

SUMMARY
We have reported a method to target lentiviral vectors to specific cell types. This method requires the incorporation of two distinct molecules on the viral vector surface: one is an antibody that renders the targeting specificity for the engineered vector, and the other is a fusogenic protein that allows the engineered vector to enter the target cell. However, the molecular mechanism that controls the targeted infection needs to be defined. In this report, we tracked the individual lentiviral particles by labeling the virus with the GFP-Vpr fusion protein. We were able to visualize the surface-displayed proteins on a single virion as well as antibody-directed targeting to a desired cell type. We also demonstrated the dynamics of virus fusion with endosomes and monitored endosome-associated transport of viruses in target cells. Our results suggest that the fusion between the engineered lentivirus and endosomes takes place at the early endosome level, and that the release of the viral core into the cytosol at the completion of the virus-endosome fusion is correlated with the endosome maturation process. This imaging study sheds some light on the infection mechanism of the engineered lentivirus and can be beneficial to the design of more efficient gene delivery vectors.
Keywords: lentiviral vector, intracellular trafficking, targeted gene delivery
INTRODUCTION
Gamma-retroviral and lentiviral vectors are currently the most commonly used gene delivery vehicles due to their ability to permanently integrate a therapeutic transgene into a target cell chromosome.1-4 Lentiviral vectors have the unique feature of being able to transduce nondividing cells, making it particularly attractive for certain gene therapy applications.5-7 Sometimes, in order to achieve a desirable therapeutic effect, the viral vectors must be capable of precisely delivering a gene of interest to specific cells without influencing non-target cells.8-10 Many efforts have been made to develop such targeting viral vector systems mostly by altering the viral envelope glycoprotein.11-16 Although certain envelope glycoproteins are structurally plastic enough to allow insertion of a new molecular recognition unit (such as peptide, single chain antibody, growth factor, etc.) for targeting, this manipulation can adversely affect the delicate coupling interactions of the binding and fusion domains of glycoproteins, resulting in enveloped vectors with decreased infectivity to the target cells.8,15,17-19
We have previously developed an efficient method to target lentivirus-mediated gene transduction to a desired cell type.10 Our engineering approach involved the incorporation of a targeting antibody and pH-dependent fusogenic protein as two distinct molecules on the lentiviral surface. Our hypothesis for targeted transduction was that the antibody binding induces endocytosis, and then the virus is brought into an endosomal compartment where the low pH environment causes the fusogenic molecule to trigger membrane fusion and release the viral core into the cytosol. In order to understand the interactions between the engineered lentivirus and the targeted cells and the underlying mechanisms of viral transduction at a molecular level, we intended to develop assays to directly visualize the intracellular behavior of the virus in living cells.
Improved understanding of virus-host cell interactions can provide crucial insights for enhancing the efficacy of virus-mediated gene delivery as well as preventing virus-triggered diseases. Insight into the dynamics of the trafficking of viral particles in living cells is fundamental to understanding a variety of the viral infection mechanisms. Many enveloped viruses enter their host cells via receptor-mediated endocytosis. The endocytosed viruses are internalized through endocytic compartments, and the viruses fuse with the endosomal membrane to release viral genome into host cells.20-23 During these processes, viruses utilize microtubule networks for movement towards the perinuclear regions.24,25 Recent studies have shown that intracellular virus trafficking is critically involved in the endosome-mediated sorting and transport of influenza virus, vesicular stomatitis virus (VSV), and semliki forest virus (SFV).26-28 The endocytic pathways used by some viruses have been explored, but some specific features of the entry mechanisms of engineered recombinant lentiviruses remain largely unknown.
In this study, we analyzed the intracellular trafficking of the targeting lentiviral vectors by utilizing dynamic imaging of single viruses within target cells. We visualized the incorporated molecules on a single virion and the targeting of antibody-displaying virus to a CD20-expressing cell line. We also imaged viral fusion and detected the endosome-associated transport of the engineered lentivirus. Our results suggest that virus-endosome fusion takes place at the early endosome stage, and that viral fusion is independent of microtubule- or actin-associated transport. We also observed the process of the dissociation of the viral core from the fused endosome. Our results shed some light on the infection model of the targeting lentiviral vector incorporated with two separated binding and fusion proteins on the surface.
RESULTS
Co-incorporation of antibody and fusogenic protein on the engineered virion
We sought out imaging methods to characterize a previously engineered lentivirus reported to be able to selectively transduce human B cells via CD20 as the viral receptor.10 Co-display of an anti-CD20 antibody and a fusogenic protein on the same virion is thought to be essential for this engineered lentivirus to infect the target cell. To image the virus, we constructed lentiviral particles harboring GFP fused to the N-terminus of the HIV accessory protein Vpr (designated GFP-Vpr, Figure 1A). GFP-Vpr-labeled lentivirus enveloped with both anti-CD20 antibody and fusogenic protein were produced as described10 except with the use of lentiviral backbone plasmid FUW lacking the GFP transgene (Figure 1A) instead of FUGW and co-transfection of an additional plasmid that expresses GFP-Vpr. During virus synthesis, GFP-Vpr provided in trans can be incorporated into the virion via the interaction between Vpr and the P6 region of the gag protein.29 To determine whether αCD20 (anti-CD20 antibody) and SINmu (fusogenic protein)10 were incorporated on the same virion, we indirectly immunofluorescent-stained the GFP-Vpr-tagged virions by a triple labeling method (Figure 1B). As controls, we also included the staining of the GFP-Vpr-labeled lentiviral particles bearing various surface proteins (FUW-GFPVpr/αCD20, FUW-GFPVpr/SINmu, or FUW-GFPVpr/VSVG); VSVG (vesicular stomatitis viral glycoprotein) is a widely used envelope glycoprotein with broad tropism. Confocal images of the individual FUW-GFPVpr/αCD20+SINmu particles showed that ∼70% of the GFP-Vpr-labeled virions colocalized with both αCD20 and SINmu (Figure 1C). This indicated that both the antibody and the fusogenic protein were indeed displayed on a single virus particle. The detection of a few GFP-negative and dye-positive spots for FUW-GFPVpr/αCD20+SINmu suggested that some of the intact virions lacked the GFP-Vpr protein, which is consistent with the previous report by McDoland et al.;29 some spots that were positive for SINmu only could be virions that lacked the incorporation of the GFP-Vpr protein and αCD20. As expected, colocalizations of the GFP-labeled virions with only αCD20 (FUW-GFPVpr/αCD20) or with only SINmu (FUW-GFPVpr/SINmu) were observed, while no colocalization of the GFP-labeled virions with either protein was detected for FUW-GFPVpr/VSVG.
Figure 1.

Co-incorporation of antibody and fusogenic protein on the single lentivirus particle. (A) The schematic representation of the labeling (GFP-Vpr) and viral (FUW and FUGW) constructs. CMV: cytomegalovirus immediate-early gene promoter; GFP: green fluorescence protein; Vpr: viral protein R; Ubi: human ubiqutin-C promoter; WPRE: woodchuck hepatitis virus posttranscriptional regulatory element. (B) The schematic representation of the virus-staining method for visualizing individual viruses. Two antibodies were used to detect the presence of αCD20 and fusogenic molecule (SINmu). (C) Colocalization of GFP-Vpr-labeled viral particles with the αCD20 antibody (red) and fusogenic molecule SINmu (blue). GFP-Vpr-labeled viruses psuedotyped by either both αCD20 and SINmu, or αCD20 antibody only, SINmu only, or VSVG protein were strained with anti-human IgG and anti-HA tag antibodies against αCD20 and SINmu. Overlapping green, red, and blue signals appears as white in a merged image. Scale bar represents 2 μm. (D) 293T/CD20 cells (2 × 105) were transduced with 2 ml of fresh unconcentrated FUGW/αCD20+SINmu virus (∼ 1 × 106 TU/ml) with or without GFP-Vpr labeling. The resulting GFP expression was analyzed by FACS. Solid line, analysis of the infected 293T/CD20; Shaded area, analysis of the non-infected 293T/CD20 (as a control).
To test whether the GFP-Vpr-labeling of lentiviruses could affect the viral infectivity, we made viruses bearing both αCD20 and SINmu (FUGW/αCD20+SINmu); FUGW is a lentiviral backbone that contains a human ubiquitin-C promoter driving the expression of a GFP transgene (Figure 1A).30 The target 293T/CD20 cells were exposed to FUGW/αCD20+SINmu with or without the incorporation of GFP-Vpr, and the percentage of GFP-expressing cells was measured by FACS three days post-infection. As shown in Figure 1D, a similar transduction efficiency was obtained, indicating that the GFP-Vpr-labeling did not markedly affect viral infectivity.
Antibody directs lentivirus to target cells
To examine whether the engineered lentiviral particles could efficiently recognize the desired cell type, we analyzed the virus-cell binding complex using a confocal microscope. A 293T cell line stably expressing the CD20 protein (293T/CD20) was used as the target cell line, and its parental cell line 293T was used as a negative control (Supplementary Figure 1A). Neither GFP nor the αCD20 signal was detected in the control 293T cells lacking CD20 expression (Supplementary Figure 1A, upper). In contrast, significant GFP and αCD20 signals were detected on the surface of 293T/CD20 cells (Supplementary Figure 1A, lower). This result suggests that our engineered lentivirus can specifically bind to a CD20-expressing cell line.
To further confirm that the virus-cell binding was induced by the viral αCD20, the lentiviral particles bearing various surface proteins (FUW-GFPVpr/αCD20+SINmu, FUW-GFPVpr/αCD20, or FUW-GFPVpr/SINmu) were incubated with 293T/CD20 cells, followed by extensive washing. The imaging results showed that the lentiviral particles bearing the αCD20 antibody (FUW-GFPVpr/αCD20+SINmu and FUW-GFPVpr/αCD20) were able to bind to the target cells, but no GFP signal was detected in the cells incubated with the viral particles bearing only the fusogenic protein SINmu (FUW-GFPVpr/SINmu, Supplementary Figure 1B). These results demonstrate that the virus-cell binding is mediated by a specific interaction between the CD20 antigen on the cell surface and the αCD20 antibody on the viral surface. We quantified the number of viral particles bound to the target cells by examining more than 20 cells and counting the bound viruses. The result showed that FUW-GFPVpr/αCD20+SINmu (15.4 particles/cell) and FUW-GFPVpr/αCD20 (16.3 particles/cell) exhibited similar binding to 293T/CD20 cells, while FUW-GFPVpr/SINmu (0.1 particles/cell) was rarely detected on the cell surface.
Incorporated fusogenic protein triggers virus-endosome fusion
To visualize the actual fusion event of internalized viruses within the endosomes, we used lipophilic dye (DiD) that can be spontaneously incorporated into viral membrane for a double-labeling of GFP-Vpr-tagged lentiviruses. The incorporation of DiD dye at a high concentration on the viral membrane can result in self-quenching of DiD fluorescence.31,32 Viral fusion with the endosome membrane can then cause a de-quenching that is due to the dispersion of DiD and can be seen by the increase of fluorescence (Figure 2A). We labeled GFP-Vpr-tagged lentiviruses (FUW-GFPVpr/αCD20+SINmu) with DiD and incubated them with 293T/CD20 cells at 37°C for various time periods. The acquired images of both the green and red fluorescence signals are shown in Figure 2C (upper), and the boxed regions are enlarged and represented as panels below. At 0 min, the GFP-Vpr-labeled viral particles were detected on the cell surface. After 10 min of incubation, viral particles appeared to be internalized inside the cell but remained detectable solely by the green signal, suggesting that although viruses were endocytosed, fusion had not occurred. The image after 30 min of incubation showed that many particles were fused with the endosomes, as indicated by the appearance of the red signal. After 60 min, more endosome-fused viral particles and brighter fusion signals were observed. These imaging results suggest that the majority of viral fusion occurs between 30 min to 60 min after incubation. In addition, viral fusion was observed in the peripheral region at earlier time points (Figure 2C, upper, 30min), but the fusion signals at later time points were mostly distributed around the perinuclear region of the cell (Figure 2C, upper, 60min).
Figure 2.

Detection of virus-endosome fusion at different time points. (A) Schematic representation of the visualization assay for virus-endosome fusion by fluorescence dequenching. (B) The time course study of virus penetration (FUGW/VSVG and FUGW/αCD20+SINmu) by using bafilomycin A1 (see Materials and Methods) (C) GFP-Vpr-labeled VSVG or both αCD20 and SINmu displaying viruses (green) were labeled with DiD (red) for 1 h at room temperature. Double-labeled viruses were incubated with 293T/CD20 cells at 37°C for 0, 10, 30, or 60 min, fixed and imaged. The boxed regions are magnified and shown in separated panels below. Yellow particles indicate viral particles fused to endosomes. Scale bar represents 5 μm. (D & E) Tracking of endosomal fusion of individual viruses. (D) Selected images obtained from a time series study starting 30 min after incubation. The arrow indicates the viral particle monitored in the live cell. The GFP-Vpr+ (green) signal and GFP-Vpr+DiD+ (yellow as the merged color of green and red) signals mark the viral particle before and after endosomal fusion, respectively. Scale bar represents 2 μm. (E) Kinetics of the fluorescence intensity of the GFP-Vpr (green) and DiD (red) signals of the virion. The fluorescence intensity was measured within the regions of interests around viral particle using the software package for the Zeiss LSM 510.
We compared the kinetics of viral fusion of our engineered lentivirus with that of conventional VSVG-pseudotyped lentivirus. It is known that VSV is endocytosed to early endosomes, where the low pH triggers endosomal fusion.33 Using the same fluorescence de-quenching assay, we observed the viral fusion of FUW-GFPVpr/VSVG. The acquired images showed that virus-endosome fusion could be detected at 10 min after incubation (Figure 2C, lower), indicating that viral fusion of VSVG-pseudotyped lentivirus occurs faster than that of the engineered targeting lentivirus.
In order to compare the timing of acidic pH-activated and fusion-involved penetration of the engineered and VSVG-pseudotyped lentiviruses, we used bafilomycin A1, the specific inhibitor of vacuolar proton ATPases, to block low pH endosomal fusion.34 Viruses were pre-bound to 293T/CD20 cells at 4°C for 1 h, and entry was initiated by shifting cells to 37°C. At different time points thereafter, bafilomycin A1 (25 nM) was added to block further viral fusion. The percentage of viral entry was normalized based on the signal at 4.5 h, when no further effect of bafilomycin A1 was observed and fusion was therefore considered to be unrestricted by the treatment. These results suggest that acid-induced penetration of VSVG-pseudotyped lentivirus occurred ∼5 min after warming, and a half-maximal penetration (50%) was reached by ∼40 min, whereas the penetration of engineered lentivirus (FUGW/αCD20+SINmu) took place ∼25 min after warming and reached a half maximal level by ∼120 min (Figure 2B). Consistent with the viral fusion kinetics determined by the de-quenching assay (Figure 2C), we found that the engineered lentivirus has slower kinetics of viral penetration than that of VSVG-pseudotyped lentivirus.
To further characterize viral fusion, we performed real-time imaging of GFP-Vpr/DiD-labeled lentiviruses in 293T/CD20 cells. We first incubated the doubly labeled viruses with 293T/CD20 cells at 37°C for 30 min to allow the internalization of viral particles, and then began imaging. Confocal time-lapse images were collected cautiously to make sure that the same viral particle was tracked at ∼15-second (s) intervals over a period of 10 min and selected images are shown in Figure 2D (see also Supplementary Movie 1). The first image (at 334.7 s) showed that before endosomal fusion, the viral particle was identified only by green color. At 418.8 s, viral fusion with the endosome was visualized by the dramatic increase of fluorescence signal as the result of DiD de-quenching (Figures 2D and 2E). Approximately 80% of the viral particles we observed (n=34) showed this pattern of de-quenching. Interestingly, the virus-endosome fusion signal was initially detected around the peripheral region (418.8 s) and then migrated towards the nucleus over ∼3 min (Figure 2D). We observed the movement of fusion signals towards nucleus in 60% of the videos recorded (n=14), while in the other 40% of cases, it appeared that the fusion signals were either stationary or bidirectional. As shown in Figure 2E, the quantification of fluorescence intensity of the GFP and the DiD signals showed that although some fluctuations of the GFP signal were detected during the imaging, only the DiD signal increased dramatically at certain moments of the recording, supporting our assumption that the observed fluorescence change is an indication of viral fusion.
Tracking of viral transport through endosomes
To visualize the distribution of endosomal compartments, we used antibodies against EEA126-28 and CI-MPR26,35 as the early and late endosomal markers, respectively. As shown in Figure 3A, the early and the late endosomal markers were largely separated. The early endosomal markers were distributed in the peripheral region, whereas the late endosomal markers were primarily found in the perinuclear region of cells. However, a close examination of the images identified several endosomes that were both EEA1- and CI-MPR-positive (Figure 3A, arrow), which have been interpreted as maturing intermediates towards the late endosomes.36,37 These intermediate endosomes containing both markers were mostly observed in the perinuclear region of the cells.
Figure 3.

Internalization and transport of viruses through endosomes. (A) Distribution of endocytic compartments in living cells. Early and late endosomes were detected by EEA1 and CI-MPR, respectively. 293T/CD20 cells were immunostained with EEA1 (red) and CI-MPR (green) and counterstained with DAPI (blue). Arrows indicate individual endosomes positive for both early and late endosomal markers. (B) GFP-Vpr-labeled viruses (FUW-GFPVpr/αCD20+SINmu) were incubated with 293T/CD20 cells (MOI ∼30) at 37°C for various time points of 0, 30, 60, or 120 min. Then these cells were fixed, permeabilized, and immunostained with EEA1 (red) and CI-MPR (blue) and counterstained with DAPI (white). The boxed regions are magnified and shown in separated panels below. The bottom panels show the localization of GFP-Vpr-labeled viral particles in the two different endosomal stages. Scale bar represents 5 μm. (C) Quantification of GFP-Vpr-labeled lentiviruses colocalized with EEA1+ (black), CI-MPR+ (white), or EEA1+CI-MPR+ (gray) endosomes at different incubation times. The result shown is the collective data from three experiments.
We conducted a colocalization experiment to examine the endosomal transport of the engineered lentivirus and to analyze the virus-fused endosomes. As shown in Figure 3B, at 0 min, the viral particles did not colocalize with either of the endosomal markers. After 30 min of incubation, particles were colocalized with EEA1 in the early endosomes, and after 60 min, many viral particles were observed in endosomes positive for both EEA1 and CI-MPR. At 120 min, most of the viral particles did not colocalize with either marker, and a small fraction was seen in the late endosomes that stained positive only for CI-MPR. We quantified this colocalization by viewing more than 20 cells (∼100 viruses) for each time point (Figure 3C) and the results were consistent with the observation seen in Figure 3B. Coupled with the previous evidence that virus-endosome fusion signals were detected between 30 and 60 min after incubation (Figure 2C), this study suggests that the virus-endosome fusion started before the late endosome stage.
To confirm the trafficking of the viral particles through various endosomes, we further examined the colocalization of engineered viral particles with red fluorescent protein-tagged Rab protein, DsRed-Rab5 and DsRed-Rab7, as the early and late endosomal markers, respectively.36,38,39 The time course images showed that the majority of viral particles were first located in organelles that were positive for DsRed-Rab5 (Supplementary Figure 2A, 30min), and were later observed in Rab7-positive and Rab5-positive organelles (Supplementary Figure 2A, 60min). The quantification of colocalization suggested that at 30 min, 62% of viruses were located in Rab5-positive organelles and 6% in Rab7-positive organelles, and at 60 min, 68% in Rab5-positive organelles and 61% in Rab7-positive organelles. The colocalization of viral particles with Rab5 was rarely detected at 120 min, but 20% was observed in Rab7-positive organelles.
Next, we compared the trafficking of our engineered lentivirus with transferrin, which is known to be trafficked from early to recycling endosomes.26 At 60 min, most of the viral particles were colocalized with both EEA1 and transferrin, and a small fraction (9%) of the viruses were observed in transferrin-positive only organelles, which could be considered as recycling endosomes (Supplementary Figure 2B, 60 min). However, at 120 min, 30% of the viruses were colocalized with transferrin-positive, EEA1-negative organelles, indicating that some fractions of viruses were trafficked to recycling endosomes (Supplementary Figure 2B, 120 min).
Microtubule-mediated virus transport
We further characterized the endosomal compartment where the viral fusion occurs by disrupting the microtubules. It has been reported that the microtubule network can facilitate the migration of viruses to the nucleus and promote the transport of endosomes in cells.24,25,29 Our colocalization study using the tubulin-specific antibody suggested that the engineered lentiviruses travel along microtubule networks (Figure 4A). In order to examine whether virus-endosome fusion occurs before or after microtubule-dependent transport, microtubules were damaged by the drug nocodazole. The reduced tubulin-staining confirmed the disruption of microtubule networks (Figure 4B). We next studied the effect of microtubule disruption on virus-endosome fusion and the confocal image showed that the viral fusion signals could be detected (Figure 4C), and quantification indicated that the viral fusion is independent of microtubule assistance (Figure 5A), suggesting that the viral fusion with endosomes occurs before microtubule-associated movement.
Figure 4.

Microtubule-associated transport of viruses. (A) Colocalization of GFP-Vpr-labeled lentiviruses with microtubule networks 1 h after infection. Microtubules were immunostained with the monoclonal antibody to α-tubulin (red). The boxed region is enlarged in right panel. Arrows indicate viruses on the microtubules. (B) Microtubule staining in a nocodazole-treated cell. Cells were preincubated with nocodazole at 37°C for 30 min to disrupt microtubules, then immunostained with the monoclonal antibody to α-tubulin (red). (C) The fixed image of GFP-Vpr/DiD-labeled viral particles in a nocodazole-treated cell (see Materials and Methods). Arrows indicate the viral particles fused to endosomes. (D) Localization of GFP-Vpr-labeled viral particles (FUW-GFPVpr/αCD20+SINmu) with the two endosomal markers after 60 min of incubation in a nocodazole pre-treated cell. (E) Microtubule staining of siRNA-treated cell. 293T/CD20 cells were transfected with α-tubulin-specific siRNA- or control siRNA. After 72 h, transfected cells were seeded and immunostained with anti- α-tubulin antibody (green). (F) The virus fusion for siRNA-treated cells. α-tubulin siRNA or control siRNA-treated cells were incubated with GFP-Vpr/DiD-labeled viruses at 37°C for 60 min and then fixed. Yellow particles denoted by arrows indicate the virus particles fused to endosomes. (G) Localization of GFP-Vpr-labeled viral particles with the two endosomal markers after 60 min of incubation with α-tubulin siRNA-treated cells. The boxed regions are enlarged and shown in separated panels. Scale bar represents 5 μm.
Figure 5.

The effects of inhibitory drugs or siRNA treatment on viral fusion, infection, and endosome maturation. (A) GFP-Vpr/DiD-labeled viruses were incubated with drug- or siRNA-treated cells at 37°C for 60 min, and then fixed. The viral particles with the fusion signal (black) or without the fusion signal (gray) were quantified. The viral particles both GFP-Vpr+ and DiD+ were considered to be fused with endosomes, while particles that were only GFP-Vpr+ were considered to be unfused virus. For quantification, 60 viral particles were examined for no drug-treatment, 64 particles for nocodazole treatment, 72 particles for α-tubulin siRNA treatment, and 68 particles for cyto-D treatment. The results were collected from three independent experiments. (B) The role of microtubules and actin filaments in the virus infection. 293T/CD20 cells which were preincubated with nocodazole or cytochalasin-D (cyto-D) were transduced with 2 ml of fresh unconcentrated FUGW/αCD20+SINmu virus. The resulting GFP expression was analyzed by FACS. (C) Quantification of GFP-Vpr-labeled viruses colocalized with EEA1+ (black), CI-MPR+ (white), or both EEA1+ and CI-MPR+ (gray) endosomes at 60 min of incubation in drug- or siRNA-treated cells. (D) The effect of α-tubulin knockdown on virus infection. Control siRNA or α-tubulin siRNA transfected cells were transduced with 2 ml of fresh unconcentrated FUGW/αCD20+SINmu virus. The percentage of GFP+ cells was analyzed by FACS.
To further investigate the effects of the microtubule-disrupting drug on the localization of virus in endosomal compartments, we pretreated cells with nocodazole for 30 min and quantified the viruses colocalizing with the different endosomal markers after 60 min of incubation. The results showed that the viral population in EEA1- and CI-MPR-double positive endosomes in nocodazole-treated cells was significantly lower than that of untreated cells, whereas the population positive only for EEA1 was higher in the treated cells (Figure 4D and Figure 5C). This indicated that viral particles were mostly located in the early endosomes devoid of CI-MPR upon nocodazole treatment and that the treatment restricted the further transport and maturation of early endosomes to EEA1- and CI-MPR-double positive endosomes. To investigate whether endosome maturation was relevant to productive infection of our engineered lentiviruses, nocodazole-pretreated cells were transduced by FUGW/αCD20+SINmu. The FACS analysis showed that the transduction rate decreased by 31%, suggesting that productive infection was affected markedly by the nocodazole inhibition of the endosome maturation (Figure 5B).
To confirm the role of microtubules in viral infection, we used siRNA to down-regulate the expression of α-tubulin in cells. 293T/CD20 cells were transfected with α-tubulin-specific siRNA or control siRNA, and the knockdown of α-tubulin expression was validated by microtubule staining (Figure 4E). The virus-endosome fusion experiment on the siRNA-treated cells showed that the down-regulation of α-tubulin did not significantly affect the efficiency of viral fusion (Figure 4F and Figure 5A). Similar to the result of the nocodazole treatment, we also observed that the maturation of the lentivirus-containing endosomes was dependent on microtubule-associated transport, as most of these endosomes were retained at the early endosome stage after the siRNA-treatment (Figure 4G and Figure 5C). As compared to treatment of the control siRNA, lower transduction was obtained for 293T/CD20 cells transfected with α-tubulin-specific siRNA and exposed to FUGW/αCD20+SINmu (Figure 5D).
Actin-mediated virus transport
We further investigated the role of actin cytoskeleton in trafficking the engineered viral particles. The colocalization study using rhodamine-conjugated phalloidin suggested that our engineered lentiviruses travel along actin-filaments (Supplementary Figure 3A). To characterize actin-mediated virus trafficking, 293T/CD20 cells were pretreated with 20 μM of cytochalasin D (cyto-D) at 37°C for 30 min to disrupt actin filaments, and then we examined the viral fusion and virus localization in endosomal compartments. We first confirmed the disruption of actin in cyto-D-treated cells by phalloidin labeling (Supplementary Figure 3B). The viral fusion assay in the cyto-D-treated cells showed that the majority of viral particles (denoted by arrows) were fused with the endosomes (Figure 5A and Supplementary Figure 3C). The viral population in endocytic markers showed that viral particles were mostly located in the early endosomes (Figure 5C and Supplementary Figure 3D). Compared to the results of microtubule disruption by either nocodozale or siRNA, actin-disruption using cyto-D resulted in greater inhibition of the viral transduction (Figure 5B). This further confirmed that the inhibition of the maturation process of lentivirus-containing endosomes could affect productive infection.
Virus release from endosomes
It has been reported previously that Vpr remains largely associated with the pre-integration complex after fusion;29 thus, GFP-Vpr labeling can be used for visualizing the release of the viral core from fused endosomes. GFP-Vpr/DiD-labeled viruses (FUW-GFPVpr/αCD20+SINmu) were incubated at 37°C for 60 min to induce virus-endosome fusion; based on our study of fusion kinetics (Figure 2C), most of the viruses should be retained in the endosomes at this stage. Confocal time-lapse images were then acquired every ∼10 s over a time period of 20 min (Supplementary Movie 2). At 0 s, the viral particle appeared to be yellow (a merged color of green and red), indicating that fusion had already taken place (Figure 6). At 461 s, the GFP-Vpr-labeled viral core (green) moved away from the DiD-labeled endosome (red), suggesting that the virus is being released into the cytosol. We recorded ∼50 videos and 12 of them showed the similar process of virus release illustrated in Figure 6. This seemingly low yield (24%) could be partially due to photo-bleaching as a result of long-time exposure to the laser source during the recording. No virus release signal was detected for the control virus lacking SINmu (FUW-GFPVpr/αCD20) (Supplementary Figure 4). This live cell imaging suggests that the completion of virus membrane fusion to release the viral core, which is required for the delivery of viral genome to the cell nucleus, is relatively long-term and possibly a sequential multi-step process.40,41
Figure 6.
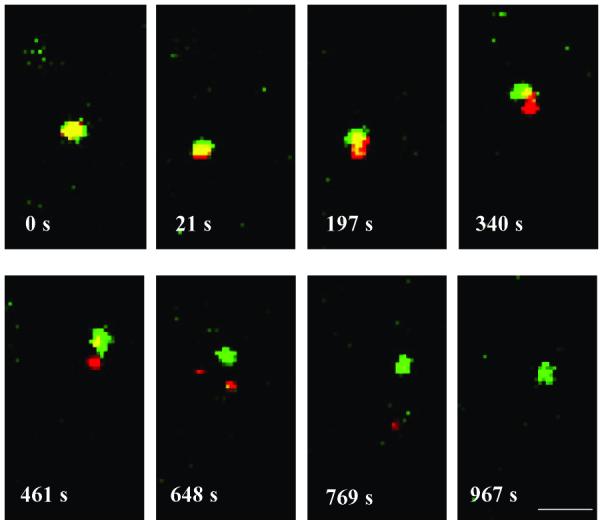
Time series images of the viral core release from an endosome starting 60 min after incubation. GFP-Vpr/DiD-labeled viruses (FUW-GFPVpr/αCD20+SINmu) were incubated with 293T/CD20 cells at 37°C for 60 min to initiate virus fusion, and then time-series images were obtained at every ∼10 seconds over a time period of 20 min. Scale bar represents 2 μm.
DISCUSSION
In this study, we have visualized individual engineered lentiviruses and have analyzed the intracellular behavior of viruses in the target cells using single viral particle tracking via confocal microscope. GFP-Vpr labeling along with other immunostaining methods has allowed us to confirm the co-incorporation of an antibody and fusogenic molecule on a single virion, separating the functions of binding and fusion, respectively. We have confirmed by the direct visualization of virus-host cell interactions that the antibody displayed on the virion surface targets a specific cell type and we have shown that viral particles were efficiently endocytosed upon antibody binding. Subsequently, the surface-displayed fusogenic molecule, SINmu,10 which is an engineered glycoprotein derived from the Sindbis virus that is binding-deficient and fusion-competent, mediated fusion in the acidic endosomal environment. By observing the trafficking of single lentivirus intracellularly, we have shed some light on the endocytic mechanisms of engineered lentivirus such as virus-endosome fusion and endosomal transport of viral particles in target cells.
By using a fluorescence de-quenching assay, we have detected the actual fusion event of the GFP-Vpr/DiD double-labeled viral particle and monitored the dynamics of fusion in living cells. Colocalization studies using endocytic markers suggested that the endosomal fusion of the engineered lentivirus started at an early stage of the endocytic pathway. Inhibitory drug treatments showed that the transport and maturation steps of the virus-endosome complex from the early to the intermediate endosome require microtubule- or actin-assisted transport. However, virus-endosome fusion was not restricted by the inhibition of microtubule- or actin-associated transport, suggesting that the majority of viral fusion begins at the early endosome level.
We also studied the fusion kinetics of the engineered lentivirus and compared it to VSVG-pseudotyped lentivirus, which is known to require low pH to mediate fusion.42 It has been shown that the fusion of VSV occurs before the late endosome stage.27 Our data suggests that it takes 70∼80 min more for the engineered lentivirus to reach a half-maximum of fusion-involved penetration than it does for VSVG-pseudotyped virus. We suspect that one possible reason for this delay could be due to the separation of the functions of binding and fusion into two individual molecules on our targeting lentivirus system. The different requirements for fusion of different viruses could be another cause for the slower entry kinetics of the engineered lentivirus. For example, the Sindbis virus requires cholesterol for the endosomal fusion, while the fusion of VSV is independent of cholesterol.43 We are conducting experiments to investigate the effects of such membrane components on the entry efficiency of the engineered lentivirus.
Using the real-time imaging of single viruses, we were able to observe the dynamics of virus-endosome fusion and transport. After the viral fusion signal was seen, we observed that many virus-containing endosomes (60%) moved towards the nucleus. Since the lipophilic membrane dye is initially incorporated onto the outer membrane of the virus, the lipid mixing (i.e. hemifusion) between the viral outer membrane and the endosomal inner membrane is initially visualized by the increase of DiD-fluorescence. The fusion-pore formation and enlargement of the pore is believed to be required for the completion of the virus membrane fusion in order to release the viral core into the cytosol.41,44 Our live-cell imaging of viral release seems to support this hypothesis. From the experiments of the drug and siRNA treatments (Figure 5A), we have learned that the hemifusion (i.e. lipid mixing) takes place before the late endosome stage. The reduced infectivity upon the drug and siRNA treatments (Figure 5B & 5D) indicates that the release of the viral core, which is a necessary step for a productive infection, is associated with the maturation process from the early to the intermediate endosomal compartments. This suggests that the virus is at least transported to the intermediate endosome to complete infection.
In summary, we demonstrated that the direct visualization of individual viruses can allow us to determine the endocytic pathway of the engineered lentivirus in living cells. The single-virus tracking techniques confirmed our hypothesis of the mechanism of targeted transduction for the engineered lentivirus. Our observations, based on the kinetics of entry of engineered lentivirus, indicate that the fusogen-mediated membrane fusion is a long process, which is markedly slower than that of VSVG-pseudotyped lentivirus, suggesting that this might be the rate limiting step of virus transduction. Therefore, the development of fusogenic molecules which induce more efficient and stable fusion could be a promising step in enhancing the lentivirus-mediated gene delivery efficacy.
MATERIALS AND METHODS
Cell lines and antibodies
The 293T/CD20 cell line was generated previously.10 293T and 293T/CD20 cells were maintained in a 5% CO2 environment in Dulbecco’s modified Eagle medium (Mediatech, Inc.) with 10% FBS (Sigma), and 2 mM L-glutamine (Hyclone). Mouse monoclonal antibody specific to the human CD20 antigen were purchased from Caltag Laboratories. Anti-HA-biotin to stain SINmu was obtained from Miltenyi Biotec Inc. Mouse monoclonal antibody against the early endosome antigen 1 (EEA1), rabbit polyclonal antibody specific to the Mannose 6 phosphate receptor (CI-MPR), and Cy5-conjugated goat anti-rabbit IgG antibody were purchased from Abcam. Cy5-conjugated streptavidin was purchased from Zymed Laboratories. Texas red-labeled goat anti-mouse IgG and AlexaFluor 594-labeled goat anti-human IgG antibodies were obtained from Molecular Probes. Taxol, nocodazole, cytochalasin D, and bafilomycin A1 were purchased from Sigma.
Plasmids
Assembly PCR was employed to fuse the GFP to the N-terminus of viral protein R (Vpr). The PCR product was then inserted into pcDNA3 (Invitrogen) to form pcDNA3-GFPVpr. The cDNAs for Rab5 and Rab7 were PCR-amplified and cloned into the pDsRed-monomer-C1 (Clontech) to form DsRed-Rab5 and DsRed-Rab7, respectively.
Virus production
GFP-Vpr-labeled lentiviruses were produced by transfecting 293T cells by a calcium phosphate precipitation method. 293T cells at 80% confluence in 6 cm culture dishes were transfected with 5 μg of the lentiviral plasmid FUW, together with 2.5 μg each of pcDNA3-GFPVpr, pαCD20 (encodes a mouse/human chimeric anti-CD20 antibody), pIgαβ (encodes human Igα and Igβ, two immunoglobulin associated proteins that are required for surface expression of antibodies), pSINmu, and the packaging vector plasmids.10 Cells were washed at 4 h posttransfection, and then medium was replaced. The viral supernatant was collected after 48 h posttransfection and filtered through a 0.45-μm pore size filter. For high-titer lentivectors, the viral supernatant was concentrated by ultracentrifugation (Optima L-90 K ultracentrifuge, Beckman Coulter) for 90 min at 82,700 × g and resuspended in an appropriate volume of Hank’s balanced salt solution (HBSS, Hyclone).
Viral transduction
For viral transduction, 293T/CD20 cells (0.2 × 106 per well) were plated in a 24-well culture dish and spin-infected with viral supernatants of FUGW/αCD20+SINmu (2 ml per well) at 2,500 rpm and 30°C for 90 min by using a Sorval Legend centrifuge. Then, the medium was removed and replaced with fresh medium and cultured for 3 days further before FACS analysis of GFP+ cells. For viral transduction with drug- or siRNA-treated cells, 293T/CD20 cells were preincubated with drugs (nocodazole (60 μM), cytochalasin D (20 μM), and bafilomycin A1 (25 nM)) or transfected with siRNAs, and then the cells (0.2 × 106 per well) were spin-infected with 2 ml of viral supernatants in a 24-well culture dish.
Time course study of fusion inhibition
293T/CD20 cells (0.4 × 105 per well) were seeded in a 96-well culture dish overnight. Viruses were added to cells and incubated at 4°C for 1 h and the resulting cells were washed with cold PBS to remove unbound viruses. The viral entry was initiated by shifting cells to 37°C. At the indicated time points, D10 media containing 25 nM of bafilomycin A1 was added to inhibit the endosome acidification. The drug was removed 3 h later and replaced with fresh D10 media. Cells were further incubated for 72 h, and the percentage of GFP-positive cells was analyzed by FACS.
Fluorescent labeling
For the detection of individual viral particles, fresh viral supernatant was overlaid upon poly-lysine-coated NO.1 coverslips in a six-well culture dish and centrifuged at 3,700 × g and 4°C for 2 h in a Sorval Legend RT centrifuge. The coverslips were rinsed with cold PBS twice, the adhered viruses were immunostained by Alexa 594 anti-human IgG and anti-HA-biotin antibodies, and they were then incubated with Cy5-streptavidin. The coverslips were mounted in Vectashield (Vector Laboratories), which is an antifade mounting medium. For imaging virus-cell binding, 5 × 105 cells were seeded onto a 35 mm glass-bottom culture dish (MatTek Corporation) and grown at 37°C overnight. The seeded cells were rinsed with cold PBS twice and incubated with the concentrated viruses for 1 h at 4°C to allow for binding. Cells were washed with cold PBS to remove unbound viruses and then fixed with 4% formaldehyde on ice for 10 min. To co-label the viral particles bound to the cell surfaces, Alexa594 anti-human IgG antibody was used to stain against the αCD20 heavy chain. Fluorescent images were taken by a Zeiss LSM 510 laser scanning confocal microscope equipped with filter sets for fluorescein, rhodamine or Texas red, and Cy5. A plan-apochromat 63×/1.4 oil immersion objective was used for imaging. Images were analyzed with the use of the Zeiss LSM 510 software version 3.2 SP2.
Imaging virus fusion and transport through endosomes
The concentrated viruses were incubated with 100 μM of 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine (DiD) (Molecular Probes) for 1 h at room temperature. For imaging virus-endosome fusion, double labeled viruses were incubated with 293T/CD20 cells at 37°C for various time periods and then fixed with 4% formaldehyde. GFP-Vpr and DiD were excited simultaneously with a 488 nm Argon and a 633 nm HeNe laser, respectively, and the emitted light was separated through the corresponding emission filter sets. All samples were scanned under the same conditions for magnification, laser intensity, brightness, gain, and pinhole size. For live cell imaging of virus fusion, 293T/CD20 cells were preincubated on a glass bottom culture dish at 37°C overnight. Viruses were incubated with the cells at 37°C for 30 min to initiate virus internalization. Images were then collected using the confocal microscope. Fluorescence intensity vs. time within the regions of interest around the virus particles were measured by using the Zeiss LSM 510 software package.
For observation of the colocalization of the virus with different endosomal markers, GFP-Vpr-labeled viruses were incubated with 293T/CD20 cells at 37°C and fixed after different time periods. Cells were then permeabilized with 0.1% Triton X-100 and immunostained with EEA1 and CI-MPR for early endosome and late endosome markers, respectively. Texas red-conjugated anti-mouse IgG and Cy5-conjugated goat anti-rabbit IgG antibodies were used as the secondary antibodies. To remove viral aggregates, virus-containing media was filtered by a 0.45 μm pore size centrifuge tube filter (Costar, NY) just before the experiments were conducted.
For the viral trafficking studies using Rab5 and Rab7 constructs, 293T/CD20 cells were transfected with DsRed-Rab5 or DsRed-Ran7 plasmid. At 48 h posttransfection, cells were seeded onto the glass-bottom culture dish and grown at 37°C overnight. GFP-Vpr-labeled viruses at MOI ∼30 were added and incubated at 37°C for the different time points and then fixed.
For the colocalization study with transferrin, GFP-Vpr-labeled viruses and 2 μM of Alexa647-conjugated transferrin (Molecular Probes) were mixed and then incubated with 293T/CD20 cells at 37°C for 60 or 120 min. Cells were fixed, permeabilized, and immunostained with EEA1 to compare trafficking of the engineered lentivirus with transferring through early to recycling endosomes.
For the real-time observation of the release of the virus from the endosome, GFP/DiD-labeled viruses were incubated with cells at 37°C for 1 h to induce virus-endosome fusion, and confocal time-lapse images were then recorded.
Microtubule-mediated transport
To observe viruses on microtubule networks, the cells were incubated with GFP-Vpr-labeled viruses at 37°C for 1 h, fixed, and then permeabilized with 0.1% Triton X-100 containing 20 μM of taxol. Microtubules were then immunostained with anti-α-tubulin mAb (Sigma) and Texas red-conjugated anti-mouse secondary antibody. For the microtubule-disrupting assay, cells were preincubated with D10 media containing 60 μM of nocodazole at 37°C for 30 min, and then viruses were added and incubated for further studies.
For the inhibition assay using small interfering RNA (siRNA), we purchased α-tubulin siRNA and the negative control siRNA from Santa Cruz Biotechnology, Inc. The transfection of siRNA was performed as described by the manufacture’s protocol. At 72 h posttransfection, equal numbers of α-tubulin siRNA and control siRNA treated 293T/CD20 cells were used for further studies.
Actin-mediated transport
To visualize viruses on actin filaments, the cells were incubated with GFP-Vpr-labeled viruses at 37°C for 1 h, fixed, and permeabilized. Actin filaments were labeled with rhodamine-conjugated phalloidin (Molecular Probes). For the actin-disrupting assay, cells were preincubated with D10 media containing 20 μM of cytochalasin-D at 37°C for 30 min, and then viruses were added and incubated for further studies.
Supplementary Material
ACKNOWLEDGEMENTS
We thank April Tai and Lili Yang for critical reading of the manuscript, and USC Norris Center Cell and Tissue Imaging Core. This work was supported by a National Institute of Health grant AI068978.
REFERENCES
- 1.Daly G, Chernajovsky Y. Recent developments in retroviral-mediated gene transduction. Mol. Ther. 2000;2:423–434. doi: 10.1006/mthe.2000.0211. [DOI] [PubMed] [Google Scholar]
- 2.Mountain A. Gene therapy: the first decade. Trends Biotechnol. 2000;18:119–128. doi: 10.1016/s0167-7799(99)01416-x. [DOI] [PubMed] [Google Scholar]
- 3.Somia N, Verma IM. Gene therapy: Trials and tribulations. Nat. Rev. Genet. 2000;1:91–99. doi: 10.1038/35038533. [DOI] [PubMed] [Google Scholar]
- 4.Verma IM, Somia N. Gene therapy-promises, problems and prospects. Nature. 1997;389:239–242. doi: 10.1038/38410. [DOI] [PubMed] [Google Scholar]
- 5.Bukrinsky MI, Sharova N, Dempsey MP, Stanwick TL, Bukrinskaya AG, Haggerty S, et al. Active nuclear import of human immunodeficiency virus type 1 preintegration complexes. Proc. Natl. Acad. Sci. USA. 1992;89:6580–6584. doi: 10.1073/pnas.89.14.6580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lewis P, Hensel M, Emerman M. Human immunodeficiency virus infection of cells arrested in the cell cycle. EMBO J. 1992;11:3053–3058. doi: 10.1002/j.1460-2075.1992.tb05376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weinberg JB, Matthews TJ, Cullen BR, Malim MH. Productive human immunodeficiency virus type 1 (HIV-1) infection of nonproliferating human monocytes. J. Exp. Med. 1991;174:1477–1482. doi: 10.1084/jem.174.6.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cronin J, Zhang XY, Reiser J. Altering the tropism of lentiviral vectors through pseudotyping. Curr. Gene Ther. 2005;5:387–398. doi: 10.2174/1566523054546224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waehler R, Russell SJ, Curiel DT. Engineering targeted viral vectors for gene therapy. Nat. Rev. Genet. 2007;8:573–587. doi: 10.1038/nrg2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang L, Baiely L, Balimore D, Wang P. Targeting lentiviral vectors to specific cell types in vivo. Proc. Natl. Acad. Sci. USA. 2006;103:11479–11484. doi: 10.1073/pnas.0604993103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gollan TJ, Green MR. Redirecting retroviral tropism by insertion of short, nondisruptive peptide ligands into envelope. J. Virol. 2002;76:3558–3563. doi: 10.1128/JVI.76.7.3558-3563.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lavillette D, Russell SJ, Cosset FL. Retargeting gene delivery using surface-engineered retroviral vector particles. Curr. Opin. Biotech. 2001;12:461–466. doi: 10.1016/s0958-1669(00)00246-9. [DOI] [PubMed] [Google Scholar]
- 13.Maurice M, Verhoeyen E, Salmon P, Trono D, Russell SJ, Cosset FL. Efficient gene transfer into human primary blood lymphocytes by surface-engineered lentiviral vectors that display a T cell-activating polypeptide. Blood. 2002;99:2342–2350. doi: 10.1182/blood.v99.7.2342. [DOI] [PubMed] [Google Scholar]
- 14.Nguyen TH, Pages JC, Farge D, Briand P, Weber A. Amphotropic retroviral vectors displaying hepatocyte growth factor-envelope fusion proteins improve transduction efficiency of primary hepatocytes. Hum. Gene Ther. 1998;9:2469–2479. doi: 10.1089/hum.1998.9.17-2469. [DOI] [PubMed] [Google Scholar]
- 15.Sandrin V, Russell SJ, Cosset FL. Targeting retroviral and lentiviral vectors. Curr. Top. Microbiol. Immunol. 2003;281:137–178. doi: 10.1007/978-3-642-19012-4_4. [DOI] [PubMed] [Google Scholar]
- 16.Somia NV, Zoppe M, Verma IM. Generation of targeted retroviral vectors by using single-chain variable fragment: an approach to in vivo gene delivery. Proc. Natl. Acad. Sci. USA. 1995;92:7570–7574. doi: 10.1073/pnas.92.16.7570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Desmaris N, Bosch A, Salaun C, Petit C, Prevost M, Tordo N, et al. Production and neurotropism of lentivirus vectors pseudotyped with lyssavirus envelope glycoproteins. Mol. Ther. 2001;4:149–156. doi: 10.1006/mthe.2001.0431. [DOI] [PubMed] [Google Scholar]
- 18.Mochizuki H, Schwartz JP, Tanaka K, Brady RO, Reiser J. High-titer human immunodeficiency virus type 1-based vector systems for gene delivery into nondividing cells. J. Virol. 1998;72:8873–8883. doi: 10.1128/jvi.72.11.8873-8883.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schnierle BS, Stitz J, Bosch V, Nocken F, Merget-Millitzer H, Engelstadter M, et al. Pseudotyping of murine leukemia virus with the envelope glycoproteins of HIV generates a retroviral vector with specificity of infection for CD4-expressing cells. Proc. Natl. Acad. Sci. USA. 1997;94:8640–8645. doi: 10.1073/pnas.94.16.8640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson JL, Hope TJ. Intracellular trafficking of retroviral vectors:obstacles and advances. Gene Ther. 2005;12:1667–1678. doi: 10.1038/sj.gt.3302591. [DOI] [PubMed] [Google Scholar]
- 21.Gruenberg J. Membrane traffic in endocytosis: insights from cell-free assays. Nat. Rev. Mol. Cell Biol. 2001;2:721–730. doi: 10.1146/annurev.cb.05.110189.002321. [DOI] [PubMed] [Google Scholar]
- 22.Klasse P, Bron R, Marsh M. Mechanisms of enveloped virus entry into animal cells. Adv. Drug. Delivery Rev. 1998;34:65–91. doi: 10.1016/S0169-409X(98)00002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin K, Helenius A. Transport of incoming influenza-virus nucleocapsids into the nucleus. J. Virol. 1991;65:232–244. doi: 10.1128/jvi.65.1.232-244.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Apodaka G. Endocytic traffic in polarized epithelial cells: Role of the actin and microtubule cytoskeleton. Traffic. 2001;2:149–159. doi: 10.1034/j.1600-0854.2001.020301.x. [DOI] [PubMed] [Google Scholar]
- 25.Mallik R, Gross SP. Molecular motors: Strategies to get along. Curr. Biol. 2004;14:R971–R982. doi: 10.1016/j.cub.2004.10.046. [DOI] [PubMed] [Google Scholar]
- 26.Lakadamyali M, Rust MJ, Zhuang X. Ligands for clathrin-mediated endocytosis are differentially sorted into distinct populations of early endosomes. Cell. 2006;124:997–1009. doi: 10.1016/j.cell.2005.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sieczkarski SB, Whittaker GR. Differential requirements of rab5 and rab7 for endocytosis of influenza and other enveloped viruses. Traffic. 2003;4:333–343. doi: 10.1034/j.1600-0854.2003.00090.x. [DOI] [PubMed] [Google Scholar]
- 28.Vonderheit A, Helenius A. Rab7 associates with early endosomes to mediate sorting and transport of semliki forest virus to late endosomes. PLos Bio. 2005;3:1225–1238. doi: 10.1371/journal.pbio.0030233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McDonald D, Vodicka MA, Lucero G, Svitkina TM, Borisy GG, Emerman M, et al. Visualization of the intracellular behavior of HIV in living cells. J. Cell Biol. 2002;159:441–452. doi: 10.1083/jcb.200203150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–872. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- 31.Lakadamyali M, Rust MJ, Babcock HP, Zhuang X. Visualizing infection of individual influenza viruses. Proc. Natl. Acad. Sci. USA. 2003;100:9280–9285. doi: 10.1073/pnas.0832269100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sakai T, Ohuchi M, Imai M, Mizuno T, Kawasaki K, Kuroda K, et al. Dual wavelength imaging allows analysis of membrane fusion of influenza virus inside cells. J. Virol. 2006;80:2013–2018. doi: 10.1128/JVI.80.4.2013-2018.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marsh M, Bolzau E, Helenius A. Penetration of Semliki Forest virus from acidic prelysosomal vacuoles. Cell. 1983;32:931–940. doi: 10.1016/0092-8674(83)90078-8. [DOI] [PubMed] [Google Scholar]
- 34.Bowman EJ, Siebers A, Altendorf K. Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc Natl Acad Sci U S A. 1988;85:7972–7976. doi: 10.1073/pnas.85.21.7972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Urayama AJHG, Sly WS, Banks WA. Developmentally regulated mannose 6-phosphate receptor-mediated transport of a lysosomal enzyme across the blood-brain barrier. Proc. Natl. Acad. Sci. USA. 2004;101:12658–12663. doi: 10.1073/pnas.0405042101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rink J, Ghigo E, Kalaidzidis Y, Zerial M. Rab conversion as a mechanism of progression from early to late endosomes. Cell. 2005;122:735–749. doi: 10.1016/j.cell.2005.06.043. [DOI] [PubMed] [Google Scholar]
- 37.Stoorvogel W, Strous GJ, Geuze HZ, Oorschot V, Schwartz AL. Late endosomes derive from early endosomes by maturation. Cell. 1991;65:417–427. doi: 10.1016/0092-8674(91)90459-c. [DOI] [PubMed] [Google Scholar]
- 38.Chavrier P, Parton RG, Hauri HP, Simons K, Zerial M. Localization of low molecular weight GTP binding proteins to exocytic and endocytic compartments. Cell. 1990;62:317–329. doi: 10.1016/0092-8674(90)90369-p. [DOI] [PubMed] [Google Scholar]
- 39.Zerial M, McBride H. Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2001;2:107–117. doi: 10.1038/35052055. [DOI] [PubMed] [Google Scholar]
- 40.Jahn R, Lang T, Sudhof TC. Membrane fusion. Cell. 2003;122:519–533. doi: 10.1016/s0092-8674(03)00112-0. [DOI] [PubMed] [Google Scholar]
- 41.Melikyan GB, Barnard RJO, Abrahamyan LG, Mothes W, Young JAT. Imaging individual retroviral fusion events: from hemifusion to pore foramation and growth. Proc. Natl. Acad. Sci. USA. 2005;102:8728–8733. doi: 10.1073/pnas.0501864102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun X, Yau VK, Briggs BJ, Whittaker GR. Role of clathrin-mediated endocytosis during vesicular stomatitis virus entry into host cells. Virology. 2005;338:53–60. doi: 10.1016/j.virol.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 43.Lu YE, Cassese T, Kielian M. The cholesterol requirement for sindbis virus entry and exit and characterization of a spike protein region involved in cholesterol dependence. J. Virol. 1999;73:4272–4278. doi: 10.1128/jvi.73.5.4272-4278.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Markosyan RM, Cohen FS, Melikyan GB. Time-resolved imaging of HIV-1 env-mediated lipid and content mixing between a single virion and cell membrane. Mol. Biol. Cell. 2005;16:5502–5513. doi: 10.1091/mbc.E05-06-0496. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.