

Summary
Ethylene is an important regulator of plant growth, development and responses to environmental stresses. Arabidopsis perceives ethylene through five homologous receptors that negatively regulate ethylene responses. RTE1, a novel gene conserved in plants, animals and some protists, was recently identified as a positive regulator of the ETR1 ethylene receptor. Here, we genetically analyze the dependence of ETR1 on RTE1 in order to obtain further insight into RTE1 function. The function of RTE1 was found to be independent and distinct from that of RAN1, which encodes a copper transporter required for ethylene receptor function. We tested the ability of an rte1 loss-of-function mutation to suppress 11 etr1 ethylene-binding domain mis-sense mutations, all of which result in dominant ethylene insensitivity due to constitutive signaling. This suppression test uncovered two classes of etr1 mutations – RTE1-dependent and RTE1-independent. The nature of these mutations suggests that the ethylene-binding domain is a possible target of RTE1 action. Based on these findings, we propose that RTE1 promotes ETR1 signaling through a conformational effect on the ethylene-binding domain.
Keywords: RTE1, ETR1, ethylene, receptor, signaling, Arabidopsis
Introduction
Ethylene is a gaseous plant hormone that is responsible for many aspects of growth and development, including fruit ripening, organ abscission, senescence, seed germination, root hair formation and flowering (Abeles et al., 1992). Ethylene is also involved in the induction of responses to abiotic and biotic stresses (Abeles et al., 1992). In Arabidopsis thaliana, dark-grown seedlings display a specific ethylene response known as the triple response, which consists of inhibition of hypocotyl and root elongation, radial swelling of the hypocotyl, and exaggeration of the apical hook (Bleecker et al., 1988; Guzmán and Ecker, 1990). The signaling pathway leading to the triple response and other ethylene responses has been dissected based on genetic screens for triple-response mutants (Li and Guo, 2007).
In Arabidopsis, ethylene is perceived by five homologous ethylene receptors (ETR1, ERS1, EIN4, ETR2 and ERS2) that are related to two-component histidine kinase receptors (Chang et al., 1993; Hua et al., 1995, 1998; Sakai et al., 1998). The receptors share a similar domain structure, consisting of three or four N-terminal transmembrane domains containing the ethylene-binding site, followed by a GAF domain of unknown function (GAF is present in phytochromes and cGMP-specific phosphodiesterases) and a C-terminal signaling output domain(s). The five receptors exhibit functional redundancy and are negative regulators of ethylene responses (Hua and Meyerowitz, 1998; Qu et al., 2007). Null mutants of the receptor genes have no detectable phenotypes, with the exception of etr1 and ers1, which exhibit hypersensitivity to ethylene (Cancel and Larsen, 2002; Hua and Meyerowitz, 1998; Qu et al., 2007). Removing several ethylene receptor genes at once results in a constitutive triple-response phenotype, even in the absence of ethylene (Hua and Meyerowitz, 1998).
Ethylene binding (Schaller and Bleecker, 1995) requires a copper co-factor (Rodriguez et al., 1999) provided by RAN1, a homolog of the mammalian Menkes/Wilson P-type ATPase copper transporter (Hirayama et al., 1999; Woeste and Kieber, 2000). RAN1 appears to be required for the proper conformation and/or activity of all the ethylene receptors; reduced RAN1 function alters the ligand specificity of the receptors (Hirayama et al., 1999), and a severe ran1 loss-offunction mutation confers a constitutive triple-response phenotype in dark-grown seedlings (Woeste and Kieber, 2000). The constitutive phenotype can be partially rescued by growing the seedlings on copper-supplemented medium (Woeste and Kieber, 2000). The molecular mechanism of ethylene receptor signaling is much less clear (Hall et al., 2007). In vitro phosphorylation assays show that ETR1 has histidine autokinase activity (Gamble et al., 1998), whereas ETR2, EIN4 and ERS2 have serine/threonine kinase activity and ERS1 has both activities (Moussatche and Klee, 2004). However, these kinase activities do not have a primary role in ethylene signaling in vivo (Gamble et al., 2002; Moussatche and Klee, 2004; Wang et al., 2003).
In the current model of ethylene receptor signaling, the receptors signal to repress ethylene responses in the absence of ethylene binding. When ethylene is bound, transmission of the signal occurs via a conformational change in the receptors, turning signaling off and allowing responses to proceed (Wang et al., 2006). This model is supported by dominant gain-of-function mutations in the receptor genes. Importantly, all known gain-of-function mutations lie within the hydrophobic transmembrane region comprising the ethylene-binding domain. For example, the etr1-1 mutation substitutes a tyrosine for a cysteine residue that is required for binding of the copper co-factor (Rodriguez et al., 1999). Therefore, the mutant ETR1-1 receptor cannot bind ethylene (Schaller and Bleecker, 1995) and the receptor is locked into a signaling conformation that cannot be turned off, resulting in ethylene insensitivity. In addition to mutations that disrupt ethylene binding, a class of gain-of-function mutations has been identified that result in insensitivity without disrupting ethylene binding (Wang et al., 2006). Such alleles are thought to specifically prevent transmission of the ethylene signal to the signaling domains of the receptor (Wang et al., 2006), and include the etr1-2 allele (Hall et al., 1999).
A negative regulator of ethylene responses, REVERSIONTO- ETHYLENE SENSITIVITY 1 (RTE1), was identified in Arabidopsis based on suppression of etr1-2 (Resnick et al., 2006). RTE1 encodes a novel integral membrane protein of unknown function found in plants, animals and some protists. The RTE1 protein co-localizes with ETR1 at the Golgi apparatus and the endoplasmic reticulum in Arabidopsis (Dong et al., 2008). RTE1 is thought to be a positive regulator of ETR1 receptor function based on several lines of evidence (Resnick et al., 2006; Zhou et al., 2007). rte1 loss-offunction mutants display ethylene hypersensitivity identical to that of the etr1-7 null mutant, whereas over-expression of RTE1 results in ethylene insensitivity that is largely dependent on ETR1 (Resnick et al., 2006; Zhou et al., 2007). Similarly, over-expression of GREEN-RIPE (GR), a tomato RTE1 homolog, prevents tomato fruit ripening (Barry and Giovannoni, 2006). Interestingly, the loss of RTE1 function suppresses etr1-2, but is unable to suppress the etr1-1 allele (Resnick et al., 2006). etr1-1 is a strong allele resulting in complete ethylene insensitivity, whereas etr1-2 results in a partial ethylene response (Hall et al., 1999). The RTE1/GR family has conserved cysteine and histidine residues, which could potentially bind a metal, leading to speculation that RTE1 and GR might play a role in the binding or transport of copper (Barry and Giovannoni, 2006; Resnick et al., 2007).
In this study, we investigated the RTE1-dependent nature of ETR1, including placement of RTE1 in the ethylenesignaling pathway with respect to RAN1 and ETR1. Our results indicate that RTE1 acts upstream of ETR1, independently of and functionally distinct from RAN1. Suppression analysis of various dominant etr1 alleles suggests a model in which RTE1 is involved in promoting or stabilizing the ‘on’ signaling state in ETR1 via the ethylene-binding domain.
Results
RTE1 and RAN1 have independent and distinct functions with respect to ETR1
RTE1 and RAN1 both play a role in wild-type ETR1 function, as a severe loss of function of either gene results in greatly reduced ETR1 function (Hirayama et al., 1999; Resnick et al., 2006; Woeste and Kieber, 2000). The ETR1 protein is still expressed in the ran1-3 severe loss-of-function mutant (Zhao et al., 2002), indicating that ETR1 is present but nonfunctional in ran1-3. To determine whether the ETR1 protein is similarly present in the rte1-2 mutant, we stably transformed the rte1-2 etr1-7 double mutant with a transgene construct encoding ETR1 fused to a 5xMyc epitope at the C-terminus. Expression of the transgene was driven by the native ETR1 promoter. This same construct is capable of rescuing the etr1-7 null mutation (Dong et al., 2008). Using Western blot analysis, we easily detected the ETR1–5xMyc fusion in the etr1-7 rte1-2 mutant (Figure 1). This indicates that the rte1-2 phenotype is not due to an absence of ETR1, but more likely due to disruption of ETR1 function.
Figure 1.
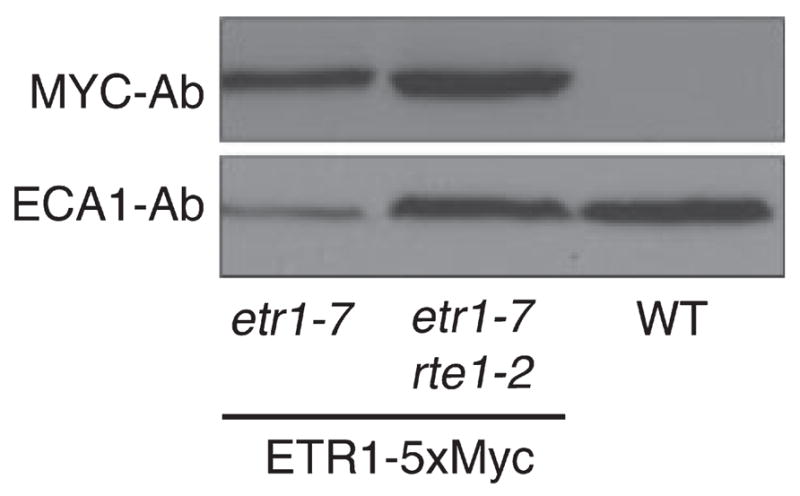
Presence of the ETR1 protein in the rte1-2 loss-of-function mutant. Western blot showing the ETR1–5xMyc monomer isolated from the microsomal membrane fraction of Arabidopsis seedlings run on denaturing PAGE and detected by an anti-c-myc antibody (MYC-Ab). The ETR1–5xMyc construct transformed into etr1-7 and the etr1-7 rte1-2 double mutant gives a band of approximately 80 kDa (left and center lanes) that is absent in the untransformed wild-type (WT, right lane). ECA1, an ER membrane protein detected by the ECA1-Ab antibody (Liang et al., 1997), was used as a loading control.
Because of the parallels between RTE1 and RAN1, we were interested in positioning RTE1 in the ethyleneresponse pathway in relation to RAN1 and ETR1. We found that over-expression of RTE1 in ran1-3 could not compensate for the loss of ran1 either in the dark (with and without copper) or in the light (Figure S1). Thus, RTE1 does not appear to be functionally redundant with RAN1. Overexpression of RTE1 is known to confer ethylene insensitivity in the wild-type (Resnick et al., 2006), but transgenic ran1-3 seedlings carrying the RTE1 over-expression construct did not show reduced sensitivity to ethylene. Instead, the seedlings exhibited essentially the same phenotype as the untransformed ran1-3 mutant, with and without ethylene treatment (data not shown). The absence of ethylene insensitivity suggests that the RTE1 over-expression phenotype is dependent on RAN1, raising the possibility that RTE1 acts in the same pathway as RAN1. Alternatively, the absence of ethylene insensitivity may have been a consequence of the effect of ran1 on ethylene receptor function, such that the RTE1 over-expression phenotype was largely blocked by the loss of ETR1 function. This possibility is consistent with the dependence of the RTE1 over-expression phenotype on ETR1 (Resnick et al., 2006; Zhou et al., 2007; J.S.R. and C.C., unpublished data). Moreover, when we constructed a double mutant of ran1-3 and rte1-2, the double mutant displayed an ethylene-response phenotype that appeared to be additive (Figure 2a), which suggests that RAN1 and RTE1 act in independent pathways rather than in the same pathway. rte1-2 is a severe loss-of-function mutant, phenotypically equivalent to the rte1-3 null mutant and closely resembling the etr1-7 null mutant (Resnick et al., 2006). It is also possible that ran1-3 and rte1-2 have minor inhibitory effects on hypocotyl elongation unrelated to ethylene signaling.
Figure 2. RTE1 action is independent and distinct from that of RAN1. Comparisons of 4-day-old dark-grown seedlings.

(a) The ran1-3 rte1-2 double mutant has an additive phenotype, suggesting that RTE1 and RAN1 act in separate pathways. Shown are representative seedlings of the indicated mutant genotypes grown in the presence and absence of 50 μM CuSO4, with wild-type (WT) as a control.
(b) Double and triple mutant phenotypes indicate that ran1-3 cannot suppress etr1-2, and suppression of etr1-2 by rte1-3 is independent of RAN1. Shown are representative seedlings of the indicated genotypes grown in the presence and absence of the ethylene precursor ACC.
(c) Genetic model of RTE1 and RAN1 action with respect to ETR1 and etr1-2 in ethylene signaling. RTE1 and RAN1 are both required for wild-type ETR1 signaling, which represses ethylene responses, but only RTE1 is required for etr1-2.
In order to determine whether RAN1 and RTE1 act through similar mechanisms to regulate ETR1, we tested whether ran1-3, like rte1, could suppress the dominant etr1-2 mutation. We found that the ran1-3 etr1-2 double mutant displayed an ethylene-insensitive phenotype identical to that of the etr1-2 single mutant, demonstrating that ran1-3 does not suppress etr1-2 (Figure 2b). This result is in sharp contrast to the ability of rte1 mutations to suppress etr1-2 (Resnick et al., 2006), and indicated that RTE1 and RAN1 have distinct effects on ETR1. The results also revealed that the etr1-2 mutation prevents inactivation of the signaling domain independently of copper availability (and therefore of ethylene binding), even though the ETR1-2 mutant protein is known to bind wild-type levels of ethylene (Hall et al., 1999). Thus, etr1-2 behaves differently from wild-type ETR1, whose function is substantially reduced in ran1-3 (Woeste and Kieber, 2000).
We next tested whether rte1-3 suppression of etr1-2 is independent of RAN1, by determining whether rte1-3 is able to suppress the ethylene insensitivity of the ran1-3 etr1-2 double mutant. We constructed a ran1-3 etr1-2 rte1-3 triple mutant, and observed that it was identical in phenotype to the etr1-2 rte1-3 double mutant when both were treated with ethylene (using the ethylene precursor 1-aminocyclopropane- 1-carboxylic acid, ACC), thus demonstrating that etr1-2 was suppressed (Figure 2b). In the absence of ACC treatment, the triple mutant displayed a constitutive ethyleneresponse phenotype as expected for the ran1-3 mutation (Figure 2b). As the constitutive ethylene-response phenotype of ran1-3 was not exhibited by the ran1-3 etr1-2 double mutant, the appearance of the constitutive-response phenotype in the triple mutant was probably due to suppression of etr1-2 by rte1-3 (Figure 2b). The fact that rte1-3 was able to suppress etr1-2 to the same extent in the presence and absence of RAN1 provided further evidence that RTE1 function is independent and distinct from RAN1 function.
The above findings are summarized in the genetic model in Figure 2(c). In the case of wild-type ETR1, both RTE1 and RAN1 are required, and when either gene is absent, ETR1 is largely non-functional. In contrast, the dominant etr1-2 mutation depends on RTE1 but not RAN1 to confer insensitivity (i.e. maintain the signaling state). Interestingly, the strong etr1-1 allele is independent of RTE1 (Resnick et al., 2006). etr1-1 is also presumably independent of RAN1, as the etr1-1 mutation blocks the binding of copper. In addition, ran1-3 does not suppress etr1-3 (Woeste and Kieber, 2000), another dominant allele that disrupts ethylene binding (similar to etr1-1) (Hall et al., 1999).
Loss of rte1 function suppresses a subset of dominant mutations in the ETR1 ethylene-binding domain
The etr1-2 mutation results in a copper-independent conformational defect in the ETR1-2 receptor that is suppressed by the loss of rte1 function. Similarly, the etr1-1 mutation is thought to lock the ETR1-1 receptor into a copper-independent signaling conformation (Hirayama et al., 1999; Woeste and Kieber, 2000), but the loss of rte1 has no effect on etr1-1. To gain insight into the basis for this difference, we examined the ability of the severe loss-of-function mutation, rte1- 2, to suppress additional dominant ethylene-insensitive etr1 alleles. We used existing etr1 mutant transgenes that had been characterized with respect to the degree of ethylene insensitivity conferred in stably transformed plants and the ability of the encoded proteins to bind ethylene in a yeastbased assay (Wang et al., 2006). Each transgene carries a mis-sense mutation located in the ethylene-binding region of ETR1 (Figure 3a). Wang et al. (2006) showed that complete disruption of ethylene binding results in strong ethylene insensitivity (as in the case of etr1-1), but that some strong ethylene-insensitive alleles encode receptors that can still bind ethylene to varying degrees. The alleles that confer insensitivity without disrupting ethylene binding (such as etr1-2) are thought to prevent the conformational switch that occurs upon ethylene binding, and result in either strong or weak ethylene insensitivity (Wang et al., 2006).
Figure 3. Effect of etr1 dominant mutant transgenes in wild-type and rte1-2 seedlings.

(a) The three transmembrane regions of the ETR1 ethylene-binding domain showing the relative positions of etr1 mutations tested for suppression in the rte1-2 background. Vertical lines indicate the approximate locations of the specified amino acid substitutions carried by etr1 transgenes transformed into the wild-type and rte1-2 to test for ethylene insensitivity and suppression of ethylene insensitivity, respectively. In place of transgenes, the etr1-1 (C65Y) and etr1-2 (A102T) mutations were tested for suppression using the etr1-1 rte1-2 and etr1-2 rte1-2 double mutants respectively.
(b) Measurements of hypocotyl length conferred by the mutant etr1 transgenes in wild-type versus rte1-2 seedlings showing the degree of ethylene insensitivity and suppression, respectively. Hypocotyl lengths (mean ± SE for 15–20 seedlings) were measured in representative homozygous lines of 4- day-old dark-grown seedlings on 20 μM ACC. Untransformed seedlings are indicated as ‘no transgene’.
In order to investigate both types of mutants, we tested 11 mutations comprising alleles that are known to confer varying degrees of ethylene binding and signaling strengths (Table 1). The mutations are distributed among the three hydrophobic segments of the ETR1 ethylene-binding domain (Figure 3a). Eight of the mutations result in strong ethylene insensitivity (D25A, Y32A, E38A, F58A, T94M, T101A, L105A, I108A) and three result in weak ethylene insensitivity (F61A, L64A, M104A). Among the strong mutations, D25A, Y32A and T94M are known to abolish or nearly abolish ethylene binding (Wang et al., 2006; W. Wang, University of Wisconsin, Madison, WI, personal communication), while T101A, L105A and I108A result in reduced ethylene binding (Wang et al., 2006). The two remaining strong mutations (E38A and F58A) are thought to specifically block the conformational switch in the receptor, as these mutant receptors retain the ability to bind ethylene (Wang et al., 2006). Among the three weak mutations, only F61A causes a reduction in ethylene binding (approximately 20% that of the wild-type), whereas the other two (L64A and M104A) are thought to disrupt the conformational switch, as neither are deficient in ethylene binding (Wang et al., 2006).
Table 1.
Summary of the ability of rte1-2 to suppress a variety of dominant etr1 mutant alleles
| Mutation | Ethylene binding (%)a | Signaling strength (%)b | Suppressed by rte1-2? (%)c |
|---|---|---|---|
| etr1-2 (A102T) | 150 | Weak (74) | Yes (36) |
| Y32A | <5 | Strong (100) | Yes (42) |
| F61A | 20 | Weak (73) | Yes (43) |
| E38A | 155 | Strong (99) | Yes (45) |
| L64A | 110 | Weak (70) | Yes (45) |
| M104A | 90 | Weak (74) | Yes (45) |
| F58A | 110 | Strong (87) | Yes (46) |
| I108A | 70 | Strong (98) | No (80) |
| D25A | 0 | Strong (100) | No (82) |
| etr1-1 (C65Y) | 0 | Strong (97) | No (93) |
| T101A | 50 | Strong (99) | No (95) |
| T94M | 1.6d | Strong (97) | No (98) |
| L105A | 50 | Strong (100) | No (98) |
Data on percentage ethylene binding with respect to wild-type obtained by Wang et al. (2006).
Values are the percentage hypocotyl length on 20 μM ACC with respect to that on no ACC in the wild-type background.
Values are the percentage hypocotyl length in the rte1-2 background with respect to that in the wild-type background on 20 μM ACC (Figure 3b).
Result provided by W. Wang (personal communication).
We transformed each of the 11 mutant transgenes into wild-type plants to assess the degree of ethylene insensitivity conferred by each transgene. In parallel, each transgene was transformed into rte1-2 mutant plants to test for the ability of rte1-2 to suppress the insensitivity conferred by each transgene. In each background, we screened 5–8 independently transformed lines in the T2 generation for each transgene, and measured hypocotyl length in the tripleresponse assay for three or four of these lines (Figure S2). In addition, hypocotyl length was measured for homozygous lines in the T3 generation exposed to 0 or 20 μM ACC (Figure 3b and Table S1). The strength of each mutant transgene (strong or weak) was determined on the basis of hypocotyl length in the wild-type background (Figure 3b and Table 1). We arbitrarily classified mutations as ‘strong’ if the mean hypocotyl length on ACC was greater than 86% of that seen without ACC. Mutations were classified as ‘weak’ if they had a mean hypocotyl length on ACC between 70-74% of that seen without ACC. None of the mutants fell within the 75–86% range.
An etr1 mutation was considered to be suppressed if the mean hypocotyl length was less than 50% of that conferred by the same mutant transgene in the wild-type background on ACC, and non-suppressed if the mean hypocotyl length was 80% or more of that conferred by the same transgene in the wild-type background on ACC. None of the mutants fell within the 50–79% range. For suppressed mutations, we measured hypocotyl length in four or five independent transformed lines (Figure 3b, Figure S2 and Table S1). On the basis of these criteria, we found that the rte1-2 mutation was able to suppress 6 of the 11 etr1 mutant transgenes. Including previous data for the etr1-2 and etr1-1 mutant alleles (Resnick et al., 2006), there were a total of seven suppressed and six non-suppressed etr1 alleles, as summarized in Figure 3(b) and Table 1 (see also Figure S2 and Table S1). None of the suppressed lines were identical to the untransformed rte1-2 mutant; instead the lines showed variation in the extent of suppression.
There was no clear pattern of features that distinguished the suppressed alleles from the non-suppressed alleles, such as ethylene-binding ability, strength of signaling (i.e. degree of insensitivity) or the location of the mutation in the predicted protein sequence. Of the seven suppressed alleles, five had been determined by Wang et al. (2006) to block the conformational switch between the binding and signaling domains of ETR1 (E38A, F58A, L64A, A102T (etr1-2) and M104A), as they result in ethylene insensitivity without affecting ethylene binding; two of these resulted in strong ethylene insensitivity (E38A and F58A) while the other three resulted in weak insensitivity. The two remaining suppressed alleles, Y32A and F61A, block ethylene binding and are thought to be located on adjacent helical faces that form the ethylene-binding pocket (Wang et al., 2006). Y32A displays strong ethylene insensitivity, but F61A has a weak effect. Six alleles were not suppressed by rte1-2, and all are strong alleles (Table 1). Three (D25A, C65Y (etr1-1) and T94M) severely block ethylene binding, while three (T101A, L105A and I108A) retain a degree of ethylene-binding ability, and were therefore considered to be defective primarily in transmitting conformational changes to the ETR1 signaling domain (Wang et al., 2006).
Discussion
Previous work has identified RTE1 as a novel regulator of ETR1. Here, we carried out detailed genetic studies to analyze the dependence of ETR1 on RTE1, including the placement of RTE1 in the ethylene-signaling pathway in relation to ETR1 and RAN1. While the molecular function of RTE1 remains unknown, it has been speculated that RTE1 serves as a copper chaperone, perhaps acting downstream of RAN1 (Barry and Giovannoni, 2006; Resnick et al., 2007), even though the rte1 mutant is not rescued by copper treatment (Resnick et al., 2006). RAN1 encodes a copper transporter that is thought to be essential for the formation of functional ethylene receptors (Hirayama et al., 1999; Woeste and Kieber, 2000). Our genetic analyses, however, indicate that RTE1 and RAN1 act in independent pathways and have distinct mechanisms with respect to ETR1 function. This is evident from phenotypic analyses of double and triple mutant combinations of severe loss-of-function mutations for rte1 and ran1 and the dominant ethylene-insensitive mutation etr1-2. In particular, the ran1-3 rte1-2 double mutant appears to have an additive phenotype, and only rte1-2 (not ran1-3) is capable of suppressing etr1-2. Furthermore, we found that suppression of etr1-2 by rte1-2 is independent of RAN1.
Notably, the etr1-2-encoded receptor retains wild-type ethylene binding ability (Hall et al., 1999), but does not behave like wild-type ETR1, which is rendered non-functional by ran1-3. Our finding that the etr1-2 phenotype is unaffected by ran1-3, despite the severe limitation in copper availability, suggests that the etr1-2-encoded receptor has a conformational defect that bypasses the requirement for copper.
For greater insight into the role of RTE1 in ETR1 signaling, we tested the ability of rte1-2 to suppress a variety of dominant ethylene-insensitive etr1 mutations, which all lie within the ethylene-binding domain of ETR1 (defined as residues 1–128). Our results reveal a class of mutations that requires RTE1 in order to confer ethylene insensitivity, as well as a class that is independent of RTE1. The underlying basis for these two classes is unclear, as there are no obvious characteristics that are exclusive to the suppressed versus non-suppressed alleles; rte1-2 can suppress both strong and weak etr1 alleles, independent of their effects on ethylene binding, and can suppress mutations located in any of the three ETR1 transmembrane domains, while other mutations with a similar range of effects on ethylene binding and similar distribution in the three transmembrane domains are not suppressed.
A characteristic shared by all the etr1 alleles tested is that they are each likely to produce a unique structural defect within the ETR1 ethylene-binding domain that consequently inhibits, to varying degrees, the conformational transition required to turn ETR1 signaling off (Wang et al., 2006). Therefore, the RTE1-dependence of certain etr1 alleles, including the wild-type ETR1 allele, may be related to particular conformations of the ethylene-binding domain. Accordingly, we propose that RTE1 has an effect on the conformation of the ethylene-binding domain that results in promotion or stabilization of the ETR1 signaling ‘on’ state. This is based on several lines of reasoning. As proposed by Wang et al. (2006), a conformational shift in the ETR1 ethylene-binding domain upon ethylene binding is responsible for transmission of the signal to the transmitter (signaling) domain in order to shut signaling off. Wang et al. (2006) found that a large majority of mutations in the ethylene-binding domain cause strong constitutive activation of ETR1 transmitter signaling, suggesting that the general conserved function of the ethylene-binding domain is to control the signaling conformation of the transmitter. It appears that only subtle changes in steric structure are required for transition between the ‘on’ and ‘off’ states, because so many of the mutations that impair the signaling transition in ETR1 do not disrupt ethylene binding (Wang et al., 2006). In addition, there are no known gain-of-function mutations in the transmitter itself, consistent with the view that control of transmitter ‘on’/’off’ activity lies primarily in the ethylene-binding domain.
It is unlikely that RTE1 acts as a co-factor for signaling output by the transmitter, because, as we have shown, some etr1 alleles have a strong signal output even in the absence of RTE1. The fact that certain etr1 alleles are susceptible to rte1, while others are not, is more likely to be related to the particular mutant conformation of the ethylene-binding domain, which in turn dictates the signaling output, i.e. certain mutant conformations would be stabilized by RTE1, while other mutant steric structures might be stable enough to be independent of RTE1. Consistent with this hypothesis, over-expression of RTE1 enhances ethylene insensitivity in etr1-2 (Resnick et al., 2006) but has no effect on etr1-1 (J.S.R. and C.C., unpublished results). Moreover, Zhou et al. (2007) showed that loss of the RTE1 over-expression phenotype in the etr1-7 null mutant background is rescued by co-expressing a truncated form of ETR1 consisting of residues 1–349 (Zhou et al., 2007), which confers signaling in etr1-7 through interactions with the ERS1 ethylene receptor (Xie et al., 2006). Thus, it appears that RTE1 acts through the N-terminal portion of ETR1.
The ETR1 receptor normally exists in several different states, including the ‘on’/’off’ states and a transitional state that lies between the ‘on’/’off’ states, as shown in Figure 4. RTE1 may promote or stabilize the ETR1 signaling ‘on’ state at either of two transitions. One involves the formation of functional ETR1 (‘A’ in Figure 4), while the other inhibits transition from the signaling ‘on’ state to the signaling ‘off’ state (‘B’ in Figure 4). We propose that, in the wild-type situation, RTE1 helps to promote the ETR1 signaling ‘on’ state from an undefined ground state that is phenotypically equivalent to the etr1 null mutant (Figure 4); ethylene binding turns ETR1 signaling off, which is a state that is known to be phenotypically different from the etr1 null mutant. We favor the model in which RTE1 acts at ‘A’ in Figure 4, based on the finding that rte1 loss-of-function results in an etr1 null-like phenotype (ethylene hypersensitivity), although we cannot rule out that this null-like phenotype includes non-ethylene-related effects, as the rte1-2 and rte1-3 mutants do not phenocopy the etr1 null mutant in every respect (Resnick et al., 2006; Zhou et al., 2007). Involvement of RTE1 in ETR1 biogenesis would be consistent with the ability of rte1-2 to suppress the Y32A mutant receptor. The RTE1-dependent Y32A mutant binds a negligible amount of ethylene, and so would rarely achieve the ethylene-bound, signaling ‘off’ state in the absence of RTE1. Similarly, the etr1-2 mutation in the ran1-3 mutant background is unable to bind ethylene in the absence of copper, but is still suppressed by rte1-2 in the presence of ran1-3. Presumably, the RTE1-independent etr1 alleles are capable of bypassing the requirement for RTE1 in step ‘A’ of Figure 4 in order to reach the signaling ‘on’ state.
Figure 4. Model for the promotion of ETR1 signaling by RTE1.
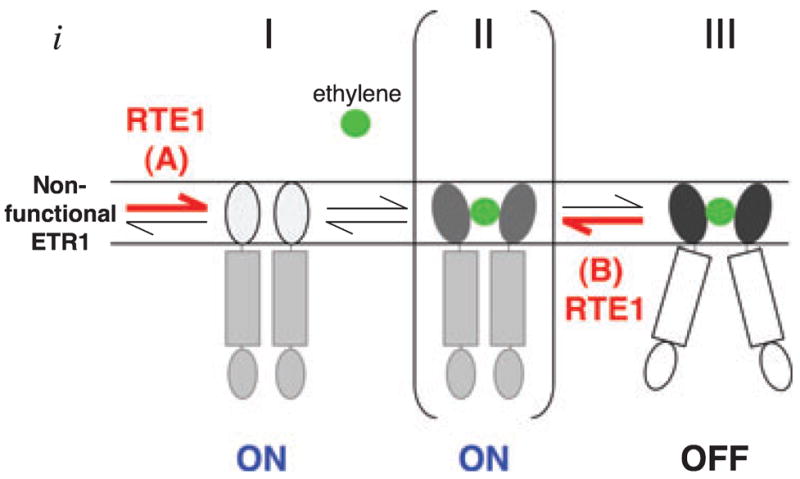
The three-state model of ethylene receptor signaling proposed by Wang et al. (2006) is adapted here for the ETR1 receptor by incorporating RTE1 at two possible points, ‘A’ and ‘B’, and including an additional state ‘i’. At both ‘A’ and ‘B’, RTE1 acts on the ethylene-binding domain to promote the signaling ‘on’ state. In state ‘i’, the nascent non-functional ETR1 protein requires the action of RTE1 at ‘A’ to allow transition to the functional ‘on’ state. In state ‘I’, ethylene is not yet bound and the signaling state of ETR1 is ‘on’; when ethylene binds, the receptor moves into a quasi-stable state (‘II’), where ethylene is bound but the receptor is not yet off (Wang et al., 2006). To enter state ‘III’, the ethylene-binding domain undergoes a conformational shift as a result of binding ethylene, and this conformational change is transmitted to the signaling domain to turn signaling off (Wang et al., 2006). RTE1 at ‘B’ inhibits transition of ETR1 from the signaling ‘on’ state (II) to the signaling ‘off’ state (III). The RTE1-dependent mutant forms of ETR1 are held in either state I (‘on’) or state II (‘on’), depending on whether they bind ethylene or not.
A notable aspect of our findings is the quantitative nature of rte1 suppression. None of the RTE1-dependent alleles appears to be completely suppressed by rte1-2, as the suppressed phenotypes are not identical to the rte1-2 single mutant. Instead, varying degrees of incomplete suppression are observed (Figure 3b). This might reflect allele differences with respect to rates of transition between ETR1 signaling ‘on’/’off’ states or between the ETR1 ground state (‘i’ in Figure 4) and the signaling ‘on’ state. Similarly, weak or strong ethylene insensitivity in the dominant mutants might result from equilibrium shifts between the ‘on’/’off’ signaling states influenced by RTE1 acting on the ethylene-binding domain. RTE1 may act at both points ‘A’ and ‘B’ in Figure 4, promoting the ETR1 ‘on’ state by altering the equilibrium between the various states.
The molecular mechanism by which RTE1 promotes ETR1 signaling remains unknown. RTE1 may play an indirect role by exerting an effect on the membrane environment, which in turn affects the conformation of the ethylene-binding domain within the membrane. Alternatively, RTE1 may play a more direct role in the folding of the ethylene-binding domain, perhaps acting as a molecular chaperone for ETR1. RTE1 does not have any detectable effect on ethylene binding, nor does RTE1 itself bind ethylene (A. Michiels and C.C., unpublished results). The specificity of rte1 for certain etr1 alleles suggests a close physical association between the RTE1 and ETR1 proteins. RTE1 and ETR1 co-localize in the endomembrane system of Arabidopsis root cells (Dong et al., 2008), but it has yet to be shown whether the two proteins physically interact. The basis for the apparent specificity of RTE1 for ETR1 versus the other ethylene receptor family members in Arabidopsis is also not yet understood.
The role of RTE1 in promoting ETR1 signaling is interesting in light of the fact that the RTE1 gene is highly conserved in organisms that do not possess ethylene receptors. Animals carry a single copy of the RTE1 gene, but the function of RTE1 in animals is unknown. The extensive genetic tools provided by the ethylene-signaling pathway in Arabidopsis may help to elucidate the conserved function of RTE1 in other organisms.
Experimental procedures
Plant strains and growth conditions
Arabidopsis thaliana wild-type ecotype Columbia (Col-0) was used throughout this study. For all seedling analyses, seeds were sown on Murashige and Skoog (MS) medium containing 0.9% agar. Following 3-day stratification at 4°C, seeds were incubated at 20°C for 4 days in either complete darkness or 24 h light. Plants were grown in soil under a 16 h light/8 h dark cycle in controlled environment chambers under fluorescent lights. For the triple-response assay, seedlings were germinated on medium containing ACC (Sigma- Aldrich, http://www.sigmaaldrich.com/) at the stated concentrations. To measure hypocotyl lengths, seedlings were digitally photographed and measurements were made using ImageJ software (http://rsb.info.nih.gov/ij/). For copper-response assays, seedlings were germinated on medium containing 50 μM CuSO4.
Genetic analysis
To create double mutants with ran1-3, a ran1-3/+ heterozygous single mutant plant was crossed separately with rte1-2 and etr1-2 single mutants. F1 plants were confirmed to be heterozygous for ran1-3 by genotyping and allowed to self-pollinate. Double mutants were identified in the F2 generation by genotyping. The rte1-2 ran1-3 and etr1-2 ran1-3 double mutant lines were maintained as heterozygotes for the ran1-3 locus, due to the adult lethality of homozygous ran1-3.
To create the ran1-3/+ rte1-3 etr1-2 triple mutant, the ran1-3/+ etr1- 2 double mutant was crossed with the rte1-3 etr1-2 double mutant. F1 plants confirmed by genotyping to be heterozygous for ran1-3 were allowed to self-fertilize, and rte1-3 etr1-2 ran1-3/+ plants were identified in the F2 generation.
For genotyping, the rte1-2, rte1-3 and etr1-2 alleles were detected using CAPS markers (Konieczny and Ausubel, 1993) as previously described (Resnick et al., 2006). The ran1-3 allele was detected by CAPS using primers 5′-CTTTGACAAAACAGGCACCTT-3′ and 5′-ACCATTATGGGTCGAACACC-3′ to amplify a DNA fragment that is cleaved by the restriction enzyme HphI if the fragment is from the mutant ran1-3 allele, but not if it is from wild-type RAN1.
Transgenic constructs and plant transformation
For RTE1 over-expression in ran1-3, a construct containing the RTE1 coding sequence driven by the CaMV 35S promoter (described by Resnick et al., 2006) was transformed by the floral dip method (Clough and Bent, 1998) using Agrobacterium tumefaciens strain GV3101. Homozygous ran1-3 plants are lethal at the rosette stage, so are not viable for transformation. Therefore, ran1-3 heterozygous plants were identified by genotyping. Transformants were selected using the herbicide Finale™ (Bayer, http://www.bayercropscience.com) and were genotyped for the ran1-3 allele. Five independent transformants, which were homozygous for 35S:RTE1 and heterozygous for ran1-3, were analyzed in the T3 generation. T3 seedlings showing a ran1-3 phenotype were examined and selected using Finale™ to confirm the presence of the 35S:RTE1 transgene.
All mutant etr1 transgenes (in plasmid pPZP211) were kindly provided by the laboratory of Anthony Bleecker (University of Wisconsin, Madison, WI, USA). The etr1 mutant transgenes were transformed into wild-type and rte1-2 plants using the floral dip method. Transformed T1 individuals were selected on MS medium containing 100 mg l−1 kanamycin. Five to eight independent transgenic lines were screened in the T2 generation on MS plates with and without 20 μM ACC, with a total of 100–200 seedlings observed per line. For each transgene, measurements were made of 15–25 seedlings for three or four segregating T2 populations, as well as for one or two homozygous T3 lines.
Western blotting
The ETR1–5xMyc construct described by Dong et al. (2008) was transformed into both the etr1-7 null mutant and the etr1-7 rte1-2 double mutant described by Resnick et al. (2006) using the floral dip method. Membrane proteins were isolated from 8–10-day-old lightgrown seedlings, followed by SDS–PAGE and Western blotting as described by Dong et al. (2008).
Acknowledgments
We are grateful to Wuyi Wang and Brad Binder (Botany Department, University of Wisconsin, Madison, WI, USA) for providing the etr1 mutant transgenes, and Heven Sze (University of Maryland, College Park, MD, USA) for the anti-ECA1 antibody. We thank Steve Mount, Zhongchi Liu, and members of the Chang lab for critical comments on the manuscript. This work was supported by grants from the National Institutes of Health (1R01GM071855) and the US Department of Energy (DE-FG02-99ER20329). C.C. was supported in part by the University of Maryland Agricultural Experiment Station, and M.R. was supported in part by the Bamford Fellowship from the University of Maryland College of Chemical and Life Sciences.
Footnotes
Supporting Information Additional supporting information may be found in the online version of this article.
Please note: Blackwell Publishing are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Abeles FB, Morgan PW, Saltveit ME., Jr . Ethylene in Plant Biology. 2. New York: Academic Press; 1992. [Google Scholar]
- Barry CS, Giovannoni JJ. Ripening in the tomato Green-ripe mutant is inhibited by ectopic expression of a protein that disrupts ethylene signaling. Proc Natl Acad Sci USA. 2006;103:7923–7928. doi: 10.1073/pnas.0602319103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleecker AB, Estelle MA, Somerville C, Kende H. Insensitivity to ethylene conferred by a dominant mutation in Arabidopsis thaliana. Science. 1988;241:1086–1089. doi: 10.1126/science.241.4869.1086. [DOI] [PubMed] [Google Scholar]
- Cancel JD, Larsen PB. Loss-of-function mutations in the ethylene receptor ETR1 cause enhanced sensitivity and exaggerated response to ethylene in Arabidopsis. Plant Physiol. 2002;129:1–11. doi: 10.1104/pp.003780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C, Kwok SF, Bleecker AB, Meyerowitz EM. Arabidopsis ethylene-response gene ETR1: similarity of product to two-component regulators. Science. 1993;262:539–544. doi: 10.1126/science.8211181. [DOI] [PubMed] [Google Scholar]
- Clough SJ, Bent AF. Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 1998;16:735–743. doi: 10.1046/j.1365-313x.1998.00343.x. [DOI] [PubMed] [Google Scholar]
- Dong CH, Rivarola M, Resnick JS, Maggin BD, Chang C. Subcellular co-localization of Arabidopsis RTE1 and ETR1 supports a regulatory role for RTE1 in ETR1 ethylene signaling. Plant J. 2008;53:275–286. doi: 10.1111/j.1365-313X.2007.03339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamble RL, Coonfield ML, Schaller GE. Histidine kinase activity of the ETR1 ethylene receptor from Arabidopsis. Proc Natl Acad Sci USA. 1998;95:7825–7829. doi: 10.1073/pnas.95.13.7825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamble RL, Qu X, Schaller GE. Mutational analysis of the ethylene receptor ETR1. Role of the histidine kinase domain in dominant ethylene insensitivity. Plant Physiol. 2002;128:1428–1438. doi: 10.1104/pp.010777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzmán P, Ecker JR. Exploiting the triple response of Arabidopsis to identify ethylene-related mutants. Plant Cell. 1990;2:513–523. doi: 10.1105/tpc.2.6.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AE, Chen QG, Findell JL, Schaller GE, Bleecker AB. The relationship between ethylene binding and dominant insensitivity conferred by mutant forms of the ETR1 ethylene receptor. Plant Physiol. 1999;121:291–299. doi: 10.1104/pp.121.1.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BP, Shakeel SN, Schaller GE. Ethylene receptors: ethylene perception and signal transduction. J Plant Growth Regul. 2007;26:118–130. [Google Scholar]
- Hirayama T, Kieber JJ, Hirayama N, Kogan M, Guzmán P, Nourizadeh S, Alonso JM, Dailey WP, Dancis A, Ecker JR. RESPONSIVE-TO-ANTAGONIST1, a Menkes/Wilson disease-related copper transporter, is required for ethylene signaling in Arabidopsis. Cell. 1999;97:383–393. doi: 10.1016/s0092-8674(00)80747-3. [DOI] [PubMed] [Google Scholar]
- Hua J, Meyerowitz EM. Ethylene responses are negatively regulated by a receptor gene family in Arabidopsis thaliana. Cell. 1998;94:261–271. doi: 10.1016/s0092-8674(00)81425-7. [DOI] [PubMed] [Google Scholar]
- Hua J, Chang C, Sun Q, Meyerowitz EM. Ethylene insensitivity conferred by Arabidopsis ERS gene. Science. 1995;269:1712–1714. doi: 10.1126/science.7569898. [DOI] [PubMed] [Google Scholar]
- Hua J, Sakai H, Nourizadeh S, Chen QG, Bleecker AB, Ecker JR, Meyerowitz EM. EIN4 and ERS2 are members of the putative ethylene receptor gene family in Arabidopsis. Plant Cell. 1998;10:1321–1332. doi: 10.1105/tpc.10.8.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konieczny A, Ausubel FM. A procedure for mapping Arabidopsis mutations using co-dominant ecotype-specific PCRbased markers. Plant J. 1993;4:403–410. doi: 10.1046/j.1365-313x.1993.04020403.x. [DOI] [PubMed] [Google Scholar]
- Li H, Guo H. Molecular basis of the ethylene signaling and response pathway in Arabidopsis. J Plant Growth Regul. 2007;26:106–117. [Google Scholar]
- Liang F, Cunningham KW, Harper JF, Sze H. ECA1 complements yeast mutants defective in Ca2+ pumps and encodes an endoplasmic reticulum-type Ca2+-ATPase in Arabidopsis thaliana. Proc Natl Acad Sci USA. 1997;94:8579–8584. doi: 10.1073/pnas.94.16.8579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussatche P, Klee HJ. Autophosphorylation activity of the Arabidopsis ethylene receptor multigene family. J Biol Chem. 2004;279:48734–48741. doi: 10.1074/jbc.M403100200. [DOI] [PubMed] [Google Scholar]
- Qu X, Hall BP, Gao Z, Schaller GE. A strong constitutive ethylene-response phenotype conferred on Arabidopsis plants containing null mutations in the ethylene receptors ETR1 and ERS1. BMC Plant Biol. 2007;7:3. doi: 10.1186/1471-2229-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnick JS, Wen CK, Shockey JA, Chang C. REVERSION-TO-ETHYLENE SENSITIVITY1, a conserved gene that regulates ethylene receptor function in Arabidopsis. Proc Natl Acad Sci USA. 2006;103:7917–7922. doi: 10.1073/pnas.0602239103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnick JS, Rivarola M, Wen C-K, Shockey JA, Chang C. A novel membrane protein conserved in plants and animals is important for ethylene receptor function in Arabidopsis thaliana. In: Ramina A, Chang C, Giovannoni J, Klee H, Perata P, Woltering E, editors. Advances in Plant Ethylene Research: Proceedings of the 7th International Symposium on the Plant Hormone Ethylene. Berlin: Springer; 2007. pp. 9–14. [Google Scholar]
- Rodriguez FI, Esch JJ, Hall AE, Binder BM, Schaller GE, Bleecker AB. A copper cofactor for the ethylene receptor ETR1 from Arabidopsis. Science. 1999;283:996–998. doi: 10.1126/science.283.5404.996. [DOI] [PubMed] [Google Scholar]
- Sakai H, Hua J, Chen QG, Chang C, Medrano LJ, Bleecker AB, Meyerowitz EM. ETR2 is an ETR1-like gene involved in ethylene signaling in Arabidopsis. Proc Natl Acad Sci USA. 1998;95:5812–5817. doi: 10.1073/pnas.95.10.5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller GE, Bleecker AB. Ethylene-binding sites generated in yeast expressing the Arabidopsis ETR1 gene. Science. 1995;270:1809–1811. doi: 10.1126/science.270.5243.1809. [DOI] [PubMed] [Google Scholar]
- Wang W, Hall AE, O’Malley R, Bleecker AB. Canonical histidine kinase activity of the transmitter domain of the ETR1 ethylene receptor from Arabidopsis is not required for signal transmission. Proc Natl Acad Sci USA. 2003;100:352– 357. doi: 10.1073/pnas.0237085100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Esch JJ, Shiu S, Agula H, Binder BM, Chang C, Patterson SE, Bleecker AB. Identification of important regions for ethylene binding and signaling in the transmembrane domain of the ETR1 ethylene receptor of Arabidopsis. Plant Cell. 2006;18:3429–3442. doi: 10.1105/tpc.106.044537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woeste KE, Kieber JJ. A strong loss-of-function mutation in RAN1 results in constitutive activation of the ethylene response pathway as well as a rosette-lethal phenotype. Plant Cell. 2000;12:443–455. doi: 10.1105/tpc.12.3.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie F, Liu Q, Wen CK. Receptor signal output mediated by the ETR1 N terminus is primarily subfamily I receptor dependent. Plant Physiol. 2006;142:492–508. doi: 10.1104/pp.106.082628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao XC, Qu X, Mathews DE, Schaller GE. Effect of ethylene pathway mutations upon expression of the ethylene receptor ETR1 from Arabidopsis. Plant Physiol. 2002;130:1983–1991. doi: 10.1104/pp.011635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Liu Q, Xie F, Wen CK. RTE1 is a Golgiassociated and ETR1-dependent negative regulator of ethylene responses. Plant Physiol. 2007;145:75–86. doi: 10.1104/pp.107.104299. [DOI] [PMC free article] [PubMed] [Google Scholar]