

Abstract
A genetic mouse model (designated cardiomyocyte-Cpr-null) with cardiomyocyte-specific deletion of the cytochrome P450 reductase (Cpr) gene was generated in this study. CPR protein levels, as well the enzyme activity of P450s, were greatly reduced in heart microsomes from the null mice, compared to wild-type mice, whereas CPR expression in other organs remained unchanged. Nonetheless, homozygous null mice were normal in appearance, gross anatomy, tissue morphology, and general cardiac functional parameters, and there was no indication of embryonic lethality or premature mortality, in contrast to the recognized role of CPR in embryonic development. Thus, this new mouse model should be useful for determination of the in vivo roles of cardiomyocyte CPR and CPR-dependent enzymes, including microsomal P450s, not only in the metabolism and toxicity of numerous xenobiotic compounds, but also in cardiac pathophysiology. As a first application, we studied the role of cardiomyocyte CPR and CPR-dependent enzymes in doxorubicin (Dox)-mediated acute cardiotoxicity. Wild-type and null mice were treated with a single intraperitoneal dose of Dox at 5, 10, or 20 mg/kg. The Dox treatment caused apoptosis and vacuolization in cardiomyocytes at the dose of 20 mg/kg, and a significant increase in the levels of serum creatine kinase at 10 and 20 mg/kg in both wild-type and null mice. However, there was no significant difference in the extent of Dox-induced cardiac injury, between the two strains; this lack of difference suggests that cardiomyocyte CPR and CPR-dependent enzymes do not play critical roles in the acute cardiotoxicity induced by Dox.
Cytochrome P450 (P450) enzymes metabolize numerous exogenous and endogenous compounds. Multiple P450 enzymes are expressed in the heart, where they may participate in the metabolism of therapeutic agents and environmental toxicants (Wang et al., 2002; Williams et al., 2003), as well as endogenous arachidonic acid (AA) (Wu et al., 1996). Cardiac P450 enzymes are believed to play critical roles in cardiac ischemia-reperfusion injury, through either increases in the production of free radicals, or else alterations in the metabolism of AA (Granville et al., 2004; Nithipatikom et al., 2004; Seubert et al., 2004). However, the proposed in vivo functions of endogenous cardiac P450s, either in xenobiotic metabolism and/or toxicity, or else in cardiac pathophysiology, remain largely unconfirmed.
The activity of microsomal P450 enzymes is dependent on the participation of NADPH-cytochrome P450 reductase (CPR), which provides electrons needed for P450-catalyzed monooxygenase reactions, as well as for the reactions catalyzed by several other enzymes, including heme oxygenases (HOs), cytochrome b5, squalene monooxygenase, and fatty acid elongases (cf. Gu et al., 2003). Mice with germline inactivation of the Cpr gene die at the embryonic stage, as a result of abnormal development of the brain, eye, heart, and limbs(Shen et al., 2002). Recently, transgenic mouse strains in which the Cpr gene was flanked by two loxP sites (designated Cprlox mouse) were generated (Wu et al., 2003); this genetic engineering permits tissue-specific Cpr knockout in postnatal mice.
To facilitate efforts to determine the biological functions of cardiac P450 enzymes in adults, and the roles of cardiac CPR and CPR-dependent enzymes in drug-mediated cardiotoxicity, we have now generated a cardiomyocyte-specific Cpr–null mouse model, designated the cardiomyocyte-Cpr-null mouse. This new mouse model was generated by cross-breeding the Cprlox mouse with a transgenic mouse (named αMHC-Cre) that affords cardiomyocyte-specific expression of the Cre recombinase (Cre) (Abel et al., 1999). In the αMHC-Cre mouse, the expression of Cre was driven by the promoter of the a-myosin heavy chain gene. The new cardiomyocyte-Cpr-null mouse was characterized for potential abnormality in appearance, gross anatomy, heart morphology, and heart weight; potential occurrence of embryonic lethality or premature mortality; potential changes in general cardiac function; and potential compensatory expression of microsomal P450s and HOs. As the first application of this new mouse model, we further determined whether the null and wild-type mice differ in their sensitivities to toxic doses of doxorubicin (Dox), a widely used anti-cancer drug; our purpose was to assess whether target-tissue CPR/P450-mediated Dox metabolism plays an important role in Dox-induced acute cardiotoxicity.
The clinical use of Dox is often limited by the dose- and time-dependent Dox-mediated acute and chronic cardiotoxicity (Singal and Iliskovic, 1998; Menna et al., 2008). Although the mechanisms of Dox-mediated cardiotoxicity are not fully understood, oxidative stress seems to play an important role, as indicated by the occurrence of cardiac lipid peroxidation (Myers et al., 1977), glutathione depletion (Doroshow et al., 1981), and decreases in glutathione peroxidase expression and activity (Li and Singal, 2000) in Dox-treated animals. Dox-induced oxidative stress and cardiotoxicity can be alleviated by pretreatment of animals with antioxidants, such as N-acetylcysteine and probucol (Doroshow et al., 1981; Li and Singal, 2000). Dox can undergo one-electron reduction by one of several enzymes, including the CPR; the reaction produces a semiquinone free radical, which can either directly attack cellular macromolecules or else indirectly (through reactions with oxygen and redox cycling) yield superoxide anion, hydrogen peroxide, and hydroxyl radicals, with consequent cellular damage to the cardiac tissue (Xu et al., 2005). Biotransformation of Dox in humans involves primarily the formation of doxorubicinol, in a reaction catalyzed by carbonyl reductase (CBR1), although Dox can also be metabolized by CPR, through reductive deglycosylation to a nontoxic metabolite (Niitsu et al., 2000; Riddick et al., 2005). The critical role of CBR1 in Dox-induced cardiotoxicity has been demonstrated with transgenic and knockout mouse models(Forrest et al., 2000; Olson et al., 2003); nevertheless, the in vivo function of CPR and P450 in Dox-mediated cardiotoxicity remained to be demonstrated. Our findings establish that cardiomyocyte CPR and CPR-dependent enzymes do not play critical roles in the acute cardiotoxicity induced by Dox.
Materials and Methods
Generation of the cardiomyocyte-Cpr-null mice
Heterozygous αMHC-Cre mice on the FVB genetic background (Abel et al., 1999) were first crossed with Cprlox/lox mice, on a mixed C57BL/6 and 129/Sv background (Wu et al., 2003); the resultant αMHC-Cre+/−/Cprlox/lox progeny were crossed with Cprlox/lox mice again, yielding pups with one of four possible genotypes: αMHC-Cre+/−/Cprlox/lox (null) and αMHC-Cre−/−/Cprlox/lox (wild-type littermate), as well as αMHC-Cre+/−/Cprlox/− and αMHC-Cre−/−/Cprlox/− (neither of the latter two was used for this study). Cre and Cpr genotypes were determined by PCR analysis of tail DNA, as previously described (Wu et al., 2003). All animal studies were approved by the Institutional Animal Care and Use Committee of the Wadsworth Center.
Immunoblot assays
Protein samples were fractionated on 10% polyacrylamide gels and transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA). For immunodetection, the following rabbit or goat antibodies were used with the indicated dilution: anti-CPR (1:1000; Stressgen, San Diego, CA), anti-HO-1 (1:500; Stressgen), anti-HO-2 (1:5000; Stressgen), anti-CYP1A1/2 (1:750; BD Gentest, Woborn, MA), anti-CYP2B1/2 (1:1000; BD Gentest), anti-human CYP2C9 (1:1000; BD Gentest), anti-human CYP2E1 (1:1000; Oxford Biomedical Research, Oxford, MI), anti-CYP3A2 (1:1000; BIOMOL International, Plymouth Meeting, PA), and anti-CYP4A (1:1000; BD Gentest). All antigens were derived from the rat, unless indicated otherwise. The blots were analyzed with use of an ECL kit (Amersham Pharmacia Biotech), and the signal intensity was quantified with a densitometer.
Tissue preparation
Mice were sacrificed by CO2 asphyxiation. For histological analysis, hearts were dissected, fixed in 10% formalin for 24 h, embedded in paraffin, and cut into 5-μm sections at the coronal plane. Sections were stained with hematoxylin and eosin (H&E). For immunohistochemical analysis, mice were first perfused with 20 ml of phosphate buffered saline (PBS) (pH 7.4), followed by 50 ml of freshly prepared, ice-cold 4% paraformaldehyde (PFA) in PBS. The hearts were dissected, and post-fixed in 4% PFA for 2 h. Cryoprotection was conducted by placing fixed hearts in 30% sucrose in PBS for 24 h. The hearts were then flash-frozen in TissueTek OCT (Sakura Finetek, Torrance, CA) and sectioned at 10 μm with a cryostat (Sakura Finetek). Frozen sections were mounted on Superfrost Plus microscope slides (Fisher Scientific, Springfield, NJ) and stored at −80°C until use.
Immunohistochemical analysis
Frozen sections were brought to room temperature for 30 min and rehydrated using PBS, followed by incubation with Triton X-100 (0.3% in PBS) for 30 min, in order to increase the permeability of the tissue sections. Nonspecific sites on the sections were blocked by two sequential incubation steps, first with Image-iT FX signal enhancer (Molecular Probes, Eugene, OR) for 30 min, and then with a blocking solution (3% BSA/0.1% Triton X-100 in PBS) for 1 h. Sections were incubated with rabbit anti-CPR antibody (Stressgen, diluted at 1:100 in the blocking solution), overnight at room temperature; then, after washing with PBS for 30 min, they were incubated with Alexa-594-conjugated chicken anti-rabbit antibody (1:200 dilution; Molecular Probes) for 1 h at room temperature. Sections were mounted with Prolong Gold anti-fade reagent with DAPI (Molecular Probes). The primary antibody was omitted for negative control slides. Sections were observed using a Nikon Eclipse i50 fluorescence microscope (Nikon, Tokyo, Japan).
Isolation of cardiomyocyte
Ventricular cardiomyocytes were isolated according to a previously reported method (Cheng et al., 2004). Briefly, adult mice were injected with heparin (100 units/mouse) 30 min before surgical operation. Mice were anesthetized with a mixture of ketamine (10 mg) and xylazine (0.7 mg). The heart was quickly removed, placed in ice-cold perfusion buffer (Cheng et al., 2004), and then cannulated to a 22-gauge needle with a blunt tip. Perfusion was started immediately, at a constant flow rate (3 ml/min), with pre-warmed perfusion buffer (37°C). After perfusion for 3–4 min, the perfusion buffer was replaced with pre-warmed (37°C) digestion buffer, which consisted of the perfusion buffer containing 0.25 mg/ml Liberase blendzyme I (Roche, Indianapolis, IN), 0.14 mg/ml trypsin (Invitrogen, Carlsbad, CA), and 12.5 μM CaCl2. After 10–12 min, the perfusion with digestion buffer was stopped. The heart was cut into pieces of ~1 mm3 in size, in a 60-mm cell culture dish. Tissue pieces were pipetted 5–10 times with a wide-opening plastic pipette, and they were then quickly transferred to a 15-ml tube containing 5 ml of a stopping buffer (perfusion buffer containing 10% calf serum and 12.5 μM CaCl2). Cells in the tissue pieces were dissociated by gentle pipetting; the resulting cell suspension was filtered through a metal sieve (300-μm mess), and the cells were allowed to settle by gravity for 10–15 min. The supernatant was decanted, whereas the cell pellet was collected and stored at −80°C until use.
Measurement of microsomal P450 activity
P450-catalyzed dealkylase activity was measured for cardiac microsomes with a sensitive fluorescence-based method (Ekins et al., 1998). The substrate, 3-cyano-7-ethoxycoumarin (non-fluorescent), can be dealkylated by several P450 enzymes, including CYP1A1, 1A2, and 2B1, to form 3-cyano-7-hydroxycoumarin, a fluorescent metabolite (White, 1988). Cardiac microsomes were prepared from pooled ventricles (from 5–7 adult mice for each group). The reaction was initiated by the addition of 2.0 mg of freshly prepared microsomal protein to a mixture containing 400 μl of an NADPH-regeneration system (Zhuo et al., 2004), 2 μl of 3-cyano-7-ethoxycoumarin (Molecular Probes, Eugene, OR; dissolved in dimethyl sulfoxide) for a final concentration of 9.3 μM, and water, to give a final reaction volume of 1.0 ml. After 45 min at 37°C, with constant shaking, the reaction was stopped by the addition of 2.0 ml of acetonitrile. The mixture was centrifuged at 12,000 g for 5 min. The supernatant was filtered through a 13-mm syringe filter (0.45 μm in diameter). Product fluorescence was measured using a Perkin Elmer LS50B Luminescence Spectrometer, with excitation at 408 nm (slit width: 5 nm) and emission at 450 nm (slit width: 10 nm). Standard curves were prepared with authentic 3-cyano-7-hydroxycoumarin (Molecular Probes). The rates of product formation were linear with incubation time, up to 90 min under the reaction conditions described above.
Doxorubicin-induced acute cardiac toxicity
Two-month-old male null and wild-type mice were injected i.p. with doxorubicin hydrochloride (Sigma, St. Louis, MO) at 5, 10, or 20 mg/kg (n = 5 in each group), while the control null and wild-type groups were injected with saline (n = 4). Four days after injection, mice were sacrificed with CO2, and blood samples were collected from the right ventricle. Paraffin sections of the hearts were stained with H&E for histological examination.
Analysis of apoptosis in mouse heart
Cardiomyocyte apoptosis was analyzed in situ by terminal deoxyneucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay, with the ApopTag Fluorescein in Situ Apoptosis Detection Kit (Chemicon, Temecula, CA). The assay was conducted with 4-μm paraffin sections according to the manufacturer’s instructions, with DAPI as a counter-stain. Sections of rat mammary gland (at 4 days after weaning of her pups) were used as positive controls for apoptosis. Three-month-old male null and wild-type mice were injected i.p. with doxorubicin at 20 mg/kg (n = 4 in each group), while the control null and wild-type groups were injected with saline (n = 4). Two days after the treatment (Liu et al., 2005), mice were sacrificed by CO2 asphyxiation and transcardially perfused, sequentially, with PBS and 10% formalin solution. The heart was removed and immersed in 10% formalin for 24 h; it was then halved, processed and embedded in paraffin block for tissue sectioning. The total number of apoptotic cells per section was counted under a fluorescence microscope with a FITC filter (Nikon Eclipse i50). Concurrently, the digital image of each section was taken by a fluorescence stereomicroscope (Olympus SZX12, Olympus, Tokyo, Japan) with DAPI/UV filter, with the area of section measured by SPOT software version 4.6 (Diagnostic Instruments, Sterling Heights, MI). The index of apoptosis was expressed as the number of apoptotic cells per mm2. For each heart, two sections were quantified, and the results were averaged.
Other methods
Serum creatine kinase (CK) activity was measured with a commercial kit (Diagnostic Chemicals, Oxford, CT), using 40 μl of serum for each measurement. A noninvasive blood pressure acquisition system (CODA 6, Kent Scientific, Torrington, CT) was used for measuring parameters of cardiac function. Systolic blood pressure, diastolic blood pressure, mean blood pressure, and heart rate were measured for conscious, 2-month-old, male null and wild-type mice (n = 5 in each group). Microsomes from the heart and other tissues were prepared according to previously described methods (Gu et al., 2003). Protein concentration was determined by the bicinchoninic acid assay (Pierce, Rockford, IL).
Results
Cardiomyocyte-specific Cpr deletion
Tissue-specific deletion of the Cpr gene in the cardiomyocyte-Cpr-null mouse was demonstrated by immunoblot analysis of microsomal preparations from various tissues of adult null mice and their wild-type littermates. As shown in Figure 1A for 2–4 month old male mice, microsomal CPR protein levels were decreased in both the ventricular and atrial samples from the null mice, compared to age-matched wild-type controls. Densitometric analysis indicated that the extents of decrease in CPR protein expression in the null mice were 70% and 50%, in ventricular and atrial samples, respectively (Fig. 1B). The occurrence of significant levels of residual CPR protein in the null mice was not unexpected, given the fact that CPR and/or P450 is known to be expressed not only in cardiomyocytes, but also in cardiac fibroblast and endothelial cells (Brittebo, 1994; Borlak et al., 2003). In experiments not shown, CPR activity toward cytochrome c was detected in ventricular microsomes from wild-type, but not in those from the null, mice; however, the low overall rates detected did not permit an accurate determination of the extent of decrease in the null mice. In contrast, the levels of CPR protein were not significantly changed in other organs examined, including liver, lung, kidney, and brain (Fig. 1C).
Fig. 1.

Immunoblot analysis of CPR expression in wild-type and cardiomyocyte-Cpr-null mice. A. Immunoblot analysis of CPR expression in cardiac microsomes. CPR protein was detected with a polyclonal anti-rat CPR antibody. Microsomes were prepared from tissues pooled from 5 (for ventricle) or 10 (for atrium) 2- to 4-month-old, male mice. For cardiomyocytes, the microsomes were prepared from cells isolated from 6 adult male mice. Each lane was loaded with 10 μg (for ventricle and cardiomyocytes) or 6 μg (for atrium) of microsomal protein. Samples were analyzed in duplicate. B. Densitometric analysis of the CPR band detected in panel A. The data shown represent the ratios of the averaged band intensities for the null mice versus the intensity for the wild-type littermates. C. Immunoblot analysis of CPR levels in microsomal preparations from liver, kidney, brain, and lung. Three microsomal preparations, each from one adult male mouse, were analyzed for each mouse strain. Each lane was loaded with 5 μg (for liver) or 10 μg (for the other tissues) of protein.
As expected, deletion of the Cpr gene in the cardiomyocyte was permanent, as indicated by the fact that a decreased CPR expression was found for cardiac microsomal samples from 13-month-old null mice, compared to age-matched wild-type littermates (data not shown). To confirm the occurrence of Cpr deletion in cardiomyocytes, we compared CPR protein levels in isolated cardiomyocytes from adult null and wild-type mice. Microscopic inspection of the isolated cell preparations indicated minimal contamination by cardiac fibroblasts (data not shown). As shown in Figure 1, panels A and B, the level of microsomal CPR protein in the isolated cardiomyocytes was ~85% lower in the null mice than that in wild-type littermates; this extent of decrease in CPR expression, which may partly reflect the presence of contaminating, CPR-expressing cardiac fibroblasts, is similar to the known extents (80-90%) of Cre-mediated gene recombination in cardiomyocytes of other αMHC-Cre+ mice (Duan et al., 2005).
The cellular distribution of CPR expression in the hearts of the null and wild-type mice was analyzed by immunohistochemistry (Fig. 2). In the wild-type mice, CPR protein was detected in the cardiomyocytes, the cell type that makes up cardiac muscle fibers, and also in other cell types, including cardiac fibroblasts and endothelial cells. In the null mice, the intensity of immunoreactive signals in the cardiomyocytes was reduced to the level seen in the negative control section, whereas CPR staining in other cell types remained unchanged, compared to the staining intensity in the wild-type mice (Fig. 2). These results further demonstrated that cardiomyocyte-specific Cpr deletion was achieved in the cardiomyocyte-Cpr-null mice.
Fig. 2.
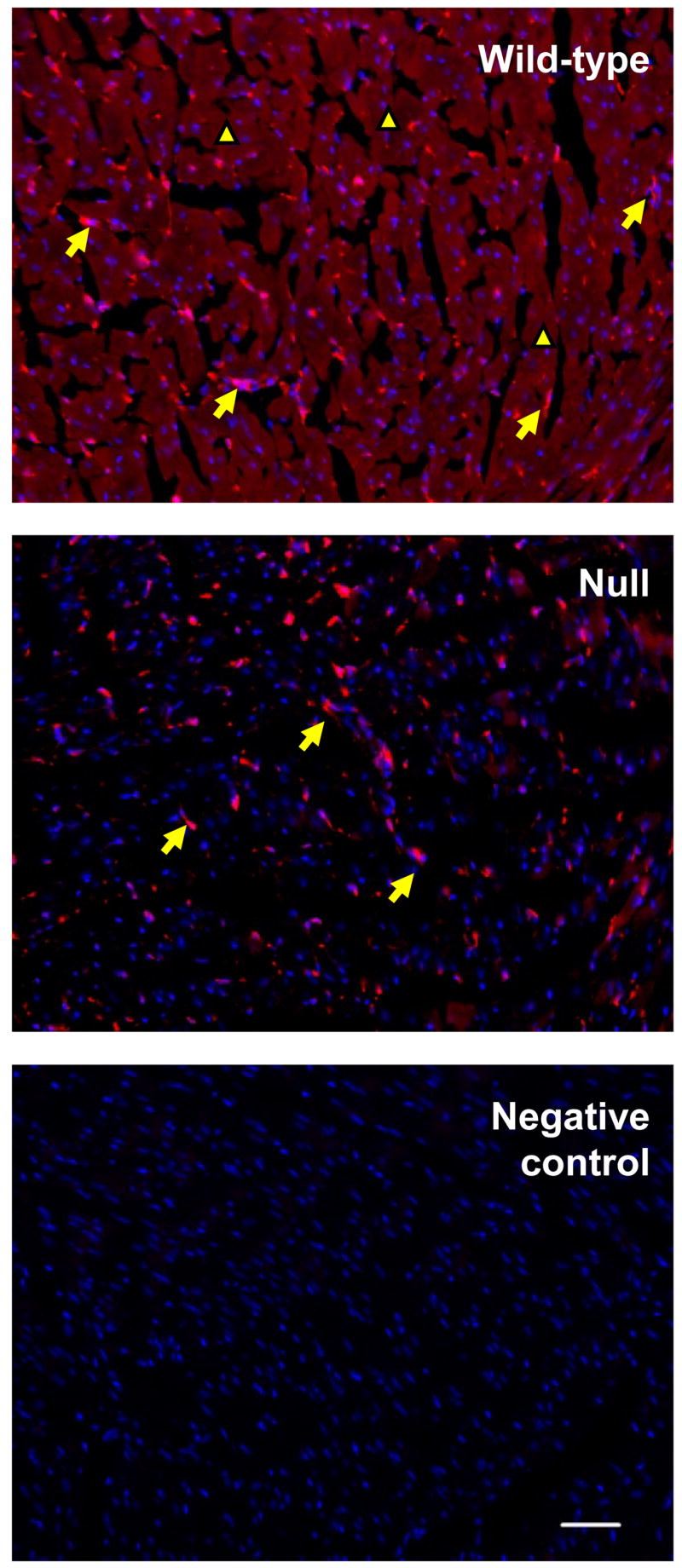
Immunofluorescent detection of CPR in the wild-type and cardiomyocyte-Cpr-null hearts. Frozen sections (10-μm thick) from 2-month-old male mice were incubated with a rabbit anti-CPR antibody. CPR expression was visualized by a chicken anti-rabbit secondary antibody conjugated with Alexa 594 (red fluorescence). The sections were counterstained with DAPI (blue fluorescence). The anti-CPR antibody was omitted in the negative control section. CPR immunostaining was found in cardiomyocytes (small arrowheads) as well as other cell types, including cells that appear to be cardiac fibroblasts (large arrowheads). Bar = 50 μm. The results shown are typical of sections from seven mice for each strain.
General characterization of the cardiomyocyte-Cpr-null mice
The null mice were indistinguishable from the wild-type mice in gross appearance, daily behavior, body weight, and mortality rates at various ages, ranging from 2 to 18 months. The ratios of heart weight to body weight, in the male (Table 1) and female (data not shown) null mice, were not different from the ratios in wild-type littermates, indicating the absence of cardiac hypertrophy. Similarly, no difference was found in the weights of other major organs, relative to body weight, between the null and the wild-type mice (data not shown). Hemodynamic parameters of the adult mice, including systolic blood pressure, diastolic blood pressure, and heart rate, did not show significant difference between the wild-type and the null mice (Table 1). These results indicated that the hearts of the null mice are functionally normal. In addition, both male and female null mice were fertile, and an analysis of genotype distribution among pups resulting from intercrossed heterozygotes indicated no sign of embryonic lethality (data not shown).
Table 1. Parameters of cardiac function in cardiomyocyte-Cpr-null mice.
Body and heart weights were measured in 2-month-old male mice (n = 10–11 for each strain). Systolic and diastolic blood pressure and heart rate were measured by a noninvasive blood pressure acquisition system (n = 4–5 for each strain). The values presented are means ± SD. There was no significant difference between the wild-type and null groups for any of the parameters measured (p > 0.05, Student’s t-test).
| Body weight (g) | Heart weight (mg) | Heart/body weight (mg/g) | Systolic blood pressure (mm Hg) | Diastolic blood pressure (mm Hg) | Heart rate (b/min) | |
|---|---|---|---|---|---|---|
| Wild-type | 29.4 ± 2.2 | 189 ± 18 | 6.4 ± 0.3 | 151 ± 8 | 112 ± 8 | 543±111 |
| Null | 29.2 ± 4.3 | 196 ± 38 | 6.7 ± 0.5 | 159 ± 21 | 124 ± 22 | 551 ± 91 |
The impact of cardiomyocyte Cpr deletion on cardiac microsomal P450 activity
Cardiac microsomes prepared from pooled ventricles from 5–7 adult wild-type or null mice were assayed for activity in the O-deethylation of 3-cyano-7-ethoxycoumarin. With the substrate at 9.3 μM, the initial rates of 3-cyano-7-hydroxycoumarin formation for the null mice (0.09 ± 0.06 pmol/min/mg protein; means ± S.D., n = 4) were significantly lower (P < 0.05) than the rates found for the wild-type mice (0.16 ± 0.07 pmol/min/mg protein; n = 4), a result indicating decreases in cardiac microsomal P450 activity concomitant with decreases in the level of CPR protein.
The impact of Cpr deletion on cardiac P450 and HO expression in the null mice
Given the previously reported compensatory gene expression changes in the liver of the liver-Cpr-null mice (Gu et al., 2003; Henderson et al., 2003), we performed immunoblot analysis to detect potential compensatory increases in the levels of CPR-dependent enzymes, including P450 and HO, in the hearts of the cardiomyocyte-Cpr-null mice. As shown in Figure 3, small increases in the levels of CYP1A1/2 and HO-2 proteins, but no increases in the levels of CYP2C, CYP2J, and CYP3A proteins, were observed in ventricular microsomes from the null mice, compared to the levels in the wild-type mice. Densitometric analysis (not shown) of the immunoblots revealed that the extent of increase was ~40% for both CYP1A1/2 and HO-2. The cardiac microsomal levels of CYP2B and HO-1 proteins were near the detection limit for both mouse strains, thus making it difficult to compare the relative expression levels between the two groups. The expression of CYP2E1 and CYP4A was not detected in either type of microsomal sample (data not shown).
Fig. 3.
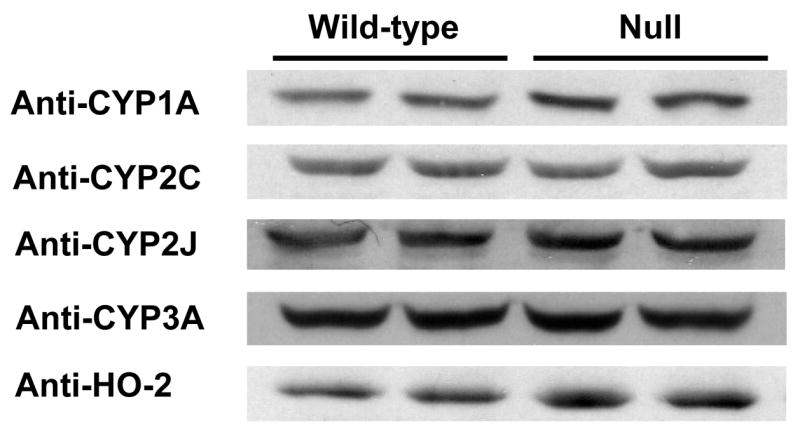
The impact of cardiomyocyte-specific Cpr deletion on cardiac P450 and HO expression. Microsomes prepared from pooled ventricles from five adult male mice were analyzed for each strain. Samples were loaded in duplicate. Each lane contained 10 μg of protein. The antibodies used are described in Materials and Methods.
The impact of cardiomyocyte Cpr deletion on Dox-induced acute cardiotoxicity
The extent of Dox-induced acute cardiotoxicity was determined by three different indices: serum levels of CK, numbers of apoptotic cells, and degrees of histological abnormality. The serum levels of CK activity were determined for adult, male, wild-type and null mice, at 4 days after treatment of the animals with a single i.p. injection of Dox (at 5, 10, or 20 mg/kg, respectively) or saline. Dox treatment caused a dose-dependent increase of the CK levels in both wild-type and null mice, compared to saline-treated groups; however, there was no significant difference in the CK levels either between saline-treated wild-type and null mice, or between Dox-treated wild-type and null mice, at any of the tested dosages (Fig. 4); the latter result is indicative of similar extents of Dox-induced acute cardiotoxicity in the two mouse strains.
Fig. 4.
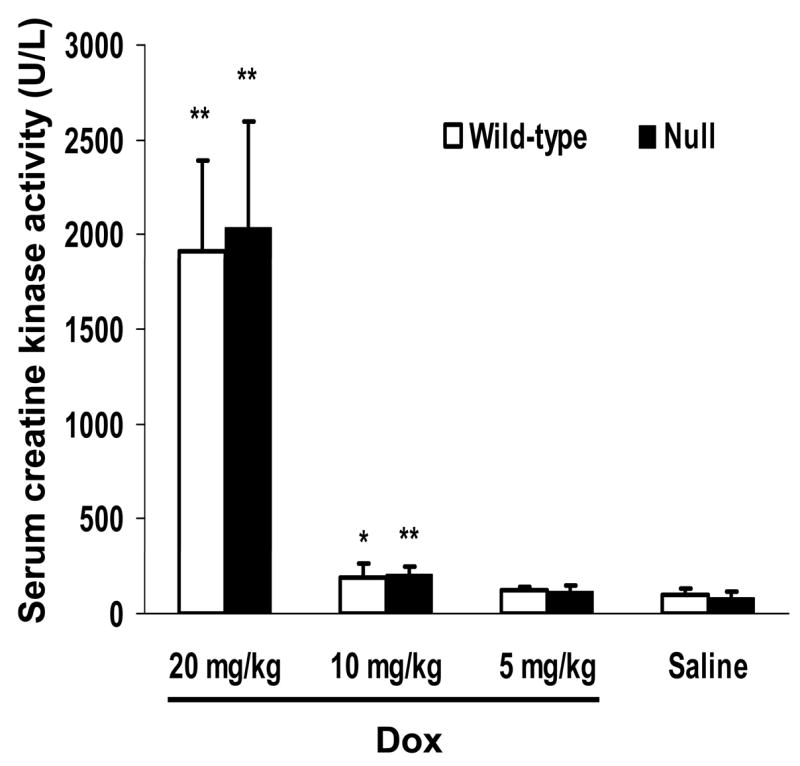
Dox-induced increase in serum CK activity. Three-month-old, male mice were given a single i.p. injection of Dox (at 5, 10, or 20 mg/kg; n = 5) or saline (n = 4). Four days after Dox treatment, serum samples were obtained, and CK activity was determined as described in Materials and Methods. The enzyme activity (rate of formation of NADPH) was expressed as U/L, which was calculated as [changes in absorbance per min] × 4180. *, P < 0.05; **, P < 0.01, compared to saline-treated mice of the same strain (Student’s t-test).
Apoptosis in the heart was analyzed by TUNEL assay in adult, male wild-type and null mice, at 2 days after treatment with doxorubicin (at 20 mg/kg) or saline. An earlier time point (2-day), compared to the time point chosen for CK determination and histological analysis (4-day), was selected, on the basis of previous reports indicating that cardiomyocyte apoptosis is an early event after Dox treatment (Arola et al., 2000). The numbers of apoptotic cells per mm2 of tissue section in saline treated wild-type and null groups were similar, at ~1/mm2 (Fig. 5). Dox-treatment caused a 3-fold increase in apoptotic cells in both wild-type and null groups (P < 0.01, versus saline-treated wild-type and null groups, respectively). However, there was no significant difference between Dox-treated wild-type and null mice.
Fig. 5.
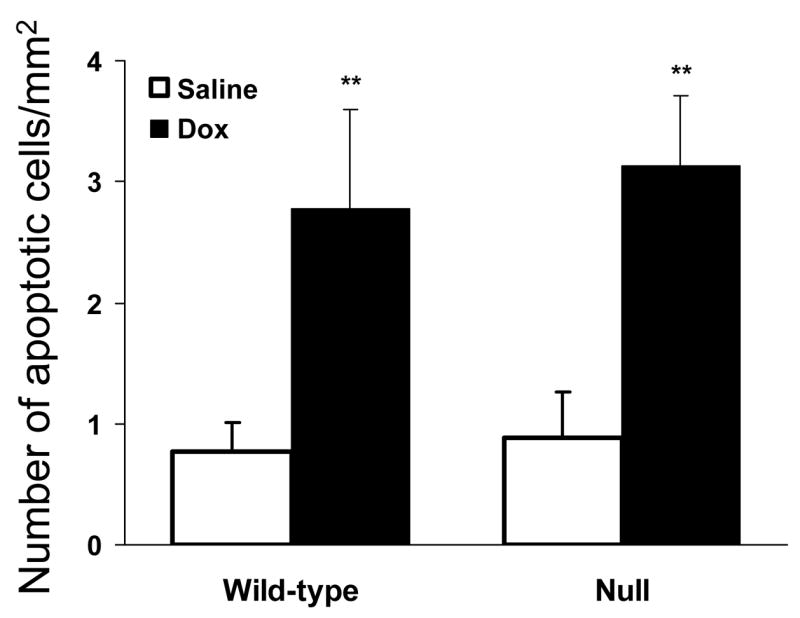
Dox-induced cardiac apoptosis. Three-month-old, male mice were given a single i.p. injection of Dox (at 20 mg/kg; n = 5) or saline (n = 4). Two days after treatment, apoptotic cells were detected by TUNEL assay in paraffin-embedded heart sections as described in Materials and Methods. **, P < 0.01, compared to saline-treated mice of the same strain (Student’s t-test).
Histological analysis of H&E-stained heart sections from saline-treated mice indicated that the hearts of wild-type and null mice are indistinguishable (Fig. 6A and 6B). At 4 days after Dox treatment, hearts from both wild-type and null mice showed widely scattered foci of one to several cardiomyocytes, with loss of striations, and granular, fragmented cytoplasm or cytoplasmic vacuolization. These foci of myocardial degeneration, which were absent in saline-treated mice, were found in ventricular as well as atrial myocardium in the Dox-treated mice from both strains (Fig. 6). These results further indicate that the deletion of Cpr in the cardiomyocytes does not protect the hearts from Dox-induced acute toxicity.
Fig. 6.

Dox-induced cardiac pathologic changes. Hearts were obtained 4 days after Dox (at 20 mg/kg) or saline treatment. Coronal sections of the heart were stained with H&E. A, saline-treated wild-type mice; B, saline-treated null mice; C, Dox-treated wild-type mice; D, Dox-treated null mice. The results shown are typical of four mice examined in each group. Arrows indicate sites of myocardial degeneration. Bar = 50 μm
Discussion
In this study, we have generated and characterized a mouse model with cardiomyocyte-specific deletion of the Cpr gene. The cardiomyocyte-Cpr-null mouse joins a growing list of conditional knockout mouse models that target the Cpr gene; these models can have valuable research applications in drug metabolism and toxicology. For examples, Cpr was deleted specifically in hepatocytes in liver-specific Cpr-null mouse models, through the use of an albumin-Cre transgenic mouse. Studies using the liver-Cpr-null mice, in which the liver microsomal CPR levels were reduced by ~95% from wild-type levels, revealed significant roles of hepatic CPR-dependent enzymes, not only in cholesterol biosynthesis, but also in systemic xenobiotic disposition and hepatic chemical toxicity (Gu et al., 2003). In a more recent study, a lung-Cpr-null mouse model was generated, in which Cpr was specifically deleted in a large fraction of type II alveolar epithelial cells and Clara cells (Weng et al., 2007). Studies using the lung-Cpr-null mouse indicated that pulmonary P450-mediated metabolic activation plays an essential role in the lung tumorigenicity of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), a tobacco-specific carcinogen.
In the present study, we find that cardiomyocyte CPR is dispensable for cardiac function in adult mice, given that the cardiac histology and hemodynamics seen in the cardiomyocyte-Cpr-null mice were normal. This result was in contrast to the documented critical function of CPR during embryonic development. Germline deletion of Cpr caused embryonic death in mid-gestation, accompanied by severe structural defects in multiple fetal organs, including the heart(Shen et al., 2002). In our cardiomyocyte-Cpr-null model, the expression of Cre recombinase, controlled by the promoter of αMHC, is expected to occur as early as E8.5, and Cre-mediated homologous recombination in the cardiomyocytes can take place at E11.5 (Gaussin et al., 2002). The functional impact of the Cpr deletion on embryonic metabolism of endogenous P450 substrates, such as retinoic acids, may not be significant until E13 or later, when sufficient amounts of the preexisting CPR protein in targeted cardiac cells have been degraded. By then, the role of CPR and CPR-dependent enzymes in cells targeted for Cpr deletion may be no longer critical for development. Alternatively, the metabolic deficiency in the Cpr-null cells could be overcome by surrounding cells with normal CPR expression; for example, retinoic acids accumulated in the Cpr-null cells could be transported to neighboring Cpr+ cells for P450-mediated degradation. Notably, no developmental or structural defect was found in the lungs of our recently reported lung-Cpr-null mouse model, in which Cre-mediated Cpr deletion was also expected to occur in mid- to late gestation (Weng et al., 2007).
The cardiomyocyte-Cpr-null model can be used to determine the biological functions of cardiomyocyte microsomal P450 enzymes in the heart. Although the overall level of P450 expression in the heart is quite low, compared to the level in the liver and other portal-of-entry organs, specific P450 enzymes, such as CYP2J and CYP2C, are relatively abundant in the cardiovascular system, where they are thought to regulate various biological and/or pathological processes through the metabolism of pertinent endogenous substrates (Wu et al., 1996; Enayetallah et al., 2004). For example, a number of P450 enzymes, including CYP2Js and CYP2Cs, can metabolize AA to epoxyeicosatrienoic acids (EETs) and hydroxyeicosatetraenoic acids (HETEs) (Fleming, 2001). The EETs could play a protective role by dilating coronary arteries during ischemia and reperfusion (Campbell and Harder, 1999), as demonstrated by findings from a cardiomyocyte-specific CYP2J2-transgenic mouse, in which microsomal AA epoxygenase activity was 3-fold higher than in the wild-type mouse, and in which post-ischemic recovery of the left ventricular function was significantly improved (Seubert et al., 2004). On the other hand, 20-HETE seems to play a major role in exacerbating cardiac postischemic damage (Nithipatikom et al., 2004). Nonetheless, definitive evidence has yet to be obtained for a role of endogenous P450 enzymes of the cardiomyocyte, as opposed to enzymes of cardiac vascular tissues (Enayetallah et al., 2004; Delozier et al., 2007) or elsewhere, in the post-ischemic cardiac injury. This knowledge gap can now be filled at least partly, through studies using the cardiomyocyte-Cpr-null mice. In this context, we recently described another transgenic mouse model, termed the Cpr-low (or Cprlow) mouse, in which the expression of CPR was greatly decreased (although not abolished) in all organs and cell types examined (Wu et al., 2005). In the hearts of the Cpr-low mice, CPR expression at 2 months of age was decreased by ~90%, compared to wild-type C57BL/6 and 129/Sv mice. The combined use of the Cpr-low and the cardiomyocyte-Cpr-null mouse models will likely be useful for distinguishing the relative roles of cardiomyocytes and the cardiac vascular system in post-ischemic reperfusion injury.
Findings of previous in vitro studies had suggested that CPR is involved in Dox-mediated cardiotoxicity, through its catalysis of a one-electron reduction of Dox; the semiquinone radical formed in this process can induce increased oxidative stress, through the generation of superoxide anion, and then hydrogen peroxide and reactive hydroxyl radicals (Riddick et al., 2005; Xu et al., 2005). However, the results from the current in vivo study do not support the notion that CPR plays a major role in Dox-mediated acute cardiotoxicity. Besides CPR, there are several other enzymes, including flavin reductase, NADH dehydrogenase, and xanthine oxidase, that could catalyze the one-electron reduction of Dox (Doroshow, 1983). The activities of these other redox enzymes in the cardiac tissue appear to be sufficiently high for mediation of Dox-induced toxicity, even in the absence of contributions from CPR. Notably, our finding is consistent with that of Ramji and co-workers, who studied the role of CPR in Dox-induced toxicity in cultured cells (Ramji et al., 2003). They showed that, although microsomal CPR activities in CPR-overexpressing human lymphoblastoid cells and human liver microsomes correlated well with the rates of Dox reduction, the susceptibility of human breast cancer cells to Dox-mediated cytotoxicity could not be enhanced through overexpression of CPR (Ramji et al., 2003). However, it remains to be determined whether CPR plays a major role in Dox-induced chronic cardiotoxicity, when animals are treated continually with the drug at doses lower than those used in the present study; the various enzymes capable of forming the Dox semiquinone radical may differ in their enzyme kinetic parameters with respect to Dox, or in their propensity to be inactivated by the oxidants produced in Dox-induced redox cycling.
It will also be interesting to determine whether the Cpr-low mouse model is protected against Dox-induced acute and/or chronic cardiotoxicity, given the potentially important role of CPR in the cardiac vascular system. In the Cpr-low mouse, the contribution of CPR to Dox-mediated toxicity would be substantially decreased in both cardiac muscles and cardiac vasculature. In that regard, although CPR expression is also decreased in the liver and elsewhere in the Cpr-low mice, systemic Dox clearance is unlikely to differ between the Cpr-low mice and wild-type mice. This assumption is supported by findings of a recent study using a liver-specific Cpr-null mouse model; there, hepatic CPR/P450 did not play a critical role in Dox clearance (Henderson et al., 2006).
In summary, we have generated and characterized a transgenic mouse model with cardiomyocyte-specific Cpr deletion. The null mouse exhibits normal tissue morphology and cardiac function. An initial application of this mouse model led to the finding that cardiomyocyte CPR and CPR-dependent enzymes do not play a critical role in the acute cardiotoxicity induced by Dox in vivo. This new knockout mouse model will be valuable for exploring potential functions of cardiomyocyte P450 enzymes in various physiological and pathological processes, such as post-ischemic reperfusion injury and xenobiotic-induced cardiotoxicity.
Acknowledgments
We gratefully acknowledge the use of the services of the Pathology Laboratory, and the Biochemistry and Molecular Genetics Core Facilities, of the Wadsworth Center. We thank Dr. Adriana Verschoor for reading the manuscript.
This study was supported in part by NIH grants ES07462 (to X.D.) and HL70070 (to E.D.A., who is an established investigator of the American Heart Association).
ABBREVIATIONS
- AA
arachidonic acid
- P450
cytochrome P450
- CPR
cytochrome P450 reductase
- Dox
doxorubicin
- CK
creatine kinase, and CBR1, carbonyl reductase
References
- Abel ED, Kaulbach HC, Tian R, Hopkins JC, Duffy J, Doetschman T, Minnemann T, Boers ME, Hadro E, Oberste-Berghaus C, Quist W, Lowell BB, Ingwall JS, Kahn BB. Cardiac hypertrophy with preserved contractile function after selective deletion of GLUT4 from the heart. J Clin Invest. 1999;104:1703–1714. doi: 10.1172/JCI7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arola OJ, Saraste A, Pulkki K, Kallajoki M, Parvinen M, Voipio-Pulkki LM. Acute doxorubicin cardiotoxicity involves cardiomyocyte apoptosis. Cancer Res. 2000;60:1789–1792. [PubMed] [Google Scholar]
- Borlak J, Walles M, Levsen K, Thum T. Verapamil: metabolism in cultures of primary human coronary arterial endothelial cells. Drug Metab Dispos. 2003;31:888–891. doi: 10.1124/dmd.31.7.888. [DOI] [PubMed] [Google Scholar]
- Brittebo EB. Metabolic activation of the food mutagen Trp-P-1 in endothelial cells of heart and kidney in cytochrome P450-induced mice. Carcinogenesis. 1994;15:667–672. doi: 10.1093/carcin/15.4.667. [DOI] [PubMed] [Google Scholar]
- Campbell WB, Harder DR. Endothelium-derived hyperpolarizing factors and vascular cytochrome P450 metabolites of arachidonic acid in the regulation of tone. Circ Res. 1999;84:484–488. doi: 10.1161/01.res.84.4.484. [DOI] [PubMed] [Google Scholar]
- Cheng L, Ding G, Qin Q, Xiao Y, Woods D, Chen YE, Yang Q. Peroxisome proliferator-activated receptor delta activates fatty acid oxidation in cultured neonatal and adult cardiomyocytes. Biochem Biophys Res Commun. 2004;313:277–286. doi: 10.1016/j.bbrc.2003.11.127. [DOI] [PubMed] [Google Scholar]
- Delozier TC, Kissling GE, Coulter SJ, Dai D, Foley JF, Bradbury JA, Murphy E, Steenbergen C, Zeldin DC, Goldstein JA. Detection of Human CYP2C8, CYP2C9, and CYP2J2 in Cardiovascular Tissues. Drug Metab Dispos. 2007;35:682–688. doi: 10.1124/dmd.106.012823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doroshow JH. Anthracycline antibiotic-stimulated superoxide, hydrogen peroxide, and hydroxyl radical production by NADH dehydrogenase. Cancer Res. 1983;43:4543–4551. [PubMed] [Google Scholar]
- Doroshow JH, Locker GY, Ifrim I, Myers CE. Prevention of doxorubicin cardiac toxicity in the mouse by N-acetylcysteine. J Clin Invest. 1981;68:1053–1064. doi: 10.1172/JCI110328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan SZ, Ivashchenko CY, Russell MW, Milstone DS, Mortensen RM. Cardiomyocyte-specific knockout and agonist of peroxisome proliferator-activated receptor-gamma both induce cardiac hypertrophy in mice. Circ Res. 2005;97:372–379. doi: 10.1161/01.RES.0000179226.34112.6d. [DOI] [PubMed] [Google Scholar]
- Ekins S, Vandenbranden M, Ring BJ, Gillespie JS, Yang TJ, Gelboin HV, Wrighton SA. Further characterization of the expression in liver and catalytic activity of CYP2B6. J Pharmacol Exp Ther. 1998;286:1253–1259. [PubMed] [Google Scholar]
- Enayetallah AE, French RA, Thibodeau MS, Grant DF. Distribution of soluble epoxide hydrolase and of cytochrome P450 2C8, 2C9, and 2J2 in human tissues. J Histochem Cytochem. 2004;52:447–454. doi: 10.1177/002215540405200403. [DOI] [PubMed] [Google Scholar]
- Fleming I. Cytochrome p450 and vascular homeostasis. Circ Res. 2001;89:753–762. doi: 10.1161/hh2101.099268. [DOI] [PubMed] [Google Scholar]
- Forrest GL, Gonzalez B, Tseng W, Li X, Mann J. Human carbonyl reductase overexpression in the heart advances the development of doxorubicin-induced cardiotoxicity in transgenic mice. Cancer Res. 2000;60:5158–5164. [PubMed] [Google Scholar]
- Gaussin V, Van de Putte T, Mishina Y, Hanks MC, Zwijsen A, Huylebroeck D, Behringer RR, Schneider MD. Endocardial cushion and myocardial defects after cardiac myocyte-specific conditional deletion of the bone morphogenetic protein receptor ALK3. Proc Natl Acad Sci U S A. 2002;99:2878–2883. doi: 10.1073/pnas.042390499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granville DJ, Tashakkor B, Takeuchi C, Gustafsson AB, Huang C, Sayen MR, Wentworth P, Jr, Yeager M, Gottlieb RA. Reduction of ischemia and reperfusion-induced myocardial damage by cytochrome P450 inhibitors. Proc Natl Acad Sci U S A. 2004;101:1321–1326. doi: 10.1073/pnas.0308185100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Weng Y, Zhang QY, Cui H, Behr M, Wu L, Yang W, Zhang L, Ding X. Liver-specific deletion of the NADPH-cytochrome P450 reductase gene: impact on plasma cholesterol homeostasis and the function and regulation of microsomal cytochrome P450 and heme oxygenase. J Biol Chem. 2003;278:25895–25901. doi: 10.1074/jbc.M303125200. [DOI] [PubMed] [Google Scholar]
- Henderson CJ, Otto DM, Carrie D, Magnuson MA, McLaren AW, Rosewell I, Wolf CR. Inactivation of the hepatic cytochrome P450 system by conditional deletion of hepatic cytochrome P450 reductase. J Biol Chem. 2003;278:13480–13486. doi: 10.1074/jbc.M212087200. [DOI] [PubMed] [Google Scholar]
- Henderson CJ, Pass GJ, Wolf CR. The hepatic cytochrome P450 reductase null mouse as a tool to identify a successful candidate entity. Toxicol Lett. 2006;162:111–117. doi: 10.1016/j.toxlet.2005.10.016. [DOI] [PubMed] [Google Scholar]
- Li T, Singal PK. Adriamycin-induced early changes in myocardial antioxidant enzymes and their modulation by probucol. Circulation. 2000;102:2105–2110. doi: 10.1161/01.cir.102.17.2105. [DOI] [PubMed] [Google Scholar]
- Liu FF, Stone JR, Schuldt AJ, Okoshi K, Okoshi MP, Nakayama M, Ho KK, Manning WJ, Marchionni MA, Lorell BH, Morgan JP, Yan X. Heterozygous knockout of neuregulin-1 gene in mice exacerbates doxorubicin-induced heart failure. Am J Physiol Heart Circ Physiol. 2005;289:H660–666. doi: 10.1152/ajpheart.00268.2005. [DOI] [PubMed] [Google Scholar]
- Menna P, Salvatorelli E, Minotti G. Cardiotoxicity of Antitumor Drugs. Chem Res Toxicol. 2008 doi: 10.1021/tx800002r. [DOI] [PubMed] [Google Scholar]
- Myers CE, McGuire WP, Liss RH, Ifrim I, Grotzinger K, Young RC. Adriamycin: the role of lipid peroxidation in cardiac toxicity and tumor response. Science. 1977;197:165–167. doi: 10.1126/science.877547. [DOI] [PubMed] [Google Scholar]
- Niitsu N, Kasukabe T, Yokoyama A, Okabe-Kado J, Yamamoto-Yamaguchi Y, Umeda M, Honma Y. Anticancer derivative of butyric acid (Pivalyloxymethyl butyrate) specifically potentiates the cytotoxicity of doxorubicin and daunorubicin through the suppression of microsomal glycosidic activity. Mol Pharmacol. 2000;58:27–36. doi: 10.1124/mol.58.1.27. [DOI] [PubMed] [Google Scholar]
- Nithipatikom K, Gross ER, Endsley MP, Moore JM, Isbell MA, Falck JR, Campbell WB, Gross GJ. Inhibition of cytochrome P450omega-hydroxylase: a novel endogenous cardioprotective pathway. Circ Res. 2004;95:e65–71. doi: 10.1161/01.RES.0000146277.62128.6f. [DOI] [PubMed] [Google Scholar]
- Olson LE, Bedja D, Alvey SJ, Cardounel AJ, Gabrielson KL, Reeves RH. Protection from doxorubicin-induced cardiac toxicity in mice with a null allele of carbonyl reductase 1. Cancer Res. 2003;63:6602–6606. [PubMed] [Google Scholar]
- Ramji S, Lee C, Inaba T, Patterson AV, Riddick DS. Human NADPH-cytochrome p450 reductase overexpression does not enhance the aerobic cytotoxicity of doxorubicin in human breast cancer cell lines. Cancer Res. 2003;63:6914–6919. [PubMed] [Google Scholar]
- Riddick DS, Lee C, Ramji S, Chinje EC, Cowen RL, Williams KJ, Patterson AV, Stratford IJ, Morrow CS, Townsend AJ, Jounaidi Y, Chen CS, Su T, Lu H, Schwartz PS, Waxman DJ. Cancer chemotherapy and drug metabolism. Drug Metab Dispos. 2005;33:1083–1096. doi: 10.1124/dmd.105.004374. [DOI] [PubMed] [Google Scholar]
- Seubert J, Yang B, Bradbury JA, Graves J, Degraff LM, Gabel S, Gooch R, Foley J, Newman J, Mao L, Rockman HA, Hammock BD, Murphy E, Zeldin DC. Enhanced postischemic functional recovery in CYP2J2 transgenic hearts involves mitochondrial ATP-sensitive K+ channels and p42/p44 MAPK pathway. Circ Res. 2004;95:506–514. doi: 10.1161/01.RES.0000139436.89654.c8. [DOI] [PubMed] [Google Scholar]
- Shen AL, O’Leary KA, Kasper CB. Association of multiple developmental defects and embryonic lethality with loss of microsomal NADPH-cytochrome P450 oxidoreductase. J Biol Chem. 2002;277:6536–6541. doi: 10.1074/jbc.M111408200. [DOI] [PubMed] [Google Scholar]
- Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med. 1998;339:900–905. doi: 10.1056/NEJM199809243391307. [DOI] [PubMed] [Google Scholar]
- Wang JF, Yang Y, Sullivan MF, Min J, Cai J, Zeldin DC, Xiao YF, Morgan JP. Induction of cardiac cytochrome p450 in cocaine-treated mice. Exp Biol Med (Maywood) 2002;227:182–188. doi: 10.1177/153537020222700305. [DOI] [PubMed] [Google Scholar]
- Weng Y, Fang C, Turesky RJ, Behr M, Kaminsky LS, Ding X. Determination of the role of target tissue metabolism in lung carcinogenesis using conditional cytochrome P450 reductase-null mice. Cancer Res. 2007;67:7825–7832. doi: 10.1158/0008-5472.CAN-07-1006. [DOI] [PubMed] [Google Scholar]
- White IN. A continuous fluorometric assay for cytochrome P-450-dependent mixed function oxidases using 3-cyano-7-ethoxycoumarin. Anal Biochem. 1988;172:304–310. doi: 10.1016/0003-2697(88)90449-6. [DOI] [PubMed] [Google Scholar]
- Williams DP, O’Donnell CJ, Maggs JL, Leeder JS, Uetrecht J, Pirmohamed M, Park BK. Bioactivation of clozapine by murine cardiac tissue in vivo and in vitro. Chem Res Toxicol. 2003;16:1359–1364. doi: 10.1021/tx034035z. [DOI] [PubMed] [Google Scholar]
- Wu L, Gu J, Cui H, Zhang QY, Behr M, Fang C, Weng Y, Kluetzman K, Swiatek PJ, Yang W, Kaminsky L, Ding X. Transgenic mice with a hypomorphic NADPH-cytochrome P450 reductase gene: effects on development, reproduction, and microsomal cytochrome P450. J Pharmacol Exp Ther. 2005;312:35–43. doi: 10.1124/jpet.104.073353. [DOI] [PubMed] [Google Scholar]
- Wu L, Gu J, Weng Y, Kluetzman K, Swiatek P, Behr M, Zhang QY, Zhuo X, Xie Q, Ding X. Conditional knockout of the mouse NADPH-cytochrome p450 reductase gene. Genesis. 2003;36:177–181. doi: 10.1002/gene.10214. [DOI] [PubMed] [Google Scholar]
- Wu S, Moomaw CR, Tomer KB, Falck JR, Zeldin DC. Molecular cloning and expression of CYP2J2, a human cytochrome P450 arachidonic acid epoxygenase highly expressed in heart. J Biol Chem. 1996;271:3460–3468. doi: 10.1074/jbc.271.7.3460. [DOI] [PubMed] [Google Scholar]
- Xu X, Persson HL, Richardson DR. Molecular pharmacology of the interaction of anthracyclines with iron. Mol Pharmacol. 2005;68:261–271. doi: 10.1124/mol.105.013383. [DOI] [PubMed] [Google Scholar]
- Zhuo X, Gu J, Behr MJ, Swiatek PJ, Cui H, Zhang QY, Xie Y, Collins DN, Ding X. Targeted disruption of the olfactory mucosa-specific Cyp2g1 gene: impact on acetaminophen toxicity in the lateral nasal gland, and tissue-selective effects on Cyp2a5 expression. J Pharmacol Exp Ther. 2004;308:719–728. doi: 10.1124/jpet.103.060301. [DOI] [PubMed] [Google Scholar]