

Abstract
Strong evidence exists for interactions of zwitterionic phosphate and amine groups in Sphingosine-1 phosphate (S1P) to conserved R and E residues present at the extracellular face of transmembrane-3 (TM3) of S1P receptors. The contribution of R120 and E121 for high affinity ligand-receptor interactions is essential, as single-point R120A or E121A S1P1 mutants neither bind S1P nor transduce S1P function. Because S1P receptors are therapeutically interesting, identifying potent selective agonists with different binding modes and in vivo efficacy is of pharmacological importance. Here we describe a modestly water-soluble highly-selective S1P1 agonist (CYM-5442) that does not require R120 or E121 residues for activating S1P1-dependent p42/p44 MAPK phosphorylation, which defines a new hydrophobic pocket in S1P1. CYM-5442 is a full agonist in vitro for S1P1 internalization, phosphorylation and ubiquitination. Importantly, CYM-5442 was a full agonist for induction and maintenance of S1P1-dependent lymphopenia, decreasing B-lymphocytes by 65% and T-lymphocytes by 85% of vehicle. Induction of CYM-5442 lymphopenia was dose and time-dependent, requiring serum concentrations in the 50 nM range. In vitro measures of S1P1 activation by CYM-5442 were non-competitively inhibited by a specific S1P1 antagonist (W146), competitive for S1P, FTY720-P and SEW2871. In addition, lymphopenia by CYM-5442 was reversed by W146 administration or upon pharmacokinetic agonist clearance. Pharmacokinetics in mice also indicated that CYM-5442 partitions significantly in central nervous tissue. These data show that CYM-5442 activates S1P1-dependent pathways in vitro and to levels of full efficacy in vivo through a hydrophobic pocket, separable from the orthosteric site of S1P binding that is headgroup dependent.
Introduction
S1P is a circulating lipid that binds to five G protein-coupled receptors (GPCRs) termed S1P1-5. S1P1 selectively regulates physiological functions in the immune and cardiovascular systems, including immune cell trafficking (Goetzl and Rosen, 2005, Rosen et al., 2007) and maintaining endothelial integrity (Lee et al, 1999; Sanchez et al., 2004; Dudek et al., 2004; Sanna et al., 2006; Foss et al., 2007). Pharmacological studies with the sphingosine analog immunosuppressant prodrug, FTY720, indicated that administration of FTY720 or S1P decreased lymphocyte counts in the mouse draining lymph node, resulting in lymphopenia (Mandala et al., 2002). Moreover, FTY720 was shown to be rapidly phosphorylated, with the phosphorylated species (FTY720-P) acting as a potent agonist on S1P1, 3-5 subtypes. Discovery of SEW2871, a selective agonist for S1P1 that replicated the lymphopenic actions of FTY720, plus development of selective S1P1 antagonists with in vivo activity, later demonstrated that S1P1 was the primary mediator of lymphocyte sequestration in secondary lymphoid organs and lymphopenia.
Mechanistic insights into S1P1 mediated lymphopenia came from combining genetics, pharmacological tools and two-photon imaging (Wei et al., 2005; Sanna et al., 2006). The latter has allowed studying real-time lymphocyte dynamics in the intact lymph node, where SEW2871 infusion reduces lymphocyte egress in the medulla, while a selective S1P1 antagonist (W146) fully reverses agonist actions.
In the thymus, S1P1 agonism enhances late thymocyte maturation, yet inhibits subsequent thymocyte egress (Rosen et al., 2003; Alfonso et al., 2006; Weinreich and Hogquist, 2008). Consequently, S1P agonist administration reduces naïve blood CD4+ and CD8+ thymocytes and B-cells by nearly 90% and 70%, respectively.
Regulation of lymphocyte trafficking with S1P1 agonists has opened the possibility for clinical modulation of lymphocyte dynamics, exemplified by the positive results reported in multiple sclerosis (MS) clinical trials with FTY720 (Hiestand et al., 2008). Although the mechanism of FTY720 in ameliorating MS symptoms is not clear, it may encompass multiple targets, both systemic, including S1P1 mediated lymphopenia, and local, by actions on neurons (Kataoka et al., 2005; Belatoni et al., 2007; Miron et al., 2008). Recently, a report has also highlighted that vascular integrity of the brain blood barrier (BBB) likely participates on the mechanism of FTY720's efficacy in MS (Foster et al., 2008). Thus, understanding the contribution of systemic and local effects of S1P1 modulation would be relevant for developing efficacious therapies for autoimmune disease.
Previous studies aimed at dissecting the S1P-S1P1 binding pocket have provided strong evidence for a model of S1P ligand interaction with three charged residues on S1P1 (Parrill et al., 2000; Wang et al., 2001). Two residues, R120 and E121, which are present on TM3 of all S1P receptors, interact with the phosphate headgroup and the ammonium moiety of S1P, respectively. The importance of R120 and E121 interactions with S1P has been demonstrated by mutagenesis. Substitution of R120 or E121 with alanine results in total loss of [P33]-S1P binding to S1P1, and consistent with the lack of binding, neither of the TM3 mutants internalize following an S1P challenge, nor stimulate S1P-mediated [35S]-GTPγS binding. Thus, the strong zwitterionic nature of S1P requires hydrophilic headgroup interactions on S1P1 TM3 RE residues to achieve high affinity binding and receptor functions.
We have shown that besides S1P, other S1P1 agonists including AFD-R (an FTY720-P analog), and SEW2871, also require R120 and E121 for full activation of intracellular pathways (Jo et al., 2005). Interesting, although SEW2871 lacks structurally charged headgroups, its binding model to S1P1 and activation of P42/p44 MAPK and AKT pathways is dependent on R120 and E121, likely by replacing the salt-bridge polar interactions by ion-dipole interactions. SEW2871 has recently shown to make additional hydrophobic interactions deep in S1P1 TM5, suggesting that it overlaps both the hydrophilic pocket and a hydrophobic pocket on S1P1 (Fujiwara et al., 2007).
Because the RE residues are invariant across S1P receptors, developing efficacious ligands that bind solely in deep hydrophobic pockets could provide advantages for selectivity, mechanism-based toxicity, and the ability of these molecules to potentially traverse impermeant barriers, like the BBB.
Here we report development and characterization of a potent and moderately water-soluble small molecule S1P1-agonist (CYM-5442) that does not require either of the critical RE S1P-S1P1 headgroup interactions, yet, it is fully active in vivo for inducing lymphopenia. Importantly, CYM-5442 partitions significantly into brain tissue. Development of in vivo-active S1P1 agonists that do not require headgroup interactions, like CYM-5442, reveal a discrete novel hydrophobic pocket on S1P1 to be further explored in therapeutics.
Materials and Methods
Reagents
S1P was purchased from Biomol. The selective S1P1 agonist SEW2871 was purchased from Maybridge. The selective S1P1 antagonist W146 was from Avanti Polar Lipids. The selective S1P2 antagonist JTE-013 was from Cayman Medical Company. P32-orthophosphate was purchased from Perkin Elmer. The MEK1 inhibitor U0126 was from Calbiochem.
Cell lines
The generation of stable CHO-K1-S1P receptor cell clones and the conditions for CRE and NFAT reporter assays have been recently documented (Schurer et al., 2008). The TANGO® S1P4 and TANGO® S1P5 stable cell lines were obtained from Invitrogen and assayed according to Invitrogen's protocols with 1 μM S1P as positive control (Schurer et al., 2008). HEK293 cells stably expressing human S1P1 tagged with C-terminal GFP (S1P1-GFP) were a gift from Timothy Hla (U. Conn. Health Center, Farmington, CT). S1P1-GFP cells were grown as reported (Gonzalez-Cabrera et al., 2007). Parental CHO-K1 cells used in transient transfection experiments were purchased from ATCC (Manassas, VA).
Chemical synthesis of CYM-5442
The schematic steps (i-iv) of CYM-5442 synthesis are indicated in Figure 2, and are as follows:
Figure 2.
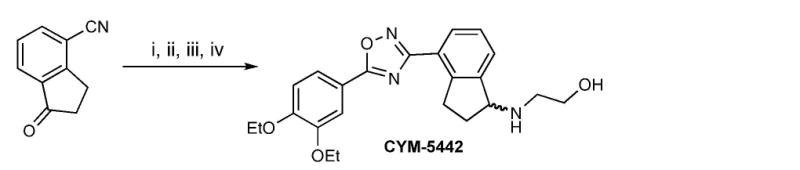
Chemical synthesis of CYM-5442.
i.- NaBH4, SiO2, EtOH, 0 °C to r.t., 2h. ii.- NH2OH.HCI, Na2CO3, EtOH, reflux, 12h. iii.- 3,4-diethoxybenzoic acid, EDCI, HOBt, DMF, 130 °C, m.w., 30min. iv.- (a) SOCl2, Py, r.t., 2h. (b) Aminoethanol, DIPEA, DMF, 50 °C, 48h.
i 1-hydroxy-2,3-dihydro-1H-indene-4-carbonitrile
To a stirred suspension of 1-oxo-2,3-dihydro-1H-indene-4-carbonitrile (1.0 equiv, 0.4M) and silica gel (catalytic) in ethanol at 0 °C was added NaBH4 (0.33 equiv). The reaction was allowed to warm up to room temperature and stirred for 2 h. The Solvent was removed under reduced pressure and the product purified by C.C. in Hexane/EtOAc (5:5) to offer 1-hydroxy-2, 3-dihydro-1H-indene-4-carbonitrile as white solid in 80% yield. 1H NMR (300 MHz, CDCl3): δ 7.62 (d, J = 7.5 Hz, 1H), 7.54 (d, J = 7.8 Hz, 1H), 7.33 (t, J = 7.8 Hz, 1H), 5.28 (t, J = 6.3 Hz, 1H), 3.28-3.18 (m, 1H), 3.02-2.92 (m, 1H), 2.63-2.52 (m, 1H), 2.06-1.99 (m, 1H).
ii 1,N-dihydroxy-indan-4-carboxamidine
To a stirred suspension of hydroxylamine hydrochloride (1.1 equiv) and Na2CO3 (1.1 equiv) in ethanol was added, in one portion, the benzonitrile prepared in previous step (1 equiv). The mixture was refluxed for 6 h followed by addition of another portion of hydroxylamine hydrochloride (1.1 equiv) and Na2CO3 (1.1 equiv), the reaction was refluxed for additional 6 h. The suspension was cooled to room temperature and filtered. The solid was washed with ethanol and the filtrate was concentrated under reduced pressure. The amidoxime-crude was recrystallized from EtOAc/Hexanes and used without further purification.
iii 4-[5-(3,4-Diethoxy-phenyl)-[1,2,4]oxadiazol-3-yl]-indan-1-ol
In a microwave vial, a stirring solution of 3, 4-diethoxybenzoic acid (1 equiv, 0.2M) in DMF was treated with HOBt (1.3 equiv) and EDCI (1.3 equiv) at room temperature. The reaction was stirred for 20 min followed by addition, in a single portion, of amidoxime (prepared in previous step) (1.1 equiv). The reaction was stirred for additional 30 min at room temperature and then heated to 130 °C for 30 min. The reaction was diluted using a saturated solution of NaCl and extracted with EtOAc (X3). The organic phase was dried over Na2SO4 anhydrous and concentrated under reduced pressure. The product was purified by C.C. using CH2Cl2:MeOH (9:1) to offer diaryloxadiazole as white solid in good yield.
1H NMR (400 MHz, CDCl3): δ 8.10 (d, J = 7.6, 1H), 7.78 (dd, J1 = 1.6 Hz, J2 = 8 Hz, 1H), 7.67 (d, J = 1.6 Hz, 1H), 7.56 (d, J = 7.6 Hz, 1H), 7.39 (t, J = 7.6 Hz, 1H), 6.97 (d, J = 8.0 Hz, 1H), 5.29 (t, J = 6.4 Hz, 1H), 4.19 (q, J = 7.2 Hz, 2H), 4.18 (q, J = 7.2 Hz, 2H), 3.51-4.43 (m, 1H), 3.22-3.14 (m, 1H), 2.59-2.51 (m, 1H), 2.04-1.97 (m, 1H), 1.5 (t, J = 7.2 Hz, 3H), 1.49 (t, J = 7.2, 3H); 13C NMR (100 MHz, CDCl3): δ 175.2, 168.9, 152.8, 148.9, 146.6, 143.3, 128.9, 127.4, 127.0, 123.8, 122.2, 116.7, 112.7, 112.4, 76.2, 64.9, 64.8, 35.7, 31.5, 14.9, 14.8. MS (EI) m/z 367 (M+), HRMS (EI) for C21H22N2O4 (M+): calcd 367.1652, found 367.1653.
iv 2-{4-[5-(3,4-Diethoxy-phenyl)-[1,2,4]oxadiazol-3-yl]-indan-1-ylamino-}-ethanol (1)
A solution of alcohol (1 equiv), at 0 °C, was treated with SOCl2 (1.1 equiv) and pyridine (1.1 equiv) in CH2Cl2. The reaction was stirred at room temperature for 2h. The reaction was diluted with CH2Cl2 and washed with NaHCO3 (2X). The organic phase was dried over sodium sulfate and concentrated under reduced pressure. The crude was dissolved in DMF and treated with the ethanolamine (2 equiv) and DIPEA (2.0 equiv). The reaction was stirred at 50 °C for 48h. The reaction was diluted with H2O and the product extracted with EtOAc (3X). The product was purified by C.C. using CH2Cl2/MeOH (9:1) to offer aminoethanol CYM-5442 as white solid in 60% yield.
1H NMR (500 MHz, CDCl3): δ 8.12 (d, J = 7.5 Hz, 1H), 7.79 (dd, J1 = 2.0 Hz J2 = 8.5 Hz, 1H), 7.68 (d, J = 2.0 Hz, 1H), 7.63 (d, J = 7.5 Hz, 1H), 7.40-7.37 (m, 1H), 4.49-4.47 (m, 1H), 4.23-4.16 (m, 4H), 3.78-3.70 (m, 1H), 3.53-3.46 (m, 1H), 3.29-3.22 (m, 1H), 2.96-2.94 (m, 4H), 2.56-2.50 (m, 1H), 2.09-2.03 (m, 1H), 1.52-1.49 (m, 6H); 13C NMR (125 MHz, CDCl3): δ 175.03, 168.66, 152.71, 148.87, 143.76, 128.71, 127.11, 123.89, 122.04, 116.67, 112.63, 112.49, 104.66, 64.84, 64.61, 62.70, 60.34, 47.98, 31.90, 29.69, 14.72, 14.64. MS (EI) m/z 410 (M+), HRMS (EI) for C23H27N3O4 (M+): calcd 410.2074, found 410.2077.
Evaluation of CYM-5442 agonist properties
Details of the screening CRE reporter assays can be found in Schurer et al., (2008). For the CRE reporter assay studies, forskolin was included at a 2 μM final concentration prior to addition of the agonists. Agonist-mediated inhibition of forskolin induced cAMP accumulation was read on an Envision fluorescent plate reader (384-well format) after 2 h agonist incubation, and with SEW2871 as the positive control. Agonist-induced S1P1-GFP internalization and ubiquitination assays have been documented previously (Gonzalez-Cabrera et al., 2008). Description of agonist-mediated S1P1-GFP phosphorylation experiments can be found in Schurer et al., (2008).
ELISA determination of p42/p44 MAPK activity
CYM-5442 and S1P-mediated p42/p44 MAPK activation was measured using an ELISA kit (Cell Signaling Technologies) in CHO-K1 cells transiently transfected with either the WT S1P1 receptor cDNA or the single-point S1P1 mutant (R120A or E121A) cDNAs (gifts from Abby Parrill, University of Memphis, TN). For transfections, cells were plated on 10-cm dishes at 80% confluence and transfected with 12 μg of each plasmid using Fugene HD (Roche) in Opti-MEM. After 16 h, each pool transfected dish was split into a 6-well plate and allowed to incubate for additional 24 h. The cells were next incubated for 4 h in serum-free DMEM prior to addition of the agonist. In the antagonist (W146) experiments or the MEK1 inhibitor (U0126), W146 or U0126 were incubated for 30 min (U0126) or 1 h (W146) at 10 μM prior to agonist treatment. Cells were then stimulated for 5 min (determined empirically to be maximal for both agonists) with increasing concentrations of CYM-5442 or S1P and activation of p42/p44 MAPK phosphorylation was assayed according to manufacturer instructions. For each condition (WT, R120A or E121A), both CYM-5442 and S1P agonists were tested in parallel, in presence and absence of W146. In addition, for each of the conditions tested, the concentration response curves for agonist-mediated activation of p42/p44 MAPK phosphorylation were plotted as a percent of the agonist eliciting the maximal response, and the potency (EC50), maximal response (Emax) and goodness of fit (R2) values were determined using Graph Pad Prism (San Diego, CA).
Pharmacokinetic studies
Pharmacokinetics of CYM-5442 was assessed in Sprague Dawley rats. The compound was formulated at 1 mg/ml (10:10:80, DMSO: tween 80: water, vol:vol:vol) and dosed at 1 mg/kg intravenous (i.v.) into the jugular or 2 mg/kg by oral gavage (P.O.). Blood was obtained at t= 5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 6 h, and 8 h into EDTA containing tubes and plasma was generated by standard centrifugation methods. Separate studies to evaluate brain exposure were done in C57Bl6 mice where CYM-5442 was dosed at 10 mg/kg by intraperitoneal (i.p.) route. Blood and brains were obtained at t= 2 h to assess the amount of compound in the target organ. All procedures and handling were according to standard operating procedures approved by the Institutional Animal Care and Use Committee (IACUC).
In order to assess in vivo pharmacokinetic parameters an LC-MS/MS bioanalytical method was developed where 25 μl of plasma was treated with 125 μl of acetonitrile containing an internal standard in a Millipore Multiscreen Solvinter 0.45 micron low binding PTFE hydrophilic filter plate (#MSRLN0450) and allowed to shake at room temperature for five minutes. The plate was then centrifuged for 5 minutes at 4,000 rpm in a tabletop centrifuge and the filtrate was collected in a polypropylene capture plate. The filtrate (10 μl) was injected using an Agilent 1200 HPLC equipped with a Thermo Betasil C18 HPLC column 5 μ (50 × 2.1 mm; # 70105-052130). Mobile Phase A was water with 0.1% formic acid. Mobile Phase B was acetonitrile with 0.1% formic acid. Flow rate was 375 μl/min using a gradient of 90% A/10% B from 0-0.5 min,, ramped to 5%A/95%B at 2 min, held at 5%A/95%B until 3.0 min, ramped to 90% A/10% B at 4 min, and held at 90% A/10% B until 7 min. An API Sciex 4000 equipped with a turbo ion spray source was used for all analytical measurements. Instrument settings had curtain gas set to 10, gas 1 and gas 2 set to 45, ionization energy set to 5500 Volts, drying gas temperature 550°C, resolution was set to unit, and dwell time was set to 100 msec. A positive ion MRM method was developed. CYM-5442 was quantitated from 2 to 1000 ng/mL. Peak areas of the m/z°410.1→193.1 product ion of CYM-5442 (DP = 45, CE = 30) were measured against the peak areas of the internal standard (sunitinib) m/z 399→283 product ion (DP = 86, CE = 41). Data was fit using WinNonLin (Pharsight Corporation, Mountain View, CA).
Similar conditions were used to determine brain levels of CYM-5442 except the brain samples were frozen upon collection. When analyzed the frozen samples were weighed and acetonitrile was added (10× weight: vol). The samples were sonicated to extract the compound from the brain matrix and then filtered as described above. Brain samples were analyzed against a standard curve generated in blank brain matrix.
Measurement of CYM-5442 mediated lymphopenia
Male C57BL6 mice weighing 30 g were used for all experiments. All animal studies were approved by the IACUC. Animals were injected i.p. with a volume of 300 μl with the indicated dose of CYM-5442 or vehicle. For W146 experiments, mice were administered with 20 mg/kg W146 or vehicle for 30 min prior to CYM-5442 administration. Vehicle consisted of sterile water (CYM-5442) or 10% DMSO, 25% Tween-20 in sterile water (W146). Following incubation at the indicated times, animals were euthanized and blood collected into tubes containing EDTA. Blood white blood cell count and lymphocyte counts were obtained using an automated veterinary Hemoanalyzer (Hospitex Diagnostics, TX) by reading 30 μl blood samples. The remaining blood was used for determination of drug serum levels (see below), or to determine, by FACs, the number of B-cells (using a FITC conjugated B220 antibody marker, BD Diagnostics, 1:100 dilution) and T-cells (using Pacific Blue CD4+ and Per-CP-Cy5.5 CD8+ antibodies; BD Diagnostics, both at 1:100). FACS analysis was done in FlowJo (Ashland, OR).
CYM-5442 and W146 serum concentration
Blood samples used in lymphopenia studies were further processed for measuring serum CYM-5442 and W146 concentrations. Samples (100 μl plasma) were extracted with 400 μl ice cold methanol (stored at -20°C), vortexed for 1 min, allowed to sit at 4°C for 30 min, and centrifuged at 16,400 rpm for 5 min. The supernatant was evaporated to a volume near dryness and methanol was added to bring the final volume to 50 μl. For calibration standards, clean plasma was spiked with increasing concentrations of CYM-5442 or W146, and processed the same way as the samples. Samples were analyzed on an Agilent LC-MS/MS system using an 1100 LC stack mated with a 6410 triple quadrapole mass spectrometer. For CYM-5442 quantification, the transition of m/z 410 -> 349.2 was monitored, and for W146, the transition of m/z 343 -> 138 was monitored. The column used was an Agilent Zorbax SB-C18 (2.1 mm × 75 mm). Flow-rate = 250 ml per min. Mobile phases were A = water: 0.1% formic acid, B = acetonitrile: 0.1% formic acid. Gradient: T=0, 15% B ->T = 8, 98% B; 5 μl injected.
Results
CYM-5442 is a potent S1P1 selective agonist
CYM-5442 (Figure 1) is a chemically optimized version of an original hit (CYM-5181) from a screen aimed to discover novel S1P receptor agonists (Schurer at al., 2008). The chemical synthesis of CYM-5442 is depicted in Figure 2. Even though CYM-5442 contains the privileged oxadiazole-based scaffold reported to fit S1P receptors (Schurer et al., 2008), CYM-5442 is selective for S1P1, as measured by high throughout agonist-antagonist formats across S1P receptors (Table 1). In high throughput agonist format with SEW2871 as the positive control, CYM-5442 inhibited forskolin-stimulated CRE transcription in a concentration dependent manner (Figure 3), being a full agonist compared to SEW2871, but of higher potency (20-fold and 100-fold) relative to CYM-5181 and SEW2871, respectively.
Figure 1.
Chemical structure of ligands used in this study.
Table 1. Selectivity of CYM-5442 for the cloned human S1P receptors.
In agonist format, CYM-5442 was tested in 12-point (up to 10 μM) concentration response curves. In antagonist format, CYM-5442 was included in the assay at 10 μM final concentration and tested for its potential to block an EC80 concentration of S1P.
| Receptor subtype | Agonist format
(Emax) (EC50) |
Antagonist format
(IC50) |
|---|---|---|
| S1P1a | (100%) (1.35 ± 0.25 nM) | N.A. |
| S1P2b | N.A. | N.A. |
| S1P3c | N.A. | N.A |
| S1P4d | N.A. | N.D. |
| S1P5e | 20% at 10 μM | N.D. |
N.A. refers to no activity; N.D., not determined
Agonist format measured forskolin-stimulated CYM-5442 inhibiton of cAMP CRE transcription on stable S1P1-CHO-K1-CRE-bla cells, with SEW2871 as positive control. W146 inhibition of SEW2871 was used as a positive control in the antagonist format
Agonist format measured CYM-5442-stimulated cAMP accumulation on stable CHO-K1-S1P2-CRE cells with S1P as positive control. JTE-013 inhibition of S1P was used as positive control in antagonist format
Agonist format measured CYM-5442-stimulated cAMP accumulation in stable S1P3-NFAT-bla-CHO-K1 cells with S1P as positive control.
Agonist format measured CYM-5442-stimulated β-arrestin activation in stable TANGO® S1P4-bla U2OS cells with S1P as positive control.
Agonist format measured CYM-5442-stimulated cAMP accumulation in stable TANGO® S1P5-bla U2OS cells with S1P as positive control.
Figure 3. Concentration dependent inhibition of CRE transcription in stable S1P1.CHO cells by S1P1 agonists.
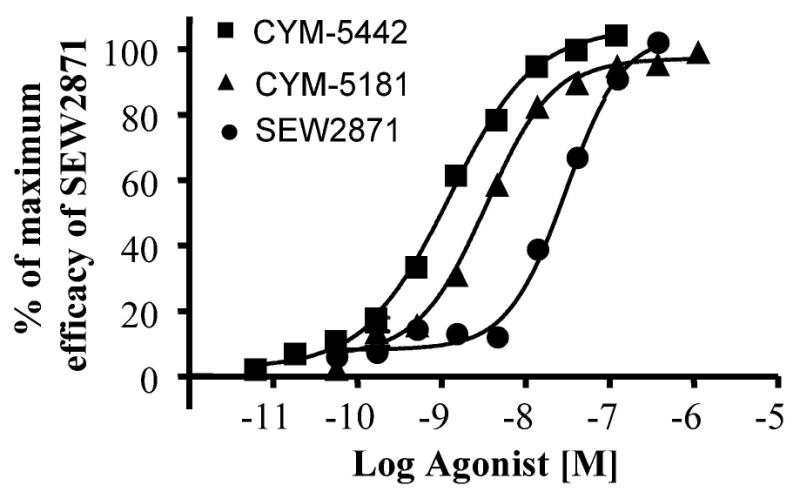
Cells stably expressing a CRE-beta-lactamase reporter and human S1P1 were incubated with increasing concentrations of SEW2871, CYM-5442 or CYM5181. The ability of the agonists to inhibit forskolin-stimulated beta-lactamase expression was measured as described (Schurrer et al., 2008). The potency (pEC50) values were determined to be -7.54, -8.47 and -8.91 for SEW2871, CYM5181 and CYM-5442, respectively.
Further characterization of the agonist properties of CYM-5442 was done using a receptor immunoprecipitation protocol previously validated for studying trafficking and fate of S1P1-GFP during agonist stimulation (Gonzalez-Cabrera et al., 2007). Using this protocol and HEK293 cells stably expressing S1P1 fused to GFP on the carboxy-terminus, we probed CYM-5442 for stimulating three agonist-S1P1 activated steps: receptor phosphorylation, receptor internalization and receptor-ubiquitin recruitment. Figure 4A indicates that incubation of 500 nM CYM-5442 with P32-orthophosphate labeled cells stimulated S1P1 phosphorylation in a time-dependent manner, similar to that obtained with 500 nM of S1P (at 30 min). CYM-5442 led to rapid S1P1 phosphorylation which was sustained throughout the analysis. To confirm the involvement of S1P1 for CYM-5442 phosphorylation, the S1P1 selective antagonist W146 was used. Pre-incubation with 10 μM of W146 for 30 min prior to CYM-5442 treatment completely abolished CYM-5442-mediated S1P1 phosphorylation, while antagonist alone had no effect on the response. In addition, silver staining of the same gel used to measure receptor phosphorylation indicated that the differences in S1P1 phosphorylation across conditions were not due to differences in immunoprecipitated S1P1 protein loading.
Figure 4. CYM-5442 activates three S1P1 dependent pathways in stable S1P1-GFP 293 cells.
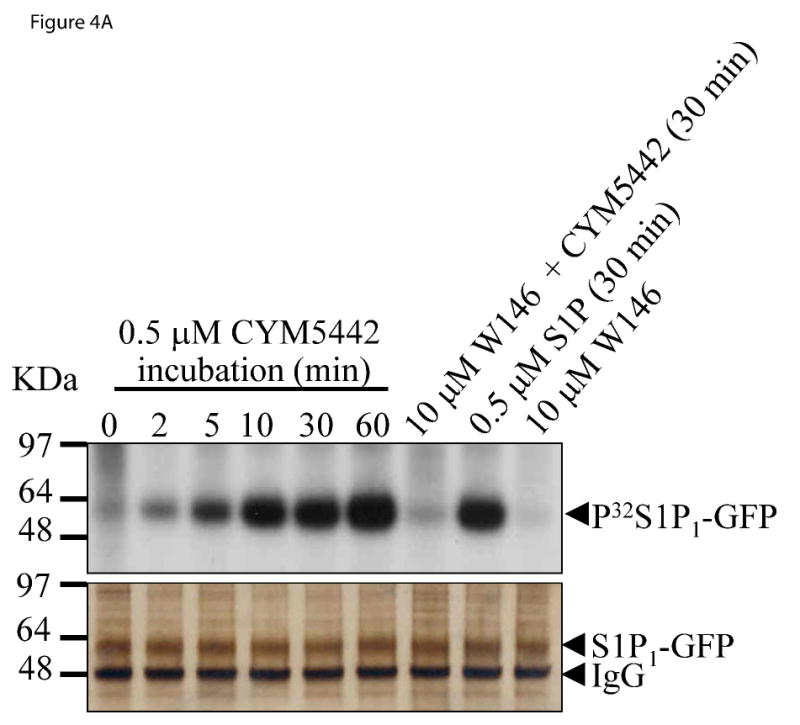
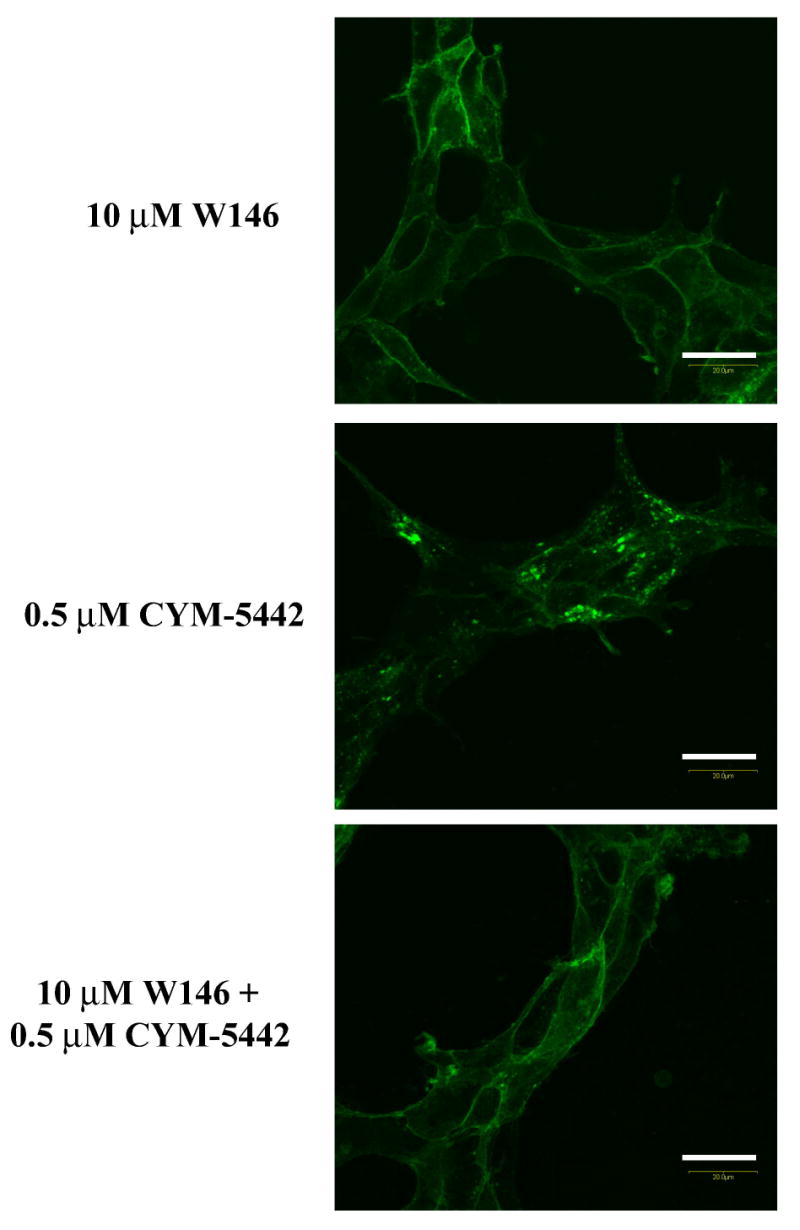
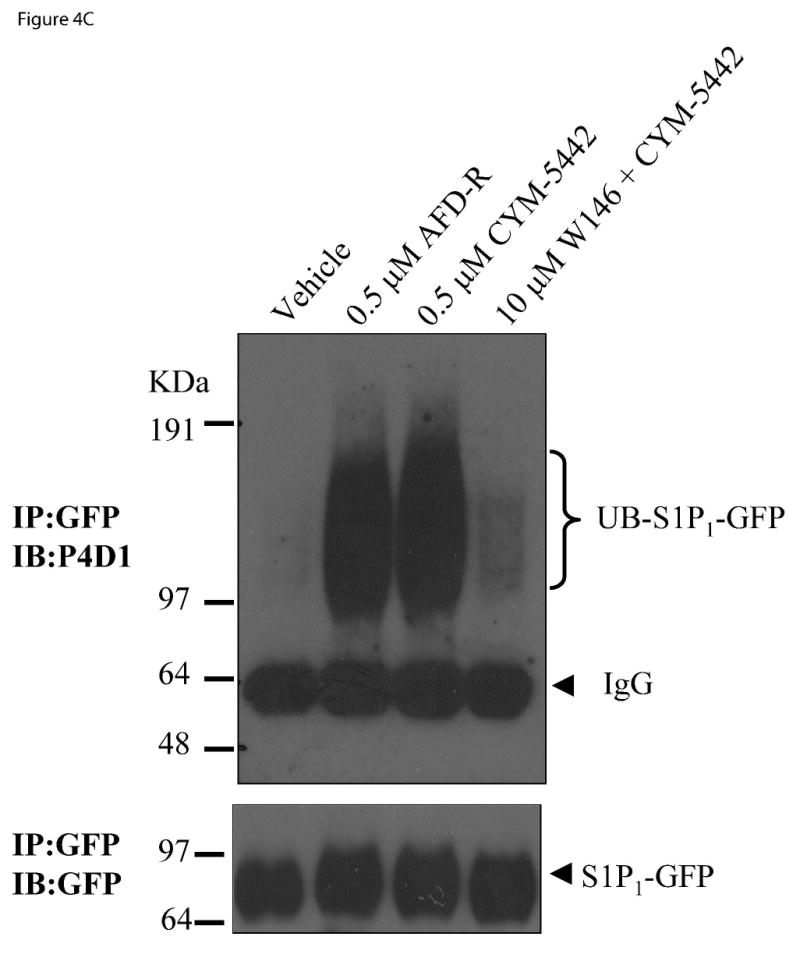
The ability of CYM-5442 to stimulate S1P1 phosphorylation, S1P1 internalization and S1P1 ubiquitylation was measured in HEK293 cells expressing human S1P1 tagged to C-terminal GFP. (A) Time-dependent induction of S1P1 phosphorylation by CYM-5442 is abolished by the S1P1 antagonist W146. Orthophosphate-P32 labeled cells were incubated with CYM-5442 for the indicated times, followed by S1P1 immunoprecipitation using GFP. Immunoprecipitates were then run by SDS-PAGE, and exposed to determine whole receptor phosphorylation status. The silver stained gel (bottom panel) confirmed equal loading of immunoprecipitated S1P1-GFP across conditions. The mass ladder is indicated in KDa. (B) CYM-5442 stimulates internalization of S1P1-GFP from the plasma membrane into cytoplasmic vesicles (at 45 min incubation, middle panel), while W146 preincubation (30 min, lower panel) blocks CYM-5442-mediated internalization. W146 alone had no effect on internalization (upper panel). Representative micrographs are shown (n=3); 20 μm scale bar; 40X. (C) CYM-5442 induces S1P1 ubiquitylation (UB-S1P1-GFP) by similar magnitude vs. the full, non-selective S1P receptor agonist, AFD-R (top immunoblot). Note that CYM-5442 ubiquitylation is abolished by W146 preincubation. For this experiment, AFD-R and CYM-5442 were incubated for 1 h and W146 was included 30 min prior to CYM-5442 stimulation. Equal loading of immunoprecipitated S1P1 receptor was confirmed by reblotting for GFP (bottom immunoblot). The mass ladder is indicated in KDa. This experiment was repeated twice with similar results.
Membrane-associated GPCRs are usually internalized into cytosolic vesicles upon agonist stimulation. We and others have shown that S1P and S1P agonist analogs internalize S1P1-GFP from the plasma membrane to cytoplasmic vesicles (Liu et al., 1999; Gonzalez-Cabrera et al., 2007; Oo et al., 2007). Figure 4B shows that incubation of S1P1-GFP cells with 500 nM CYM-5442 stimulated the internalization of S1P1-GFP receptor from a membrane-associated localization to an intracellular, multivesicular compartment. Similar internalization pattern was obtained by 0.5 μM S1P incubation (not shown). Internalization of S1P1-GFP by CYM-5442 was completely blocked by preincubation with 10 μM W146, and consistent with the phosphorylation data, W146 alone did not have an effect on internalization.
S1P1 agonism with some ligands, termed supraphysiological because of their ability to alterreceptor signaling reserve, results in degradation of S1P1-GFP in lysosomes and proteasomes, and the magnitude of agonist-dependent receptor ubiquitination has been reported to influence receptor fate (Gonzalez-Cabrera et al, 2007; Oo et al, 2007). Thus, agonist recruitment of ubiquitin to S1P1 can be used as a measure of ligand receptor activation. The ability of AFD-R, a highly efficient ubiquitinator (Gonzalez-Cabrera et al, 2007; Oo et al, 2007), and CYM-5442 to recruit ubiquitin onto immunoprecipitated S1P1-GFP is shown in Figure 4C. CYM-5442 stimulated receptor ubiquitination (UB-S1P1-GFP) to AFD-R levels. Ubiquitination by the agonists resulted in high mass-weight smears (approximately from 97 KDa to 180 KDa) of ubiquitin-receptor complexes running immediately above the immunoprecipitated receptor. Pre-incubation with the selective antagonist W146 was shown to nearly abolish CYM-5442-stimulated S1P1 ubiquitination. Overall, the in vitro data indicates that CYM-5442 is a potent and selective S1P1 agonist.
CYM-5442 does not require ionic S1P-receptor headgroup interactions for activating S1P1-mediated p42/p44 MAPK phosphorylation
S1P and synthetic S1P1 agonists have been shown to activate the mitogenic p42/p44-MAPK pathway across many cell types (Sorensen et al., 2003; Jo et al., 2005; Ahmad et al., 2006). The ability of CYM-5442 and S1P to activate p42/p44-MAPK was studied by ELISA, using lysates of CHO-K1 cells transiently expressing either the human wild-type S1P1 (WT) or the two separate single-point S1P1 mutants (R120A and E121A), known to abolish binding and activity of S1P on S1P1. Time-course studies demonstrated that maximal p42/p44-MAPK activity was achieved at 5 minutes in CHO-K1 cells expressing S1P1 (Supplemental Figure 1A). The time-course also showed that treatment with 500 nM S1P for 5 min did not lead to measurable p42/p44 MAPK activity in mock-transfected CHO-K1 cells. S1P and CYM-5442 stimulated P42/P44 MAPK phosphorylation was completely blocked by pre-incubation with 10 μM of the MEK1 inhibitor U0126 (Supplemental Figure 1B).
Using the 5 min incubation time, we sought to determine the requirement of S1P1 R120 and E121 residues for CYM-5442 and/or S1P activation of P42/P44 MAPK. For these experiments, concentration response curves with the agonists were performed on cells expressing each of the single point mutants (R120A and E121A), and these responses were compared to those in the WT receptor, both in the presence and absence of the selective S1P1 antagonist W146. Figure 5 shows the plots of the average concentration responses in the three transfectants, while the potency (EC50), maximal response (Emax) and goodness of fit (R2) value estimates derived from curve fitting are included in Table 2. In WT cells, both S1P and CYM-5442 led to the concentration dependent activation of P42/P44 MAPK phosphorylation, with EC50 vales of 2.3 nM and 46 nM, respectively. In WT transiently transfected CHO-K1 cells, S1P was a full agonist (Emax of 1.0 or 100%) while CYM-5442 was a partial agonist (Emax of 0.7 or 70% of S1P). Incubation of 10 μM W146 prior to addition of S1P resulted in a significant rightward shift (60-fold, EC50 of 140 nM) in potency of S1P vs. S1P alone (Figure 5). On the other hand, preincubation with W146 in WT cells led to the complete inhibition of CYM-5442-mediated p42/p44 MAPK phosphorylation (Figure 5). These results indicate that W146 is a competitive antagonist with S1P and a non-competitive antagonist against CYM-5442.
Figure 5. CYM-5442 does not require two essential S1P headgroup receptor interactions for activating p42/p44 MAPK.

Wild-type (WT) human S1P1 and two single-point S1P1 receptor mutants (R120A and E121A), reported to independently disrupt S1P-S1P1 binding and/or S1P-S1P1 function, were transiently transfected into CHO-K1 cells. Following 48 h, cells were stimulated for 5 min with increasing concentrations of S1P or CYM-5442, and agonist-dependent S1P1 activation of p42/p44 MAPK activity was determined using ELISA studies. Parallel agonist concentration responses were also performed with cells that had been preincubated with 10 μM of W146 for 30 min. For each of the three conditions, the fitted curves are the mean ± SD of three independent experiments. The potency (pEC50), intrinsic activity (Emax) and goodness of fit (R2) derived from the agonists' activation curves were determined for WT, R120A and E121A transfected cells, both in the absence (-) or presence (+) of the S1P1 antagonist W146. These values are shown in Table 1.
Table 2. CYM-5442 does not require essential S1P headgroup-receptor interactions for activating p42/p44 MAPK.
Mean potency (EC50), intrinsic activity (Emax) and goodness of fit (R2) values for the S1P and CYM-5442 response curves depicted in Figure 4. Minus (-) and plus (+) refer to the presence or absence of 10 μM W146 in the assay.
| WT | R120A | E121A | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S1P | 5442 | S1P | 5442 | S1P | 5442 | |||||||
| - | + | - | + | - | + | - | + | - | + | - | + | |
| LogEC50 | -8.6 | -6.8 | -7.3 | -5.7 | -7.2 | -7.7 | -7.2 | -6.8 | -6.8 | -7.2 | -6.9 | -5.9 |
| Emax (%) | 100 | 90 | 70 | 7 | 25 | 17 | 100 | 86 | 27 | 27 | 100 | 80 |
| R2 | 0.98 | 0.98 | 0.95 | NF | NF | NF | 0.98 | 0.99 | NF | NF | 0.99 | 0.79 |
NF: no fit
As expected, substitution of S1P1 R120 for alanine (R120A) resulted in a near loss of p42/p44 MAPK activity for S1P. Curve fitting the S1P data in this mutant indicated relatively poor fits (R2<0.4). Interestingly, the R120A mutant was still able to maintain p42/p44 MAPK activity when incubated with CYM-5442, and as a result, the data was plotted relative to CYM-5442 maximum. The EC50 for CYM-5442 in the R120A mutant was determined to be not-significantly different to that of WT CYM-5442 cells (R120A EC50, 67 nM; WT EC50, 46 nM). Unlike WT cells, preincubation of 10 μM W146 in mutant R120A cells did not significantly alter the p42/p44 MAPK activity by CYM-5442, and led to a modest (3-fold) rightward shift in EC50 (67 nM), suggesting that in the absence of headgroup localization, W146 is a weak competitive antagonist of the hydrophobic site.
Consistent with the notion that S1P makes a functional headgroup interaction with E121, S1P did not lead to significant p42/p44 MAPK activation in E121A S1P1 mutant cells. On the other hand, activation of p42/p44 MAPK by CYM-5442 in E121A S1P1 cells was concentration dependent, with a mean EC50 value of 134 nM. W146 preincubation led to a 10-fold rightward shift in potency of CYM-5442 for activating p42/p44 MAPK phosphorylation in E121A transfected cells. These results indicate that for S1P1 dependent p42/p44 MAPK activation, CYM-5442 does not require the R120 and E121 S1P1 residues that make up functional S1P-S1P1 headgroup interactions.
CYM-5442 pharmacokinetics
We next evaluated the pharmacokinetics of CYM-5442 in rats. Measures included are depicted in Table 3. Overall, CYM-5442 was modestly orally bioavailable (F=26%). Routes of delivery influenced the half-lives (t1/2) of 50 min. (i.v.) and 3 h (P.O.), which supported its use in vivo. Notably, in mice, CYM-5442 administration was highly central nervous system penetrant. An i.p. dose of 10 mg/kg for 2 h resulted in a 13.7 ± 2.9 μM concentrations in brain compared to 1.08 ± 0.3 μM in plasma. This brain to plasma ratio of approximately 13:1 suggests that CYM-5442 may be a useful tool for studying the roles of S1P1 in the central nervous system.
Table 3.
Pharmacokinetic parameters of CYM-5442 in rats.
| Parameters | CYM-5442
(2 mg/kg; POa) |
CYM-5442
(1 mg/kg; i.v.a) |
|---|---|---|
| t1/2 | 4.00 ± 1.45 | 3.05 ± 0.37 |
| MRT (h) | 6.35 ± 2.10 | 3.54 ± 0.38 |
| Cl (ml/min/kg) | 360.1 ± 73.3 | 72.90 ± 11.70 |
| Vss (L) | ND | 15.60 ± 3.50 |
| AUC (μM.h) | 0.23 ± 0.053 | 0.57 ± 0.10 |
| AUC (% extrap,ob) | 27.80 ± 14.20 | 11.35 ± 14.20 |
| Cmax (nM) | 36 ± 7.2 | 220.0 ± 18.0 |
| Tmax | 1.17 ± 0.76 | 0.083 ± 0.00 |
| F% | 20.50 ± 4.67 | ND |
Formulation (% v/v): 10/10/80 (DMSO/Tween-20/ water)
ND, not determined
CYM-5442 induces and maintains lymphopenia in mice through S1P1 activation
Administration of S1P1 agonists like SEW2871 or FTY720-P induces rapid and reversible lymphopenia in mice (Mandala et al., 2002, Sanna et al., 2004). To determine whether CYM-5442 could lead to the induction of lymphopenia, we treated mice i.p. with a dose of 10 mg/kg and compared whole blood white blood cell (WBC) count and the number of circulating B-cells and T-cells, against vehicle treated mice. A five hour treatment protocol was chosen based on the pharmacokinetic data (Table 3). WBC counts in CYM-5442 animals were decreased by 64 % vs. vehicle at 5 h (Table 4). FACS analyses of whole blood from treated animals indicated that CYM-5442 decreased B220+ B-cells by 63% compared to vehicle, whereas CD4+ and CD8+ T-cell counts were decreased by 83% and 84% of vehicle, respectively. This data indicates that a single dose of CYM-5442 induces acute lymphopenia in mice.
Table 4. Acute CYM-5442 administration induces lymphopenia in mice.
Mice (three per group) were injected i.p. with a dose of 10 mg/kg in a 300 μl volume. Vehicle control mice received sterile water injections. Five hours post treatment, animals were euthanized and blood was collected into tubes containing EDTA. Blood lymphopenia was assessed by comparing absolute white blood cell (WBC) number between CYM-5442-treated and vehicle-treated animals, as measured with an automated hemoanalyzer. In addition, FACS analyses were used to measure blood B220-positive cells (B220+, a marker for B-cells), and from the B220- population, the percent of single-positive CD4 (CD4+) and CD8 (CD8+) T-cells.
| Parameters | Vehicle | CYM-5442 |
|---|---|---|
| WBC (K/mm3) | 11.9±0.2 | 4.3±0.4* |
| % B220+ | 40.5±0.6 | 14.7±0.5* |
| % CD4+ | 11.5±0.2 | 1.9±0.2* |
| % CD8+ | 11.9±0.3 | 1.9±0.4* |
P<0.05 vs. vehicle treated
To ascertain whether the effects of CYM-5442 were dose-dependent, we employed a 5 h treatment and a dosing range of 0.3-10 mg/kg. The dose-response of CYM-5442 for inhibiting CD4+ and CD8+ cell populations is shown in Figure 6A, and a representative example of the FACS scatter plots in individual mice is shown in Figure 6B. The data indicates that CYM-5442 decreased CD4+ and CD8+ cell counts in a dose-dependent manner, attaining near-maximal effects on inhibition of these cell populations at serum CYM-5442 levels that ranged between 50 nM to 100 nM. Overall, the CYM-5442 mediated inhibition of B-cell and T-cell number had an estimated ED50 of 0.5, 2.0 and 1.0 mg/kg for CD4+, CD8+ and B220+, respectively (Figure 6C).
Figure 6. CYM-5442 induction of lymphopenia in mice is dose- and time-dependent.
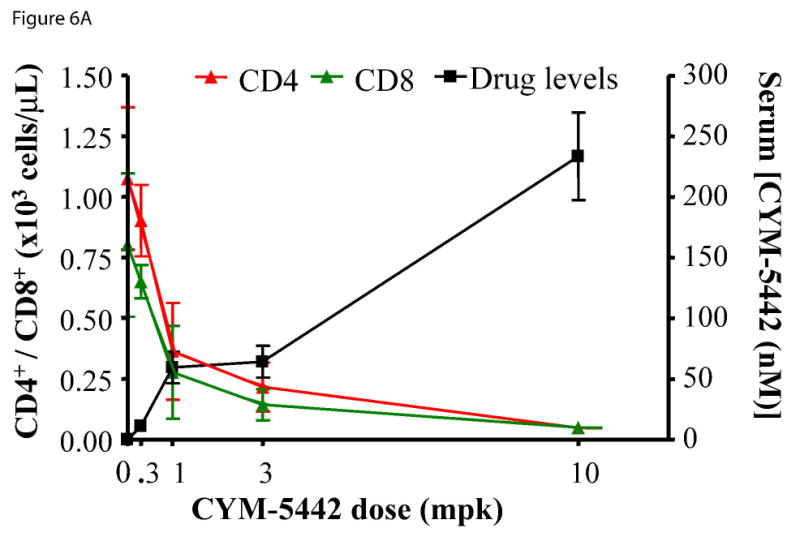
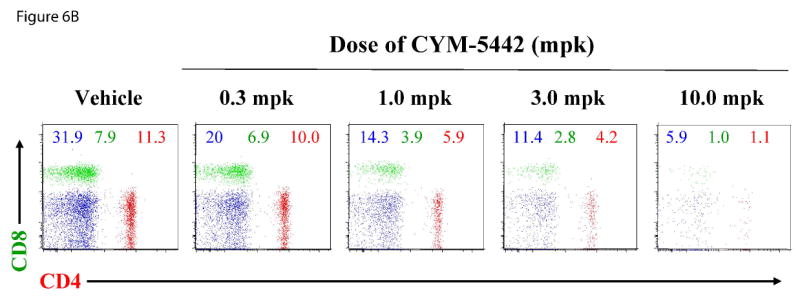
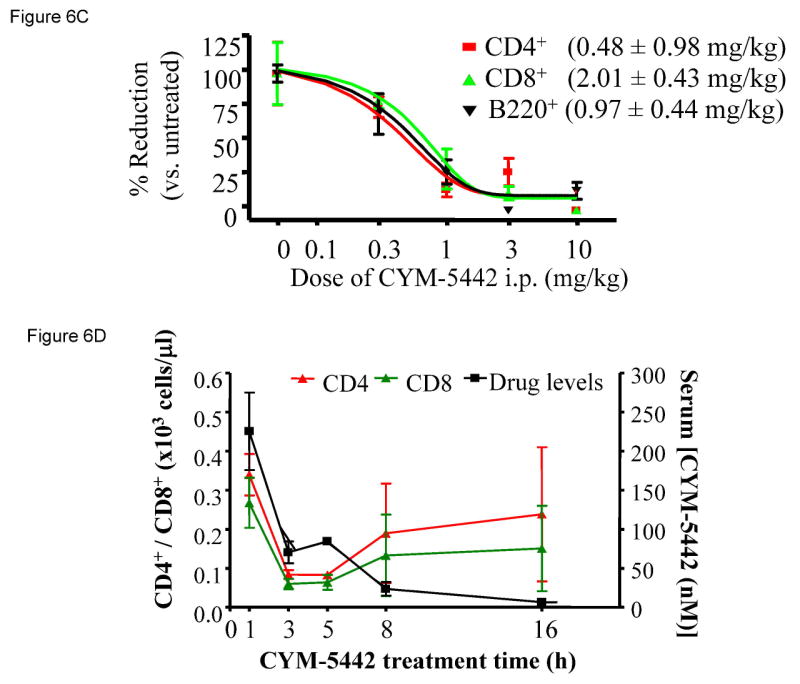
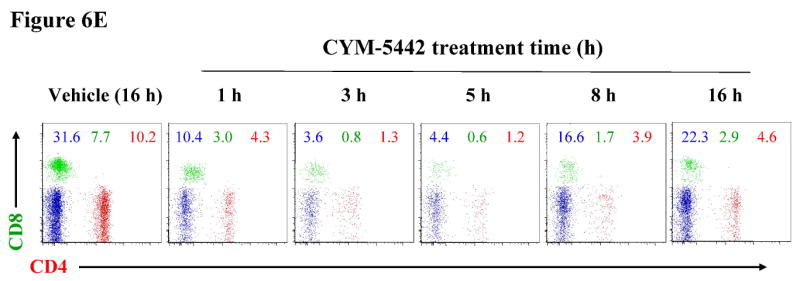
C57BL/6J male mice (three mice for group) were injected i.p. with either increasing doses of CYM-5442 or vehicle (water) to determine the dose dependence of CYM-5442 for induction of lymphopenia, or with a fixed dose (10 mg/kg) of CYM-5442 or vehicle for time-course studies. For these studies, blood was extracted from each animal and used to measure B220+, CD4+ and CD8+ cell counts, as well as CYM-5442 serum concentration. (A) The average inhibition of single-positive CD4 (CD4+, red) and CD8 (CD8+, green) T-cell number is shown at the indicated dose of CYM-5442. The mean serum CYM-5442 concentration reveals that a 50-100 nM free CYM-5442 concentration range is required for induction of lymphopenia at 5 h. (B) Representative plots of B220+ (blue), CD4+ (red) and CD8+ (green) percent decreases are shown at the indicated CYM-5442 dose. (C) The mean dose-response for CYM-5442 mediated inhibition of B220+, CD4+ and CD8+ cell counts was used to calculate the ED50s (D) Time-course of inhibition of CD4+ and CD8+ T-cell number following 10 mg/kg CYM-5442. The mean CYM-5442 serum concentration is also plotted. (E) Characteristic scatter plots of CYM-5442 percent inhibition of B-cell (B-220+, blue) and T-cell (CD4+, red and CD8+, green) numbers are shown following 1, 3, 5, 8 and 16 h post CYM-5442 administration.
Next, we measured the time-course for CYM-5442 mediated lymphopenia, and whether CYM-5442 effects could be reversed upon agonist clearance from plasma. These studies were performed employing a 16 h window, and both B-cell and T-cell counts were determined in conjunction with measures of serum CYM-5442 drug levels from the same animals (Figure 6D). A representative example of individual animal FACS scatter plots is shown in Figure 6E. With a fixed dose of 10 mg/kg, shown in Figure 5A to be maximal for CYM-5442 induction of B-cell and T-cell lymphopenia, the data indicates that CYM-5442 CD4+ and CD8+ counts dropped significantly during the first 3 h of administration, and appeared to be maintained at these low levels from 3 h to 5 h. After five hours, the B-cell and T-cell counts began to reverse towards basal levels; consistent with the time in which CYM-5442 began to disappear from plasma. Consistent with the dose-response data on Figure 5A, CYM-5442 induction and maintenance of lymphopenia in the time-course study occurred at serum levels of CYM-5442 ranging between 50-100 nM. The direct relationship between B-cell and T-cell recovery and loss of plasma serum CYM-5442 content thus suggested that CYM-5442 mediated lymphopenia can be reversed upon CYM-5442 clearance from the organism.
The minimal signal for lymphocyte sequestration and induction of lymphopenia by S1P1 agonists depends on activation of S1P1. To ascertain whether the in vivo effects of CYM-5442 were due to S1P1 receptor activation, we used the selective S1P1 antagonist, W146, shown here to block all of the measures of CYM-5442 activation in vitro. For antagonist studies, we used a 5 h window and a dose of CYM-5442 of 2 mg/kg, which is close to the ED50 for CYM-5442 induction of B-cell and T-cell lymphopenia (Figures 6A and 6C). A competing dose of W146 of 20 mg/kg was first administered to mice and allowed to equilibrate for 30 min prior to CYM-5442 administration. Table 5 shows that at near ED50, CYM-5442 led to the reduction of CD4+, CD8+ and B220+ cell counts by approximately 50% of vehicle, while W146 was able to partially recover CYM-5442 effects on B-220+, CD4+ and CD8+ cell counts. Importantly, neither administration of W146 alone nor of vehicle had measurable effects on cell counts (data not shown). This indicates that CYM-5442 mediated lymphopenia is dependent on S1P1 receptor activation.
Table 5. Lymphopenia by CYM-5442 administration is S1P1 dependent.
Mice (three per group) were first injected i.p. with a 20 mg/kg W146 in a 300 μl volume. W146 vehicle control mice received equal volume of 10 mM sodium carbonate: 2% β-cyclodextrin (vol/vol). Thirty minutes later, animals were injected with either 2 mg/kg CYM-5442 or vehicle (sterile water). Five hours post CYM-5442 treatment, animals were euthanized and blood was collected into tubes containing EDTA. The number of B220-positive cells (B220+, a marker for B-cells) and single-positive CD4 (CD4+) and CD8 (CD8+) T-cells were determined by FACS analysis and expressed as a percent change relative to values obtained in animals receiving both drug vehicles. A sample aliquot of the blood was used to measure the serum drug concentration levels by LC-MS/MS. Drug concentrations are included in parentheses in μM (± SD).
| W146 alone
(1.75±0.56) |
CYM-5442
(0.18±0.05) |
W146 + CYM-5442
(1.76±0.31)(0.19±0.04) |
|
|---|---|---|---|
| B220+ | 114.7±5.9 | 46.0±18.1* | 92.4±31.2 |
| CD4+ | 108.1 ±17.4 | 53.5 ± 6.5* | 89.1±18.7 |
| CD8+ | 112.2±19.6 | 47.4±7.5* | 92.2±15.1 |
p<0.05 relative to W146 alone
Discussion
Using a chemical approach, we have characterized a small molecule compound, CYM-5442, as a selective and efficacious S1P1 agonist for inducing and maintaining lymphopenia in vivo. It is of interest that the pharmacological properties of CYM-5442 for activating S1P1-dependent pathways are distinct from S1P, in that the ligand has no groups capable of mimicking the head-groups of S1P. The data strongly suggest that CYM-5442 interacts with S1P1 in a binding pocket separate from key S1P1 residues R120 and E121 essential for high affinity S1P binding and receptor activation (Parrill et al., 2000; Wang et al., 2001; Jo et al., 2005). Alternative residues of the putative hydrophobic pocket interacting with oxadiazole series of compounds, such as the original S1P1 agonist screening hit CYM-5181, have been recently published by Schurer et al., (2008). Because CYM-5442 has an EC50 within a factor of 3 of CYM-5181, the binding free energies are within experimental error of each other, and thus the docking of the ligand CYM-5442 into the receptor pocket is the same as the model shown for CYM-5181 (Figures 6, Schurer et al, (2008)) and distinct from the head-group constrained interactions of S1P (Supplementary Figure 5, Schurer et al (2008)). These differences between S1P and CYM-5442 find expression quantitatively by the retention of CYM-5442 activation of the E121A and R120A receptor mutants which fail to respond to S1P, as well as by the different mode of inhibition for CYM-5442 compared to S1P using the S1P1 antagonist W146. Mutagenesis of either R120 or E121 residues, present on the extracellular face of TM3, to alanine, has been shown to abolish S1P-S1P1 binding and subsequent receptor activation (Parrill et al., 2000; Wang et al., 2001; Jo et al., 2005), due to the inability of the charged phosphate and ammonium headgroups in S1P to make polar interactions with neutral alanine on the R120A and E121A mutants. While S1P stimulated MEK1 dependent p42/p44 MAPK phosphorylation in WT cells in a concentration-dependent manner, and with a potency consistent with literature values (Mandala et al., 2002; Sanna et al., 2004), neither of the mutant receptors was able to elicit significant signal when treated with S1P. On the other hand, the same single-point mutants, shown to be unresponsive to S1P, were still able to fully activate MEK1-dependent p42/p44 MAPK activity when treated with CYM-5442, with potency values found to be not significantly different between WT and R120A receptors, although the E121A receptors displayed slightly lower potency than WT. These data are consistent with different receptor binding requirements for the amphiphilic orthosteric agonist, S1P, and the hydrophobic selective agonist, CYM-5442.
W146 has proved to be a reliable chiral-selective S1P1 antagonist in vitro and in vivo (Yoon et al., 2008; Gonzalez-Cabrera et al., 2007; Sanna et al., 2006). As mentioned above, W146 antagonism of p42/p44 MAPK activity in these studies revealed two modes of action, competitive for S1P and non-competitive for CYM-5442. W146 antagonism of S1P1 signaling is therefore independent of whether S1P1 activation is occurring through the orthosteric site or an adjacent hydrophobic site. Evidence for the spatial proximity of the orthosteric pocket to the hydrophobic one is provided by the competitive antagonist shift of W146 in the R120A or E121A mutated receptors in response to activation by CYM-5442. Since W146 is an amino-phosphonate, capable of making RE headgroup interactions, these data suggest that W146 is no longer constrained in the single point mutants thus allowing more flexible binding within the hydrophobic pocket. Consistent with these data, non-competitive inhibition of CYM-5442 function was also observed with W146 in U2OS cells coupled to TANGO® S1P1-bla, while W146 in the same assay was a competitive inhibitor against S1P (not shown). Other S1P1 selective agonists with similar structures to CYM-5442 also display non-competitive W146 inhibition (not shown), suggesting that diaryl-oxadiazole agonists like CYM-5442 interact with a common hydrophobic pocket on S1P1 (Schurer et al., 2008).
The in vivo data with CYM-5442 demonstrates its usefulness for inducing and maintaining S1P1-dependent lymphopenia, with a minimal serum concentration for achieving maximal lymphopenia around 50 nM. Since systemic S1P1 antagonism with W146 reversed CYM-5442-induced lymphopenia, and W146 alone had no effect on B-cell and T-cell counts, the data indicates that S1P1 agonist signals are minimal and obligatory for lymphopenia induction. These data provide additional evidence against the functional antagonism hypothesis for S1P1-mediated immunomodulation (Matloubian et al., 2004; Schwab and Cyster, 2007).
All effects of CYM-5442 both in vitro and in vivo are reversed or abolished by the selective S1P1 receptor antagonist W146. These data restrict the pharmacological effects of CYM-5442 to this single receptor subtype. Though CYM-5442 is an enantiomeric mixture of 2 isomers, this mixture does not appear to contibute to the pharmacology or selectivity based upon our selective antagonist studies. Work is still ongoing to resolve the enantiomers. Differential crystallization using chiral salt forms such as L-tartaric acid have thus far been unsuccessful. Separation using chiral columns by HPLC is currently ongoing with no positive results yet and an enantio-specific synthesis may be formally required.
CYM-5442 possesses additional properties that make it a useful S1P1 chemical agonist. CYM-5442 was found to be 10,000-fold selective for S1P1 over S1P5. Besides selectivity, CYM-5442 is approximately 10-50-fold more potent than SEW2871 in vitro, depending on which agonist format was used, and induces lymphopenia at approximately 5 to 10-fold lower doses than SEW2871. Favorable pharmacokinetics and moderate water solubility would make CYM-5442 a useful tool for studying S1P1 function in tissues where drug formulation or penetrance may present challenges (i.e., lung and central nervous system). The levels of CYM-5442 in brain tissue, with a brain-plasma ratio of 13:1 following a bolus dose in mice, are notable. The beneficial effects of FTY720 in ameliorating MS symptoms have been associated, in part, with the inhibition of BBB leakiness occurring during disease progression. CNS-penetrant and efficacious non-prodrug S1P1 agonists such as CYM-5442, that are also antagonist reversible in the periphery, may be good proof-of-concept chemical tools for investigating the contributions of S1P1 to MS therapy. These allow the contributions of BBB integrity, lymphocyte sequestration and glial and neuronal S1P1 function to be assessed. Certainly, selective S1P1 agonists and antagonists have shown to inversely modulate the integrity of endothelial barriers in vivo (Sanna et al., 2006; Foss et al., 2007). We have data indicating that endothelial integrity is tonically regulated by the S1P-S1P1 axis (Sanna et al., 2006). This servo mechanism results in agonist enhancement of barrier function by inhibiting leakage induced by pro-angiogenic stimuli, including VEGF and thrombin. Antagonism alone leads to barrier opening effects. These actions have been well documented in lung and skin vasculature, and having a matched agonist-antagonist pair like CYM-5442 and W146 would be optimal for further studying the hypothetical endothelial S1P-S1P1 rheostat.
Highly selective and potent chemical tools in general have characteristically slow off-rates, and have been of great interest in recent crystallographic studies (Cherezov et al., 2007; Rosenbaum et al., 2007) to define both orthosteric and spatially distinct hydrophobic binding pockets within this family of Class A GPCRs, and the minimalist changes in receptor structure necessary for full pharmacological efficacy. Such chemical tools, when deeply characterized and broadly available to the field (Rosen et al., 2008), can impact on the progress of mechanistic understanding in physiology, pathology and structural biology and ultimately on therapeutics.
Supplementary Material
(A) Time-course of S1P and CYM-5442 for stimulating p42/p44 MAPK activation in CHO-K1 cells transiently transfected with WT human S1P1. Plotted on the same graph is the effect of S1P incubation in mock-transfected CHO-K1 cells. (B) S1P and CYM-5442-mediated p42/p44 MAPK activation is blocked by the MEK1 inhibitor U0126 (1 μM, 30 min).
Acknowledgments
The authors are grateful to Bill Webb (Ctr. for Mass Spectrometry, TSRI) for measuring serum drug levels and Aaron Semana for expert technical assistance.
Abbreviations
- CYM-5442
2-(4-(5-(3,4-diethoxyphenyl)-1,2,4-oxadiazol-3-yl)-2,3-dihydro-1H-inden-1-yl amino) ethanol
- SEW2871
5-[4-phenyl-5-(trifluoromethyl)-2-thienyl]-3-[3-(trifluoromethyl) phenyl]-1, 2, 4-oxadiazole
- W146
(R)-3-Amino-(3-hexylphenylamino)-4-oxobutylphosphonic acid
- GTPγS
guanosine 5′-O-[gamma-thio]triphosphate
- EDTA
ethylenediaminetetraacetic acid
- MAPK
Mitogen activated protein kinase
- FTY720
2-amino-2-(4-octylphenethyl)propane-1,3-diol
Footnotes
Supported by National Institutes of Health grants AI-055509, MH-074404 and AI-074564).
References
- Ahmad M, Long JS, Pyne NJ, Pyne S. The effect of hypoxia on lipid phosphate receptor and sphingosine kinase expression and mitogen-activated protein kinase signaling in human pulmonary smooth muscle cells. Prostaglandins Other Lipid Mediat. 2006;79:278–286. doi: 10.1016/j.prostaglandins.2006.03.001. [DOI] [PubMed] [Google Scholar]
- Alfonso C, McHeyzer-Williams MG, Rosen H. CD69 down-modulation and inhibition of thymic egress by short and long-term selective chemical agonism of S1P1 receptors. Eur J Immunol. 2006;36:149–159. doi: 10.1002/eji.200535127. [DOI] [PubMed] [Google Scholar]
- Balatoni B, Storch MK, Swoboda EM, Schönborn V, Koziel A, Lambrou GN, Hiestand PC, Weissert R, Foster CA. FTY720 sustains and restores neuronal function in the DA rat model of MOG-induced experimental autoimmune encephalomyelitis. Brain Res Bull. 2007;74:307–316. doi: 10.1016/j.brainresbull.2007.06.023. [DOI] [PubMed] [Google Scholar]
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek SM, Jacobson JR, Chiang ET, Birukov KG, Wang P, Zhan X, Garcia JG. Pulmonary endothelial cell barrier enhancement by sphingosine 1-phosphate: roles for cortactin and myosin light chain kinase. J Biol Chem. 2004;279:24692–24700. doi: 10.1074/jbc.M313969200. [DOI] [PubMed] [Google Scholar]
- Foss FW, Jr, Snyder AH, Davis MD, Rouse M, Okusa MD, Lynch KR, Macdonald TL. Synthesis and biological evaluation of gamma-aminophosphonates as potent, subtype-selective sphingosine 1-phosphate receptor agonists and antagonists. Bioorg Med Chem. 2007;15:663–677. doi: 10.1016/j.bmc.2006.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster CA, Mechtcheriakova D, Storch MK, Balatoni B, Howard LM, Bornancin F, Wlachos A, Sobanov J, Kinnunen A, Baumruker T. FTY720 Rescue Therapy in the Dark Agouti Rat Model of Experimental Autoimmune Encephalomyelitis: Expression of Central Nervous System Genes and Reversal of Blood-Brain-Barrier Damage. Brain Pathology. 2008 doi: 10.1111/j.1750-3639.2008.00182.x. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara Y, Osborne DA, Walker MD, Wang DA, Bautista DA, Liliom K, Van Brocklyn JR, Parrill AL, Tigyi G. Identification of the hydrophobic ligand binding pocket of the S1P1 receptor. J Biol Chem. 2007;282:2374–2385. doi: 10.1074/jbc.M609648200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Cabrera PJ, Hla T, Rosen H. Mapping Pathways Downstream of Sphingosine 1-Phosphate Subtype 1 by Differential Chemical Perturbation and Proteomics. J Biol Chem. 2007;282:7254–7264. doi: 10.1074/jbc.M610581200. [DOI] [PubMed] [Google Scholar]
- Hiestand PC, Rausch M, Meier DP, Foster CA. Ascomycete derivative to MS therapeutic: S1P receptor modulator FTY720. Prog Drug Res. 2008;66:363–81. doi: 10.1007/978-3-7643-8595-8_8. [DOI] [PubMed] [Google Scholar]
- Jo E, Sanna MG, Gonzalez-Cabrera PJ, Thangada S, Tigyi G, Osborne DA, Hla T, Parrill AL, Rosen H. S1P1-Selective In Vivo-Active Agonists from High-Throughput Screening: Off-the-Shelf Chemical Probes of Receptor Interactions, Signaling, and Fate. Chem Biol. 2005;12:703–715. doi: 10.1016/j.chembiol.2005.04.019. [DOI] [PubMed] [Google Scholar]
- Kataoka H, Sugahara K, Shimano K, Teshima K, Koyama M, Fukunari A, Chiba K. FTY720, sphingosine 1-phosphate receptor modulator, ameliorates experimental autoimmune encephalomyelitis by inhibition of T cell infiltration. Cell Mol Immunol. 2005;2:439–448. [PubMed] [Google Scholar]
- Lee MJ, Thangada S, Claffey KP, Ancellin N, Liu CH, Kluk M, Volpi M, Sha'afi RI, Hla T. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1 -phosphate. Cell. 1999;99:301–312. doi: 10.1016/s0092-8674(00)81661-x. [DOI] [PubMed] [Google Scholar]
- Liu CH, Thangada S, Lee MJ, Van Brocklyn JR, Spiegel S, Hla T. Ligand-induced trafficking of the sphingosine-1-phosphate receptor EDG-1. Mol Biol Cell. 1999;10:1179–1190. doi: 10.1091/mbc.10.4.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Chen W, Hale J, Lynch CL, Mills SG, Hajdu R, Keohane CA, Rosenbach MJ, Milligan JA, Shei G, Chrebet G, Parent SA, Bergstrom J, Card D, Forrest M, Quackenbush EJ, Wickham LA, Vargas H, Evans RM, Rosen H, Mandala S. Discovery of potent 3,5-diphenyl-1,2,4-oxadiazole sphingosine-1-phosphate-1 (S1P1) receptor agonists with exceptional selectivity against S1P2 and S1P3. J Med Chem. 2005;48:6169–6173. doi: 10.1021/jm0503244. [DOI] [PubMed] [Google Scholar]
- Mandala S, Hajdu R, Bergstrom J, Quackenbush E, Xie J, Milligan J, Thornton R, Shei GJ, Card D, Keohane C, Rosenbach M, Hale J, Lynch CL, Rupprecht K, Parsons W, Rosen H. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science. 2002;296:346–349. doi: 10.1126/science.1070238. [DOI] [PubMed] [Google Scholar]
- Miron VE, Schubart A, Antel JP. Central nervous system-directed effects of FTY720 (fingolimod) J Neurol Sci. 2008 doi: 10.1016/j.jns.2008.06.031. In press. [DOI] [PubMed] [Google Scholar]
- Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, Allende ML, Proia RL, Cyster JG. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427:355–360. doi: 10.1038/nature02284. [DOI] [PubMed] [Google Scholar]
- Oo ML, Thangada S, Wu MT, Liu CH, Macdonald TL, Lynch KR, Lin CY, Hla T. Immunosuppressive and anti-angiogenic sphingosine 1-phosphate receptor-1 agonists induce ubiquitinylation and proteasomal degradation of the receptor. J Biol Chem. 2007;282:9082–9089. doi: 10.1074/jbc.M610318200. [DOI] [PubMed] [Google Scholar]
- Parrill AL, Wang D, Bautista DL, Van Brocklyn JR, Lorincz Z, Fischer DJ, Baker DL, Liliom K, Spiegel S, Tigyi G. Identification of Edg1 receptor residues that recognize sphingosine 1-phosphate. J Biol Chem. 2000;275:39379–39384. doi: 10.1074/jbc.M007680200. [DOI] [PubMed] [Google Scholar]
- Rosen H, Alfonso C, Surh CD, McHeyzer-Williams MG. Rapid induction of medullary thymocyte phenotypic maturation and egress inhibition by nanomolar sphingosine 1-phosphate receptor agonist. Proc Natl Acad Sci. 2003;100:10907–10912. doi: 10.1073/pnas.1832725100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen H, Goetzl EJ. Sphingosine 1-phosphate and its receptors: An autocrine and paracrine network. Nat Rev Immunol. 2005;5:560–570. doi: 10.1038/nri1650. [DOI] [PubMed] [Google Scholar]
- Rosen H, Sanna MG, Cahalan SM, Gonzalez-Cabrera PJ. Tipping the gatekeeper: S1P regulation of endothelial barrier function. Trends Immunol. 2007;28:102–107. doi: 10.1016/j.it.2007.01.007. [DOI] [PubMed] [Google Scholar]
- Rosen H, Gonzalez-Cabrera PJ, Marsolais D, Cahalan SM, Don A, Sanna MG. Modulating tone: the overture of S1P receptor immunotherapeutics. Immunol Rev. 2008;223:221–35. doi: 10.1111/j.1600-065X.2008.00645.x. [DOI] [PubMed] [Google Scholar]
- Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, Kobilka BK. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- Sanna MG, Liao J, Jo E, Alfonso C, Ahn MY, Peterson MS, Webb B, Lefebvre S, Chun J, Gray N, Rosen H. Sphingosine 1-phosphate (S1P) Receptor Subtypes S1P1 and S1P3, Respectively, Regulate Lymphocyte Recirculation and Heart Rate. JBiol Chem. 2004;279:13839–13848. doi: 10.1074/jbc.M311743200. [DOI] [PubMed] [Google Scholar]
- Sanna MG, Wang SK, Gonzalez-Cabrera PJ, Don A, Marsolais D, Matheu MP, Wei SH, Parker I, Jo E, Cheng WC, Cahalan MD, Wong CH, Rosen H. Enhancement of capillary leakage and restoration of lymphocyte egress by a chiral S1P1 antagonist in vivo. Nat Chem Biol. 2006;2:434–441. doi: 10.1038/nchembio804. [DOI] [PubMed] [Google Scholar]
- Sanchez T, Estrada-Hernandez T, Paik JH, Wu MT, Venkataraman K, Brinkmann V, Claffey K, Hla T. Phosphorylation and action of the immunomodulator FTY720 inhibits vascular endothelial cell growth factor-induced vascular permeability. J Biol Chem. 2003;278:47281–47290. doi: 10.1074/jbc.M306896200. [DOI] [PubMed] [Google Scholar]
- Schurer SC, Brown SJ, Gonzalez-Cabrera PJ, Schaeffer MT, Chapman J, Jo E, Chase P, Spicer T, Hodder P, Rosen H. ACS Chem Biol. Vol. 3. 2008. Ligand-Binding Pocket Shape Differences between Sphingosine 1-Phosphate (S1P) Receptors S1P1 and S1P3 Determine Efficiency of Chemical Probe Identification by Ultrahigh-Throughput Screening; pp. 486–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab SR, Cyster JG. Finding a way out: lymphocyte egress from lymphoid organs. Nat Immunol. 2007;8:1295–1301. doi: 10.1038/ni1545. [DOI] [PubMed] [Google Scholar]
- Sorensen SD, Nicole O, Peavy RD, Montoya LM, Lee CJ, Murphy TJ, Traynelis SF, Hepler JR. Common signaling pathways link activation of murine PAR-1, LPA, and S1P receptors to proliferation of astrocytes. Mol Pharmacol. 2003;64:1199–209. doi: 10.1124/mol.64.5.1199. [DOI] [PubMed] [Google Scholar]
- Wang DA, Lorincz Z, Bautista DL, Liliom K, Tigyi G, Parrill AL. A single amino acid determines lysophospholipid specificity of the S1P1 (EDG1) and LPA1 (EDG2) phospholipid growth factor receptors. J Biol Chem. 2001;276:49213–49220. doi: 10.1074/jbc.M107301200. [DOI] [PubMed] [Google Scholar]
- Wei SH, Rosen H, Matheu MP, Sanna MG, Wang SK, Jo E, Wong CH, Parker I, Cahalan MD. Sphingosine 1-phosphate type 1 receptor agonism inhibits transendothelial migration of medullary T cells to lymphatic sinuses. Nat Immunol. 2005;6:1228–1235. doi: 10.1038/ni1269. [DOI] [PubMed] [Google Scholar]
- Yan L, Huo P, Doherty G, Toth L, Hale JJ, Mills SG, Hajdu R, Keohane C, Rosenbach MJ, Milligan MA, Shei G, Chrebet G, Bergstrom J, Card D, Quackenbush E, Wickham A, Mandala SM. Discovery of 3-arylpropionic acids as potent agonists of sphingosine-1-phosphate receptor-1 (S1P1) with high selectivity against all other known S1P receptor subtypes. Bioorg Med Chem Lett. 2006;16:3679–3683. doi: 10.1016/j.bmcl.2006.04.084. [DOI] [PubMed] [Google Scholar]
- Yoon CM, Hong BS, Moon HG, Lim S, Suh PG, Kim YK, Chae CB, Gho YS. Sphingosine-1-phosphate promotes lymphangiogenesis by stimulating S1P1/Gi/PLC/Ca2+ signaling pathways. Blood. 2008 doi: 10.1182/blood-2007-11-125203. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Time-course of S1P and CYM-5442 for stimulating p42/p44 MAPK activation in CHO-K1 cells transiently transfected with WT human S1P1. Plotted on the same graph is the effect of S1P incubation in mock-transfected CHO-K1 cells. (B) S1P and CYM-5442-mediated p42/p44 MAPK activation is blocked by the MEK1 inhibitor U0126 (1 μM, 30 min).