

Abstract
All trans retinoic acid (atRA) has been shown to inhibit the growth of CAOV3 ovarian carcinoma cells and to elevate the level of p27 cyclin dependent kinase inhibitor. We report here that phosphorylation at S10 residue is an important event in mediating p27 role in atRA induced growth arrest. atRA treatment of atRA sensitive CAOV3 cells increases the levels of S10 phospho p27 in both nuclear and cytoplasmic cell compartments. This increase is accompanied by a decrease in the levels of skp2 protein. This effect was not observed in SKOV3 cells which are resistant to atRA growth inhibitory effect. An A10p27 mutant that cannot be phosphorylated at S10 induces a dominant negative effect on the atRA effect on the levels and activity of endogenous p27. Overexpression of A10p27 mutant renders CAOV3 cells more resistant to atRA treatment and reverses the effect that atRA has on p27 binding to CDKs, on CDK activity, and on the expression of S phase genes.
Keywords: retinoic acid, ovarian cancer, growth arrest, p27, phosphorylation
Introduction
All trans retinoic acid (atRA) is the biologically active derivative of vitamin A. atRA has been considered a potentially promising chemotherapeutic agent, due to its pro-apoptotic and growth suppressing activity. The growth of a variety of solid tumors and tumor derived cell lines, including breast, ovarian and colon, has been shown to be inhibited by atRA (Kaleagasioglu et al, 1993, Wu et al, 1997).
Previous results from our laboratory have demonstrated that atRA treatment of the ovarian carcinoma cell line CAOV3 results in >50% growth suppression. This results from arrest of the cell cycle during the G1 phase (Wu et al, 1997). The G1 check point is regulated by a multitude of molecules, including the retinoblastoma family of proteins, cyclin dependent kinases (Cdks), and cyclin dependent kinase inhibitors (Cdki) (Stiegler and Giordano, 1999). P27 is a well studied Cdki whose altered expression has been linked to a number of neoplastic conditions, including ovarian cancer (Lloyd et al, 1999). In an attempt to understand the molecular mechanisms by which atRA suppresses ovarian carcinoma cell growth we previously compared the expression of a variety of cell cycle regulatory proteins following atRA treatment of atRA sensitive CAOV3 cells versus atRA resistant SKOV3 cells. We observed a striking increase in the levels of p27 protein following atRA treatment of CAOV3 cells but not SKOV3 cells (Vuocolo et al., 2004).
P27 expression and function is regulated by transcriptional (Servant et al., 2000) and translational (Agrawal et al., 1996; Hengst and Reed, 1996; Millard et al., 1997) mechanisms and through degradation by the 26S proteosome and miRNA (le Sage et al., 2007). Serine 10 (S10) is the major phosphorylation site on p27 (Ishida et al., 2000). Phosphorylation at this residue increases the stability of p27 in vitro (Nakayama et al., 2000) and in vivo (Borriello, 2006) and signals the nuclear export of p27 to the cytoplasm upon cell cycle re-entry (Rodier et al., 2001). S10 phosphorylation has also been shown to be required for T157 phosphorylation by AKT and subsequent protein localization in the cytoplasm (Shin et al., 2005).
In these studies we investigated the role of p27 phosphorylation in mediating atRA induced growth inhibition. Our results show that atRA treatment of atRA sensitive CAOV3 cells leads to an increase in the levels of S10 phosphorylation of p27 in both nuclear and cytoplasmic cell compartments. This increase was accompanied by a decrease in the levels of skp2 protein. Similar results were not observed in SKOV3 cells which are not growth inhibited by atRA treatment. Finally, we demonstrated that overexpression of a mutant of p27 that cannot be phosphorylated on S10 induces a dominant negative effect on the endogenous p27 activity. This dominant negative effect reverses the atRA effect on p27 binding to CDKs, on inhibition of CDK activity, on the expression of S phase genes and ultimately on the inhibition of growth of ovarian carcinoma cells. Thus, phosphorylation of serine 10 in p27 is critical for the inhibition of growth by atRA.
Materials and Methods
Cell Culture
The human ovarian carcinoma cell lines, CAOV3 and SKOV3 were obtained from the American Type Culture Collection (Rockville, MD). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 2mM L-glutamine, 100 units/ml penicillin and 100 μg/ml streptomycin.
Antibodies and Reagents
Anti-p27 (sc-528), anti-S10-P-p27 (sc-12939-R), anti-p21 (sc-756), anti-p57 (sc-1040), anti-p16 (sc-468), anti-cyclin A (sc-751), anti-cyclin D (sc-718), anti-cyclin E (sc-481), anti-CDK1 (sc-54), anti-CDK2 (sc-163), anti-CDK4 (sc-260), anti-CDK6 (sc-177), anti-Rb2/p130 (sc-317) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-T157-P-p27 was purchased from Cell Signaling. Anti-enolase (sc-7455, Santa Cruz Biotechnology), anti-lamin B (sc-6217, Santa Cruz Biotechnology), anti-actin (sc-1616 Santa Cruz Biotechnology) antibodies were used for normalization. atRA was obtained from Hoffman La Roche (Nutley, NJ) as a generous gift and used at a concentration of 1μM in ethanol.
Western Blotting and Immunoprecipitation
Whole cell extracts (WCE) were prepared by lysing the cell pellets in 1×TNE buffer (0.5M tris ph 8.0, 1.5 M NaCl, 10% Nonidet P-40, 20mM EDTA ph 8.0) for 30 min at 4°C then centrifuged at 20,000 g for 15 min. Cytoplasmic extracts were prepared by lysing the cells in hypotonic buffer (10 mM Hepes ph 7.9, 1.5 mM MgCl2, 10mM KCl, 0.2mM PMSF 0.5mM DTT) followed by mechanical lysing through a 22G syringe needle and spun at 2600 g. The supernatant was stored as cytoplasmic protein extract. The remaining pellet was washed with wash buffer (10mM hepes, 1mM EDTA, 60mM KCl) and centrifuged at 2600 g for 10 min. The new pellet was resuspended in nuclear resuspension buffer (60mM KCl, 250mM Tris ph 7.8). After a 15 min spin at 20,000 g the supernantant was stored as nuclear protein extract. Protein concentration was measured by the Bradford assay using bovine serum albumin as a standard (Bio-Rad, Hercules, CA). Proteins were loaded on 7 - 15% SDS-PAGE gels, electrophoresed and transferred to PDVF membrane. The membrane was blocked in 5% nonfat dry milk, incubated with a 1:500 - 1:1000 dilutions of primary antibody. Horse-radish peroxidase-conjugated secondary antibodies (Amersham Biosciences, Buckinghamshire, England) were used at a dilution of 1:1500, and immunoreactive proteins were visualized with the ECLTM or ECL-plusTM enhanced chemiluminescence detection system (Amersham Biosciences). For immunoprecipitation, 500 μg of WCE was incubated with 4 μg of antibody for 3 hours at 4°C and then incubated with BSA-blocked A/G PLUS agarose beads (sc-2003, Santa Cruz Biotechnology) for 3 hours at 4°C. After washing, imunoprecipitates were loaded on SDS-PAGE gel and Western blot was performed.
Plasmid constructs and generation of stable cell lines
P27 cDNA flanked by BamHI and XhoI restriction sites was obtained by amplification of mRNA from CAOV3 cells using the primers: sense 5′AGGGATCCATGTCAAACGTGGGAGTC 3′ and anti sense 5′AGCTCGAGTTACGTTTGACGTCTTCT 3′. p27 coding region was cloned into pCDNA3.1 His/C (Invitrogen, San Diego, CA). “QuikChange site-directed mutagenesis kit” from Stratagene (La Jolla, CA) was used to prepare site-directed mutations of wt p27. S10 sequence AGC was changed to GCC for the alanine mutant (A10p27) and to GAG for the glutamic acid mutant (E10p27). Mutations were confirmed by DNA sequencing. Wild type and mutant p27 plasmids were transfected into CAOV3 and SKOV3 cells and stable clones were selected with 800ug/ml G418.
Kinase assay
CDK1, CDK2, CDK4 and CDK6 activity was measured using the IQTM system from Pierce (Rockford, IL). The assay was performed on immunoprecipitates from CAOV3 cells and CAOV3 over-expressing the wild type and A10 mutant p27. 500ug total protein was immunoprecipitated with 4ug antibody and 30 ul protein A/G beads (Santa Cruz). After 3 hour incubation at 4°C the beads were washed 5 times with TNE buffer and incubated in kinase buffer with 1ul of the synthetic dye labeled substrate provided for 2 hours. The reaction was stopped and equilibrated with Stop buffer. Fluorescence intensity was measured with a Millipore (Billerica, MA) fluorometer.
Real time PCR
Total RNA was isolated using the RNA-Bee reagent (TelTest, Inc, Friendswood, TX) following the manufacturer’s protocol. 1ug of RNA was reverse transcribed into cDNA using a RT for PCR kit (Clontech, Mountain View, CA) following the manufacturer’s protocol. Real time quantitative PCR was performed using SYBR green PCR Master Mix (Applied Biosystems, Foster City, CA). The PCR reaction was incubated at 95°C for 10min followed by 40 amplification cycles that consisted of a 45sec denaturation step at 95°C, a 45 sec annealing step at 60°C and a 1.5 min extension step at 72°C. Following primers were used: TK: forward 5′CCTGAGGATGGCCTGGATTCA3′, reverse: 5′ATTTCATAAGCTACAGCAGAG3′, DHFR: forward 5′CTGTTTATAAGGAAGCCATGAATC3′, reverse 5′ACACCTGGGTATTCTGGCAG3′, CDC2: forward 5′GGGGATTCAGAAATTGATCA3′, reverse 5′TGTCAGAAAGCTACATCTTC3′, CDC6: forward 5′TCAAACCCGATCCCAGGCACAGGC3′, reverse 5′ACCGCTGCTGGATCTGAG3′.
Results
atRA treatment results in increased levels of total and S10 phospho p27 but not T157 phospho p27 in atRA sensitive CAOV3 cells, but not atRA resistant SKOV3 cells
Our studies have utilized two well characterized ovarian carcinoma cell lines, CAOV3 and SKOV3. As we have previously reported, Figure 1A shows that treatment with 1uM atRA results in ∼50% inhibition of growth of CAOV3 cells but not of SKOV3 cells (Wu et al, 1997). Also, consistent with previous results from our laboratory, Figure 1B shows that atRA treatment for 5 days induces a ∼5-8 fold increase in the levels of p27 protein in CAOV3 cells but not in SKOV3 cells. (Vuocolo et al., 2004). Since accumulation of p27 protein appears to correlate with atRA inhibition of ovarian carcinoma cell growth, and the accumulation and functional activity of p27 is determined by phosphorylation of specific residues, we wished to investigate the effect of atRA treatment on p27 phosphorylation.
Figure 1. atRA treatment increases the levels of p27 and S10 phospho p27 in both the cytoplasmic and the nuclear cell compartment.

A.CAOV3 and SKOV3 cells were treated for the indicated time with ethanol or 1uM atRA. Live cells were counted using trypan blue staining. B. Western blot analysis of CAOV3 and SKOV3 cells treated for 1,3 and 5 days with 1uM atRA. Cell lysates were subjected to western blotting to detect the p27, S10 phospho p27 and T157 phospho p27. Actin was used as loading control.
C. Cytoplasmic, nuclear and whole cell protein extracts were obtained from CAOV3 cells treated for 5 days with ethanol or 1uM atRA and subjected to western blot analyses. Enolase and Lamin B were used as cytoplasmic and nuclear markers.
Levels of p27 have been shown to be regulated mostly at the posttranslational level. Serine 10 is the major site of phosphorylation and has been shown to be required for subsequent phosphorylation at T157 which determines p27 localization in the cell (Shin et al, 2005). In order to determine if atRA treatment affected the phosphorylation status of p27, we used phospho-specific p27 antibodies to evaluate the effect of atRA treatment on the levels of these two phospho-p27 forms. Figure 1B shows that upon 3 and 5 day atRA treatment of the sensitive cell line CAOV3 resulted in an increase in the amount of p27 phosphorylated at the S10 residue. This increase is proportional and occurs at the same time point as the atRA induced increase in the levels of total p27. The amount of p27 phosphorylated at T157 was not altered by atRA treatement. The atRA resistant cell line SKOV3 did not exhibit any significant modulation of phospho-p27 levels upon atRA treatment.
It has been reported (Rodier et al, 2001) that one mechanism by which p27 protein levels and function are down regulated is by export of the protein from the nucleus to the cytoplasm where it is degraded via the ubiquitin/26S proteasome system. We hypothesized that since atRA treatment leads to an increase in p27 levels, atRA could actually induce the nuclear import of p27 where it would be protected from degradation and where it would bind to cdks and inhibit their kinase activity. However, western blot results shown in Figure 1C comparing the levels of p27 in nuclear versus cytoplasmic extracts from cells treated with atRA and cells treated with the ethanol vehicle show that p27 and S10-phospho p27 levels increase in both the nuclear and cytoplasmic compartment. T157-phospho p27 levels remain the same in ethanol and atRA treated CAOV3 cells in the cytoplasmic extracts. As expected, there was no T157-phospho p27 detected in the nuclear extracts. We treated CAOV3 cells with leptomycin B to inhibit this process. P27 localization within the cell proved to be leptomycin B independent (data not shown). In light of the western blot results and previous data from our laboratory (Vuocolo et al, 2004) it would appear that the atRA induced accumulation of p27 is not a result of an alteration in nuclear trafficking.
Phosphorylation of p27 at residue S10 is a critical event in mediating atRA sensitivity in CAOV3 cells
atRA treatment induces an increase in the levels of both total p27 and S10-phospho p27. We next wished to determine if this phospho form is critical in mediating atRA induced growth inhibition. To address this question we created a p27 mutant that cannot be phosphorylated at the S10 residue because the serine has been mutated to alanine (A10p27). We transfected this histidine-tagged A10p27 mutant plasmid into CAOV3 cells. As a control we also transfected CAOV3 cells with a histidine-tagged wild type p27 encoding plasmid. Stable single cell clones were selected and expanded. It should be noted that the his-tagged wild type recombinant p27 is a functional protein as proven by its ability to interact with CDK 1,2,4 and 6 (figure 6). Expression of the exogenous p27 was confirmed by western blot as shown in Figure 2A. The wild type p27 CAOV3 clone expressed levels of exogenous p27 significantly higher than endogenous levels. Out of the five CAOV3 clones expressing the A10 mutant p27 protein, three (Clones 17, 19, and 22) expressed it at levels similar or slightly higher than endogenous p27.
Figure 6. A10p27 does not bind CDK2 and has a dominant negative effect on the p27 kinase inhibitory activity.

Interaction between p27 and CDKs was evaluated in vivo using immunoprecipitation. 500ug whole cell extract from CAOV3-WTp27 cl.26 and CAOV3-A10p27 cl.17 cells were immunoprecipitated using p27, CDK1, 2, 4, 6, or a non-specific antibody and then subjected to western blot analysis using specified antibodies.
Figure 2. Overexpression of A10p27 mutant alters the sensitivity of CAOV3 ovarian cancer cells to atRA induced growth suppression.

A. Western blot analysis of whole cell lysates from CAOV3 cells and CAOV3 clones over expressing WTp27 and A10p27. B. CAOV3 clones over expressing wild type or mutant p27 were treated with ethanol or 1uM atRA for 5 days. Live cells were counted using trypan blue staining. Data are shown as percent inhibition of ethanol. Experiment was repeated at least 4 times, each time in triplicate. Student t test was performed. C. Expression of exogenous and endogenous p27 was analyzed using an antibody against total p27. Expression of S10-phospho p27 was analyzed with a S10-phospho p27specific antibody.
atRA treatment of the CAOV3 cells over expressing the wild type p27 results in 40-60% growth inhibition, a response comparable to the parental CAOV3 cells. In contrast, Figure 2B shows that CAOV3 clones that over express the mutant A10 form of p27 are more resistant to atRA treatment, exhibiting only 15-25% growth arrest. It should be noted that upon ethanol treatment the CAOV3 clone over expressing WTp27 exhibited a growth rate comparable to that of the CAOV3 clones over expressing the A10p27 mutant, suggesting that the increased atRA resistance in the A10p27 clones is not due to a lower growth rate. Also Figure 2C shows that atRA treatment of the CAOV3 clones over expressing the mutant A10 form of p27, does not result in an increase in the endogenous or the exogenous total p27 levels but rather leads to decreased levels of both endogenous and exogenous total p27. A similar pattern is observed for the S10-phospho p27. Thus, prevention of phosphorylation at S10 alters the sensitivity of CAOV3 ovarian cancer cells to atRA induced growth suppression. It is possible that this occurs as a result of reducing the stability of p27.
Mutant A10p27 does not change the cytoplasmic-nuclear localization of p27 or S10-phospho p27, but does increase susceptibility to proteasome mediated degradation
As stated previously, p27 levels are regulated mainly at the post transcriptional level, through either mislocalization within the cell or through phosphorylation followed by degradation (Pagano et al, 1995). Phosphorylation of p27 at the S10 site has been shown to increase p27 stability at the posttranslational level (Borriello et al, 2006). It is possible that the A10 mutant p27 can compete with WT p27 for molecules in the cell that mediate localization and/or degradation.
To determine the mechanism through which A10p27 exercises its effect, we next determined if over expression of the A10p27 mutant has any effect on the localization of endogenous or exogenous p27 in the cell. Figure 3 shows that in CAOV3 cells overexpressing WT p27, both endogenous and exogenous wild type p27 localized to both cytoplasm and nucleus. atRA treatment did not change the pattern of localization and increased the levels of both exogenous and endogenous p27 in both cell compartments. In contrast, in the CAOV3 cells over expressing the mutant A10p27, while the exogenous p27 localized in both nucleus and cytoplasm, atRA treatment resulted in a reduction in levels of both the endogenous and exogenous p27 in both the cytoplasm and in the nucleus. Clearly, overexpression of the A10p27 mutant reduces the accumulation and presumably the stability of p27 in both the nucleus and cytoplasm following atRA treatment.
Figure 3. The dominant negative effect of the A10-p27 mutant is due to the degradation of both endogenous and exogenous p27.

A. Western blot analysis of cytoplasmic, nuclear and whole cell lysates from CAOV3 or CAOV3-WTp27 and CAOV3-A10p27 clones upon 5 day atRA treatment. Enolase and Lamin B were used as cytoplasmic and nuclear markers.
Next we determined the levels of skp2, a protein known to play an important role in the degradation of p27 through the proteasome pathway. We performed western blot analysis of skp2 levels in CAOV3 cells overexpressing either wild type or A10 mutant p27. Figure 4 shows that upon atRA treatment, skp2 levels decrease in CAOV3 cells which overexpress the WTp27 clone, but increase in the A10p27 clones. This result is consistent with the effect of atRA treatment on p27 levels in these different stable transfectant cell clones. This suggests that overexpression of the A10p27 mutant reduces levels of p27 upon atRA treatment by increasing levels of skp2 leading to a decrease in the stability of p27.
Figure 4. atRA treatment increases the levels of skp2 and leads to a decrease in the stability of p27 in CAOV3 clones overexpressing A10p27.

Skp2 protein levels were analyzed by western blot analysis of whole cell extracts from CAOV3 clones over expressing wild type or mutant A10 p27 treated for 1,3 and 5 days with 1uM atRA.
These data suggest that the mutant A10p27 has a dominant negative effect on the levels of endogenous p27 and also on the function of endogenous p27 in mediating RA growth suppression.
Overexpression of A10p27 mutant in CAOV3 reverses the RA effect on cell cycle regulatory proteins
In order to examine the effect of overexpression of the A10p27 mutant on cell cycle progression we next performed western blot analysis on whole cell protein extracts from cells treated for 5 days with atRA to determine changes in the levels of a variety of cell cycle regulatory gene products including CDKi proteins, cyclins, CDKs and Rb2/p130 protein. Figure 5 shows the results of this study. P16 was the only member of the Ink4 family of CDKi we were able to detect. Upon atRA treatment p16 levels increase in CAOV3 and CA-WTp27 clones and decrease in the CA-A10p27 clones. We were able to detect all three members of the Cip/Kip family. P57 levels were not modulated by atRA treatment while p21 showed a pattern of expression similar to p16 and p27. Following atRA treatment, p21 levels increased in empty vector CAOV3 cells and wt-p27 overexpressing CAOV3 cells but decreased in each of the A10p27 overexpressing CAOV3 clones (figure 5A). Likewise, Rb2/p130 levels increased upon atRA treatment in wild type p27 expressing cells but decreased in A10p27 overexpressing cells (figure 5B). This effect of atRA treatment on p21, p16 and Rb2/p130 expression in these p27 clones is consistent with the fact that overexpression of this p27 mutant blocks the growth inhibitory response to atRA treatment.
Figure 5. Overexpression of A10p27 mutant into CAOV3 reverses the atRA effect on cell cycle regulatory proteins.
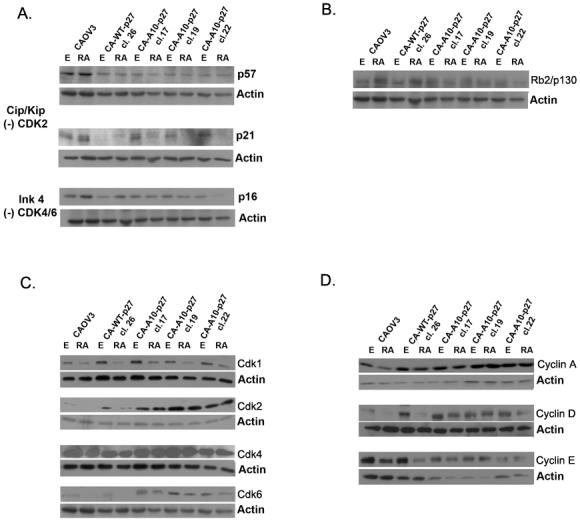
P57, p21, p16 (A.), Rb2/p130 (B.), cyclin A, D and E (C) and CDK1, 2, 4, and 6 (D) levels were analyzed by Western Blot in extracts obtained from CAOV3 and CAOV3 clones treated for 5 days with either ethanol or 1uM RA. Actin was used as loading control
Western blot analysis revealed significantly higher levels of CDK1, CDK2 and CDK6 protein in the clones over expressing the A10p27 compared to wild type CAOV3 cells or the clone over expressing wild type p27 (figure 5C). atRA treatment results in decreased CDK1 levels in CAOV3 and in all p27 clones, suggesting that this is a nonspecific atRA effect, independent of the atRA effect on growth. CDK2 and CDK6 levels decrease upon atRA treatment in CAOV3 cells and in WT-p27 clone, but not in A10p27 clones. CDK4 protein levels do not appear to change significantly upon atRA treatment of either the parental CAOV3 cells, the WT p27 CAOV3 cells or the A10p27 CAOV3 cell clones. A similar pattern was observed for the corresponding cyclins. As shown in Figure 5D, atRA treatment decreased the levels of cyclin A, D and E in the cell lines expressing wild type p27 but did not have any significant effect on cyclin levels in A10p27 clones. This correlates with the atRA effect on the growth of the cells.
A10-p27 clones exhibit differential CDK and cyclin binding activity upon atRA treatment when compared to wild type CAOV3
The consequence of the A10p27 overexpression in CAOV3 cells on the function of endogenous p27 has been examined by measuring p27 binding to cyclin dependent kinases as determined by coimmunoprecipitation and by evaluating the consequence of these binding patterns on CDK enzymatic activity. Figure 6 shows that endogenous and exogenous wild type p27 bind all of the CDKs and this binding is increased by atRA treatment. The A10p27 mutant binds to CDK1, CDK4 and CDK6 but surprisingly failed to bind CDK2. atRA treatment decreased the binding of endogenous p27 to CDKs in the CAOV3 clones over expressing the mutant A10p27 protein.
In an attempt to explain the absence of binding of A10p27 to CDK2 we hypothesized that that mutant p27 is not able to bind cyclin E, the cyclin partner of CDK2. Table 1 shows that WT p27 and A10p27 bind all tested cyclins, suggesting a different mechanism for the lack of binding between A10p27 and CDK2.
Tabel 1.
Endogenous and exogenous p27 binding to CDKs and cyclins
| CAOV3 | ||||
|---|---|---|---|---|
| endogenousp27 | ||||
| Ethanol | atRA | |||
| CDK1 | * | * | ||
| CDK2 | * | ** | ||
| CDK4 | * | ** | ||
| CDK6 | * | ** | ||
| Cyclin A | * | * | ||
| Cyclin D | * | ** | ||
| Cyclin E | * | ** | ||
| CA-WT p27 cl.26 | ||||
|---|---|---|---|---|
| endogenus p27 | exogenous His-p27 | |||
| Ethanol | atRA | Ethanol | atRA | |
| CDK1 | * | * | * | * |
| CDK2 | * | ** | * | ** |
| CDK4 | * | ** | * | ** |
| CDK6 | * | ** | * | ** |
| Cyclin A | * | * | * | * |
| Cyclin D | * | ** | * | ** |
| Cyclin E | * | ** | * | ** |
| CA-A10 p27 cl.17 | ||||
|---|---|---|---|---|
| endogenous p27 | exogenous His-p27 | |||
| Ethanol | atRA | Ethanol | atRA | |
| CDK1 | * | * | * | * |
| CDK2 | * | * | ||
| CDK4 | * | * | * | * |
| CDK6 | * | * | * | * |
| Cyclin A | * | * | * | * |
| Cyclin D | * | * | * | * |
| Cyclin E | * | * | * | * |
Figure 7 shows that atRA treatment caused a 3-4-fold decrease in the kinase activity of CDK2, CDK4 and CDK6 assayed in CAOV3 wild type cells as well as in the CAOV3 clone over expressing wild type p27. Overexpression of A10p27 blocked this atRA reduction of CDK2, CDK4 and CDK6 activity. CAOV3 clones that over express the mutant p27 also exhibit higher levels of basal CDK2 activity, correlating with higher basal levels of CDK2 protein in A10p27 clones. atRA had no significant effect on p27 binding to CDK1 or on its kinase activity. These results suggest that prevention of phosphorylation at S10 in p27 results in a significant alteration in the reduction in CDK kinase activity normally observed in response to atRA treatment. The atRA effect on the kinase activity correlates with the effect on p27 binding to CDKs and corresponding cyclins.
Figure 7. atRA treatment decreases CDK2, 4 and 6 activity in CAOV3 and WT-p27 clones, but not in A10-p27 clones. CDK1 does not seem to have a significant role in mediating atRA sensitivity in cells expressing WT p27.
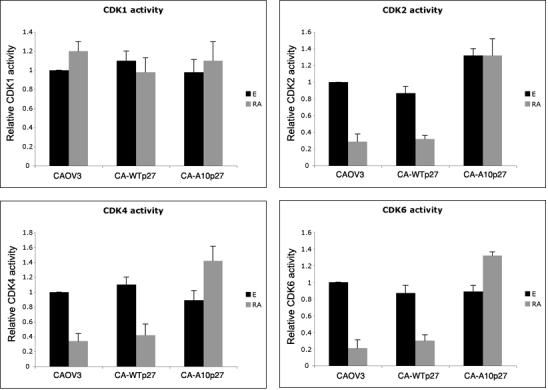
CDK1, 2, 4 and 6 activity was measured in CAOV3, CAOV3-Tp27 cl.26 and CAOV3-A10p27 cl.17 treated for 5 days with 1uM atRA, using an IQ kinase assay
Over expression of A10-p27 mutant blocked the effect of atRA on the expression of genes that are required for S phase entry
atRA treatment of A10p27 over expressing CAOV3 clones resulted in an opposite effect on cell cycle regulatory proteins and on the kinase activity of most S phase regulatory CDKs. We tested the effect of the A10p27 mutant on the expression of genes required for S phase entry. Using quantitative Real Time PCR we analyzed the expression of TK, DHFR, CDC6 and CDC2 following 5 days of atRA treatment of CAOV3, CA-WTp27 and CA-A10p27 clones. Figure 8 shows that atRA treatment of the cells that are growth inhibited by atRA results in a corresponding decrease in expression of these genes. A10p27 clones that are much less inhibited by atRA treatment did not show a significant atRA effect at the level of mRNA expression of these S phase genes. This is consistent with the fact that CAOV3 cells overexpressing A10p27 are less sensitive to the inhibition of growth by atRA.
Figure 8. Analysis of S phase genes.

RT-Q-PCR was used to measure the mRNA expression of the indicated genes in cells treated for 5 days with atRA.
Overexpression of a constitutively phosphorylated p27 (E10p27) converts SKOV3 cells from a cell line that is fully resistant to the growth inhibitory effects of atRA to a cell line that is partially sensitive
In light of our results which showed that the atRA resistant cell line SKOV3 does not exhibit increased S10 phosphorylation upon atRA treatment and that overexpression of the non-phosphorylatable A10p27 mutant converts atRA sensitive CAOV3 cells to partially resistant cells, we predicted that if S10 phosphorylation was in fact a critical event for atRA growth inhibition in ovarian carcinoma cells, increasing the phosphorylation of residue S10 of p27 in SKOV3 cells should render them sensitive to atRA growth inhibition. This was accomplished by generating a mutant p27 that mimics a constitutively phosphorylated p27 by mutating S10 to glutamic acid (E10p27). We transfected a histidine tagged-E10p27 mutant into SKOV3 cells. As a control, we transfected a histidine tagged wild type p27. Stably transfected single cell clones were selected and expanded and assayed by western blot for expression of endogenous and exogenous p27.
As can be seen in Figure 9A, three SKOV3 clones (Clones 15, 36 and 37) expressed wild type his-tagged p27 at levels significantly higher than endogenous p27. Out of five E10p27 SKOV3 clones, two (Clones 17 and 26) expressed the mutant E10p27 at levels higher than the endogenous p27 (figure 9B). Overexpression of WTp27 in SKOV3 cells resulted in a conversion of the atRA resistant SKOV3 cells to a partially atRA sensitive cell line. Figure 9C shows that SKOV3 cells which overexpress wt-p27 exhibited a 15-25% inhibition of growth following atRA treatment for 5 days. This suggests that SKOV3 cells lack the threshold p27 levels required for atRA mediated signaling leading to growth arrest. Overexpression of E10p27 in SKOV3 cells resulted in a similar level of growth inhibition following atRA treatment. This also suggests that 15-25% growth inhibition is the maximum atRA effect that can be obtained through p27 signaling in SKOV3 cells. Nevertheless, the result of this experiment demonstrates that modulation of the phosphorylation status of the S10 residue in p27 plays a major role in determination of the growth response of ovarian carcinoma cells to atRA.
Figure 9. Phospho-p27 mutants have an effect on the atRA induced growth inhibition and on the atRA increase in p27 levels.

P27 antibody was used to detect the levels of both endogenous and exogenous protein in SKOV3 clones overexpressing wild type (A) and E10p27 (B). C. SKOV3 clones over expressing wild type or mutant p27 were treated with ethanol or 1uM atRA for 5 days. Live cells were counted using the trypan blue staining. Data are shown as percent inhibition of ethanol treated cells. Experiment was repeated twice, each time in triplicate.
Discussion
Since p27 phosphorylation is an important event mediating protein stabilization (Sherr and Roberts, 1999, Hengst and Reed, 1998), localization within the cell (Liang et al, 2002, Shin et al, 2002, Viglietto et al, 2002, Fujita et al, 2002) and function of the protein and since p27 protein levels significantly increase in ovarian carcinoma cells which have been growth inhibited by atRA, we wished to investigate the role of p27 phosphoryation in mediating the effects of atRA treatment on growth . Our studies revealed the following new information about the molecular mechanism by which atRA treatment leads to growth suppression: (1) atRA treatment induces phosphorylation of p27 at residue S10 but not at residue T187; (2) atRA treatment leads to an increase in p27 protein levels in both the nucleus and cytoplasm; (3) overexpression of a mutant of p27 that cannot be phosphorylated at residue S10 (A10p27) reduces the sensitivity of ovarian carcinoma cells to atRA; (4) conversely, overexpression of a mutant of p27 that acts as if it is constitutively phosphorylated at residue S10 (E10p27) converts atRA resistant ovarian carcinoma cells to a partially sensitive phenotype exhibiting 15-25% growth inhibition; (5) overexpression of the A10p27 mutant induces a dominant negative effect on the endogenous p27 activity. This dominant negative activity reverses the atRA effect on p27 binding to CDKs, on inhibition of CDK activity, on reduction in the expression of S phase genes and ultimately on the inhibition of growth of ovarian carcinoma cells. These results provide strong evidence that modulation of phosphorylation at serine 10 in p27 determines the growth response of ovarian carcinoma cells to atRA.
Since 80% of total p27 is phosphorylated at the S10 site (Ishida et al, 2000) it is logical that S10 phosphorylation would be critical for mediating atRA growth arrest. Likewise, it is not surprising that agents which induce growth suppression such as atRA would target this residue and activate or induce the kinase responsible for its phosphorylation. The mechanisms regulating S10 phosphorylation are poorly understood. Specifically, the kinase that phosphorylates p27 on S10 and its role in the regulation of cell cycle progression has not been defined. Akt, hKIS, Dirk/Myrk and MAPK have been proposed as candidate kinases in different cell lines at different cell cycle check points (Kotake et al, 2005, Nacusi et al, 2006, Boehm et al, 2002, Deng et al, 2004). atRA treatment of CAOV3 cells arrests the cell cycle progression during the late G1 stage (Wu et al., 1997). hKIS and Dyrk/Myrk are G0 specific kinases so it is likely that these kinases are not acting in response to RA treatment. We were not able to detect any phospho Akt by western blot analysis in CAOV3 cells (data not shown). MAPK activity decreases with RA treatment, making it a less probable kinase for S10 in CAOV3 cells. Other groups hypothesize the existence of a novel kinase, different from these four that can phosphorylate p27 at the S10 site. We propose that this yet unidentified kinase binds both wild type and A10p27 and the mutant protein sequesters significant levels of the kinase, preventing it from phosphorylating the endogenous protein. This results in a less stable endogenous p27. Therefore, overexpression of A10p27 decreases the levels of wild type p27, leading to a reduction in p27 binding to the CDKs. As a result, the CDKs remain active in facilitating cell proliferation, reflected in a decrease in atRA mediated growth inhibition.
A major mechanism which regulates p27 level and stability is susceptibility to proteolysis by the ubiquitin-proteasome pathway (Pagano et al., 1995). Phosphorylation of p27 on threonine 187 (T187) by CDK2-cyclin E complex precedes ubiquitination of p27 with the help of the Skp2-containing E3 ubiquitin-protein ligase, SCF (Tsvetkov et al, 1999, Nakayama et al., 2000). Ubiquitination of p27 by SCF results in degradation of p27 by the 26S proteasome (Carrano et al., 1999; Sutterluty et al., 1999). We previously reported that atRA treatment of CAOV3 cells results in an increase in the levels of p27 protein which results from decreased ubiquitination, decreased expression of skp2 and decreased degradation of p27 by the skp2 ubiquitin ligase system (Vuocolo et al, 2004). In the current study the atRA induced decrease of A10p27 and endogenous p27 in A10 p27 clones is accompanied by an increase in the levels of skp2 and presumably an increase in ubiquitination and 26S proteosomal degradation. This is consistent with the idea that S10 phospho p27 is a more stable protein, as demonstrated in vivo and in vitro by several other groups (Ishida et al, 2000, Kotake et al, 2005). For example, Borriello et al. (2006) showed in a study in the neuroblastoma cell line LAN that atRA induced accumulation of p27 but, in contrast to our results, this was not accompanied by a decrease in the proteasome system as measured by the levels of skp2 protein. The accumulation of S10-phospho p27 was shown to take place in the nucleus and to precede the total p27 accumulation and the atRA induced growth arrest.
In our study we observed an atRA dependent accumulation of total p27 as well as S10-phospho p27 in both the cytoplasm and nucleus. Likewise, in the A10p27 mutant expressing ovarian carcinoma cells we observed a comparable decrease in p27 levels in both the nucleus and cytoplasm. While several pathways involved in p27 translocation have been proposed, the exact mechanism of translocation remains unknown. Potential mechanisms include phosphorylation of p27 on S10, T157 and/or T198 (Liang et al, 2002, Shin et al, 2002, Reed, 2002, Fujita et al, 2003, Sekimoto et al, 2004), binding of p27 to Jab1 or direct binding of p27 to the transport protein CRM1 via the nuclear export signal (Tomoda et al, 1999). Some of the confusion arises from conflicting results. For example, studies done by Ishida et al. (2002) with expression of mutants in cultured cells indicated that phosphorylation at S10 is required for p27 export into the cytoplasm and subsequent phosphorylation at T157. In contrast, analysis in mice which express the A10p27 mutant in place of WTp27 revealed that phosphorylation at S10 is not required for p27 translocation in mouse embryonic fibroblasts (Kotake et al, 2005). We did not perform an exhaustive analysis of p27 translocation in our model system. However, since we failed to see a differenential effect on p27 levels in nucleus versus cytoplasm, it would appear that the modulation of p27 levels by atRA in both wtp27 expressing ovarian carcinoma cells and in A10p27 expressing cells does not involve an alteration in nuclear trafficking.
As stated previously, we found that atRA treatment increased S10-phospho p27 in both nuclear and cytoplasmic cell compartments but did not increase the levels of T157-phospho p27. This suggests that not all S10-P-p27 is phosphorylated at T157. It has been proposed that cytoplasmic p27 can switch p27 function from that of a tumor suppressor to that of an oncogene (Reed, 2002). atRA treatment increased the levels of p27 in the cytoplasmic compartment while having a growth inhibitory effect. Since atRA treatment did not lead to phosphorylation of T157 in either the nucleus or cytoplasm, our results suggest that phosphorylation at T157 might be the important event which regulates the switch in p27 function between tumor suppressor and oncogene. Since atRA induces the tumor suppressor activity of p27, this hypothesis explains also why atRA treatment does not induce the T157-phospho p27 levels.
This hypothesis is supported by our observation that overexpression of A10p27 completely blocked the inhibition of CDK enzyme activity observed upon atRA treatment. As stated above, the mechanism responsible for this most likely involves decreased stability of wild type p27. In addition, it is possible that the A10p27 successfully competes with the reduced levels of wild type p27 for binding to CDKs. In contrast to what happens when wild type p27 binds to CDKs, when A10p27 binds CDKs, kinase activity may not be inactivated, perhaps as a result of a conformational change that does not block or affect the kinase domain. We found that while the A10p27 mutant does bind CDK1, 4 and 6, this binding is reduced upon atRA treatment. Interestingly, the A10p27 mutant does not bind to CDC2. This is contrary to a previous study by Besson et al., (2006) who showed that A10p27 binds and inhibits CDK2 activity in vitro. The CDK2 protein sequence responsible for binding p27 is very similar to the p27 binding sites on the other CDKs. Also, since A10p27 binds cyclin A, D, and E we can only assume that, either CDK2 binding to p27 is blocked by a third binding partner whose own binding to p27 is dependent on S10 phosphorylation, or A10p27 exhibits a conformational change that hinders CDK2 binding in vivo.
These results suggest that one mechanism that leads to resistance of ovarian tumor cells to atRA growth suppression may be the hypo phosphorylation of the serine10 locus of p27. It is possible that atRA resistant ovarian tumors constitute an environment that hinders S10 phosphorylation and that by modulating the activity of the kinase(s) responsible for this event the atRA resistance can be overcome.
Acknowledgments
Contract grant sponsor: National Institute of Health; Contract grant number CA64945
References
- Agrawal D, Hauser P, McPherson F, Dong F, Garcia A, Pledger WJ. Repression of p27kip1 synthesis by platelet-derived growth factor in BALB/c 3T3 cells. Mol Cell Biol. 1996;16(8):4327–36. doi: 10.1128/mcb.16.8.4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besson A, Gurian-West M, Chen X, Kelly-Spratt KS, Kemp CJ, Roberts JM. A pathway in quiescent cells that controls p27Kip1 stability, subcellular localization, and tumor suppression. Genes Dev. 2006;20(1):47–64. doi: 10.1101/gad.1384406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm M, Yoshimoto T, Crook MF, Nallamshetty S, True A, Nabel GJ, Nabel EG. A growth factor-dependent nuclear kinase phosphorylates p27(Kip1) and regulates cell cycle progression. Embo J. 2002;21(13):3390–401. doi: 10.1093/emboj/cdf343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borriello A, Cucciolla V, Criscuolo M, Indaco S, Oliva A, Giovane A, Bencivenga D, Iolascon A, Zappia V, Della Ragione F. Retinoic acid induces p27Kip1 nuclear accumulation by modulating its phosphorylation. Cancer Res. 2006;66(8):4240–8. doi: 10.1158/0008-5472.CAN-05-2759. [DOI] [PubMed] [Google Scholar]
- Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol. 1999;1(4):193–9. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- Deng X, Mercer SE, Shah S, Ewton DZ, Friedman E. The cyclin-dependent kinase inhibitor p27Kip1 is stabilized in G(0) by Mirk/dyrk1B kinase. J Biol Chem. 2004;279(21):22498–504. doi: 10.1074/jbc.M400479200. [DOI] [PubMed] [Google Scholar]
- Fujita N, Sato S, Katayama K, Tsuruo T. Akt-dependent phosphorylation of p27Kip1 promotes binding to 14-3-3 and cytoplasmic localization. J Biol Chem. 2002;277(32):28706–13. doi: 10.1074/jbc.M203668200. [DOI] [PubMed] [Google Scholar]
- Hengst L, Reed SI. Translational control of p27Kip1 accumulation during the cell cycle. Science. 1996;271(5257):1861–4. doi: 10.1126/science.271.5257.1861. [DOI] [PubMed] [Google Scholar]
- Hengst L, Reed SI. Inhibitors of the Cip/Kip family. Curr Top Microbiol Immunol. 1998;227:25–41. doi: 10.1007/978-3-642-71941-7_2. [DOI] [PubMed] [Google Scholar]
- Ishida N, Kitagawa M, Hatakeyama S, Nakayama K. Phosphorylation at serine 10, a major phosphorylation site of p27(Kip1), increases its protein stability. J Biol Chem. 2000;275(33):25146–54. doi: 10.1074/jbc.M001144200. [DOI] [PubMed] [Google Scholar]
- Ishida N, Hara T, Kamura T, Yoshida M, Nakayama K, Nakayama KI. Phosphorylation of p27Kip1 on serine 10 is required for its binding to CRM1 and nuclear export. J Biol Chem. 2002;277(17):14355–8. doi: 10.1074/jbc.C100762200. [DOI] [PubMed] [Google Scholar]
- Kaleagasioglu F, Doepner G, Biesalski HK, Berger MR. Antiproliferative activity of retinoic acid and some novel retinoid derivatives in breast and colorectal cancer cell lines of human origin. Arzneimittelforschung. 1993;43(4):487–90. [PubMed] [Google Scholar]
- Kotake Y, Nakayama K, Ishida N, Nakayama KI. Role of serine 10 phosphorylation in p27 stabilization revealed by analysis of p27 knock-in mice harboring a serine 10 mutation. J Biol Chem. 2005;280(2):1095–102. doi: 10.1074/jbc.M406117200. [DOI] [PubMed] [Google Scholar]
- le Sage C, Nagel R, Egan DA, Schrier M, Mesman E, Mangiola A, Anile C, Maira G, Mercatelli N, Ciafre SA, Farace MG, Agami R. Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. Embo J. 2007;26(15):3699–708. doi: 10.1038/sj.emboj.7601790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Zubovitz J, Petrocelli T, Kotchetkov R, Connor MK, Han K, Lee JH, Ciarallo S, Catzavelos C, Beniston R, Franssen E, Slingerland JM. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med. 2002;8(10):1153–60. doi: 10.1038/nm761. [DOI] [PubMed] [Google Scholar]
- Lloyd RV, Erickson LA, Jin L, Kulig E, Qian X, Cheville JC, Scheithauer BW. p27kip1: a multifunctional cyclin-dependent kinase inhibitor with prognostic significance in human cancers. Am J Pathol. 1999;154(2):313–23. doi: 10.1016/S0002-9440(10)65277-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millard SS, Yan JS, Nguyen H, Pagano M, Kiyokawa H, Koff A. Enhanced ribosomal association of p27(Kip1) mRNA is a mechanism contributing to accumulation during growth arrest. J Biol Chem. 1997;272(11):7093–8. doi: 10.1074/jbc.272.11.7093. [DOI] [PubMed] [Google Scholar]
- Nacusi LP, Sheaff RJ. Akt1 sequentially phosphorylates p27kip1 within a conserved but non-canonical region. Cell Div. 2006;1:11. doi: 10.1186/1747-1028-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K, Nagahama H, Minamishima YA, Matsumoto M, Nakamichi I, Kitagawa K, Shirane M, Tsunematsu R, Tsukiyama T, Ishida N, Kitagawa M, Hatakeyama S. Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. Embo J. 2000;19(9):2069–81. doi: 10.1093/emboj/19.9.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano M, Tam SW, Theodoras AM, Beer-Romero P, Del Sal G, Chau V, Yew PR, Draetta GF, Rolfe M. Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science. 1995;269(5224):682–5. doi: 10.1126/science.7624798. [DOI] [PubMed] [Google Scholar]
- Reed SI. Keeping p27(Kip1) in the cytoplasm: a second front in cancer’s war on p27. Cell Cycle. 2002;1(6):389–90. doi: 10.4161/cc.1.6.261. [DOI] [PubMed] [Google Scholar]
- Rodier G, Montagnoli A, Di Marcotullio L, Coulombe P, Draetta GF, Pagano M, Meloche S. p27 cytoplasmic localization is regulated by phosphorylation on Ser10 and is not a prerequisite for its proteolysis. Embo J. 2001;20(23):6672–82. doi: 10.1093/emboj/20.23.6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekimoto T, Fukumoto M, Yoneda Y. 14-3-3 suppresses the nuclear localization of threonine 157-phosphorylated p27(Kip1) Embo J. 2004;23(9):1934–42. doi: 10.1038/sj.emboj.7600198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant MJ, Coulombe P, Turgeon B, Meloche S. Differential regulation of p27(Kip1) expression by mitogenic and hypertrophic factors: Involvement of transcriptional and posttranscriptional mechanisms. J Cell Biol. 2000;148(3):543–56. doi: 10.1083/jcb.148.3.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13(12):1501–12. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- Shin I, Yakes FM, Rojo F, Shin NY, Bakin AV, Baselga J, Arteaga CL. PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat Med. 2002;8(10):1145–52. doi: 10.1038/nm759. [DOI] [PubMed] [Google Scholar]
- Shin I, Rotty J, Wu FY, Arteaga CL. Phosphorylation of p27Kip1 at Thr-157 interferes with its association with importin alpha during G1 and prevents nuclear re-entry. J Biol Chem. 2005;280(7):6055–63. doi: 10.1074/jbc.M412367200. [DOI] [PubMed] [Google Scholar]
- Stiegler P, Giordano A. Role of pRB2/p130 in cellular growth regulation. Anal Quant Cytol Histol. 1999;21(4):363–6. [PubMed] [Google Scholar]
- Sutterluty H, Chatelain E, Marti A, Wirbelauer C, Senften M, Muller U, Krek W. p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nat Cell Biol. 1999;1(4):207–14. doi: 10.1038/12027. [DOI] [PubMed] [Google Scholar]
- Tomoda K, Kubota Y, Kato J. Degradation of the cyclin-dependent-kinase inhibitor p27Kip1 is instigated by Jab1. Nature. 1999;398(6723):160–5. doi: 10.1038/18230. [DOI] [PubMed] [Google Scholar]
- Tsvetkov LM, Yeh KH, Lee SJ, Sun H, Zhang H. p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr Biol. 1999;9(12):661–4. doi: 10.1016/s0960-9822(99)80290-5. [DOI] [PubMed] [Google Scholar]
- Viglietto G, Motti ML, Bruni P, Melillo RM, D’Alessio A, Califano D, Vinci F, Chiappetta G, Tsichlis P, Bellacosa A, Fusco A, Santoro M. Cytoplasmic relocalization and inhibition of the cyclin-dependent kinase inhibitor p27(Kip1) by PKB/Akt-mediated phosphorylation in breast cancer. Nat Med. 2002;8(10):1136–44. doi: 10.1038/nm762. [DOI] [PubMed] [Google Scholar]
- Vuocolo S, Soprano DR, Soprano KJ. p27/Kip1 mediates retinoic acid-induced suppression of ovarian carcinoma cell growth. J Cell Physiol. 2004;199(2):237–43. doi: 10.1002/jcp.10468. [DOI] [PubMed] [Google Scholar]
- Wu S, Donigan A, Platsoucas CD, Jung W, Soprano DR, Soprano KJ. All-transretinoic acid blocks cell cycle progression of human ovarian adenocarcinoma cells at late G1. Exp Cell Res. 1997;232(2):277–86. doi: 10.1006/excr.1997.3495. [DOI] [PubMed] [Google Scholar]