

Abstract
Brugada syndrome has been linked to mutations in SCN5A. Agents that dissociate slowly from the sodium channel such as flecainide and ajmaline unmask the Brugada syndrome electrocardiogram and precipitate ventricular tachycardia/fibrillation. Lidocaine, an agent with rapid dissociation kinetics, has previously been shown to exert no effect in patients with Brugada syndrome. We characterized a novel double mutation of SCN5A (V232I in DI-S4+L1308F in DIII-S4) identified in a rare case of lidocaine (1 mg/kg)-induced Brugada syndrome. We studied lidocaine blockade of INa generated by wild-type and V232I+L1308F mutant cardiac sodium channels expressed in mammalian TSA201 cells using patch clamp techniques. Despite no significant difference in steady-state gating parameters between V232I+L1308F and wild-type sodium currents at baseline, use-dependent inhibition of INa by lidocaine was more pronounced in V232I+L1308F versus wild-type (73.0±0.1% versus 18.23±0.04% at 10 μmol/L measured at 10 Hz, respectively). A dose of 10 μmol/L lidocaine also caused a more negative shift of steady-state inactivation in V232I+L1308F versus wild-type (-14.1±0.3 mV and -4.8±0.3 mV, respectively). The individual mutations produced a much less accentuated effect. We report the first case of lidocaine-induced Brugada electrocardiogram phenotype. The double mutation in SCN5A, V232I, and L1308F alters the affinity of the cardiac sodium channel for lidocaine such that the drug assumes Class IC characteristics with potent use-dependent block of the sodium channel. Our results demonstrate an additive effect of the 2 missense mutations to sensitize the sodium channel to lidocaine. These findings suggest caution when treating patients carrying such genetic variations with Class I antiarrhythmic drugs.
Keywords: arrhythmia (mechanisms), Brugada syndrome, ion channels, Na channels, sudden death
Brugada syndrome (BrS) is an inherited cardiac disease characterized by an ST segment elevation in the right precordial electrocardiogram (ECG) leads (V1 to V3) and a high incidence of sudden death.1,2 The electrocardiographic manifestation of the syndrome is often concealed but can be unmasked using sodium channel blockers.3,4 Sodium channel blockers with slow dissociation kinetics such as flecainide and ajmaline are known to unmask the BrS ECG phenotype as well as precipitating ventricular tachycardia/fibrillation. Lidocaine, a Class IB antiarrhythmic agent, with rapid dissociation kinetics has little or no effect on the ST segment in patients with BrS.4 This study presents a rare case of lidocaine-induced BrS phenotype with a unique novel double mutation of SCN5A capable of altering the drug-receptor interaction so as to confer on lidocaine profound use-dependent sodium channel-blocking characteristics.
Methods
The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Index Patient
A 45-year-old black man with no history of cardiac disease presented to the hospital emergency department after a seizure. His initial ECG in the emergency department did not show ST segment abnormalities. He then became combative and had a run of monomorphic wide complex ventricular tachycardia. Lidocaine, 70 mg followed by a continuous infusion of 1 mg/min, was administered and led to ST segment elevation in V1 to V3. A slight right precordial ST elevation remained even 1 year after discontinuation of lidocaine.
Serial cardiac markers were negative for evidence of acute myocardial infarction. The patient did not have chest pain, and there was no evolution of ECG signs of infarction.
The wide complex ventricular tachycardia was hemodynamically destabilizing and was thus quickly shocked. The ventricular tachycardia was described as monomorphic based on single-lead ECG recordings from the emergency department and analysis by the cardiologist on duty. The lidocaine-induced ST segment elevation and the fact that the patient had a malignant arrhythmia and ST segment elevation unmasked by the Na channel blocker led to a diagnosis of BrS. Because of the unique characteristics of the case, the patient was referred for genotyping to determine the presence or absence of a channelopathy.
Mutational Analysis
The study was approved by the regional Institutional Review Board and written informed consent was obtained. Genomic DNA was isolated from peripheral blood leukocytes using a commercial kit (Gentra System; Puregene) as previously described.5 The exon and intron boundaries of the SCN5A gene were amplified and analyzed by direct sequencing. Polymerase chain reaction products were purified with a commercial reagent (ExoSAP-IT; USB) and were directly sequenced from both directions using an ABI PRISM 3100-Avant Automatic DNA sequencer.
Mutagenesis and Transfection of the TSA201 Cell Line
Mutant SCN5A channel cDNA was prepared using the megaprimer method for site-directed mutagenesis using the plasmid pcDNA3-SCN5A, which contains SCN5A cDNA cloned into pcDNA3.1+ (Invitrogen, Carlsbad, Calif).
Mutated and wild-type (WT) sodium channels were expressed in the human embryonic kidney cell line TSA201 as previously described.6 Briefly, transient transfection was carried out with the calcium phosphate precipitation method using equimolar amounts of SCN5A cDNA (WT or mutant) and SCN1B cDNA subcloned into a pRc/cytomegalovirus vector. In addition, CD8 cDNA in a 2.4:1 molar ratio with SCN5A was cotransfected as a reporter gene to visually identify transfected cells using Dynabeads (M-450 CD8 Dynal). The cells were grown on polylysine-coated 35-mm culture dishes and placed in a temperature-controlled chamber for electro-physiological study (EPS; Medical Systems, Greenvale, NY). Channel characteristics were studied 72 hours after transfection.
Electrophysiology
Membrane currents were measured using a whole-cell patch clamp technique in transfected TSA201 cells in the absence and presence of lidocaine (Sigma Co) All recordings were obtained at room temperature (22°C) using an Axopatch 200B amplifier equipped with a CV-201A head stage (Axon Instruments). Macroscopic whole-cell Na+ current was recorded using perfusing bath solution containing (in mmol/L) 140 NaCl, 5 KCl, 1.8 CaCl2, 1 MgCl2, 2.8 Na Acetate, 10 HEPES, and 10 glucose (pH 7.3 with NaOH). Tetraethylammonium chloride (5 mmol/L) was added to the buffer to block TEA-sensitive native currents. Patch pipettes were pulled from borosilicate glass (7052; Model PP-89; Narashige) to obtain resistances between 1 and 2.5 mol/LΩ when filled with a solution containing (in mmol/L) 5 NaCl, 5 KCl, 130 CsF, 1.0 MgCl2, 5 EGTA, and 10 HEPES (pH 7.2 with CsOH). Currents were filtered with a 4-pole Bessel filter at 5 kHz and digitized at 10 kHz.
Steady-state availability of the sodium channel was fitted to the Boltzmann equation, I/Imax=1/(1+exp([V-V1/2]/k) where V1/2 and k are the midpoint and the slope factor, respectively, and V is the membrane potential.
All data acquisition and analysis were performed using the suite of pCLAMP programs V9.2 (Axon Instruments, Union City, Calif), EXCEL (Microsoft), and ORIGIN 6.1 (Microcal Software, Northampton, Mass).
Statistical Analysis
Data are expressed as mean±SEM. Two-tailed Student t test was performed using statistical software in SigmaPlot 2000 (Jandel Scientific Software). Differences were considered to be statistically significant at a value of P<0.05.
Results
Lidocaine-Induced Electrocardiogram-Like Brugada Syndrome
Figure 1A shows ST segment elevation in the right precordial leads recorded from the proband after intravenous administration of 1 mg/kg lidocaine. The ECG shows a coved-type ST segment elevation >0.2 mV followed by a negative T wave after administration of lidocaine, typical for a Type 1 ECG phenotype in BrS.
Figure 1.

Molecular genetics of SCN5A linked to BrS. A, ECG showing ST segment elevation in V1 to V3 recorded after intravenous administration of lidocaine (1 mg/kg). B-C, Polymerase chain reaction-based sequence of SCN5A showing G-A transversion at codon 232 of exon 6 and a G-T transversion at codon 1308 in exon 22 resulting in amino acid substitution of valine for isoleucine (V232I) and leucine for phenylalanine yielding (L1308F). D, Location of mutations in domain III segment 4 and domain I segment 4 using the conventional transmembrane topology model.
DNA analysis revealed a novel double mutation in SCN5A (Figure 1B). The first was a single nucleotide substitution (g>a) involving codon 232 (gtc-to-atc) in exon 6 of SCN5A resulting in amino acid substitution of valine for isoleucine (V232I) located at the COOH terminus of the transmembrane segment S4 of domain I of the channel protein. The second missense mutation was a single nucleotide substitution (c>t) at 1308 (ctc-to-ttc) in exon 22 resulting in amino acid substitution of the leucine at position 1308 for a phenylalanine (L1308F). Interestingly, this amino acid is also located at the COOH terminus of the transmembrane segment S4 but in domain III (Figure 1C). The mutations were absent in over 400 references alleles from 200 ethnically matched controls.
Biophysical Characteristics of Wild-Type and Double-Mutated (V232I+L1308F) SCN5A Channels
Figure 2 illustrates the current-voltage relationship of the WT and double mutant channel expressed in TSA201 cells in the absence and presence of 30 μmol/L lidocaine. WT and V232I+L1308F channels displayed a similar current-voltage relationship with a reversal potential of +45 mV, consistent with the theoretical value for ENa expected from the Na concentration of the internal and external solutions. Lidocaine (30 μmol/L) reduced V232I+L1308F maximum current more than WT (71±3% versus 29±2%, P<0.001; Figure 2C).
Figure 2.

Effect of lidocaine on I-V and steady-state inactivation in WT and V232I+L1308F channels. Sodium currents generated in mammalian TSA201 cells in the absence (A) and presence of 30 μmol/L lidocaine (B). Lidocaine more potently blocked the mutated channels. Representative currents in A and B were elicited by sequential test pulses at 0.06 Hz between -90 mV and -15 mV in increments of 5 mV from a holding potential of -120 mV (inset). C, Current-voltage (I-V) relationship in presence of 30 μmol/L lidocaine. D, Steady-state inactivation curves for WT and mutant channels in control. Symbols represent mean±SEM for 4 to 10 cells. Boltzmann analysis showed that the slope factor (k) and the half-inactivation potential (Hinf50) did not differ significantly between groups.
Steady-state inactivation curves, obtained using a 500-ms prepulse to different voltages followed by a step to -20 mV, were similar for WT and V232I+L1308F channels (Figure 2D). Despite similar biophysical characteristics in control, 30 μmol/L lidocaine produced a much more pronounced negative shift of the steady-state inactivation curve in the V232I+L1308F versus WT channels (P<0.001): 18.8±0.3 mV for V232I+L1308F (n=8) and 6.6±0.3 for WT (n=10; Figure 3B). Lidocaine (3 to 100 μmol/L) shifted the half-inactivation potential (Hinf50) in a dose-dependent manner for both channels, but more importantly, in V232I+L1308F (Figure 3C).
Figure 3.

Effect of lidocaine tonic block on steady-sate inactivation in WT and V232I+ L1308F channels. A, Representative current recordings at -20 mV elicited from a cell held at -120 mV and preconditioned at membrane potentials between -140 mV and -60 mV (inset) at 0.06 Hz under control conditions. B, Steady-state inactivation (availability) obtained as the ration of peak currents to maximum current for each channel was plotted against the conditioning potential and fitted to a Boltzmann distribution. C, Lidocaine-induced shift in midinactivation potential (Hinf50) of WT and V232I+L1308F channels as a function of lidocaine concentration. Double mutants are significantly different from the WT channel under the same dosage of lidocaine. *P<0.05.
Tonic Block
Blockade of sodium channels by lidocaine has been explained on the basis of the modulated receptor hypothesis,7,8 which proposes that opening of the channel increases accessibility of lidocaine to its binding site, which in turn stabilizes the channel in its inactivated state. As a consequence, the channel displays a dynamic block of higher affinity when channels are activated and subsequently inactivated and a lower-affinity tonic block in its resting state. To assess lidocaine tonic block, we measured the effect of the drug on INa at rest by holding the membrane at -120 mV for 15 seconds (0.06 Hz) before each measurement. Figure 4A shows INa recordings elicited by a 20-ms pulse before and after exposure to 30 μmol/L lidocaine. Tonic block was more important in V232I+L1308F with EC50 values of 254±36 μmol/L and 17±2 μmol/L for tonic block in WT and V232I+L1308F, respectively (Figure 4C).
Figure 4.

Effect of lidocaine on tonic and use-dependent block in WT and V232I+L1308F (VI+LF) channels. A, Current recordings before and after extracellular application of 30 μmol/L lidocaine. Pulse duration was 20 ms, and the holding potential was -120 mV and given at a rate of 15s (0.06 Hz) to assess the tonic block. B, UDB in WT and mutant channels at 2 and 10 Hz under 30 μmol/L lidocaine. C, Dose-response relationship for tonic block (TB) and UDB (10 Hz) on WT and VI+LF channels. EC50 for TB was 248±28 μmol/L and 16±2 μmol/L for WT and VI+LF separately and EC50 for UDB was 57±5 μmol/L and 5.6±0.4 μmol/L for WT and VI+LF separately. D, Frequency-dependence of UDB by 30 μmol/L lidocaine on WT and V232I+L1308F expressed as the EC50 against the stimulating frequency. *P<0.05.
Use-Dependent Block
Use-dependent block (UDB) refers to potentiation of the blockade of INa by lidocaine in which the frequency of the activating stimulus is increased. It is explained by the modulated receptor hypothesis as an increased accessibility of the drug to its binding site within the channel during rapid opening and closing of the channel that increase the number of channels blocked by lidocaine. UDB prolongs the refractory period in a rate-dependent fashion and is the basis for the antiarrhythmic action of lidocaine. We next compared the lidocaine UDB at depolarization frequencies of 0.2, 1, 2, and 10 Hz in WT and V232I+L1308F channels. Pulse duration was 20 ms, and the holding potential was -120 mV. Figure 4B depicts the onset of the use-dependent block caused by 10 μmol/L lidocaine at stimulus frequencies of 2 and 10 Hz. Lidocaine caused little use-dependent block in WT channels, but potently blocked V232I+L1308F channels (Figure 4C-D). The concentration-response relationships, obtained from a standard Hill equation 1/(1+[[lidocaine]/EC50]), revealed that EC50 for UDB of V232I+L1308F channels was shifted well into the therapeutic range of EC50 for block of WT channels and remained considerably above it.
Biophysical Characteristics of Separately Mutated V232I and L1308F SCN5A Channels
Ackerman et al9 identified L1308F as a polymorphism found mostly in Americans of African descent. To test for potentiation of the lidocaine block introduced by the V232I substitution and this polymorphism, we separately tested the effects of each mutation. Blockade of V232I and L1308F peak current by 30 μmol/L lidocaine was similar to the block in WT (29±2%; Figure 2C) with 25±9% and 21±3% reduction, respectively (Figure 5A); Figure 5B shows that half-inactivation voltage for V232I and L1308F was negatively shifted by 10±1 mV (n=4) and 8±1 mV (n=5), respectively, by 30 μmol/L lidocaine. Thus, lidocaine had stronger effects on V232I channel availability compared with WT and L1308F (V232I versus WT, P<0.01; L1308F versus WT, P=not significant). The lidocaine-induced shift in V232I+L1308F channel availability (18.8±0.3 mV) was greater than each single mutated channels (V232I+L1308F versus V232I, P<0.001; V232I+L1308F versus L1308F, P<0.001), but as expected from addition of the effects of each mutation.
Figure 5.
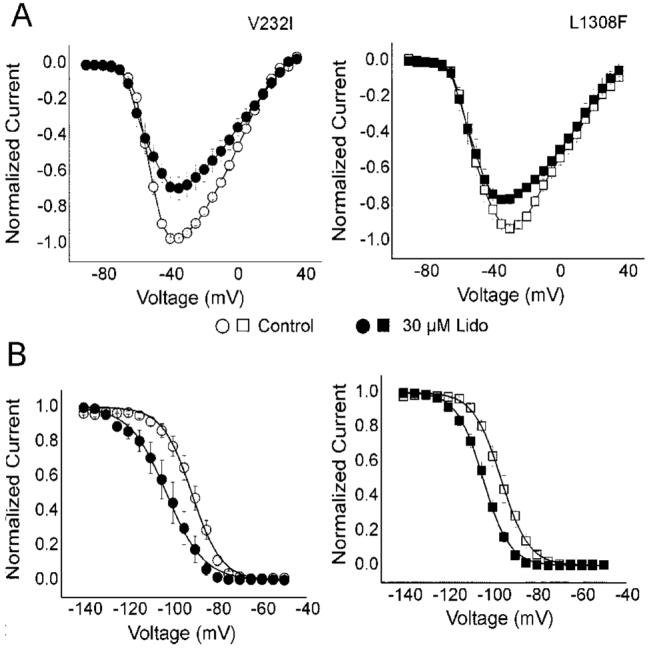
Effect of lidocaine TB on biophysical properties of V232I and L1308F. A, I-V relationship shows similar block by 30 μmol/L lidocaine at 0.06 Hz for V232I and L1308F (25.6±8.9% versus 21.8±2.9%, respectively). B, Steady-state inactivation curves of V232I and L1308F under 30 μmol/L lidocaine. Hinf50 (mV)=-91.92±0.76 (V232I in control), -102.21±0.82 (V232I with 30 μmol/L lidocaine), -96.28±1.36 (L1308F in control), -104.32±1.3 (L1308F with 30 μmol/L lidocaine).
We next compared UDB on each mutation (Figure 6). We found similar EC50 for UDB at 0.2 and 2 Hz for the single mutations and WT. The results at 0.2 Hz indicate that lidocaine affinity for UDB on the double mutant was approximately 10 times higher than what we obtained for L1308F and WT (P<0.05). At a frequency of 2.0 Hz, lidocaine’s EC50 on WT was approximately 2 times lower than what we found for the single mutations L1308F and V232I (P<0.05) and approximately 9 times that of the double mutation (P=0.001).
Figure 6.
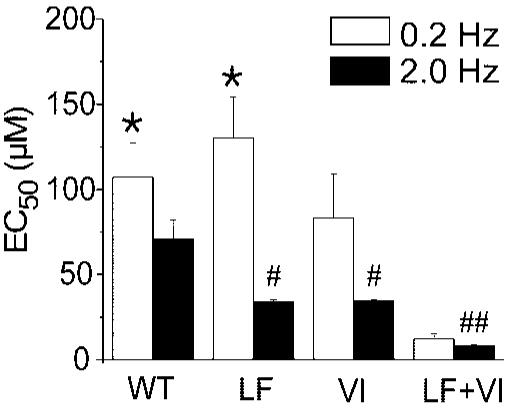
Effect of mutations L1308F (LF), V232I (VI), and V232I+L1308F (VI+LF) on lidocaine affinity for use-dependent block at 0.2 and 2Hz. UDB was assessed as described in Figure 4. EC50 were obtained by measuring the reduction of peak current amplitude during each stimulus train and normalizing it to the maximal reduction. EC50 values (μmol/L) were 107.22±20.25 (WT), 130.43±23.89 (LF), 83.30±25.79 (VI), and 12.17±3.10 (VI+LF). At 2.0 Hz, EC50 values (μmol/L) were 70.88±11.10 (WT), 33.92±1.31 (LF), 34.41±0.81 (VI), and 8.23±0.42 (VI+LF). Statistical significance: *P<0.05 at 0.2 Hz versus VI+LF; #P<0.05, and ##P<0.01 at 2.0 Hz versus WT. n=20; 11, 6 and 7 for WT, LF, VI, and VI+LF separately.
Because UDB can be due to slower recovery from inactivation, we next compared recovery between mutated and WT channels using a double pulse protocol (Figure 7). Mutated channels exhibited recovery times similar to WT channels in control conditions (Table). Lidocaine delayed recovery from inactivation of the double mutant. A 2 exponential fit to the data (Figure 8) yielded similar time constants for V232I and L1308F, respectively, but slower than WT in presence of lidocaine (Table). Recovery for VI+LF was 3-fold slower than each of the single mutation taken separately under 30 μmol/L lidocaine.
Figure 7.

Effect of the double-mutation V2321+L1308F on the influence of lidocaine on recovery from inactivation. A, Effect of 30 μmol/L lidocaine on WT. B, Same as A for the double-mutant channel. Recovery from inactivation assessed by a double-pulse protocol (P1 to P2) consisting of 20 ms conditioning (P1) and test (P2) pulses to 0 mV separated by varying recovery intervals (ΔT) at -120 mV.
Figure 8.

Modulation of the effects of lidocaine on recovery from inactivation by mutations V232I, L1308F and V232I+L1308F. Normalized peak current (P2/P1) were fitted to a double exponential function: y(t)=y0+A(1-exp[-t/τf])+B(1-exp[-t/τs]) where τf and τs represents the recovery time constant. The fitting parameters are shown in Table 1. n=11 to 16 cells per condition.
Discussion
Our results show that the V232I+L1308F double missense mutation in SCN5A produced no significant change in the current-voltage relationship, steady-state inactivation, or kinetics of INa, consistent with the lack of a disease phenotype in the patient under basal conditions. Despite the lack of any apparent alteration in gating parameters, V232I+L1308F channels displayed a larger reduction in INa (73±0.09% for V232I+L1308F) versus WT (18.2±0.1%) during lidocaine tonic block (Figure 2C). These observations are consistent with the ability of lidocaine to induce the Brugada phenotype in this particular case.
Although steady-state inactivation of V232I+L1308F was no different from WT in control, 10 μmol/L lidocaine shifted half-inactivation of V232I+L1308F channels by -14.1±0.3 mV versus -4.8±0.3 mV for WT (Figure 3C). Such reduction in availability is likely to further contribute to the Brugada phenotype.
The changes in steady-state inactivation by lidocaine on each mutation seem additive because their sum nearly matches the shift observed in the double mutant. Similarly, UDB was larger and recovery from inactivation was slower in the double mutant as expected from an equal contribution by each mutation. These observations suggest independent additive effects by both mutations to potentiate the block by lidocaine.
Our results show that polymorphism L1308F confers a greater sensitivity of the cardiac sodium channel to blockade by lidocaine. One likely mechanism to explain the appearance of the BrS phenotype in this patient would be that L1308F lowered the threshold for arrhythmias in this patient and that the second mutation V232I, through an additive effect, brought this threshold in the therapeutic range for lidocaine. This is the first study showing that polymorphism L1308F confers a greater sensitivity of sodium channels to lidocaine. It demonstrates that, as previously shown for long QT syndrome, some polymorphisms may lead to drug-induced BrS.
V232 and L1308 are located at the intracellular end of S4-DI and the middle of S4-DIII, respectively. Both are remote from amino acids known to participate to the lidocaine receptor site.10-12 The transmembrane segment S4 contains positive charges that confer voltage dependence to activation of the channel. Neither mutation is expected to disrupt the pore of the channel, but they may be implicated in the opening of the channel.13-15 The movement of segment S4 in response to changes in membrane potential is also a strong modulator of the inactivation of the channel16-18 and potential interaction between S4 and S6 may modulate accessibility of lidocaine to its receptor site.19 Alanine scanning mutagenesis indicates that residues in S6 in domains I, III, and IV contribute significantly to the lidocaine receptor site20-23 with residues F1664 and N1769 in domains III and IV of NaV1.2 being important. Analogy to potassium channels suggests that rotational movement of S6 during activation and inactivation alters access of lidocaine to its receptor.24,25 Moreover, the effect of lidocaine on the gating charge from segments S4, particularly in domain III, suggest that allosteric coupling does exist between the lidocaine binding site on S6 and the voltage sensors in S4.20,26,27 Based on these observations, we speculate that residues L1308 and V232 alter the coupling between S4 and S6 to increase the affinity for lidocaine. This hypothesis is supported by the lack of changes in steady-state inactivation by the mutations, which suggest the increased affinity for lidocaine due to enhanced inactivation.
Sheets and Hanck20 showed that lidocaine stabilizes the gating charge of S4 of domain III and IV in a depolarized configuration during activation (segment likely in outward position). Each mutation could therefore promote stabilization of S4 in the activated configuration and increase the affinity for lidocaine by prolonging exposure of the lidocaine binding site on S6. An interesting corollary emerges when we combine our results on steady-state inactivation with this hypothesis. During low-amplitude depolarizations, channels do not open but transit through series of closed, but activated, states before opening and inactivation. Because our results suggest that mutations V232I+L1308F alter the transitions between activated states and thus the binding of lidocaine, potentiation of the drug affinity for these closed states may shift the availability of the V232I+L1308F channels. It may also contribute to use-dependent block because channels stimulated at higher rates spend more time in the activated states.
Clinical Relevance
The electrocardiographic manifestations of the congenital BrS can be unmasked by sodium channel blockers and several other conditions, including alcohol and cocaine toxicity.3,28-36
Pharmacological agents capable of unmasking the congenital syndrome are sodium channel blockers such as flecainide,3,4,37,38 pilsicainide,39,40 propafenone,41 ajmaline,3,42 and procainamide,3,29 psychotropic drugs such as amitriptyline,43,44 nortriptyline,32 desipramine,30 and clomipramine31 may also induce acquired forms of the BrS. Most sodium channel blockers that act in this capacity are known to dissociate from the sodium channel with slow kinetics and induce strong use-dependent block typical of Class IC antiarrhythmic agents. In contrast, lidocaine and mexiletine (Class IB) display rapid dissociation kinetics and produce little to no ST segment elevation in patients with congenital BrS.4
We report a rare case of lidocaine-induced BrS phenotype in an individual who, to our knowledge, did not previously manifest the disease. The patient is shown to carry a double mutation in SCN5A capable of altering the interaction of lidocaine with the sodium channel so that the drug assumes characteristics of a Class IC agent, demonstrating potent use-dependent block of the sodium channel. Recently, Ackerman et al9 found mutation L1308F in one of 319 blacks but not among 295 whites. Our laboratory found it in one of 100 whites. The status of L1308F as a polymorphism remains to be determined; however, our results clearly indicate that this variant is an important determinant for the sensitivity of patients to lidocaine. Caution should therefore be exerted when treating carrier patients with Class I antiarrhythmic drugs. To our knowledge, this is the first demonstration of a compound mutation associated with an acquired or congenital form of BrS.
Acknowledgments
We thank Miss Judy Breedlove, Dr Arthur Iodice, and Mr Robert Goodrow for their technical help.
Sources of Funding
This work was supported by US National Institutes of Health grants HL47678 (CA) and HL66169 (RB) from the National Heart, Lung and Blood Institute; and grants from the American Heart Association (RB), National Heart Foundation, a program of the American Health Assistance Foundation (JMC), and NYS and Florida Grand Lodges, F. & A.M, Fondation Leducq, Québec, Canada (RB), Canadian Institutes of Health Research (CIHR) Grant #134443 (RD).
Footnotes
Disclosures
None.
References
- 1.Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome: a multicenter report. J Am Coll Cardiol. 1992;20:1391–1396. doi: 10.1016/0735-1097(92)90253-j. [DOI] [PubMed] [Google Scholar]
- 2.Antzelevitch C, Brugada P, Brugada J, Brugada R. The Brugada Syndrome: From Bench to Bedside. Blackwell Futura; Oxford: 2005. [Google Scholar]
- 3.Brugada R, Brugada J, Antzelevitch C, Kirsch GE, Potenza D, Towbin JA, Brugada P. Sodium channel blockers identify risk for sudden death in patients with ST-segment elevation and right bundle branch block but structurally normal hearts. Circulation. 2000;101:510–515. doi: 10.1161/01.cir.101.5.510. [DOI] [PubMed] [Google Scholar]
- 4.Shimizu W, Antzelevitch C, Suyama K, Kurita T, Taguchi A, Aihara N, Takaki H, Sunagawa K, Kamakura S. Effect of sodium channel blockers on ST segment, QRS duration, and corrected QT interval in patients with Brugada syndrome. J Cardiovasc Electrophysiol. 2000;11:1320–1329. doi: 10.1046/j.1540-8167.2000.01320.x. [DOI] [PubMed] [Google Scholar]
- 5.Brugada R, Hong K, Dumaine R, Cordeiro J, Gaita F, Borggrefe M, Menendez TM, Brugada J, Pollevick GD, Wolpert C, Burashnikov E, Matsuo K, Wu YS, Guerchicoff A, Bianchi F, Giustetto C, Schimpf R, Brugada P, Antzelevitch C. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation. 2004;109:30–35. doi: 10.1161/01.CIR.0000109482.92774.3A. [DOI] [PubMed] [Google Scholar]
- 6.Dumaine R, Towbin JA, Brugada P, Vatta M, Nesterenko VV, Nesterenko DV, Brugada J, Brugada R, Antzelevitch C. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res. 1999;85:803–809. doi: 10.1161/01.res.85.9.803. [DOI] [PubMed] [Google Scholar]
- 7.Hille B. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J Gen Physiol. 1977;69:497–515. doi: 10.1085/jgp.69.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hondeghem LM, Katzung BG. Time- and voltage-dependent interactions of antiarrhythmic drugs with cardiac sodium channels. Biochim Biophys Acta. 1977;472:373–398. doi: 10.1016/0304-4157(77)90003-x. [DOI] [PubMed] [Google Scholar]
- 9.Ackerman MJ, Splawski I, Makielski JC, Tester DJ, Will ML, Timothy KW, Keating MT, Jones G, Chadha M, Burrow CR, Stephens JC, Xu C, Judson R, Curran ME. Spectrum and prevalence of cardiac sodium channel variants among black, white, Asian, and Hispanic individuals: implications for arrhythmogenic susceptibility and Brugada/long QT syndrome genetic testing. Heart Rhythm. 2004;1:600–607. doi: 10.1016/j.hrthm.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 10.Sunami A, Dudley SC, Jr, Fozzard HA. Sodium channel selectivity filter regulates antiarrhythmic drug binding. Proc Natl Acad Sci U S A. 1997;94:14126–14131. doi: 10.1073/pnas.94.25.14126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sunami A, Glaaser IW, Fozzard HA. A critical residue for isoform difference in tetrodotoxin affinity is a molecular determinant of the external access path for local anesthetics in the cardiac sodium channel. Proc Natl Acad Sci U S A. 2000;97:2326–2331. doi: 10.1073/pnas.030438797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Common molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant block of voltage-gated Na+ channels. Proc Natl Acad Sci U S A. 1996;93:9270–9275. doi: 10.1073/pnas.93.17.9270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chanda B, Bezanilla F. Tracking voltage-dependent conformational changes in skeletal muscle sodium channel during activation. J Gen Physiol. 2002;120:629–645. doi: 10.1085/jgp.20028679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chanda B, Asamoah OK, Bezanilla F. Coupling interactions between voltage sensors of the sodium channel as revealed by site-specific measurements. J Gen Physiol. 2004;123:217–230. doi: 10.1085/jgp.200308971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuhn FJ, Greeff NG. Movement of voltage sensor S4 in domain 4 is tightly coupled to sodium channel fast inactivation and gating charge immobilization. J Gen Physiol. 1999;114:167–183. doi: 10.1085/jgp.114.2.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen LQ, Santarelli V, Horn R, Kallen RG. A unique role for the S4 segment of domain 4 in the inactivation of sodium channels. J Gen Physiol. 1996;108:549–556. doi: 10.1085/jgp.108.6.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cha A, Ruben PC, George AL, Jr, Fujimoto E, Bezanilla F. Voltage sensors in domains III and IV, but not I and II, are immobilized by Na+ channel fast inactivation. Neuron. 1999;22:73–87. doi: 10.1016/s0896-6273(00)80680-7. [DOI] [PubMed] [Google Scholar]
- 18.Popa MO, Alekov AK, Bail S, Lehmann-Horn F, Lerche H. Cooperative effect of S4-S5 loops in domains D3 and D4 on fast inactivation of the Na+ channel. J Physiol. 2004;561:39–51. doi: 10.1113/jphysiol.2004.065912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sheets MF, Hanck DA. Molecular action of lidocaine on the voltage sensors of sodium channels. J Gen Physiol. 2003;121:163–175. doi: 10.1085/jgp.20028651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science. 1994;265:1724–1728. doi: 10.1126/science.8085162. [DOI] [PubMed] [Google Scholar]
- 21.Wang GK, Quan C, Wang S. A common local anesthetic receptor for benzocaine and etidocaine in voltage-gated mu1 Na+ channels. Pflugers Arch. 1998;435:293–302. doi: 10.1007/s004240050515. [DOI] [PubMed] [Google Scholar]
- 22.Catterall WA. Molecular mechanisms of gating and drug block of sodium channels. Novartis Found Symp. 2002;241:206–218. [PubMed] [Google Scholar]
- 23.Yarov-Yarovoy V, McPhee JC, Idsvoog D, Pate C, Scheuer T, Catterall WA. Role of amino acid residues in transmembrane segments IS6 and IIS6 of the Na+ channel alpha subunit in voltage-dependent gating and drug block. J Biol Chem. 2002;277:35393–35401. doi: 10.1074/jbc.M206126200. [DOI] [PubMed] [Google Scholar]
- 24.del CD, Holmgren M, Liu Y, Yellen G. Blocker protection in the pore of a voltage-gated K+ channel and its structural implications. Nature. 2000;403:321–325. doi: 10.1038/35002099. [DOI] [PubMed] [Google Scholar]
- 25.Perozo E, Cortes DM, Cuello LG. Structural rearrangements underlying K+-channel activation gating. Science. 1999;285:73–78. doi: 10.1126/science.285.5424.73. [DOI] [PubMed] [Google Scholar]
- 26.Hanck DA, Makielski JC, Sheets MF. Lidocaine alters activation gating of cardiac Na channels. Pflugers Arch. 2000;439:814–821. doi: 10.1007/s004249900217. [DOI] [PubMed] [Google Scholar]
- 27.Sheets MF, Hanck DA. Outward stabilization of the S4 segments in domains III and IV enhances lidocaine block of sodium channels. J Physiol. 2007;582:317–334. doi: 10.1113/jphysiol.2007.134262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brugada P, Brugada J, Brugada R. Arrhythmia induction by antiarrhythmic drugs. Pacing Clin Electrophysiol. 2000;23:291–292. doi: 10.1111/j.1540-8159.2000.tb06751.x. [DOI] [PubMed] [Google Scholar]
- 29.Miyazaki T, Mitamura H, Miyoshi S, Soejima K, Aizawa Y, Ogawa S. Autonomic and antiarrhythmic drug modulation of ST segment elevation in patients with Brugada syndrome. J Am Coll Cardiol. 1996;27:1061–1070. doi: 10.1016/0735-1097(95)00613-3. [DOI] [PubMed] [Google Scholar]
- 30.Babaliaros VC, Hurst JW. Tricyclic antidepressants and the Brugada syndrome: an example of Brugada waves appearing after the administration of desipramine. Clin Cardiol. 2002;25:395–398. doi: 10.1002/clc.4950250809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goldgran-Toledano D, Sideris G, Kevorkian JP. Overdose of cyclic antidepressants and the Brugada syndrome. N Engl J Med. 2002;346:1591–1592. doi: 10.1056/NEJM200205163462020. [DOI] [PubMed] [Google Scholar]
- 32.Tada H, Sticherling C, Oral H, Morady F. Brugada syndrome mimicked by tricyclic antidepressant overdose. J Cardiovasc Electrophysiol. 2001;12:275. doi: 10.1046/j.1540-8167.2001.00275.x. [DOI] [PubMed] [Google Scholar]
- 33.Pastor A, Nunez A, Cantale C, Cosio FG. Asymptomatic Brugada syndrome case unmasked during dimenhydrinate infusion. J Cardiovasc Electrophysiol. 2001;12:1192–1194. doi: 10.1046/j.1540-8167.2001.01192.x. [DOI] [PubMed] [Google Scholar]
- 34.Ortega-Carnicer J, Bertos-Polo J, Gutierrez-Tirado C. Aborted sudden death, transient Brugada pattern, and wide QRS dysrrhythmias after massive cocaine ingestion. J Electrocardiol. 2001;34:345–349. doi: 10.1054/jelc.2001.26318. [DOI] [PubMed] [Google Scholar]
- 35.Nogami A, Nakao M, Kubota S, Sugiyasu A, Doi H, Yokoyama K, Yumoto K, Tamaki T, Kato K, Hosokawa N, Sagai H, Nakamura H, Nitta J, Yamauchi Y, Aonuma K. Enhancement of J-ST-segment elevation by the glucose and insulin test in Brugada syndrome. Pacing Clin Electrophysiol. 2003;26:332–337. doi: 10.1046/j.1460-9592.2003.00044.x. [DOI] [PubMed] [Google Scholar]
- 36.Araki T, Konno T, Itoh H, Ino H, Shimizu M. Brugada syndrome with ventricular tachycardia and fibrillation related to hypokalemia. Circ J. 2003;67:93–95. doi: 10.1253/circj.67.93. [DOI] [PubMed] [Google Scholar]
- 37.Fujiki A, Usui M, Nagasawa H, Mizumaki K, Hayashi H, Inoue H. ST segment elevation in the right precordial leads induced with class IC antiarrhythmic drugs: insight into the mechanism of Brugada syndrome. J Cardiovasc Electrophysiol. 1999;10:214–218. doi: 10.1111/j.1540-8167.1999.tb00662.x. [DOI] [PubMed] [Google Scholar]
- 38.Gasparini M, Priori SG, Mantica M, Napolitano C, Galimberti P, Ceriotti C, Simonini S. Flecainide test in Brugada syndrome: a reproducible but risky tool. Pacing Clin Electrophysiol. 2003;26:338–341. doi: 10.1046/j.1460-9592.2003.00045.x. [DOI] [PubMed] [Google Scholar]
- 39.Takenaka S, Emori T, Koyama S, Morita H, Fukushima K, Ohe T. Asymptomatic form of Brugada syndrome. Pacing Clin Electrophysiol. 1999;22:1261–1263. doi: 10.1111/j.1540-8159.1999.tb00612.x. [DOI] [PubMed] [Google Scholar]
- 40.Shimizu W, Aiba T, Kurita T, Kamakura S. Paradoxic abbreviation of repolarization in epicardium of the right ventricular outflow tract during augmentation of Brugada-type ST segment elevation. J Cardiovasc Electrophysiol. 2001;12:1418–1421. doi: 10.1046/j.1540-8167.2001.01418.x. [DOI] [PubMed] [Google Scholar]
- 41.Matana A, Goldner V, Stanic K, Mavric Z, Zaputovic L, Matana Z. Unmasking effect of propafenone on the concealed form of the Brugada phenomenon. Pacing Clin Electrophysiol. 2000;23:416–418. doi: 10.1111/j.1540-8159.2000.tb06774.x. [DOI] [PubMed] [Google Scholar]
- 42.Rolf S, Bruns HJ, Wichter T, Kirchhof P, Ribbing M, Wasmer K, Paul M, Breithardt G, Haverkamp W, Eckardt L. The ajmaline challenge in Brugada syndrome: diagnostic impact, safety, and recommended protocol. Eur Heart J. 2003;24:1104–1112. doi: 10.1016/s0195-668x(03)00195-7. [DOI] [PubMed] [Google Scholar]
- 43.Bolognesi R, Tsialtas D, Vasini P, Conti M, Manca C. Abnormal ventricular repolarization mimicking myocardial infarction after heterocyclic antidepressant overdose. Am J Cardiol. 1997;79:242–245. doi: 10.1016/s0002-9149(96)00727-8. [DOI] [PubMed] [Google Scholar]
- 44.Rouleau F, Asfar P, Boulet S, Dube L, Dupuis JM, Alquier P, Victor J. Transient ST segment elevation in right precordial leads induced by psychotropic drugs: relationship to the Brugada syndrome. J Cardiovasc Electrophysiol. 2001;12:61–65. doi: 10.1046/j.1540-8167.2001.00061.x. [DOI] [PubMed] [Google Scholar]