

Abstract
A novel small molecule thiocarbazate (PubChem SID 26681509), a potent inhibitor of human cathepsin L (EC 3.4.22.15) with an IC50 of 56 nM, was developed following a 57,821 compound screen of the NIH Molecular Libraries Small Molecule Repository. After a 4 hr preincubation with cathepsin L, this compound became even more potent, demonstrating an IC50 of 1.0 nM. The thiocarbazate was determined to be a slow-binding and slowly reversible competitive inhibitor. Through a transient kinetic analysis for single-step reversibility, inhibition rate constants were kon = 24,000 M-1s-1 and koff = 2.2 × 10-5 s-1 (Ki = 0.89 nM). Molecular docking studies were undertaken using the experimentally-derived X-ray crystal structure of papain/CLIK-148 (1cvz.pdb). These studies revealed critical hydrogen bonding patterns of the thiocarbazate with key active site residues in papain. The thiocarbazate displayed 7- to 151-fold greater selectivity toward cathepsin L than papain and cathepsins B, K, V, and S with no activity against cathepsin G. The inhibitor demonstrated a lack of toxicity in human aortic endothelial cells and zebrafish. Additionally, the thiocarbazate inhibited in vitro propagation of malaria parasite Plasmodium falciparum with an IC50 of 15.4 μM and inhibited Leishmania major with an IC50 of 12.5 μM.
Cathepsins are a family of ubiquitous lysosomal proteases that have demonstrated important physiopathological roles in inflammation, osteo- and rheumatoid arthritis, osteoporosis, Alzheimer’s disease, multiple sclerosis, pancreatitis, apoptosis, liver disorders, lung disorders, myocardial disorders, diabetes, muscular dystrophy, tumor invasion and metastasis (Fujishima et al., 1997; Leist and Jaattela, 2001; Nomura and Katunuma, 2005; Turk and Guncar, 2003). Cathepsin L is an endopeptidase that is catalytically the most active known lysosomal cysteine protease (Otto and Schirmeister, 1997). It is a papain-like cysteine protease of interest due to involvement in many biological processes including epidermal homeostasis, hair follicle morphogenesis and cycling, extracellular matrix degradation, and viral entry of Severe Acute Respiratory Syndrome (SARS) coronavirus, Ebola virus, and Hendra virus (Chandran et al., 2005; Nomura and Katunuma, 2005; Pager and Dutch, 2005; Simmons et al., 2005; Turk et al., 2001). Due to the roles of many cathepsin L-like proteases in diseases such as malaria (falcipain), leishmaniasis (Leishmania major cathepsin L), Chagas disease (cruzipain), African trypanosomiasis (congopain), toxoplasmosis (Toxoplasma gondii cathepsin L), amoebiasis (histolysain), and sleeping sickness (rhodesain), inhibitors of human cathepsin L may be highly valuable as therapeutic treatments against these infectious diseases.
As part of the National Institutes of Health Molecular Libraries Screening Centers Network (NIH MLSCN), the Penn Center for Molecular Discovery (PCMD)1 performs high throughput screens (HTS) against various targets, deposits data into PubChem2, and develops small molecule probes. A high throughput screen of 57,821 compounds from the NIH Molecular Libraries Small Molecule Repository (BioFocus DPI; San Francisco, CA) was performed against human cathepsin L. The average Z′ factor for the screen was 0.73, indicating good plate uniformity throughout the run (Zhang et al., 1999). Results of this screen are available on PubChem as BioAssay #460.3
The major constituent of the most potent hit from the high throughput screen was SID 861540 (Figure 1A) with an apparent IC50 of 130 nM. We subsequently determined through chemical synthesis and structure elucidation by LC-MS that the active compound was actually the ring-opened form of oxadiazole SID 861540 (Myers et al., 2008). SID 26681509 (Figure 1B), the more stable, Boc-protected S-enantiomer of the ring-opened by-product, proved to be a potent and reversible human cathepsin L inhibitor. Although thiocarbazate inhibitors of cathepsins have not yet been reported, members of the structurally similar class of aza-peptides have been shown to be effective inhibitors of cysteine proteases (Baggio et al., 1996; Magrath and Abeles, 1992; Thompson et al., 1997; Xing and Hanzlik, 1998). Herein, we report the biochemical and biological characterization and molecular docking of SID 26681509, a novel, potent, and selective thiocarbazate inhibitor of human cathepsin L.
Figure 1.
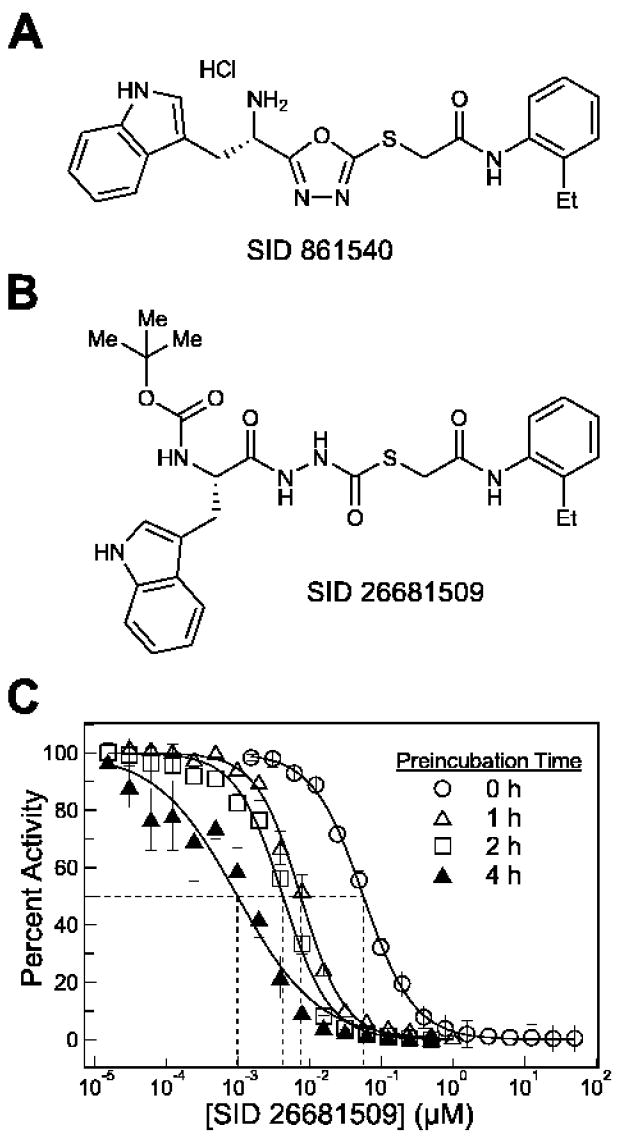
A. Oxadiazole SID 861540, the major constituent of the most potent hit from the high throughput screen. B. Thiocarbazate SID 26681509, the Boc-protected S-enantiomer of the ring-opened by-product of the original HTS hit. C. Activity of SID 26681509 against human cathepsin L after preincubation with the enzyme target for 0 h (○), 1 h (Δ), 2 h (□), and 4 h (▲).
Materials and Methods
Cathepsin L assay optimization
The cathepsin L assay was carried out using 1 μM Z-Phe-Arg-7-amido-4-methylcoumarin (Z-Phe-Arg-AMC, Sigma C9521) and 8.7 ng/mL human liver cathepsin L (Calbiochem 219402) in 100 μL reactions (96-well plate). Assay buffer consisted of 20 mM sodium acetate, 1 mM ethylenediaminetetraacetic acid (EDTA) and 5 mM cysteine, pH 5.5. Cathepsin L was incubated in assay buffer for 30 min prior to dispensing into wells to allow for efficient reduction of the active site cysteine required for full enzymatic activity. AMC dilution controls were performed and no inner filter effect quenching was observed at fluorophore concentrations as high as 50 μM.
IC50 determination
SID 26681509 was synthesized as described previously (Myers et al., 2008). A 16-point two-fold serial dilution dose response of SID 26681509 was performed to determine its relative potency against cathepsin L. Each well of a Corning 3686 96-well assay plate contained 38 μL water and 2 μL inhibitor (ranging from 2.5 mM to 76 nM) in dimethyl sulfoxide (DMSO). Positive and negative controls were present to serve as internal controls into which 2 μL DMSO was transferred in place of the inhibitor. Ten μL of 10 μM Z-Phe-Arg-AMC in five times concentrated assay buffer and 50 μL of 17.4 ng/mL cathepsin L in assay buffer were added sequentially to initiate the proteolytic reaction. A total of 50 μL of assay buffer was dispensed in place of enzyme into negative control wells. This resulted in a final dose response concentration range from 50 μM to 1.5 nM inhibitor (2% DMSO) in 100 μL final reaction volume. The fluorescence intensity of each well of the assay plates was monitored on a PerkinElmer Envision microplate reader (excitation 355 nm, emission 460 nm) to measure the AMC released by the enzyme-catalyzed hydrolysis of Z-Phe-Arg-AMC. Data was scaled using internal controls and fit to a four-parameter logistic model (IDBS XLfit equation 205; Guildford, Surrey, United Kingdom) to obtain IC50 values in triplicate.
Preincubation studies
To establish the time-dependent mechanism of inhibition, enzyme and inhibitor were preincubated for various time points in a 96-well microplate prior to the addition of substrate to initiate the enzymatic reaction. 47.5 μL of cathepsin L (18.3 ng/mL) and 47.5 μL of SID 26681509 at various concentrations in assay buffer were incubated up to 4 hrs. Five μL of Z-Phe-Arg-AMC substrate were then added and the plate was monitored for AMC hydrolysis on the Envision fluorescent microplate reader.
Reversibility
To test the reversibility of SID 26681509, cathepsin L at 100-fold its final assay concentration (870 ng/mL) and inhibitor at 10-fold its IC50 after 1 hr preincubation were combined and incubated for 1 hr at room temperature at 2 μL. This mixture was then diluted 100-fold in a Corning 3650 96-well plate with assay buffer containing 1 μM Z-Phe-Arg-AMC to a final volume of 200 uL. A rapidly reversible inhibitor should dissociate from the enzyme to restore greater than approximately 90% of enzymatic activity (Figure 2A). Fluorescence intensities of the 200 μL reaction wells were monitored continuously for AMC hydrolysis on the Envision plate reader.
Figure 2.
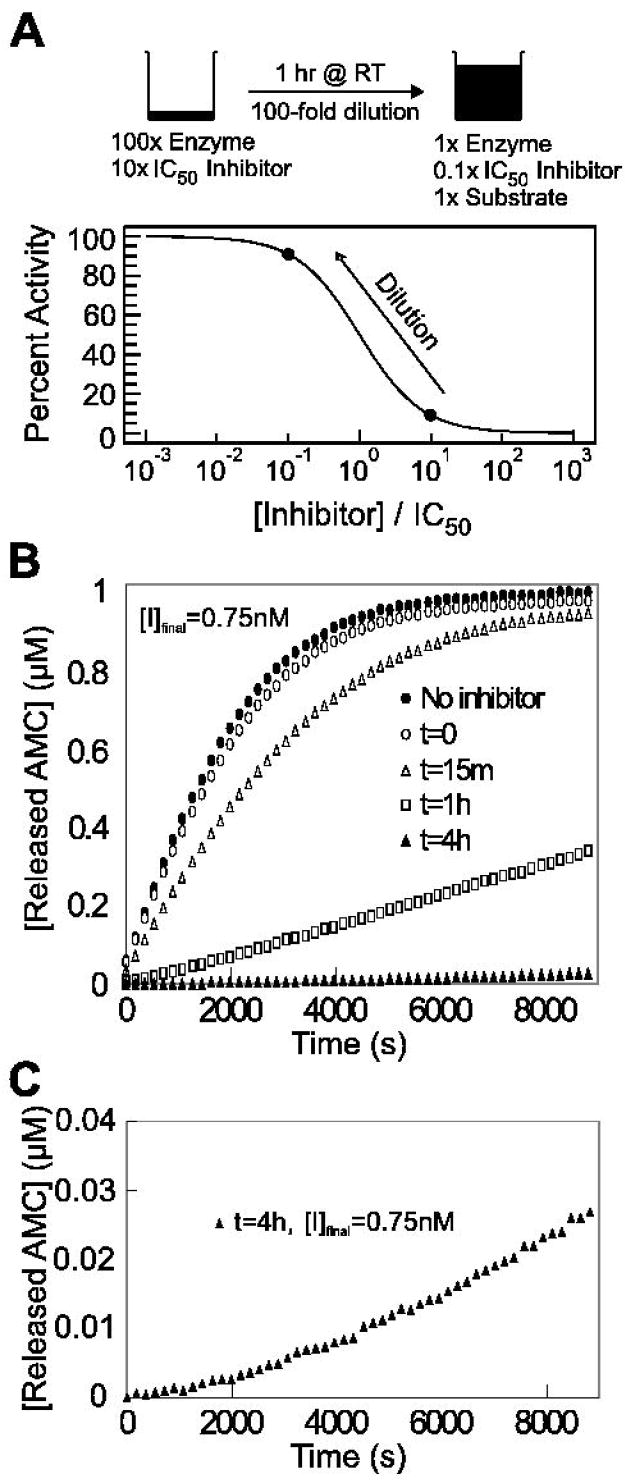
A. Dilution protocol for determination of reversibility. Cathepsin L at 100-fold its final assay concentration (870 ng/mL) and inhibitor at 10-fold its IC50 after 1 hr preincubation (75 nM) were combined and incubated for 1 hr at room temperature at 2 μL. This mixture was then diluted 100-fold with assay buffer containing 1 μM Z-Phe-Arg-AMC. A rapidly reversible inhibitor should dissociate from the enzyme to restore greater than approximately 90% of enzymatic activity. B. Reversibility data for SID 26681509 after 0 min (○), 15 min (Δ), 1 hr (□), and 4 hr (▲) preincubation with cathepsin L and upon 100-fold dilution into assay buffer containing Z-Phe-Arg-AMC. A full enzyme-substrate reaction without inhibitor (●) served as a positive control. By comparing initial substrate conversion rates of the control and 1 hr preincubation study, it can be seen that only 11% enzymatic activity is restored after 6000 s, making SID 26681509 a slowly reversible inhibitor of human cathepsin L. C. Reaction progress curve with 4 hr preincubation of cathepsin L and SID 26681509. This upward curvature demonstrates that the enzyme is recovering and the reaction is reversible. The rate of AMC release after 8820 s was 4.7 times greater than the initial release rate.
Data fitting
In the kinetic simulations, the concentrations of chemical species ([E], [S], [I], [P], [ES], [EI]) over time were calculated using a system of ordinary differential equations for each reaction step (Figure 4A). Progress curves for each inhibitor concentration were fit to a five-parameter (k1, k-1, kon, koff, kcat) kinetic inhibition model using APPSPACK optimization software. APPSPACK is a generic solver for linearly-constrained optimization problems (Griffin and Kolda, 2006).
Figure 4.
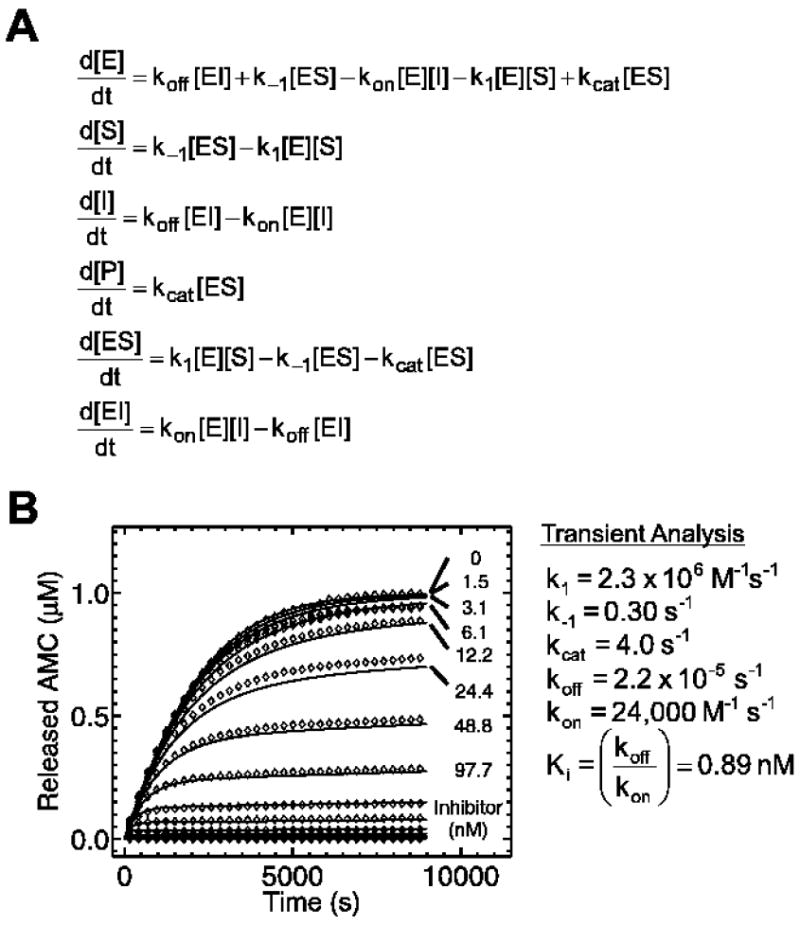
A. Ordinary differential equations governing the single-step mechanism of inhibition shown in Figure 3A. B. Reaction progress curves (◊) are shown for 8.7 ng/mL human cathepsin L enzyme and 1 μM Z-Phe-Arg-AMC substrate with varying concentrations of SID 26681509 inhibitor. They have been fit to a five-parameter inhibition kinetic model (▬) using APPSPACK optimization software with a linear least squares objective function.
Selectivity
SID 26681509 was assayed for inhibition against papain and cathepsins B, G, K, S, and V. Papain from Carica papaya (Calbiochem 5125, 11 ng/mL), human cathepsin K (Calbiochem 342001, 35 ng/mL), human spleen cathepsin S (Calbiochem 219344, 40 ng/mL), and human cathepsin V (Calbiochem 219467, 39 ng/mL) were assayed using Z-Phe-Arg-AMC substrate at 20 μM, 20 μM, 15 μM, and 1 μM, respectively. Human liver cathepsin B (Calbiochem 219362, 65 ng/mL) was assayed using 15 μM Z-Arg-Arg-AMC substrate (Bachem I-1135). Human neutrophil cathepsin G (Calbiochem 219373, 4.2 μg/mL) was assayed using 15 μM Suc-Ala-Ala-Pro-Phe-AMC substrate (Sigma S9761). All reactions were performed in 20 mM sodium acetate buffer containing 5 mM cysteine and 1 mM EDTA, pH 5.5. Reaction progress was monitored using the Envision microplate reader. IC50 values were measured in triplicate.
Cytotoxicity
Human aortic endothelial cells were seeded in a Corning 3704 384-well white sterile tissue culture-treated microplate at 1000 cells/25 μL/well. The plate was centrifuged and incubated at 37 °C for 24 hr. SID 26681509 and doxorubicin positive control were then serially diluted in EGM-2 endothelial cell media (Lonza CC4176). Five μL each of these serial dilutions were added to the cells in triplicate, resulting in final concentrations of compound from 100 μM to 156 nM (0.17% DMSO). The plate was centrifuged and incubated at 37 °C for 24 hrs. Thirty μL CellTiter-Glo™ (Promega G7570) were added to each well and centrifuged. After 10 minutes, luminescence was measured using the Envision microplate reader.
Malaria assay
Eight two-fold serial dilutions of SID 26681509 in RPMI 1640 media (Invitrogen 11875) containing L-Glutamine, 50 mg/L hypoxanthine, 6 g/L HEPES, 0.5% Albumax II bovine serum (Invitrogen 11021), 0.225% sodium bicarbonate, and 1 μg/mL gentamicin were performed in Corning 3704 microplates. Adding 30 μL red blood cells infected with synchronized ring stage luciferase-expressing Plasmodium falciparum parasites at 0.5% parasitaemia and 4% hematocrit to 10 μL compound resulted in final concentrations tested of 50 μM to 1.5 nM. In addition, 30 μL normal red blood cells and 30 μL infected red blood cells were added to two control columns containing 10 μL media. The plates were incubated at 37 °C in a 92% humidity chamber with 5% CO2, 5% O2, and 90% N2 for 48 hrs to allow for two cycles of red blood cell rupture and invasion to take place. Forty μL BrightGlo™ (Promega E2610) were added to each well and centrifuged. After 5 minutes, luminescence was measured using the Envision microplate reader.
Leishmaniasis assay
Five thousand Leishmania major promastigotes were plated per well in a 384-well microtiter plate in a 20 μL volume of promastigote growth medium. Promastigotes were treated with a concentration range from 0 μM to 50 μM of SID 26681509 for 44 hrs. Five μL Cell-Titer-Blue™ were added per well and incubated for 4 hrs. Relative fluorescence units (A560/A590) were captured on a SpectraMax M5 microtiter plate reader. DMSO concentrations were held constant at 0.5%.
Results
Kinetic characterization
With immediate mixing of enzyme, substrate and inhibitor (no preincubation of enzyme and inhibitor), SID 26681509 was found to inhibit human cathepsin L with an IC50 of 56 ± 4 nM. After preincubation with enzyme for 1, 2, and 4 hrs prior to substrate addition at t = 0, SID 26681509 displayed increasing potency with IC50 values falling to 7.5 ± 1.0 nM, 4.2 ± 0.6 nM, and 1.0 ± 0.5 nM, respectively, demonstrating a slow onset of inhibition against the target enzyme (Figure 1C).
The mechanism of inhibition, to determine whether the compound acted as a rapidly reversible, slowly reversible, or irreversible inhibitor, was evaluated using a preincubation/dilution assay (Copeland, 2005). By preincubating human cathepsin L and the compound for 1 hr at 10-fold its IC50 after 1 hr preincubation (75 nM), a condition is created whereby >90% of the enzyme should be in an enzyme-inhibitor complex (Figure 2A). Upon 100-fold dilution of the 1 hr preincubated mixture of cathepsin L and the inhibitor into assay buffer containing 1 μM Z-Phe-Arg-AMC substrate, approximately 11% enzymatic activity was returned after 6000 s into the reaction, by comparison of the substrate conversion rates of the preincubated and uninhibited reactions (Figure 2B). For the 4 hr preincubated enzyme-inhibitor reaction condition (Figure 2C), 99.8% of the reaction was inhibited immediately after addition of substrate due to almost all the enzyme being bound to small molecule inhibitor SID 26681509. After 8820 s, the rate of product formation for the 4 hr preincubated reaction was 4.7 times greater than the initial rate of product formation, showing that the inhibitor was being released from the enzyme-inhibitor complex and enzymatic activity was indeed recovering. Therefore, SID 26681509 was determined to be a very slowly reversible inhibitor of human cathepsin L.
Nonlinear regression of transient kinetics
For human cathepsin L cleavage of Z-Phe- Arg-AMC, Km and kcat were determined through initial rate analysis to be 0.77 μM and 1.5 s-1, respectively (Figure 3B). A nonlinear regression for transient dynamics was conducted based on the reaction scheme shown in Figure 3A. Here, the values of k1, k-1, kon, and koff are explicitly estimated rather than combined into the equilibrium parameters, Km and Ki, estimated by traditional kinetic analyses. The best fit parameters were k1 = 2.3 × 106 M-1s-1, k-1 = 0.30 s-1, kcat = 4.0 s-1, kon = 24,000 M-1s-1, and koff = 2.2 × 10-5 s-1 (Figure 4B). The regressed Ki = 0.89 nM was quite consistent with the measured IC50 = 1.0 ± 0.5 nM obtained after 4 hr preincubation of human cathepsin L with SID 26681509. To explore alternate models for inhibition, the data were fit to models for irreversible inhibitor binding ([E]+[I]→[EI]); two-step inhibitor binding ([E]+[I]↔[EI]1↔[EI]2), where a weak enzyme-inhibitor encounter complex is formed prior to the formation of a more tightly-bound enzyme-inhibitor complex; and uncompetitive inhibitor binding ([ES]+[I]↔[ESI]), where inhibitor binds only to the enzyme-substrate complex. These models failed to reproduce the data as well as the five-parameter model described above for reversible, single-step competitive inhibition.
Figure 3.
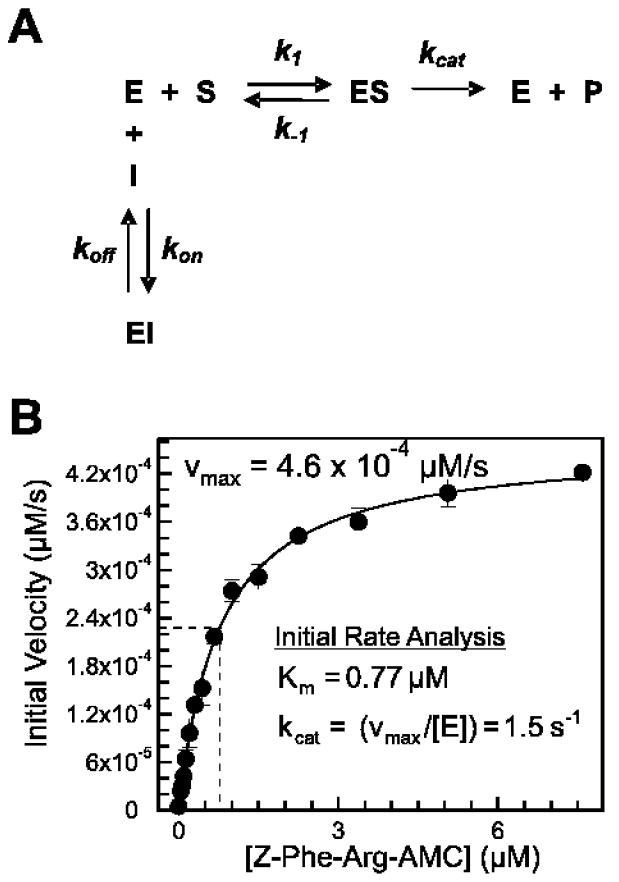
A. Single-step mechanism for simple, reversible, slow binding inhibition governed by kinetic constants kon and koff. B. Km and kcat determination for human cathepsin L enzymatic reaction with Z-Phe-Arg-AMC substrate. Km and kcat were determined to be 0.77 μM and 1.5 s-1, respectively.
Mechanism of reversibility
The return of activity shown in Figure 2C demonstrated that the thiocarbazate was a reversible inhibitor. Transient kinetic analyses (Figure 4) quantified the rate of reversibility. To investigate the mechanism of reversibility and the generation of a putative leaving group, a stoichiometric reaction between 4.5 μM cathepsin L and 4.5 μM SID 26681509 was analyzed by liquid chromatography-mass spectrometry [Shimadzu LC-MS/4.6 mm × 50 mm Premier C18 column, 1 mL/min and a step from 90:10 to 60:40 water:acetonitrile with 10 min hold time, mobile phase contained 0.05% formic acid]. A potential thiol leaving group formed by reaction of cathepsin L with the thiocarbazate carbonyl of SID 26681509 was synthesized (MW = 195) and was detectable on the LC-MS at a concentration of 100 nM with a retention time of 12.1 min (no suppression detected due to presence of human cathepsin L). However, this thiol leaving group was not detected by LC-MS after 6, 12, and 24 hrs incubation of human cathepsin L with SID 26681509. While this result argues against acylation of cathepsin L by the inhibitor, formation of a tetrahedral intermediate by attack of the active site Cys residue on the thiocarbazate carbonyl of SID 26681509 is not excluded. In fact, when the thiocarbazate sulfur in SID 26681509 is replaced by carbon, the resulting molecule is a much weaker inhibitor (IC50 > 50 μM, data not shown).
Selectivity against papain and cathepsins B, G, K, S, and V
SID 26681509 was tested for inhibitory activity against papain and human cathepsins B, G, K, L, S, and V (Table 1) with no preincubation of enzyme and inhibitor. IC50 values were calculated at time points of 10 min, 30 min, 60 min, and 90 min. The selectivity indexes of SID 26681509 (a ratio of the IC50 against the assayed protease divided by the IC50 against cathepsin L) ranged from 7 to 151 for the various papain-like cysteine proteases (Table 2). SID 26681509 inhibited papain and cathepsins B, K, S, and V with IC50 values determined after one hour ranging from 618 nM to 8.442 μM. As expected, SID 26681509 showed no inhibitory activity against the serine protease cathepsin G. The IC50 values systematically decreased with time for each protease, demonstrating the slow-binding nature of the small molecule inhibitor. The qualitative order of the selectivity index is fairly insensitive to when the measurement was taken; however, the weak trends observed in the selectivity index data likely reflect the relative rates of slowly reversible inhibition of the enzyme. Thus, it would appear that the slowly reversible reaction proceeds faster for cathepsins V and S than for cathepsin L; whereas, it proceeds more slowly for papain.
Table 1.
IC50 values of SID 26681509 against papain and human cathepsins B, G, K, L, S, and V.
| Enzyme | IC50 @ 10 min (μM) | IC50 @ 30 min (μM) | IC50 @ 60 min (μM) | IC50 @ 90 min (μM) | Selectivity Index | % Identity to Cat L |
|---|---|---|---|---|---|---|
| Cathepsin L, human liver | 0.155 ± 0.012 | 0.075 ± 0.005 | 0.056 ± 0.004 | 0.050 ± 0.003 | 1 | 100 |
| Cathepsin V, human, recombinant, NSO cells | 1.008 ± 0.146 | 0.688 ± 0.039 | 0.618 ± 0.035 | 0.576 ± 0.024 | 7-11 | 78 |
| Cathepsin S, human spleen | 1.107 ± 0.134 | 0.745 ± 0.010 | 0.724 ± 0.011 | 0.721 ± 0.029 | 7-14 | 55 |
| Papain, Carica papaya | 6.765 ± 0.367 | 2.981 ± 0.282 | 2.600 ± 0.208 | 1.562 ± 0.314 | 31-46 | 48 |
| Cathepsin B, human liver | 7.492 ± 0.693 | 2.983 ± 0.295 | 2.527 ± 0.073 | 2.512 ± 0.139 | 40-50 | 26 |
| Cathepsin K, human, recombinant, E. coli | 17.596 ± 1.040 | 8.460 ± 0.366 | 8.442 ± 0.140 | 6.857 ± 0.063 | 113-151 | 58 |
| Cathepsin G, human neutrophil | >50 | >50 | >50 | >50 | -- | -- |
Table 2.
Selectivity indexes of SID 26681509 against papain and human cathepsins B, G, K, L, S, and V.
| Enzyme | Selectivity Index @ 10 min | Selectivity Index @ 30 min | Selectivity Index @ 60 min | Selectivity Index @ 90 min |
|---|---|---|---|---|
| Cathepsin L, human liver | 1 | 1 | 1 | 1 |
| Cathepsin V, human, recombinant, NSO cells | 7 | 9 | 11 | 11 |
| Cathepsin S, human spleen | 7 | 10 | 13 | 14 |
| Papain, Carica papaya | 44 | 40 | 46 | 31 |
| Cathepsin B, human liver | 48 | 40 | 45 | 50 |
| Cathepsin K, human, recombinant, E. coli | 114 | 113 | 151 | 137 |
| Cathepsin G, human neutrophil | -- | -- | -- | -- |
Biological assays
SID 26681509 was found to be non-toxic to human aortic endothelial cells at 100 μM. The inhibitor also demonstrated a lack of toxicity to zebrafish in a live organism assay at 100 μM. SID 26681509 was active in an in vitro propagation assay against Plasmodium falciparum with an IC50 of 15.4 ± 0.6 μM (Figure 5A). Additionally, the thiocarbazate inhibitor was toxic toward Leishmania major promastigotes with an IC50 of 12.5 ± 0.6 μM (Figure 5B).
Figure 5.
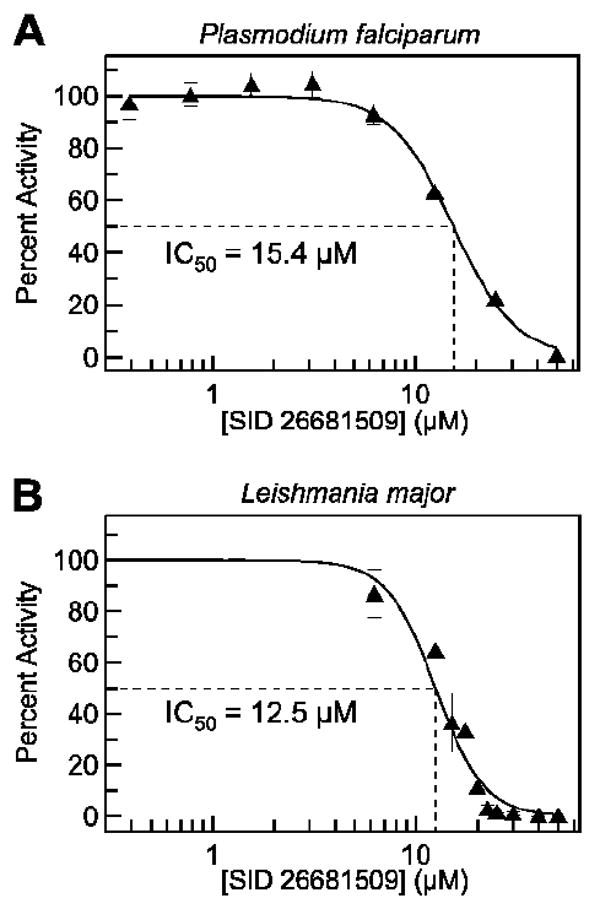
A. IC50 against Plasmodium falciparum was determined to be 15.4 ± 0.6 μM. B. IC50 against Leishmania major was determined to be 12.5 ± 0.6 μM.
Molecular docking of SID 26681509 in papain
The co-crystal structure of CLIK-148 bound to papain (1cvz.pdb) (Katunuma et al., 1999; Tsuge et al., 1999) was used as a model to study hydrogen bonding and hydrophobic interactions of the thiocarbazate inhibitor SID 26681509 within the cysteine protease binding site. The chemical structure of CLIK-148 is depicted in Figure 6A. Other researchers have used papain to design highly specific cathepsin inhibitors and CLIK-148 directly inhibits cathepsin L (Katunuma et al., 1999; LaLonde et al., 1998; Tsuge et al., 1999). An effort to construct a cathepsin L homology model based on the coordinates of 1cvz.pdb led to inconclusive docking results, particularly with respect to the arrangement of critical hydrophobic groups in the S2 subsite of the enzyme binding pocket.4 The co-crystal structure coordinates of human cathepsin L complexed with the small molecule inhibitor E-64 (Fujishima et al., 1997) would have been preferred; however, they were not publicly available. Most of the residues within the catalytic binding site are conserved between papain and cathepsin L, including those in papain that make direct hydrogen bonding contacts to the CLIK-148 inhibitor: Gln19, Cys25, Gly66, Asp158, and Trp177. Since cathepsin L is most homologous to papain within the papain superfamily of cysteine proteases, the high resolution (1.7 Å) structure of papain/CLIK-148 served as an excellent starting point for studying small molecule inhibitors of cathepsin L.
Figure 6.
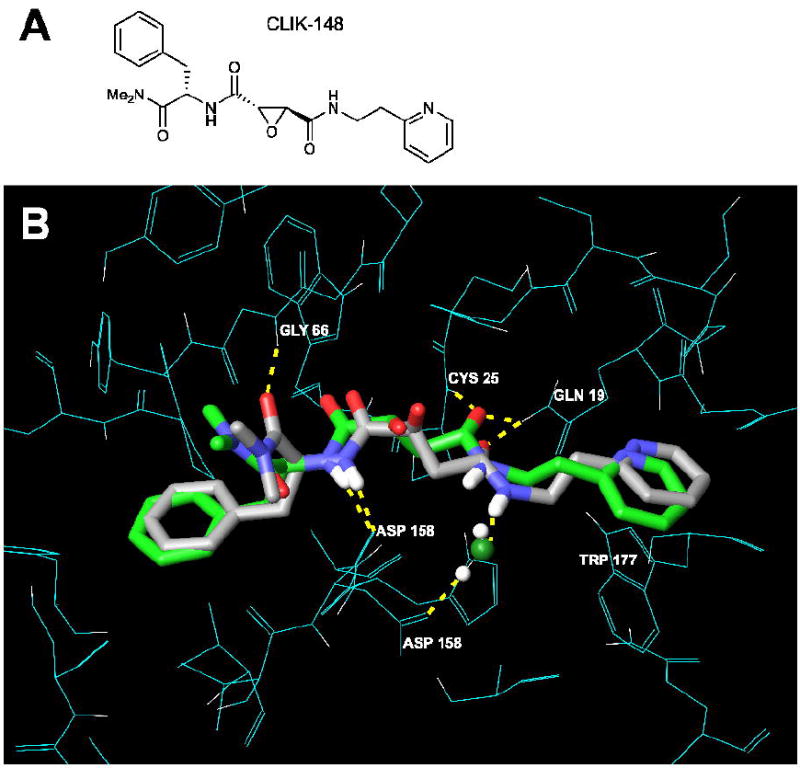
A. CLIK-148, a cathepsin L-specific inhibitor (Katunuma et al., 1999; Tsuge et al., 1999). B. Overlay of papain/CLIK-148 crystal structure (1cvz.pdb, green) with independently docked epoxide ring-opened form of CLIK-148 (gray). The XP Glide score for the top-scoring pose was -9.27 kcal/mol.
Molecular docking studies of CLIK-148 and SID 26681509 in the binding site of papain were carried out using XP (extra precision) Glide software. Predictions of accurate binding modes have been accomplished with reasonable accuracy using XP Glide docking, resulting in computationally-derived protein/ligand complexes with adequate root mean square deviations from the known experimentally-derived co-crystal structure (Perola et al., 2004). Our initial docking studies were conducted on the papain/CLIK-148 system in order to verify that XP Glide could reproduce the binding mode of CLIK-148.
To prepare this system for docking, the covalent bond between CLIK-148 and papain was broken, and the epoxide ring-opened form of CLIK-148 was independently docked into papain. The highest scoring pose for CLIK-148 obtained from this docking study overlaid very well with the experimentally-derived bound inhibitor CLIK-148 (Figure 6B). The XP Glide score for CLIK-148 in papain was -9.27 kcal/mol. With this validation, we studied the interaction of SID 26681509 with papain.
SID 26681509 was prepared for docking using LigPrep software. The highest scoring pose of SID 26681509 had an excellent score of -9.04 kcal/mol. This score was very close to the XP Glide score obtained for independently docked CLIK-148 in papain. In addition, many of the residues that made contacts between CLIK-148 and papain were also involved in making contacts between SID 26681509 and papain (Figure 7A). The backbone NH hydrogens of Gln19 and Cys25 made direct hydrogen bonding contacts to the thiocarbazate carbonyl oxygen of SID 26681509; the backbone NH hydrogen of Gly66 made a hydrogen bond to the acyl hydrazine CO oxygen of the ligand; the backbone carbonyl oxygen of Asp158 was involved in a hydrogen bonding network to both a hydrazine NH and an amide NH of SID 26681509; and finally, the Trp177 side chain NH formed a hydrogen bond to an amide carbonyl oxygen of SID 26681509. In addition, the 2-ethylanilide group of SID 26681509 made a large hydrophobic contact with the aromatic side chain of Trp177; Trp177 is located in the prime region of the enzyme binding pocket (S1′ subsite). The indole group of SID 26681509 occupies the S2 subsite of the enzyme binding pocket. When the docking poses of CLIK-148 and SID 26681509 were overlaid (Figure 7B), SID 26681509 looked remarkably like the epoxide ring-opened form of CLIK-148. This overlay illustrated that both inhibitors maintain the same critical distances between the two carbonyl groups that are disposed in a 1,4 relationship to each other. The intramolecular distance between the 1,4-dicarbonyl in both CLIK-148 and SID 26681509 was approximately 4.80 to 4.92 Å.
Figure 7.
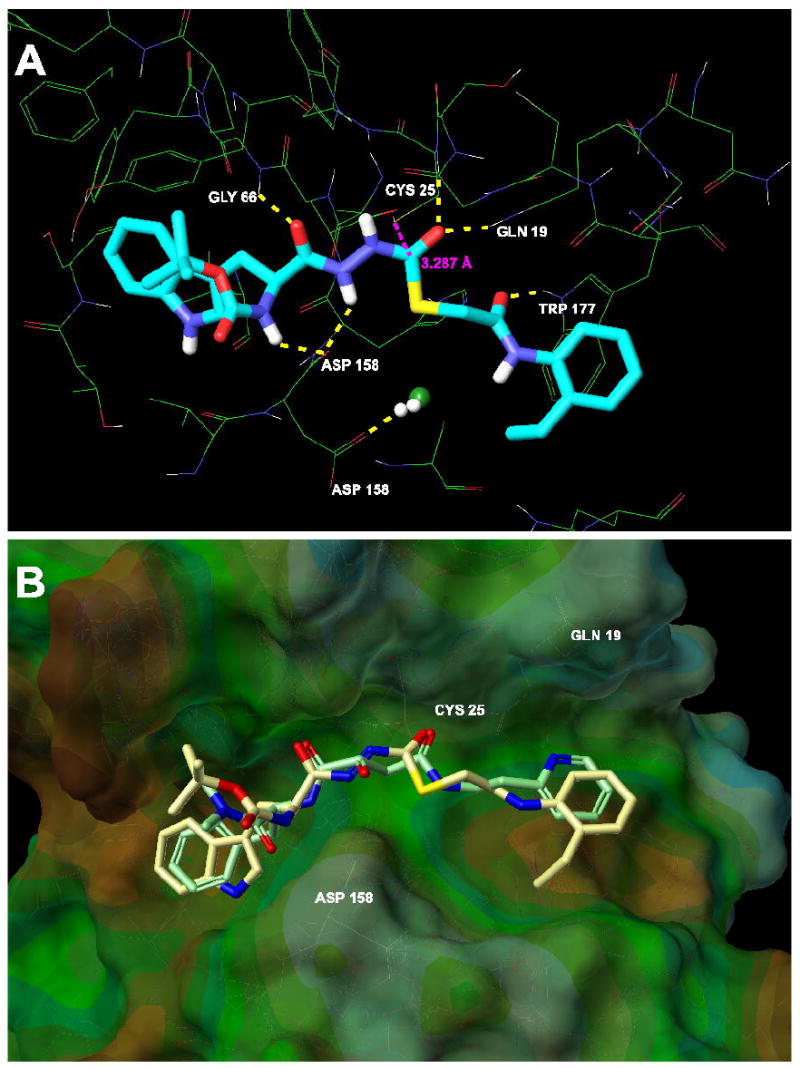
A. Hydrogen bonding interactions between SID 26681509 and papain involve catalytic residues Gln19, Cys25, Gly66, Asp158 and Trp177. The distance between the Cys25 sulfur atom and the thiocarbazate carbonyl carbon is 3.287 Å. This is the carbon likely to undergo nucleophilic attack by the sulfur. B. Overlay of papain/CLIK-148 (light green) with computational model of SID 26681509 in papain (tan). There are striking similarities between the binding interactions of the two inhibitors.
Finally, the active site Cys25 sulfur is in close proximity (3.289 Å) to the carbonyl carbon of the thiocarbazate. Although the contribution from covalent bonding between this carbon and sulfur cannot be directly assessed through our docking studies, the molecule sits in the proper orientation to achieve this covalent binding interaction (Figure 7A). As indicated above, however, we lack compelling evidence for covalent binding between the enzyme and SID 26681509.
Discussion
Our kinetic analyses demonstrate that SID 26681509 is a highly potent and selective competitive inhibitor of human cathepsin L with slow binding and slow reversibility kinetics. Molecular docking of SID 26681509 into the papain crystal structure revealed hydrophobic interactions within the S2 and S1′ subsites and hydrogen bonding interactions with Gln19, Cys25, Gly66, Asp158, and Trp177. These interactions share a high degree of similarity with the papain/CLIK-148 complex (Katunuma et al., 1999; Tsuge et al., 1999).
With the exception of cathepsin K, the range of selectivity indexes of SID 26681509 decreased as the percent identity to cathepsin L increased. This may be due to the strong P2 preference of cathepsin K for proline (Choe et al., 2006). It is interesting to note that SID 26681509 is 7 to 11 times more potent against cathepsin L than cathepsin V. Cathepsin V, which is sometimes referred to as cathepsin L2, shares 78% amino acid sequence identity with cathepsin L, and has been shown to compensate for the role of cathepsin L in epidermal homeostasis and hair follicle morphogenesis of knockout mice (Hagemann et al., 2004; Nagler and Menard, 2003; Reinheckel et al., 2001).
Using the papain/CLIK-148 coordinate system, we were able to independently dock SID 26681509 into the binding site of papain. This led to the conclusion that SID 26681509 appears to bind to papain in a manner similar to CLIK-148. Five residues that are conserved between papain and cathepsin L make direct contacts to both inhibitors. In addition, a highly hydrophobic/aromatic site involving Trp177 interacts with the hydrophobic 2-ethylanilide group of SID 26681509.
The fact that SID 26681509 inhibits both malaria and leishmaniasis suggests that it acts in a cellular system requiring transit across lipid membranes. However, the micromolar potency, as opposed to sub-nanomolar potency against purified human cathepsin L, was not surprising since i) we have no measure of the internal concentration of inhibitor achieved in these organisms and ii) the active site geometries of their cathepsin L-like cysteine proteases might differ from that of the human enzyme. Further investigations of SID 26681509 and related analogs against purified cathepsin L-like enzymes such as falcipain, congopain, cruzipain, T. gondii cathepsin L, histolysain, and rhodesain are warranted based on the findings of this study. The thiocarbazate scaffold can be readily derivatized, introducing functional groups to occupy specific binding sites in a variety of cysteine proteases, and thus holds promise as a general scaffold for the design of specific cysteine protease inhibitors.
Acknowledgments
We thank Professor Michael A. Pack (University of Pennsylvania School of Medicine) for testing SID 26681509 for zebrafish toxicity. We also thank Ms. Claire Darling and Professor Doron C. Greenbaum (University of Pennsylvania School of Medicine) for assistance in testing SID 26681509 against Plasmodium falciparum.
Financial support for this work was provided by the National Institutes of Health (5U54HG003915-02 and 5U54HG003915-03).
Nonstandard abbreviations
- AMC
7-amido-4-methylcoumarin
- DMSO
dimethyl sulfoxide
- E-64
1-[L-N-(trans-epoxysuccinyl)leucyl]amino-4-guanidinobutane
- EDTA
ethylenediaminetetraacetic acid
- HEPES
4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid
- HTS
high throughput screen
- LC-MS
liquid chromatography-mass spectrometry
- MLSCN
Molecular Libraries Screening Centers Network
- NIH
National Institutes of Health
- PCMD
Penn Center for Molecular Discovery
- SARS
severe acute respiratory syndrome
- SID
substance identifier
Footnotes
PCMD web address: http://www.seas.upenn.edu/~pcmd.
PubChem web address: http://pubchem.ncbi.nlm.nih.gov.
PCMD cathepsin L hits: http://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=460.
Homology modeling was performed using Molecular Operating Environment (MOE) software available from the Chemical Computing Group, Montreal, Canada.
References
- Baggio R, Shi YQ, Wu YQ, Abeles From good substrates to good inhibitors: design of inhibitors for serine and thiol proteases. Biochemistry. 1996;35(11):3351–3353. doi: 10.1021/bi952879e. [DOI] [PubMed] [Google Scholar]
- Chandran K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science. 2005;308(5728):1643–1645. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe Y, Leonetti F, Greenbaum DC, Lecaille F, Bogyo M, Bromme D, Ellman JA, Craik CS. Substrate profiling of cysteine proteases using a combinatorial peptide library identifies functionally unique specificities. J Biol Chem. 2006;281(18):12824–12832. doi: 10.1074/jbc.M513331200. [DOI] [PubMed] [Google Scholar]
- Copeland RA. Evaluation of enzyme inhibitors in drug discovery: a guide for medicinal chemists and pharmacologists. J. Wiley; Hoboken, N.J: 2005. [PubMed] [Google Scholar]
- Fujishima A, Imai Y, Nomura T, Fujisawa Y, Yamamoto Y, Sugawara T. The crystal structure of human cathepsin L complexed with E-64. FEBS Lett. 1997;407(1):47–50. doi: 10.1016/s0014-5793(97)00216-0. [DOI] [PubMed] [Google Scholar]
- Griffin JD, Kolda TG. Asynchronous Parallel Generating Set Search for Linearly-Constrained Optimization. Sandia National Laboratories; Livermore, CA: 2006. [Google Scholar]
- Hagemann S, Gunther T, Dennemarker J, Lohmuller T, Bromme D, Schule R, Peters C, Reinheckel T. The human cysteine protease cathepsin V can compensate for murine cathepsin L in mouse epidermis and hair follicles. Eur J Cell Biol. 2004;83(1112):775–780. doi: 10.1078/0171-9335-00404. [DOI] [PubMed] [Google Scholar]
- Katunuma N, Murata E, Kakegawa H, Matsui A, Tsuzuki H, Tsuge H, Turk D, Turk V, Fukushima M, Tada Y, Asao T. Structure based development of novel specific inhibitors for cathepsin L and cathepsin S in vitro and in vivo. FEBS Lett. 1999;458(1):6–10. doi: 10.1016/s0014-5793(99)01107-2. [DOI] [PubMed] [Google Scholar]
- LaLonde JM, Zhao B, Smith WW, Janson CA, DesJarlais RL, Tomaszek TA, Carr TJ, Thompson SK, Oh HJ, Yamashita DS, Veber DF, Abdel-Meguid SS. Use of papain as a model for the structure-based design of cathepsin K inhibitors: crystal structures of two papain-inhibitor complexes demonstrate binding to S’-subsites. J Med Chem. 1998;41(23):4567–4576. doi: 10.1021/jm980249f. [DOI] [PubMed] [Google Scholar]
- Leist M, Jaattela M. Triggering of apoptosis by cathepsins. Cell Death Differ. 2001;8(4):324–326. doi: 10.1038/sj.cdd.4400859. [DOI] [PubMed] [Google Scholar]
- Magrath J, Abeles RH. Cysteine protease inhibition by azapeptide esters. J Med Chem. 1992;35(23):4279–4283. doi: 10.1021/jm00101a004. [DOI] [PubMed] [Google Scholar]
- Myers MC, Shah PP, Diamond SL, Huryn DM, Smith AB., III Identification and Synthesis of a Unique Thiocarbazate Cathepsin L Inhibitor. Bioorg Med Chem Lett. 2008;18(1):210–214. doi: 10.1016/j.bmcl.2007.10.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagler DK, Menard R. Family C1 cysteine proteases: biological diversity or redundancy? Biol Chem. 2003;384(6):837–843. doi: 10.1515/BC.2003.094. [DOI] [PubMed] [Google Scholar]
- Nomura T, Katunuma N. Involvement of cathepsins in the invasion, metastasis and proliferation of cancer cells. J Med Invest. 2005;52(12):1–9. doi: 10.2152/jmi.52.1. [DOI] [PubMed] [Google Scholar]
- Otto HH, Schirmeister T. Cysteine Proteases and Their Inhibitors. Chem Rev. 1997;97(1):133–172. doi: 10.1021/cr950025u. [DOI] [PubMed] [Google Scholar]
- Pager CT, Dutch RE. Cathepsin L is involved in proteolytic processing of the Hendra virus fusion protein. J Virol. 2005;79(20):12714–12720. doi: 10.1128/JVI.79.20.12714-12720.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perola E, Walters WP, Charifson PS. A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance. Proteins. 2004;56(2):235–249. doi: 10.1002/prot.20088. [DOI] [PubMed] [Google Scholar]
- Reinheckel T, Deussing J, Roth W, Peters C. Towards specific functions of lysosomal cysteine peptidases: phenotypes of mice deficient for cathepsin B or cathepsin L. Biol Chem. 2001;382(5):735–741. doi: 10.1515/BC.2001.089. [DOI] [PubMed] [Google Scholar]
- Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc Natl Acad Sci U S A. 2005;102(33):11876–11881. doi: 10.1073/pnas.0505577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SK, Halbert SM, Bossard MJ, Tomaszek TA, Levy MA, Zhao B, Smith WW, Abdel-Meguid SS, Janson CA, D’Alessio KJ, McQueney MS, Amegadzie BY, Hanning CR, DesJarlais RL, Briand J, Sarkar SK, Huddleston MJ, Ijames CF, Carr SA, Garnes KT, Shu A, Heys JR, Bradbeer J, Zembryki D, Lee-Rykaczewski L, James IE, Lark MW, Drake FH, Gowen M, Gleason JG, Veber DF. Design of potent and selective human cathepsin K inhibitors that span the active site. Proc Natl Acad Sci U S A. 1997;94(26):14249–14254. doi: 10.1073/pnas.94.26.14249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuge H, Nishimura T, Tada Y, Asao T, Turk D, Turk V, Katunuma N. Inhibition mechanism of cathepsin L-specific inhibitors based on the crystal structure of papain-CLIK148 complex. Biochem Biophys Res Commun. 1999;266(2):411–416. doi: 10.1006/bbrc.1999.1830. [DOI] [PubMed] [Google Scholar]
- Turk D, Guncar G. Lysosomal cysteine proteases (cathepsins): promising drug targets. Acta Crystallogr D Biol Crystallogr. 2003;59(Pt 2):203–213. doi: 10.1107/s0907444902021479. [DOI] [PubMed] [Google Scholar]
- Turk V, Turk B, Turk D. Lysosomal cysteine proteases: facts and opportunities. Embo J. 2001;20(17):4629–4633. doi: 10.1093/emboj/20.17.4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing R, Hanzlik RP. Azapeptides as inhibitors and active site titrants for cysteine proteinases. J Med Chem. 1998;41(8):1344–1351. doi: 10.1021/jm970802d. [DOI] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4(2):67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]