new poles, i.e. new poles that are at the beginning of their respective cell cycles. In contrast, one sister cell has a brighter old pole while the other sister has a dimmer old pole. The brighter old pole is presumably the older of the two and thus was the old pole in the immediate progenitor cell (“O” of the “baby cell” in Fig. 6A); the age of this brighter/older old pole is unknown (Fig. 6A). The dimmer old pole was presumably the new pole in the progenitor cell and is now a “just-emerged” old pole (O1, Fig. 6A). For all 338 sister pairs analyzed, in all but one cell of one pair, the old pole is brighter than the new pole. This is true not only for the half of cells containing the brighter old pole but also for the half containing the dimmer old pole as shown by rank order plots of [old-new] pole brightnesses (Fig. 6Bii; top row; expansion of lowest brightness levels in middle row). Furthermore, when all old pole brightnesses and all new pole brightnesses are each arranged in rank order, it emerges that virtually every old pole, of either type (O or O1), is brighter than any new pole (Fig. 6Bii, bottom row). Correspondingly, in these just-divided cells, average pole brightness for all new poles (N) is less than that for dimmer old poles (O1) which is less than that for brighter old poles (O) (just-divided [orange]; Fig. 6D). The properties of these many just-divided cells can be compared with cells representing all of the frames of the four lineage movies. Analogous patterns are observed for all three aspects described above (Fig. 6B, compare i with ii in all three rows; Fig.6D compare orange and black).
Tsr-GFP intensities at poles in non-lineage populations
To further assess the possibility of using pole brightness to distinguish new versus old poles, we examined two different populations whose heritage was not known. One such population is provided by baby cells that have just been released from the column (“newborn”, Fig. 6A). A second such population is provided by cells in an asynchronously growing culture (Fig. 6Biii and 6Biv). In both populations, in each given cell, the dimmer and brighter poles should be the new and old poles, respectively. Accordingly: the array of [bright - dim] pole brightness differences and the arrays of total brighter and total dimmer pole brightnesses in the two non-lineage populations are very similar to the analogous [old-new] and total old and new pole brightnesses in the just-divided and lineage movie cell populations.
Accuracy of pole brightness values
As yet another way of comparing different cell populations, we examined the average brightnesses of different categories of poles (Fig. 6C). In any given cell, one pole is new (or dim) and the other is old (or bright); further, in a given test population, the old poles are all in their first generation as old poles (“O1”) or represent first plus older generations (“all old”), depending on the nature of the experiment and, for movie lineages, the particular cells chosen. Additionally, for each of the three types of poles (new, O1 or all old), those poles may be specifically in cells that just divided and thus are at the start of their cell cycle (“start cc”) or they may be present cells that represent all points in the cell cycle (“tot cc”). The considerations presented above predict that pole brightnesses for different categories of poles should occur in the following hierarchy: New-start cc < New-tot cc < O1-start cc < O1-tot cc < all old-tot cc. When the appropriate pole types are considered for the several populations analyzed, this relationship is precisely observed. Further, levels are comparable among different test populations in each category (Fig. 6C).
As another test we compared the range of brightnesses observed in the asynchronous population to that predicted based on the average rate of brightness increase observed in the lineage analysis in Fig. 5C). In the predicted pool half of the cells would have new poles; one quarter have a first-generation old pole; one eighth have a second-generation old pole, etc. (Fig. 6D left). When this theoretical population is compared to the actual asynchronous cells from Fig.6Civ there is a close alignment between the two populations (Fig. 6D right).
Can pole intensity be used to evaluate pole age in individual cells?
The fact that pole age and pole brightness are strongly correlated raises also the further question of the extent to which pole brightness can be used to reliably define pole age. This issue only arises for old poles, since the new pole of each cell is, by definition, in its first generation of life. We find, however, that there is substantial variation among brightness levels for old poles of the same age and, moreover, successive generations exhibit substantially overlapping brightness levels (e.g. Fig. 5B). Furthermore, since old pole intensities at the end of one generation and the beginning of the next are, by their nature, virtually the same, it is challenging to integrate the discrete parameter of cell generation with the continuous parameter of pole brightness. It is our impression that old pole brightness can give a rough idea of pole age. For example, an old pole with a brightness level of 100 is likely to be in its second generation as a pole but with a small probability that it is in its third; and a pole with a brightness level of 500 is almost certainly more than four or five generations old, but with a small probability that it is younger.
In accord with these findings, it has also been shown that Tsr-GFP cluster size depends strongly on protein concentration (Thiem and Sourjik, 2008), which, in turn varies dramatically due to stochastic nature of gene expression (Kollmann et al., 2005). Taken together these considerations imply that an average correlation between polar age and brightness will be smeared, because the history of protein expression levels will be very different in individual cells of the same age.
Tsr-GFP pole intensities and old-new pole differences vary according to growth medium at 30°C
We also explored the effects of varying growth conditions on our analysis. We examined populations of asynchronous cells growing at the same temperature as in the analyses above (30°C) but in two other growth media: LB (doubling time = 60 min; Fig. 6Bv) and MOPS rich medium (doubling time = 44 min; Fig. 6Bvi). In both of these conditions, Tsr-GFP intensities are generally higher, at both the brighter and dimmer poles, than in standard AB medium (compare Fig. 6Biv with 6Bvi). This is particularly true for MOPS-rich medium (note change in scale of vertical axes in Figure 6Bvi versus other graphs). Thus, in this case, intensity averages were taken for the 100 brightest pixels, rather than the brightest 50 pixels as in other cases. However, in both growth media, dimmer and brighter poles again exhibit substantial differences in Tsr-GFP fluorescence, both on a population basis and on a per-cell basis, similar to or greater than those observed in AB medium (Fig. 6Bv,vi, top and middle panels; compare with Fig. 6Biv top and middle rows). We infer that relative Tsr-GFP fluorescence intensity can reliably distinguish old and new poles of individual cells also in these richer growth media.
Conclusion
Tsr-GFP is more abundant at the old pole of a cell than at its new pole. This difference is due to the fact that Tsr-GFP accumulates progressively at old and new poles, but does not accumulate at mid-cell. In AB medium, old and new poles of individual cells can be specifically identified by their relative Tsr-GFP brightness levels, even in cells with very new (first-generation) old poles. [Old-new] pole differences appear to be robust enough that this approach can also be reliably applied to cells growing in other conditions that confer faster growth rates. Absolute pole brightness can provide a rough estimate of pole age but cannot specify exact generational age.
Experimental procedures
Strain construction
The tsr-gfp strain NK9456 was constructed by replacing the genomic copy of tsr in NK9386 with a fusion gene. NK9386 is a derivative of E. coli CM735 containing a fliC deletion (Bates et al., 2005).
Gene fusion was carried out by multiple step PCR using Pfx DNA polymerase (Invitrogen, Carlsbad, CA). The primers used are listed in Table 1. PCR products were cleaned using an in-gel purification kit (QIAGEN, Valencia, CA) before being used as template in subsequent PCR reactions or cloning. A 599 bp DNA fragment containing the 3′-end of tsr gene was amplified by primer tsrmid1055 and tsr3end1 from the genomic DNA of E. coli strain NK9386. A 721 bp fragment containing the downstream sequence of tsr was amplified by primers tsrdown4 and tsrdownhind. Gfp was amplified by PCR with primers jlf100 and jlr101 from a plasmid pNK3885. This plasmid encodes a GFP variant with mutations to improve photostability and brightness in E. coli (J.L. Ferat and N. Kleckner, unpublished data).
Table 1. Primers used in this study.
| Primer | Sequence | Target Sequence |
|---|---|---|
| tsrmid1055 | 5′-GCGATATCTCCACCAGTTCGCAG-3′ | 3′-end of tsr gene, forward |
| tsr3end1 | 5′-AAATGTTTCCCAGTTCTCCTCGCTATC-3′ | 3′-end of tsr gene, reverse |
| tsrdown4 | 5′-CATGAAAATGCCCGATAAGCAAAATG-3′ | downstream sequence of tsr gene, forward |
| tsrdownhind | 5′-AGCAAGCTTATCTCAATGCAGGTAG-3′ | downstream sequence of tsr gene, reverse |
| jlf100 | 5′-ATGAGTAAAGGAGAAGAAC-3′ | Complete gfp gene, forward |
| jlr101 | 5′-TTTGTATAGTTCATCCATG-3′ | Complete gfp gene, reverse |
| gfptd4 | 5′CATGGATGAACTATACAAATAATCGCCATGAAAATGCCCGATAAGCAAAATG-3′ | 3′-end of gfp and 5′- end of thedownstream sequence of tsr gene separated by a linker “TCGC” |
| gfpf1g6 | 5′-GGTGGCGGTGGCGGTGGCATGAGTAAAGGAGAAGAAC-3′ | 5′-end of the gfp gene proceeded by the 6-G sequence |
| t3end1g6 | 5′- GCCACCGCCACCGCCACCAAATGTTTCCCAGTTCTCCTCGCTATC-3′ | 3′-end of tsr gene without the stop codon followed by the 6-G sequence |
| tdbamh | 5′-GCAGGGATCCTTATCTCAATGCAGGTAG-3′ | 3′-end of the tsr downstream sequence followed by a BamH I site |
| tmnot1 | 5′-GCAGGCGGCCGCATATCTCCACCAGTTCGCAG-3′ | 5′-end of the tsr-end sequence proceeded by a NotI site |
Gfp was first fused 5′ of the tsr downstream sequence. Primer gfptd4 was paired with tsrdownhind to introduce a four base linker between the stop codon of gfp and the downstream sequence of tsr. Equal amounts of this PCR product were mixed with the amplified gfp as template in the next PCR reaction. Primer gfpf1g6 was paired with primer tdbamh to amplify a region encompassing gfp and the tsr downstream sequence. This reaction also introduced a BamHI site to the 3′-end of the fusion and a tandem hexameric glycine encoding sequence (6-G) as a linker before the gfp gene. The physiological significance of this linker is discussed in the main text. Primer t3end1g6 was paired with tsrmid1055 in a PCR reaction using the tsr 3′-end sequence as template to introduce the 6-G sequence to the 3′-end of tsr. The original stop codon of tsr was also removed by this step. Equal moles of this PCR product were mixed with the gfp-tsr downstream fusion. The introduced 6-G sequence mediates the annealing of these two fragments. Primer tmnot1 was paired with tdbamh in a PCR reaction using the annealed fragments as template. A NotI cutting site was introduced to the 5′-end of the construct. The final PCR product was digested with NotI and BamHI, and ligated into Bluescript vector (Stratagene, La Jolla, CA). This plasmid was named pBTG and propagated by transformation into DH5α cells (Invitrogen). The construct was confirmed by sequencing of the insert. Plasmid pBTG was digested by NotI and BamHI and the product was ligated into pKOV (a gift of Prof. G Church, Harvard Medical School). This ligation product was used to transform NK9386. The bacteria were selected on plates with chloramphenicol and sucrose (Link et al., 1997). The plates were scanned using a Molecular Imager FX (Bio-Rad, Hercules, CA) with an external 488nm laser and the recombinant colonies expressing GFP-tagged protein were picked.
The absence of genomic tsr and the presence of the tsr-gfp fusion gene in NK9456 were further confirmed by in situ PCR. Additionallly cell fractionation was performed and the presence of the GFP-tagged protein confirmed in the membrane fraction. The growth rate of NK9456 was measured in LB, AB (Bates et al., 2005), and MOPS medium (EZ Rich medium; Teknova, Hollister, CA) at 24°C, 30°C and 37°C respectively. No statistically significant difference between this strain and the mother strain has been detected.
A serine blind control strain was made using a Tn5-1a disruption of tsr from HCB268 (a gift from Prof. H. Berg, Harvard University) introduced into NK9386 by P1 transduction.
AB Medium
AB medium (Clarke and Maaloe, 1967) was used for all experiments unless otherwise noted. AB medium is a 1:1 mixture of A salts [2 g/l (NH4)2SO4, 6 g/l Na2HPO4, 3 g/l KH2PO4, 3 g/l NaCl pH 7.4] and B salts [200 mg/l MgCl2, 10 mg/l CaCl2]. A salts and B salts were autoclaved separately before being combined and supplemented with a nutrient solution consisting of 0.05% glucose, 0.2% alanine, 0.2% proline, 0.2% thiamine, 20 μg/ml methionine, 20 μg/ml histidine, 20 μg/ml tryptophan and 0.5 μg/ml FeCl3.7H2O.
Swarm assay
For the swarm assay a late log phase (OD600 = 1.0) culture in was induced with 1 mM IPTG for 1 hour to synthesize flagella. 1.5μl of the induced culture was spotted onto the middle of a soft agar plate (0.35 % agar in fresh AB medium; IPTG and nutrient solution with or without 1 mM serine were added to the agar solution as it cooled). After inoculation, the soft agar plates were incubated at 30°C for 75 hour. Images of the plates were acquired using a CCD camera (Videoscope International Inc., Washington D.C.) equipped with a Sony video graphic printer and the diameter of the concentric circles was measured.
Preparation of slides for cell visualization
Lineage analysis
0.8% agarose was dissolved in AB medium without alanine and thiamine, and melted in a microwave. Alanine and thiamine were added when the agar solution began to cool down. The microfluidic chamber was made by replica molding the agar against a poly(dimethylsiloxane) stamp made by soft lithography as described before (Takeuchi et al., 2005). Each channel was 1μm deep and 1μm wide. A schematic of the chamber is shown in Fig. 4A. E. coli in early log phase (OD600 = 0.2) were seeded into the chamber and grown at 30°C. The temperature of the chamber was maintained by heating with a hair dryer from a fixed distance. Images were taken every 20 min. In order to supply the bacteria enough nutrient, 20 to 50 μl of freshly prepared nutrient solution without the A salts and B salts were rapidly pipetted into the opening at the bottom of the chamber every 60 minutes (Fig. 4A).
Population analyses
For analysis of cells in liquid populations, a cell sample was layered on a 0.8% agar slab on a microscopic slide and a cover slip was gently placed over the sample area and sealed with grease (analogously to Fig. 4A). For asynchronous cultures and aliquots of newborn cells from a baby cell column (Bates et al., 2005), cells were photographed immediately. To confirm old/new pole brightness, an aliquot of newborn baby cells was collected and further grown in liquid AB medium at 30°C for 110 min, placed on the slide, and the slide then further incubated at 30°C for another 10 min prior to observations. Many cells in this sample undergo division during this ten-minute period. Since cell mobility was constrained by the soft agar, the relative positions of the daughter cells revealed the identities of old and new poles.
Microscopy
All images were collected and analyzed using Metamorph software (Universal Imaging, 10 Downingtown, PA) on an Axioplan 2IE motorized microscope (Zeiss, Gottingen, Germany) with an EM-CCD camera (Roper Scientific, Tuscan, AZ).
Collection of 2D images
Each cell photographed was first visualized by phase contrast microscopy and the focal plane adjusted to give maximum image sharpness. This procedure places the focal plane at or very close to the middle of the cell. 3D reconstructions with focal planes of 0.2 μm depth reveal that, normally, the 2 focal planes in the middle of the cell both give good discrimination of brightness differences between the two poles, likely because polar aggregates form a relatively symmetrical polar “cap” (Fig. 2). Thus, focusing on the basis of the phase contrast image should place the plane of focus well within this zone of good discrimination.
3D reconstruction
Cells were fixed by 2.5% paraformaldehyde. 3 dimensional analysis was performed by taking 30 planes separated by 0.2 μm. Many of the focal planes contain little or no information, however, starting well below and ending well above the cell insures no information is missed. The image stacks were deconvoluted in AutoDeblur (AutoQuant Imaging, Inc, Troy, NY). 3D reconstruction was done with Volocity (Improvision Inc, Waltham, MA.) and ImageJ (NIH, Bethesda, MD).
Supplementary Material
Acknowledgements
This research was funded by NIH grant NIH GM-RO1-025326. We are grateful to Aude Bourniquel and Brian Ho for help with baby cell column and time lapse imaging, to Doug Weibel for providing the microfluidic device used in lineage analysis, and to Victor Sourjik for providing results prior to publication.
References
- Ausmees N, Jacobsson K, Lindberg M. A unipolarly located, cell-surface-associated agglutinin, RapA, belongs to a family of Rhizobium-adhering proteins (Rap) in Rhizobium leguminosarum bv. trifolii. Microbiology. 2001;147:549–559. doi: 10.1099/00221287-147-3-549. [DOI] [PubMed] [Google Scholar]
- Banno S, Shiomi D, Homma M, Kawagishi I. Targeting of the chemotaxis methylesterase/deamidase CheB to the polar receptor-kinase cluster in an Escherichia coli cell. Mol Microbiol. 2004;53:1051–1063. doi: 10.1111/j.1365-2958.2004.04176.x. [DOI] [PubMed] [Google Scholar]
- Bates D, Epstein J, Boye E, Fahrner K, Berg H, Kleckner N. The Escherichia coli baby cell column: a novel cell synchronization method provides new insight into the bacterial cell cycle. Mol Microbiol. 2005;57:380–391. doi: 10.1111/j.1365-2958.2005.04693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates D, Kleckner N. Chromosome and replisome dynamics in E. coli: loss of sister cohesion triggers global chromosome movement and mediates chromosome segregation. Cell. 2005;121:899–911. doi: 10.1016/j.cell.2005.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon LD, Goehring N, Janakiraman A, Yan AW, Wu T, Beckwith J, Goldberg MB. IcsA, a polarly localized autotransporter with an atypical signal peptide, uses the Sec apparatus for secretion, although the Sec apparatus is circumferentially distributed. Mol Microbiol. 2003;50:45–60. doi: 10.1046/j.1365-2958.2003.03674.x. [DOI] [PubMed] [Google Scholar]
- Carlsson F, Stalhammar-Carlemalm M, Flardh K, Sandin C, Carlemalm E, Lindahl G. Signal sequence directs localized secretion of bacterial surface proteins. Nature. 2006;442:943–946. doi: 10.1038/nature05021. [DOI] [PubMed] [Google Scholar]
- Clarke DJ, Maaloe O. DNA replication and the division cycle of Escherichia coli B/r. J Mol Biol. 1967;23:99–112. [Google Scholar]
- de Pedro MA, Quintela JC, Holtje JV, Schwarz H. Murein segregation in Escherichia coli. J Bacteriol. 1997;179:2823–2834. doi: 10.1128/jb.179.9.2823-2834.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietzel I, Kolb V, Boos W. Pole cap formation in Escherichia coli following induction of the maltose-binding protein. Arch Microbiol. 1978;118:207–218. doi: 10.1007/BF00415731. [DOI] [PubMed] [Google Scholar]
- Jain S, van Ulsen P, Benz I, Schmidt MA, Fernandez R, Tommassen J, Goldberg MB. Polar localization of the autotransporter family of large bacterial virulence proteins. J Bacteriol. 2006;188:4841–4850. doi: 10.1128/JB.00326-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KK, Yokota H, Kim SH. Four-helical-bundle structure of the cytoplasmic domain of a serine chemotaxis receptor. Nature. 1999;400:787–792. doi: 10.1038/23512. [DOI] [PubMed] [Google Scholar]
- Kollmann M, Lovdok L, Bartholome K, Timmer J, Sourjik V. Design principles of a bacterial signalling network. Nature. 2005;438:504–507. doi: 10.1038/nature04228. [DOI] [PubMed] [Google Scholar]
- Laus MC, Logman TJ, Lamers GE, van Brussel AA, Carlson RW, Kijne JW. A novel polar surface polysaccharide from Rhizobium leguminosarum binds host plant lectin. Mol Microbiol. 2006;59:1704–1713. doi: 10.1111/j.1365-2958.2006.05057.x. [DOI] [PubMed] [Google Scholar]
- Liberman L, Berg HC, Sourjik V. Effect of chemoreceptor modification on assembly and activity of the receptor-kinase complex in Escherichia coli. J Bacteriol. 2004;186:6643–6646. doi: 10.1128/JB.186.19.6643-6646.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link AJ, Phillips D, Church GM. Methods for generating precise deletions and insertions in the genome of wild-type Escherichia coli: application to open reading frame characterization. J Bacteriol. 1997;179:6228–6237. doi: 10.1128/jb.179.20.6228-6237.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lybarger SR, Maddock JR. Differences in the polar clustering of the high-and low-abundance chemoreceptors of Escherichia coli. Proc Natl Acad Sci USA. 2000;97:8057–8062. doi: 10.1073/pnas.130195397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNulty C, Thompson J, Barrett B, Lord L, Andersen C, Roberts IS. The cell surface expression of group 2 capsular polysaccharides in Escherichia coli: the role of KpsD, RhsA and a multi-protein complex at the pole of the cell. Mol Microbiol. 2006;59:907–922. doi: 10.1111/j.1365-2958.2005.05010.x. [DOI] [PubMed] [Google Scholar]
- Mileykovskaya E, Dowhan W. Visualization of phospholipid domains in Escherichia coli by using the cardiolipin-specific fluorescent dye 10-N-nonyl acridine orange. J Bacteriol. 2000;182:1172–1175. doi: 10.1128/jb.182.4.1172-1175.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro L, McAdams HH, Losick R. Generating and exploiting polarity in bacteria. Science. 2002;298:1942–1946. doi: 10.1126/science.1072163. [DOI] [PubMed] [Google Scholar]
- Shiomi D, Banno S, Homma M, Kawagishi I. Stabilization of polar localization of a chemoreceptor via its covalent modifications and its communication with a different chemoreceptor. J Bacteriol. 2005;187:7647–7654. doi: 10.1128/JB.187.22.7647-7654.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sourjik V, Berg HC. Functional interactions between receptors in bacterial chemotaxis. Nature. 2004;428:437–441. doi: 10.1038/nature02406. [DOI] [PubMed] [Google Scholar]
- Stewart EJ, Madden R, Paul G, Taddei F. Aging and death in an organism that reproduces by morphologically symmetric division. PLoS Biol. 2005;3:e45. doi: 10.1371/journal.pbio.0030045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi S, DiLuzio WR, Weibel DB, Whitesides GM. Controlling the shape of filamentous cells of Escherichia coli. Nano Lett. 2005;5:1819–1823. doi: 10.1021/nl0507360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiem S, Kentner D, Sourjik V. Positioning of chemosensory clusters in E. coli and its relation to cell division. Embo J. 2007;26:1615–1623. doi: 10.1038/sj.emboj.7601610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiem S, Sourjik V. Stochastic assembly of chemoreceptor clusters in Escherichia coli. Mol Microbiol. 2008;68:1228–1236. doi: 10.1111/j.1365-2958.2008.06227.x. [DOI] [PubMed] [Google Scholar]
- Wang X, Liu X, Possoz C, Sherratt DJ. The two Escherichia coli chromosome arms locate to separate cell halves. Genes Dev. 2006;20:1727–1731. doi: 10.1101/gad.388406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzel BK, Spicer SS, Dvorak HF, Heppel LA. Cytochemical localization of certain phosphatases in Escherichia coli. J Bacteriol. 1970;104:529–542. doi: 10.1128/jb.104.1.529-542.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woldringh CL. Is Escherichia coli getting old? Bioessays. 2005;27:770–774. doi: 10.1002/bies.20271. [DOI] [PubMed] [Google Scholar]
- Zhang P, Khursigara CM, Hartnell LM, Subramaniam S. Direct visualization of Escherichia coli chemotaxis receptor arrays using cryo-electron microscopy. Proc Natl Acad Sci USA. 2007;104:3777–3781. doi: 10.1073/pnas.0610106104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.