

Summary
Addition of erythromycin (Em) to a Bacillus subtilis strain carrying the ermC gene results in ribosome stalling in the ermC leader peptide coding sequence. Using ΔermC, a deletion derivative of ermC that specifies the 254-nucleotide ΔermC mRNA, we showed previously that ribosome stalling is concomitant with processing of ΔermC mRNA, generating a 209-nucleotide RNA whose 5′ end maps to codon 5 of the ΔermC coding sequence. Here we probed for peptidyl-tRNA to show that ribosome stalling occurs after incorporation of the amino acid specified by codon 9. Thus, cleavage upstream of codon 5 is not an example of “A-site cleavage” that has been reported for Escherichia coli. Analysis of ΔermC mRNA processing in endoribonuclease mutant strains showed that this processing is RNase J1-dependent. ΔermC mRNA processing was inhibited by the presence of stable secondary structure at the 5′ end, demonstrating 5′-end-dependence, and was shown to be a result of RNase J1 endonuclease activity, rather than 5′ -to-3′ exonuclease activity. Examination of processing in derivatives of ΔermC that had codons inserted upstream of the ribosome stalling site revealed that Em-induced ribosome stalling can occur considerably further from the start codon than would be expected based on previous studies.
Introduction
The ermC gene, which encodes a ribosomal RNA methylase, expresses inducible Em resistance in B. subtilis. ermC induction occurs at the posttranslational level, whereby a ribosome that is bound by Em stalls while translating a leader peptide coding sequence (CDS). This results in a structural rearrangement of the ermC leader region RNA such that the ribosome binding site for translation of the methylase CDS becomes accessible (Dubnau, 1984, Weisblum, 1985). By mutation analysis, it was shown that the most important leader peptide amino acids required for Em-induced ribosome stalling were residues 5-9, SIFVI (Fig. 1A). A stop codon inserted at codon 10 had no effect on Em induction (Mayford & Weisblum, 1989). Mutations in codons I3 and F4 were also observed to affect ribosome stalling (Hue & Bechhofer, 1991). It was proposed that the ribosome stalls with the V8 codon in the P site and the I9 codon in the A site (Mayford & Weisblum, 1989).
Fig. 1.
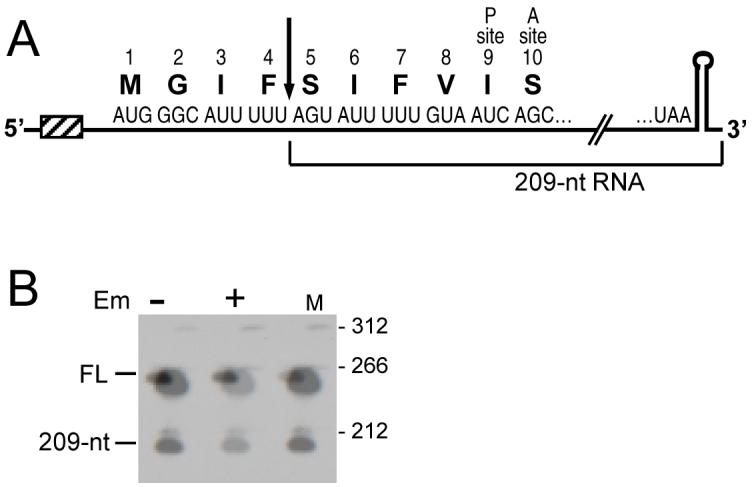
A. Schematic of ΔermC mRNA. The first 10 codons and the stop codon of ΔermC are shown. Hatched box represents the Shine-Dalgarno sequence. Downward arrow indicates the site of processing to produce the 5′ end of the 209-nt RNA. Location of codons 9 and 10 in the ribosomal P and A sites is indicated.
B. Northern blot analysis of ΔermC mRNA in the absence and presence of Em, as indicated. On the right are the sizes (in nts) of 5′-end-labeled pSE420 TaqI DNA fragments (Brosius, 1992) run in lane M. Migration of full-length (FL) and 209-nt RNA is indicated. The probe was a riboprobe complementary to nts 1-172 of ΔermC mRNA.
We have been working with ΔermC mRNA as a model for mRNA processing in B. subtilis. ΔermC mRNA, a 254-nucleotide (nt) message, is encoded by a deletion derivative of ermC. ΔermC contains a 62-amino-acid reading frame, of which the first 16 codons come from the ermC leader peptide CDS and these are fused to the C-terminal portion of the ermC methylase CDS. We reported several years ago that, upon Em-induced ribosome stalling in the 5′-proximal part of ΔermC mRNA, mRNA processing occurs, which was believed to be the result of endonucleolytic cleavage (Drider et al., 2002). The cleavage site was mapped by primer extension to +45 of the ΔermC transcript, i.e., between codons 4 and 5 of the ΔermC coding sequence (Fig. 1A). This cleavage generated a 209-nt downstream fragment, which was easily detectable on Northern blot analysis (see Fig. 1B), and a predicted 45-nt upstream fragment, which could not be detected. We hypothesized that the upstream fragment was undetectable because it bears an unprotected 3′ end and is therefore degraded rapidly by 3′-to-5′ exonuclease activity. This cleavage was observed in B. subtilis mutants lacking Bs-RNase III or RNase M5, the available B. subtilis endonuclease mutants at that time. The possibility that the 209-nt RNA fragment was generated by 5′ exonuclease activity was not considered likely at the time, as no 5′-to-3′ exonuclease was known to exist in bacteria. However, the report of Condon and colleagues that the recently-discovered B. subtilis endoribonuclease RNase J1 (Even et al., 2005) also has 5′-to-3′ exoribonuclease activity (Mathy et al., 2007) raised the possibility that the 209-nt ΔermC RNA fragment was generated not by endonucleolytic cleavage but by exonucleolytic degradation up to the +45 nucleotide.
Studies in E. coli have shown that ribosome stalling at internal mRNA sites can induce an endonucleolytic cleavage of the mRNA sequence that is in the ribosomal A site -the so-called “A-site cleavage” (Hayes & Sauer, 2003, Sunohara et al., 2004). A-site cleavage generates a 3′ end at which a ribosome is bound and cannot dissociate. This triggers the tmRNA trans-translation system (Dulebohn et al., 2007, Withey & Friedman, 2003), which results in offloading of the stalled ribosome, degradation of the prematurely-terminated polypeptide, and degradation of the upstream mRNA fragment by RNase R (Hong et al., 2005, Richards et al., 2006). A-site cleavage occurs in the absence of several bacterial toxins that have been shown to have ribonuclease activity. In some cases, RelE protein is required for A-site cleavage, but it is not clear whether RelE can act as an endonuclease in the presence of a stalled ribosome or whether RelE triggers an endogenous endonuclease function of the ribosome (Christensen & Gerdes, 2003, Pedersen et al., 2003). Isolated RelE does not show ribonuclease activity.
We are not aware of any report of A-site cleavage in B. subtilis. Thus, it was of interest to determine whether the ΔermC cleavage is an A-site cleavage, and for this we needed to map the ribosome stall site (RSS) precisely. As mentioned, Mayford and Weisblum (1989) proposed that Em-induced ribosome stalling occurs with I9 in the A site. If so, the cleavage that we observed (between codons 4 and 5) would be well upstream of the A site. The current study used a method to determine directly where the ribosome stalls. In the course of these experiments, we observed unexpected ribosome stalling in some of the ΔermC constructs, which has implications for the mechanism of translation inhibition by Em.
Another aspect of the current study was to determine the endonuclease responsible for cleavage. Since publication of our study on cleavage of ΔermC mRNA, several B. subtilis endoribonucleases have been discovered and at least partially characterized, including RNase Z (Pellegrini et al., 2003, Redko et al., 2007), RNase J1 (Britton et al., 2007, de la Sierra-Gallay et al., 2008, Even et al., 2005, Mathy et al., 2007), RNase J2 (Even et al., 2005), and EndoA (Pellegrini et al., 2005). In addition, a conditional mutant for the RNA component of RNase P was reported (Wegscheid et al., 2006). In this study we examined Em-induced mRNA processing in the currently available endoribonuclease mutant strains.
Results
Mapping of ribosomal stall site
The effect of adding Em to a strain carrying ΔermC on a plasmid is shown in Fig. 1B, where the 209-nt RNA, the result of processing of full-length ΔermC mRNA, is observed only when Em is added. Previously, we showed that ΔermC mRNA processing was dependent on Em-induced ribosome stalling (Drider et al., 2002). To map precisely where the Em-bound ribosome stalls on ΔermC mRNA, we probed tRNAs that are involved in translation of ΔermC codons in and around the RSS. Codons 3-9, the codons implicated from previous work in ribosome stalling, encode either S, I, F, or V (Fig. 1A). As such, oligonucleotide probes were designed to hybridize with the respective tRNAs (Table 1). Total RNA was isolated from a B. subtilis strain carrying ΔermC on a plasmid that bears the cop-1 mutation, which replicates at about 200 copies per cell (Villafane et al., 1987). The RNA isolation was done under conditions that have been shown previously to preserve peptidyl-tRNA species (Varshney et al., 1991). We reasoned that the high-copy number of ΔermC in this strain, combined with the strong ermC promoter, would result in a large amount of ΔermC mRNA, of which some fraction would contain a stalled ribosome when Em was added. An Em-bound ribosome would be stalled in the pre- or post-translocational state, with a peptidyl-tRNA in the A or P site (Lovmar et al., 2004, Menninger & Otto, 1982). If the number of stalled ribosomes was large enough, we would be able to detect the peptidyl-tRNA that accumulates, against a background of a much larger excess of aminoacyl-tRNA. The identity of the peptidyl-tRNA would reveal the P-site or A-site codon at which ribosome stalling occurs. The results of such an experiment are shown in Fig. 2. While we detected no difference in the tRNAser, tRNAphe, and tRNAval species with or without Em present, tRNAile showed a slightly larger band in the presence of Em. To rule out an effect of Em on tRNAile that was independent of ΔermC, a strain that did not contain ΔermC was probed with the tRNAile probe. No difference was observed with or without Em added (Fig. 2, control). We concluded that Em-induced ribosome stalling occurs with an Ile codon in the P or A site, although which one of the three Ile codons in the N-terminal portion of ΔermC could not be determined from this experiment.
Table 1.
Oligonucleotide probes for specific tRNAs
| tRNA gene(s) | probe sequencea |
|---|---|
| trnD-ser | CCTTGAGACGGAGTTGACCGCCTAC |
| trnA/O-ile | CGACCTCACGCTTATCAGGCGTGCG |
| trnB/D-phe | ACCGCCGACACACGGATTTTCAGTCCGTT |
| trnB/J/SL/D-val | CCGCCGACCCTCTGCTTGTAAGGCA |
Underlined sequence is the complement to the tRNA anticodon (i.e., same sequence as the codon)
Fig. 2.
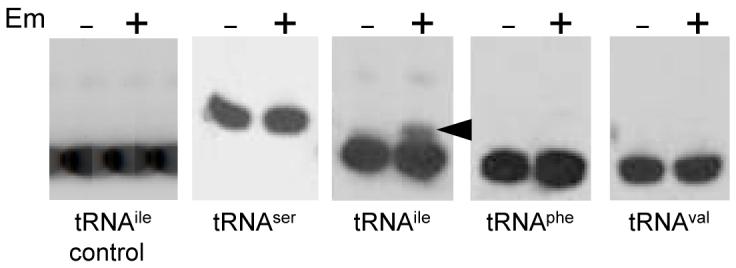
Northern blot analysis of tRNA isolated from a wild-type strain grown in the presence and absence of Em, as indicated. The specific tRNA that was probed with an antisense oligonucleotide (Table 1) is indicated below each panel. Arrowhead marks the additional band detected with the tRNAile probe, which represents peptidyl-tRNAile. First panel at left had RNA isolated from a control strain that did not carry a ΔermC plasmid.
Mutations of the Ile codons of the ΔermC N-terminal coding sequence were constructed. In the first construct, the Ile at position 3 was deleted. This did not affect the site of ribosome stalling, as judged by the size of the downstream cleavage product (Fig. 3A). Although this deletion did cause a three-fold reduction of the amount of the 209-nt fragment, as a percentage of total ΔermC RNA, the results suggested that I3 was not required for Em-induced ribosome stalling and subsequent RNA processing. In another experiment, the Ile at position 9 was changed to a Val codon. The amount of the downstream processed fragment was about half that of wild-type ΔermC (Fig. 3A). Probing for tRNAval in this strain showed the appearance of a peptidyl-tRNA species that was dependent on the presence of Em (Fig. 3B), but which was not present in the strain with the wild-type ΔermC sequence. When this blot was stripped and probed for tRNAile, the peptidyl-tRNA band was observed in the wild type but not in the strain with the Ile to Val change at codon 9. These results indicate that ribosome stalling occurs with codon 9 in the P or A site, and thus the processing of ΔermC mRNA between codons 4 and 5 is not a case of A-site cleavage. While our experiments could not differentiate whether codon 9 was in the P or A site, experiments reported in the recent paper of Mankin and colleagues (Vazquez-Laslop et al., 2008), which looked at incorporation of ermC leader peptide amino-acids in an in vitro system, allows us to conclude that the ribosome stalls with codon 9 in the P site; see Discussion.
Fig. 3.
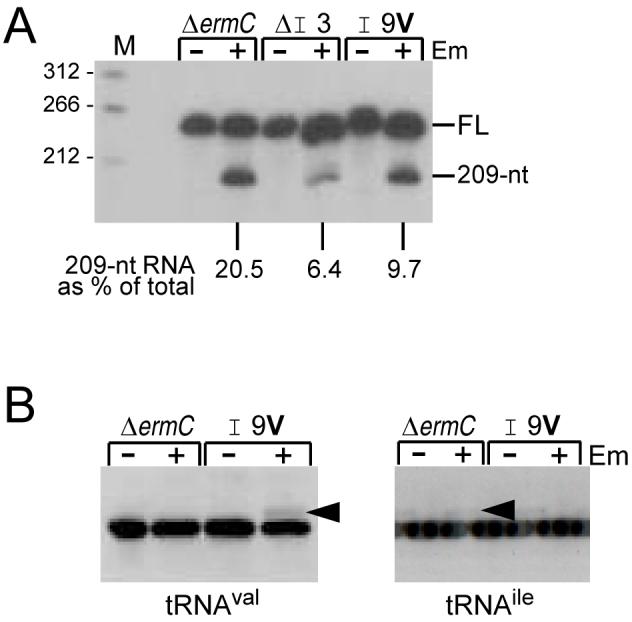
A. Northern blot analysis of ΔermC RNA encoded by mutant constructs with either deletion of codon 3 (ΔI3) or change of codon 9 from Ile to Val (I9V). Percentage of 209-nt RNA fragment, relative to total ΔermC RNA (average of two experiments), is indicated below the blot. Control for total amount of RNA loaded per lane in this Northern blot and the ones in Figs. 4-7 was by stripping the blot and probing with a 5S rRNA probe (not shown).
B. Northern blot analysis of tRNA, using the tRNAval and tRNAile probes, as indicated. Arrowheads mark peptidyl-tRNA species.
Identification of ribonuclease activity required for processing
As mentioned in the Introduction, we had shown previously that two endoribonucleases, Bs-RNase III and RNase M5, were not involved in ΔermC mRNA cleavage (Drider et al., 2002). We tested for the involvement of four other B. subtilis endoribonucleases - RNases J1, J2, P, and Z - whose genes were more recently cloned and mutant strains constructed. RNase J2 is not essential, so a knockout strain could be used. The RNase J2 mutant strain showed a similar level of ΔermC mRNA processing as the wild type (Fig. 4A, lane 4). RNases J1, P, and Z, are essential, so conditionally expressing strains, which are IPTG-dependent for growth, were used. Conditions for these latter strains were chosen such that RNA was isolated from strains grown without IPTG to a point where there was a significant reduction in the concentration of the relevant RNase, although it was probably not eliminated completely. Depletion of RNase Z and RNase P did not result in a significant decrease in ΔermC mRNA processing (Fig. 4A, lanes 11 and 12, and lanes 15 and 16). For the RNase Z mutant strain there was actually more processed product in the absence of IPTG than in its presence. This may indicate a requirement for RNase Z in the turnover of the 209-nt RNA, or, alternatively, increased stalling due to a lower level of tRNA whose maturation is RNase Z-dependent (Pellegrini et al., 2003).
Fig. 4.

Ribonuclease responsible for ΔermC mRNA processing. A. Northern blot analysis of ΔermC mRNA from various strains, grown without or with Em and - for RNase J1, Z, and P mutants - without or with IPTG to induce RNase expression. The mutated ribonuclease is indicated above each set of lanes; wt, wild type. Variation in the amount of ΔermC mRNA was consistently observed. Percentage of 209-nt RNA fragment, relative to total ΔermC RNA (average of two experiments), is indicated below the relevant lanes.
B. Primer extension analysis of ΔermC 5′ ends in wild-type and RNase J1 mutant strains. The band marked with an asterisk is observed only in the RNase J1 strain, in the presence or absence of induction of RNase J1 expression, and represents 5′ ends at about nt 10 of the ΔermC transcript. Sizes of bands in sequencing lanes (nts) are indicated at left. The sequencing ladder was generated from M13mp18 single-stranded DNA.
In the RNase J1 mutant strain, the level of ΔermC mRNA processing was almost four-fold lower when grown in the absence of IPTG than in its presence (Fig. 4A, lane 7 and 8). The actual amount of 209-nt RNA in the RNase J1 mutant grown without IPTG may have been even lower, since the 4.1% value includes some background hybridization, which was often observed in RNAs isolated from the RNase J1 mutant strain. The results from Northern blot analysis of the RNase J1 mutant strain were confirmed by primer extension analysis, using a primer whose 5′ end was complementary to a position 193 nts from the mapped ΔermC mRNA transcriptional start site. The 5′ end of the 209-nt downstream fragment was predicted to be 149 nts from the start of this primer. The results in Fig. 4B showed that hardly any reverse transcriptase product corresponding to the 5′ end of the 209-nt fragment was detected when the RNase J1 conditional strain was grown in the absence of IPTG. Interestingly, in the RNase J1 mutant strain, independent of IPTG, a 5′ end (actually, a doublet on lighter exposure) was mapped at around +10 of the ΔermC transcript (asterisk in Fig. 4B). The basis for additional RNAs with different 5′ ends in this strain is not clear.
In experiments not shown, EndoA, the recently characterized, broad-specificity endoribonuclease of B. subtilis (Pellegrini et al., 2005), was also shown not to be required for ΔermC mRNA processing. Finally, we tested whether the tmRNA system, which recognizes stalled ribosomes (Withey & Friedman, 2003) and participates in decay of RNA fragments (Hong et al., 2005, Richards et al., 2006) was involved in ΔermC mRNA processing. In a strain that was deleted for the ssrA gene, encoding tmRNA, a wild-type level of ΔermC mRNA processing was observed (data not shown).
Effect of 5′-terminal structure on processing
The involvement of RNase J1 in ΔermC mRNA processing directed our attention to the 5′ end of the message. It has been shown that RNase J1 ribonuclease activity is sensitive to the phosphorylation state of the 5′ end (de la Sierra-Gallay et al., 2008, Even et al., 2005, Mathy et al., 2007). In a previous study on the effect of 5′-terminal secondary structure on mRNA stability (Sharp & Bechhofer, 2005), a number of ΔermC derivatives were constructed that contained predicted 5′ stem-loop structures. Many of these resulted in ΔermC mRNAs that were extremely stable even in the absence of ribosome stalling. We wished to determine whether a 5′ stabilizing element would also affect RNase J1-dependent processing. Three constructs were chosen (Fig. 5A), each with a different 5′ stem-loop sequence but all of which showed an increased ΔermC mRNA half-life from 6 minutes to about 20 minutes (Sharp & Bechhofer, 2005). Strains carrying these constructs were treated with or without Em, and Northern blot analysis was performed. As can be seen in Fig. 5B, the presence of the 5′-terminal secondary structure resulted in a large reduction in the amount of ΔermC mRNA cleavage.
Fig. 5.
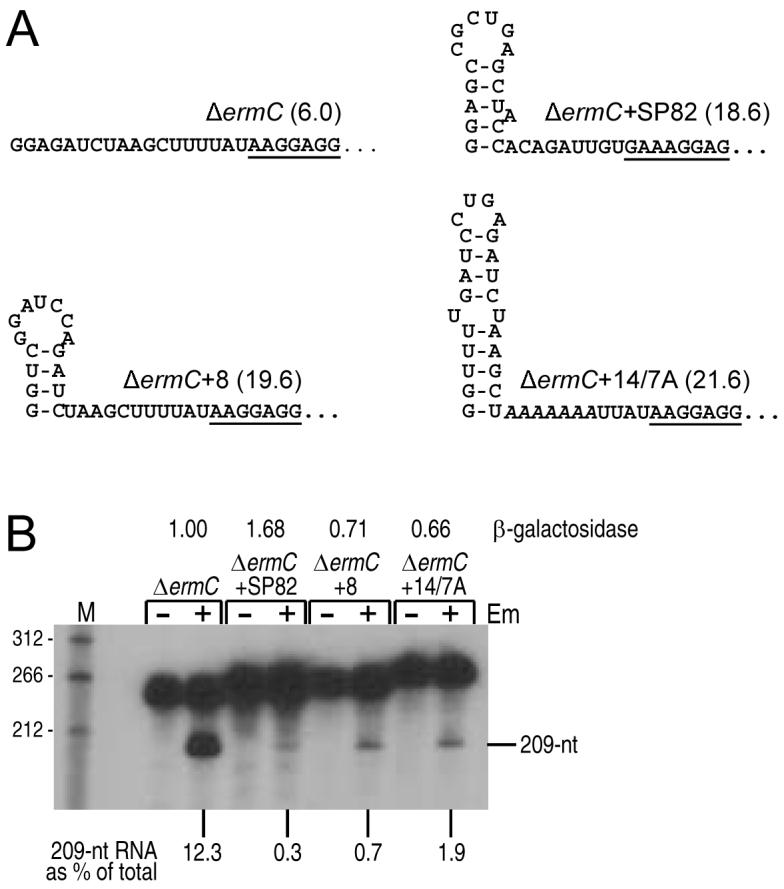
A. Predicted 5′-terminal secondary structure of ΔermC and derivatives. In parentheses is the mRNA half-life (min) of these constructs (Sharp & Bechhofer, 2005). The ribosome binding site is underlined in each case.
B. Northern blot analysis of RNA processing for the 5′ stem-loop ΔermC constructs shown in part A. Percentage of 209-nt RNA fragment, relative to total ΔermC RNA (average of two experiments), is indicated below the blot. At the top are relative values of β-galactosidase expression (average of three experiments) for ΔermC-lacZ translational fusions bearing the respective 5′-terminal structures.
It was possible that the reduction in ΔermC mRNA processing was due to a reduction in translational efficiency; that is, perhaps the presence of the 5′-terminal structure near the translation initiation region (see Fig. 5A) affected ribosome binding. Since ΔermC mRNA processing is dependent on ribosome stalling, reduced translational efficiency would result in reduced cleavage. To eliminate this possibility, translational fusions were made between ΔermC codon 47 and the lacZ coding sequence. β-galactosidase assays showed that the translational efficiencies of these fusions were all within two-fold of the wild-type, and did not reflect the extent of RNA processing (data in Fig. 5B, top). These results show that RNase J1-mediated processing of ΔermC mRNA is sensitive to the structural status of the 5′ end.
Exoribonuclease vs. endonuclease activity of RNase J1
Since RNase J1 shows both 5′-to-3′ exonuclease activity and endonuclease activity (Mathy et al., 2007), generation of the 209-nt downstream fragment could be the result of endonuclease cleavage, as we had assumed previously (Drider et al., 2002), or could be the result of 5′-to-3′ exonucleolytic processing up to the site between codons 4 and 5, where processing would be blocked by the stalled ribosome. Three experimental approaches were undertaken to resolve this issue. Codons 3-6, the site at which processing occurs, constitute a highly AU-rich region, which is characteristic of RNase J1 cleavage sites (Deikus et al., 2008, Even et al., 2005, Yao et al., 2007). This sequence was mutated to make it less AU-rich (Fig. 6A), introducing silent mutations in codons 3-5 and a conservative change in codon 6 (Ile to Leu), which has been shown previously not to affect Em-induced ribosome stalling (Mayford & Weisblum, 1989). The Northern blot result in Fig. 6B (one of multiple repeats) showed that the extent of processing to give the 209-nt RNA was reduced by about 50%. This is consistent with endonuclease cleavage being reduced by changes in the target site. If the 209-nt RNA had arisen by a block to 5′-to-3′ exonucleolytic processivity, changes in the RNA sequence would not be expected to have an effect.
Fig 6.

Demonstration of endonuclease activity of RNase J1 on ΔermC mRNA. A. Schematic diagram of changes made around the putative RNase J1 cleavage site. The changed codons are at the bottom, in italics, with the amino-acids encoded indicated below.
B. Northern blot analysis of processed RNA for ΔermC with wild-type codons 3-6 and a construct with mutated codons 3-6. Percentage of 209-nt RNA fragment, relative to total ΔermC RNA (average of three experiments), is indicated below the blot.
C. Endonuclease cleavage of wild-type and mutant ΔermC RNA in vitro. No RNase J1 was added for lanes 1 and 4. H76A mutant RNase J1, which has virtually no nuclease activity (Britton et al., 2007), was added for lanes 2 and 5. Wild-type RNase J1 was added for lanes 3 and 6.
D. Double RSS construct. Codons 1-10 of the two leader region coding sequences are shown, with codon 9 larger and in bold. Asterisks denote stop codons. The upward arrows indicate expected sites of endonuclease cleavage, with the size of processed RNAs shown below.
E. Northern blot analysis of processed RNA from the double RSS construct. To the right are expected sizes of processed RNAs, with the percent of total ΔermC mRNA indicated in parentheses for each species.
In a second experiment, the effect of the mutated RNase J1 target site was analyzed in vitro. 5′-end-labeled RNAs with wild-type sequence or the same mutant sequence in codons 3-6 were prepared to contain a triphosphate 5′ end, which has been shown to be resistant to 5′-to-3′ exonuclease activity (de la Sierra-Gallay et al., 2008, Deikus et al., 2008, Mathy et al., 2007). The labeled RNA was incubated in the presence of purified wild-type RNase J1 (Fig. 6C, lanes 3 and 6) or mutant RNase J1 (Fig. 6C, lanes 2 and 5). The wild-type RNase J1 protein gave multiple endonuclease cleavage products, some of which were more prominent. We have found previously that RNase J1 can cleave single-stranded RNA in vitro at multiple sites (Deikus et al., 2008), and this may represent non-specific cleavage. The same products were observed when the substrates were incubated with less enzyme and for shorter incubation times (data not shown). The important result was the difference between the wild-type and mutant substrates in the accumulation of a ∼45-nt product (arrowheads in Fig. 6C), which likely represents a group of cleavages at around +45. The absence of this band in the mutant substrate suggested that RNase J1 indeed recognized the sequence around codons 3-6 as an endonuclease recognition site.
The third experiment gave the best indication of RNase J1 endonuclease cleavage. A ΔermC derivative was constructed that contained two RSSs, 81 nts apart (Fig. 6D). The upstream RSS was followed by a stop codon, to ensure that ribosome transit from the upstream coding sequence would not interfere with binding at the downstream ribosome binding site. Addition of Em to a strain containing this construct should result in stalling at both RSSs. To show that this was indeed the case, the two RSSs contained different sequences at codon 9: Ile in the upstream RSS and Val in the downstream RSS. Probing for peptidyl tRNA from this strain showed that, indeed, both tRNAile and tRNAval peptidyl tRNAs were detected in the strain carrying the double ribosome stall construct (data not shown). We reasoned as follows: If the 209-nt RNA was the result of exonucleolytic processing from the 5′ end up to the stalled ribosome, in the double RSS construct we should see only (or mostly) a single Em-dependent processed product of 290 nts, representing a block to exonuclease activity at the upstream RSS. If, however, the 209-nt RNA was the result of endonuclease cleavage, then the double RSS construct should give approximately similar amounts of two Em-dependent processing products, which would be 290 nts and 209 nts in length. The results in Fig. 6E are consistent with endonuclease cleavage, as the level of the two processing products was approximately equal.
Ribosome stalling at distal sites
Before pursuing the strategy to identify the site of ribosome stalling by probing for peptidyl-tRNAs directly, we constructed a derivative of ΔermC that had a FLAG tag inserted directly after the initiation codon (boxed in Fig. 7A). Assuming Em-induced ribosome stalling still occurred in this construct, the plan was to isolate the FLAG-tagged peptide from a strain that had been treated with Em and determine its composition by mass spectrometry, thus establishing where ribosome stalling had occurred. In fact, a FLAG-tagged peptide could not be detected by Western blotting (data not shown), which could have been due to rapid degradation of the truncated peptide. In any event, we abandoned this approach to identifying the RSS. However, we noticed that the ΔermC construct with the inserted 8-codon FLAG tag was still cleaved in response to addition of Em (Fig. 7B). Although the amount of such cleavage, relative to total ΔermC in the strain, was about 40% that of the wild-type ΔermC, the size of the processing fragment appeared to be the same as wild-type ΔermC. This suggested that ribosome stalling was occurring at the same place despite the displacement by 8 codons of the ribosome stall sequence away from the N terminus, which could have implications for the mechanism of Em action (see Discussion). To determine precisely the site of ΔermC processing in the FLAG-tagged construct, primer extension was done to map the 5′ end of the downstream RNA fragment. As shown in Fig. 7C, the 5′ end mapped to the same nucleotide in the wild type and in the FLAG-tagged derivative. Probing of tRNAile in a strain carrying a high-copy version of the FLAG-tagged ΔermC plasmid showed that a peptidyl-tRNAile could be detected in response to Em addition (Fig. 7D). This suggested that ribosome stalling was occurring at the same sequence of codons for the FLAG-tagged derivative as for the wild type.
Fig. 7.

A. Coding sequence of ΔermC FLAG-tagged construct, with additional inserts of 2 or 8 amino-acids shown above and possible alternate reading frames shown below. Expected cleavage sites for alternate coding sequences are indicated by upward arrows.
B. Northern blot analysis of ΔermC FLAG-tagged constructs containing either the FLAG tag alone (boxed), or two additional codons (FLAG+2), or eight additional codons (FLAG+8). Percentage of 209-nt RNA fragment, relative to total ΔermC RNA (average of two experiments), is indicated below the blot.
C. Primer extension analysis of 5′ ends from ΔermC mRNA and ΔermC+FLAG tag mRNA. The sequencing ladder at left was as in Fig. 4B.
D. Northern blot analysis of tRNA, using the tRNAile probe. Arrowheads mark peptidyl-tRNAile.
E. Northern blot analysis of tRNA, using the tRNAval and tRNAile probes, on the FLAG-tagged construct with the Val at codon 17. Arrowhead marks peptidyl-tRNAval.
To eliminate the possibility that ribosome stalling in the FLAG-tagged construct was occurring at any of the upstream Ile codons (codons 11 and 14 are also Ile codons), the Ile codon at 17 was changed to a Val codon. The data in Fig. 7E demonstrated that tRNA probing detected accumulation of a peptidyl-tRNAval in this construct, but no accumulation of a peptidyl-tRNAile. In experiments not shown, probing of the FLAG-tagged construct (i.e., with Ile at codon 17) using the tRNAval probe did not detect any peptidyl-tRNA product. These results are consistent with ribosome stalling on the FLAG-tagged construct with codon 17 in the P site.
Em-dependent ribosome stalling at such a far distance from the start of the coding sequence was quite surprising, in view of the proposed mechanism of Em-dependent ribosome stalling. It was possible that the downstream processed RNA fragment that we observed in the case of the FLAG-tagged derivative was the result of an alternative ribosome stalling site. As can be seen in Fig. 7A, introduction of the FLAG-tag coding sequence supplies two new possible AUG start codons (double underlined in Fig. 7A). Initiation from either of these start codons would result in translation of a peptide in an alternative reading frame that has a more N-proximal stretch of hydrophobic residues at which an Em-bound ribosome could stall (FLV if starting from first AUG codon; FLVFL if starting from the second AUG codon). Three observations argue against this possibility: First, stalling of the ribosome in the FLAG-tagged construct gave a peptidyl-tRNAile (Fig. 7D), and there is no Ile codon in the alternative reading frames. Second, assuming that ΔermC processing in the FLAG-tagged construct occurs the same distance (15 nts) from the A site codon as in the wild type, then stalling with codon 10 of alternate reading frame 1 in the A site would have given a 220-nt downstream RNA fragment, and stalling with codon 10 of alternate reading frame 2 in the A site would have given a 214-nt downstream RNA fragment (up arrows in Fig. 7A). Our primer extension results comparing the wild-type and the FLAG-tagged construct (Fig. 7B) show clearly that the 5′ end of the downstream RNA fragment for the FLAG-tagged construct is either the same or no more than 1-2 nts different from the 5′ end mapped for the wild-type ΔermCRNA processing. Third, we created a FLAG-tagged derivative that used only GAC codons for the four consecutive Asp residues, thus eliminating the possibility of alternative start codons. This construct gave the same 209-nt RNA fragment upon Em addition (data not shown).
To see how far the RSS could be moved away from the N-terminus and still allow stalling, additional ΔermC derivatives were constructed that contained the FLAG tag plus 2 or 8 extra amino-acids, which were inserted between the start codon and the beginning of the FLAG-tag sequence (Fig. 7A, top). Displacement of the ribosome stalling site by two more codons resulted in ΔermC mRNA cleavage that was only 3% of total, while displacement of the ribosome stalling site by eight more codons resulted in an almost complete loss of cleavage in response to Em (Fig. 7B).
Discussion
Em-induced ribosome stalling on the ermC leader peptide CDS has been the subject of many previous studies, as it is a key step in this classic example of gene regulation by translational attenuation (Dubnau, 1984, Weisblum, 1985). Stalling of an Em-bound ribosome also serves as a model for studies of the inhibitory action of Em (Tenson et al., 2003). Our results with tRNA probing demonstrate that I9 is the final amino acid incorporated in the peptide chain before Em-dependent ribosome stalling. These experiments did not, however, distinguish whether the I9 codon is in the A site (i.e., pre-translocation), as proposed by Mayford and Weisblum (1989), or the P site (i.e., post-translocation). While this manuscript was in revision, a report by Mankin and colleagues appeared in which an in vitro system containing E. coli ribosomes was used to show that Em-dependent stalling occurs with codon 9 in the P site and codon 10 in the A site (Vazquez-Laslop et al., 2008). In addition, the data in this paper suggested that the accumulated peptidyl-tRNA was not a drop-off product. Assuming the results of the in vitro E. coli system are applicable to the in vivo situation in B. subtilis, we can conclude that Em-dependent stalling leaves a ribosome with codon I9 in the P site and codon S10 in the A site. Thus, Em-induced ΔermC mRNA processing, which occurs between codons 4 and 5, is not an example of A-site cleavage. It remains to be determined whether A-site cleavage occurs in B. subtilis.
The question of which ribonuclease activity is responsible for Em-dependent processing was addressed in these studies. Experiments showed that a reduction in cellular RNase J1 content resulted in a significant decrease in the level of ΔermC processing (Fig. 4). The fact that secondary structure located at the 5′ terminus interfered with Em-induced ΔermC processing (Fig. 5) indicated a 5′-end-dependent ribonuclease activity, of which RNase J1 is the only known example in B. subtilis (de la Sierra-Gallay et al., 2008, Even et al., 2005, Mathy et al., 2007). Note that the previous studies on the effect of 5′ secondary structure on ΔermC mRNA half-life (Sharp & Bechhofer, 2005) were performed in the absence of Em. Thus, both the turnover of ΔermC mRNA and the processing upon ribosome stalling are inhibited by 5′-terminal structure. The simplest interpretation of these results is that RNase J1 is directly responsible for initiation of ΔermC mRNA decay and for cleavage at the RSS. The effect of 5′ structure on RNase J1 activity needs to be addressed systematically.
Experiments shown in Fig. 6 suggest that RNase J1 processing of ΔermC mRNA is by endonucleolytic cleavage between codons 4 and 5, and not exonucleolytic decay from the 5′ end up to the stalled ribosome. As such, the RNase J1 recognition site may consist of specific ΔermC nucleotides around codons 4-5, and RNase J1 may cleave at this site constitutively. In the absence of the stalled ribosome, such cleavage would initiate decay of the downstream fragment, likely by RNase J1 switching to 5′-to-3′ exonuclease mode and rapidly degrading the downstream 209-nt RNA, in accordance with a model that we and others have proposed for the role of RNase J1 in B. subtilis RNA turnover (de la Sierra-Gallay et al., 2008, Deikus et al., 2008). In the presence of the stalled ribosome, the 5′ end of the 209-nt RNA would be protected from attack by RNase J1, and this RNA fragment would accumulate.
Protection of the 5′ end of the 209-nt RNA by the stalled ribosome is consistent with structural studies of mRNA:ribosome interactions. Using Thermus thermophilus ribosomes, Noller and colleagues have determined that the section of mRNA that is wrapped around the ribosome “neck,” and interacts most closely with the ribosome, stretches from 6 nts upstream to 3 nts downstream of the A site (Yusupova et al., 2001). In our case, cleavage between codons 4 and 5 would generate a 5′ end that is 15 nucleotides upstream of the A site or 9 nts upstream of the mRNA sequence that is most closely associated with the ribosome. Exonuclease progression is often blocked several nts away from a bound protein or an RNA secondary structure, so our results are consistent with the stalled ribosome constituting a block to further RNase J1-mediated 5′- to-3′ exonucleolytic degradation after an initial endonuclease cleavage. The binding cleft in RNase J1, in which the 5′ end of an RNA is thought to be situated for catalysis (de la Sierra-Gallay et al., 2008), may not be able to accommodate an RNA 5′ end that is surrounded by ribosomal structural components.
An alternative explanation for ΔermC mRNA processing is that RNase J1 does not catalyze endonuclease cleavage directly but is required to activate some other ribonuclease activity, perhaps one that is ribosome-associated. Our in vitro results (Fig. 6C), which indicate recognition of the cleavage site contained in codons 3-6, are more consistent with direct cleavage by RNase J1.
A noteworthy aspect of this study is the finding that ribosome stalling occurred in the ΔermC construct that had a FLAG tag inserted immediately after the initiation codon. This insertion moved the RSS further away from the start of translation, such that Em-induced stalling was occurring with codon 17 in the P site (Fig. 7A). This result is quite surprising in view of the work of others on the mechanism of Em inhibition of translation and ribosome stalling (Tenson et al., 2003, Vazquez-Laslop et al., 2008). Their results indicated that the RSS must be located at a particular distance from the N-terminus, optimally ending at codon 9. Translation of an mRNA, in the presence of Em, that had an RSS located further than codon 9 would likely result in peptidyl-tRNA drop-off before the stall site was reached. Our results, on the other hand, suggest that an Em-bound ribosome can synthesize a nascent peptide twice that size (17 amino-acids) before it reaches the stalling site. Further studies will be required to understand the structural basis of this finding, including the determination whether the FLAG-tag sequence is unique in allowing stalling further downstream.
Experimental procedures
Bacterial strains
The B. subtilis host was BG1, which is trpC2 thr-5. E. coli strain DH5α (Grant et al., 1990) was the host for plasmid constructions.
Standard procedures
The preparation and transformation of B. subtilis competent cell cultures were as described previously (Dubnau & Davidoff-Abelson, 1971). Supplemented minimal medium for RNA isolation, Northern blot analysis, and 5S rRNA probing as a quantitation control were as described (Oussenko et al., 2005). For induction of ribosome stalling, Em was added to a concentration of 0.04 μg/ml at mid-logarithmic phase (Klett = 80 units; green filter) and cells were harvested 20 minutes later. For the analysis of processing in ribonuclease mutants (Fig. 4), strains were grown in 2x YT medium (1% yeast extract, 2% tryptone, 1% NaCl). Since the conditional mutants for ribonucleases J1, P, and Z carry an Em-resistance gene as part of the pspac promoter construct, a higher concentration of Em (3.0 μg/ml) was used to induce ribosome stalling, and this was added at the time of inoculation from an overnight culture.
Plasmids
ΔermC mRNA was transcribed from plasmid pYH250, an E. coli/B. subtilis shuttle plasmid that carries ΔermC and a chloramphenicol resistance marker (Sharp & Bechhofer, 2005), and derivatives thereof. In pYH250, a HindIII site is present at the beginning of the ΔermC transcriptional unit and an EcoRI site is located downstream of the ΔermC transcription terminator. The HindIII site was incorporated into mutagenic PCR primers that included nucleotide sequence changes in the N-terminal ΔermC CDS. These 5′ primers were used in conjunction with a 3′ primer to amplify ΔermC, giving an amplicon that was cloned as a HindIII-EcoRI fragment into the pYH250 vector. In the FLAG+2 construct, a 6-base pair BamHI site was added immediately after the start codon. For the FLAG+8 construct, complementary oligonucleotides with BamHI-compatible overhanging ends were annealed and inserted into the FLAG+2 BamHI site. For the I9V mutation, we took advantage of the fact that codons 17-18 of the ΔermC CDS constitute a HpaI site. A mutagenic oligonucleotide including this HpaI site was used in conjunction with a 3′ primer complementary to sequences upstream of ΔermC to generate a PCR amplicon that was cloned as an XbaI-HpaI fragment back into the pYH250 vector. For the double RSS construct, the ΔermC gene containing the I9V change was amplified by PCR to generate a fragment with an upstream EcoRV site and a downstream EcoRI site. This was used to replace the HpaI-EcoRI small fragment of a ΔermC plasmid that had been constructed previously to contain a stop codon after codon 9 (Sharp & Bechhofer, 2003).
In strains where peptidyl-tRNA probing was done, it was necessary to use a high-copy-number version of the pYH250-derived plasmid. This was constructed by substituting an AvrII-SwaI fragment, which contains part of the B. subtilis replicon, with the corresponding fragment from the cop-1 mutant plasmid (Villafane et al., 1987). The single base-pair change that constitutes the cop-1 mutation lies in a BspHI recognition site (TCATGA changed to TGATGA), so digestion with BspHI was used to confirm the presence of the cop-1 mutation. For reasons that we do not understand, the presence of the cop-1 mutation on a pYH250-derived shuttle plasmid sometimes causes instability in E. coli. Therefore, we used plasmid pSD285, a ΔermC derivative of the high-copy-number plasmid pBD404 (Bechhofer & Wang, 1998), which carries only a B. subtilis replicon, for some experiments. As the quantitation of 209-nt RNA was determined based on percentage of total ΔermC RNA, differences in ΔermC copy number between strains were taken into account. Quantitation of full-length ΔermC mRNA and RNA processing products was done with a Storm 860 PhosphorImager (Molecular Dynamics) or a Typhoon TRIO variable mode imager (GE Healthcare).
tRNA analysis
For Northern blot analysis of aminoacyl- and peptidyl-tRNA, RNA was isolated under acidic conditions in the cold, using the method of Varshney et al. (1991). 15 ml cultures were grown to Klett = 80 units, with or without 0.04 μg/ml Em added at Klett = 30 units. Cells were pelleted at 1,500 × g, the cell pellet was resuspended in the remaining supernatant and was transferred to a microcentrifuge tube. 250 μl of lysis buffer (10 mg/ml lysozyme in 100 mM Na-acetate, pH 4.5) were added and lysis was allowed to continue on ice for 20 min. All subsequent steps were done at 4°C. 250 μl of 500 mM Na-acetate, pH 4.5, 20 mM EDTA were added, and the nucleic acid was extracted with an equal volume (500 μl) of phenol (equilibrated with 300 mM Na-acetate, pH 4.5, 20 mM EDTA) by vortexing 30 s, 60 s, and again 60 s, with incubation on ice for 60 s in between each vortexing. Phases were separated by centrifugation for 12 min, and the aqueous phase was transferred to a fresh tube containing 500 μl phenol solution as above. The solution was vortexed for 60 s and the phases were separated by centrifugation for 10 min. Nucleic acid was precipitated from the aqueous phase by addition of 2.5 volumes ethanol, incubation on ice for 1 hr, and centrifugation for 15 min. The pellet was dissolved in 60 μl of 300 mM Na-acetate, pH 4.5, and nucleic acid was precipitated again with 2.5 volumes ethanol. The final pellet was dissolved in 20 μl 10 mM Na-acetate, pH 4.5, 1 mM EDTA.
For gel electrophoresis, 2 μg of total RNA was added to 10 μl of loading buffer consisting of 50% urea, 10 mM Na-acetate, pH 5, 1 mM EDTA, 0.01% bromophenol blue. The gel was an 8 M urea, 9% polyacrylamide gel prepared in 100 mM Na-acetate, pH 5, 1 mM EDTA, and was run at 4°C in the same buffer using a Mini-PROTEAN II electrophoresis cell (Bio-Rad) for 3.5 hrs at 110-115 V. The gel was rinsed in water, electroblotted overnight at 30 V, and processed as usual for polyacrylamide gel Northern blotting.
RNase J1 activity in vitro
5′-end-labeled RNA substrates for in vitro experiments consisted of ΔermC nucleotides 1-200 (wild type or mutant). These were prepared by T7 RNA polymerase transcription (Ambion T7 MAXIscript kit), in the presence of [γ-32P]GTP, of a PCR fragment that contained the ΔermC sequence, with a T7 promoter included in the upstream primer. Purification of the transcription product and assay of RNase J1 endonuclease activity were as described (Deikus et al., 2008).
Acknowledgements
This work was supported by Public Health Service grant GM-48804 from the National Institutes of Health. We thank Chris Hayes for advice on tRNA analysis, Ciaran Condon for purified RNase J1 protein, and Martin Lovmar for e-mail discussions on ribosome stalling in the FLAG-tagged constructs.
References
- Bechhofer DH, Wang W. Decay of ermC mRNA in a polynucleotide phosphorylase mutant of Bacillus subtilis. J Bacteriol. 1998;180:5968–5977. doi: 10.1128/jb.180.22.5968-5977.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton RA, Wen T, Schaefer L, Pellegrini O, Uicker WC, Mathy N, Tobin C, Daou R, Szyk J, Condon C. Maturation of the 5′ end of Bacillus subtilis 16S rRNA by the essential ribonuclease YkqC/RNase J1. Mol Microbiol. 2007;63:127–138. doi: 10.1111/j.1365-2958.2006.05499.x. [DOI] [PubMed] [Google Scholar]
- Brosius J. Compilation of superlinker vectors. Methods Enzymol. 1992;216:469–483. doi: 10.1016/0076-6879(92)16043-j. [DOI] [PubMed] [Google Scholar]
- Christensen SK, Gerdes K. RelE toxins from bacteria and Archaea cleave mRNAs on translating ribosomes, which are rescued by tmRNA. Mol Microbiol. 2003;48:1389–1400. doi: 10.1046/j.1365-2958.2003.03512.x. [DOI] [PubMed] [Google Scholar]
- de la Sierra-Gallay IL, Zig L, Jamalli A, Putzer H. Structural insights into the dual activity of RNase J. Nat Struct Mol Biol. 2008;15:206–212. doi: 10.1038/nsmb.1376. [DOI] [PubMed] [Google Scholar]
- Deikus G, Condon C, Bechhofer DH. Role of Bacillus subtilis RNase J1 Endonuclease and 5′-Exonuclease Activities in trp Leader RNA Turnover. J Biol Chem. 2008;283:17158–17167. doi: 10.1074/jbc.M801461200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drider D, DiChiara JM, Wei J, Sharp JS, Bechhofer DH. Endonuclease cleavage of messenger RNA in Bacillus subtilis. Mol Microbiol. 2002;43:1319–1329. doi: 10.1046/j.1365-2958.2002.02830.x. [DOI] [PubMed] [Google Scholar]
- Dubnau D. Translational attenuation: the regulation of bacterial resistance to the macrolide-lincosamide-streptogramin B antibiotics. CRC Crit Rev Biochem. 1984;16:103–132. doi: 10.3109/10409238409102300. [DOI] [PubMed] [Google Scholar]
- Dubnau D, Davidoff-Abelson R. Fate of transforming DNA following uptake by competent Bacillus subtilis. I. Formation and properties of the donor-recipient complex. J Mol Biol. 1971;56:209–221. doi: 10.1016/0022-2836(71)90460-8. [DOI] [PubMed] [Google Scholar]
- Dulebohn D, Choy J, Sundermeier T, Okan N, Karzai AW. Trans-translation: the tmRNA-mediated surveillance mechanism for ribosome rescue, directed protein degradation, and nonstop mRNA decay. Biochemistry. 2007;46:4681–4693. doi: 10.1021/bi6026055. [DOI] [PubMed] [Google Scholar]
- Even S, Pellegrini O, Zig L, Labas V, Vinh J, Brechemmier-Baey D, Putzer H. Ribonucleases J1 and J2: two novel endoribonucleases in B.subtilis with functional homology to E.coli RNase E. Nucleic Acids Res. 2005;33:2141–2152. doi: 10.1093/nar/gki505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant SG, Jessee J, Bloom FR, Hanahan D. Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc Natl Acad Sci U S A. 1990;87:4645–4649. doi: 10.1073/pnas.87.12.4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes CS, Sauer RT. Cleavage of the A site mRNA codon during ribosome pausing provides a mechanism for translational quality control. Mol Cell. 2003;12:903–911. doi: 10.1016/s1097-2765(03)00385-x. [DOI] [PubMed] [Google Scholar]
- Hong SJ, Tran QA, Keiler KC. Cell cycle-regulated degradation of tmRNA is controlled by RNase R and SmpB. Mol Microbiol. 2005;57:565–575. doi: 10.1111/j.1365-2958.2005.04709.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hue KK, Bechhofer DH. Effect of ermC leader region mutations on induced mRNA stability. J Bacteriol. 1991;173:3732–3740. doi: 10.1128/jb.173.12.3732-3740.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovmar M, Tenson T, Ehrenberg M. Kinetics of macrolide action: the josamycin and erythromycin cases. J Biol Chem. 2004;279:53506–53515. doi: 10.1074/jbc.M401625200. [DOI] [PubMed] [Google Scholar]
- Mathy N, Benard L, Pellegrini O, Daou R, Wen T, Condon C. 5′-to-3′ exoribonuclease activity in bacteria: role of RNase J1 in rRNA maturation and 5′ stability of mRNA. Cell. 2007;129:681–692. doi: 10.1016/j.cell.2007.02.051. [DOI] [PubMed] [Google Scholar]
- Mayford M, Weisblum B. ermC leader peptide. Amino acid sequence critical for induction by translational attenuation. J Mol Biol. 1989;206:69–79. doi: 10.1016/0022-2836(89)90524-x. [DOI] [PubMed] [Google Scholar]
- Menninger JR, Otto DP. Erythromycin, carbomycin, and spiramycin inhibit protein synthesis by stimulating the dissociation of peptidyl-tRNA from ribosomes. Antimicrob Agents Chemother. 1982;21:811–818. doi: 10.1128/aac.21.5.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oussenko IA, Abe T, Ujiie H, Muto A, Bechhofer DH. Participation of 3′-to-5′ exoribonucleases in the turnover of Bacillus subtilis mRNA. J Bacteriol. 2005;187:2758–2767. doi: 10.1128/JB.187.8.2758-2767.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen K, Zavialov AV, Pavlov MY, Elf J, Gerdes K, Ehrenberg M. The bacterial toxin RelE displays codon-specific cleavage of mRNAs in the ribosomal A site. Cell. 2003;112:131–140. doi: 10.1016/s0092-8674(02)01248-5. [DOI] [PubMed] [Google Scholar]
- Pellegrini O, Mathy N, Gogos A, Shapiro L, Condon C. The Bacillus subtilis ydcDE operon encodes an endoribonuclease of the MazF/PemK family and its inhibitor. Mol Microbiol. 2005;56:1139–1148. doi: 10.1111/j.1365-2958.2005.04606.x. [DOI] [PubMed] [Google Scholar]
- Pellegrini O, Nezzar J, Marchfelder A, Putzer H, Condon C. Endonucleolytic processing of CCA-less tRNA precursors by RNase Z in Bacillus subtilis. EMBO J. 2003;22:4534–4543. doi: 10.1093/emboj/cdg435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redko Y, Li de Lasierra-Gallay I, Condon C. When all’s zed and done: the structure and function of RNase Z in prokaryotes. Nat Rev Microbiol. 2007;5:278–286. doi: 10.1038/nrmicro1622. [DOI] [PubMed] [Google Scholar]
- Richards J, Mehta P, Karzai AW. RNase R degrades non-stop mRNAs selectively in an SmpB-tmRNA-dependent manner. Mol Microbiol. 2006;62:1700–1712. doi: 10.1111/j.1365-2958.2006.05472.x. [DOI] [PubMed] [Google Scholar]
- Sharp JS, Bechhofer DH. Effect of translational signals on mRNA decay in Bacillus subtilis. J Bacteriol. 2003;185:5372–5379. doi: 10.1128/JB.185.18.5372-5379.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp JS, Bechhofer DH. Effect of 5′-proximal elements on decay of a model mRNA in Bacillus subtilis. Mol Microbiol. 2005;57:484–495. doi: 10.1111/j.1365-2958.2005.04683.x. [DOI] [PubMed] [Google Scholar]
- Sunohara T, Jojima K, Tagami H, Inada T, Aiba H. Ribosome stalling during translation elongation induces cleavage of mRNA being translated in Escherichia coli. J Biol Chem. 2004;279:15368–15375. doi: 10.1074/jbc.M312805200. [DOI] [PubMed] [Google Scholar]
- Tenson T, Lovmar M, Ehrenberg M. The mechanism of action of macrolides, lincosamides and streptogramin B reveals the nascent peptide exit path in the ribosome. J Mol Biol. 2003;330:1005–1014. doi: 10.1016/s0022-2836(03)00662-4. [DOI] [PubMed] [Google Scholar]
- Varshney U, Lee CP, RajBhandary UL. Direct analysis of aminoacylation levels of tRNAs in vivo. Application to studying recognition of Escherichia coli initiator tRNA mutants by glutaminyl-tRNA synthetase. J Biol Chem. 1991;266:24712–24718. [PubMed] [Google Scholar]
- Vazquez-Laslop N, Thum C, Mankin AS. Molecular mechanism of drug-dependent ribosome stalling. Mol Cell. 2008;30:190–202. doi: 10.1016/j.molcel.2008.02.026. [DOI] [PubMed] [Google Scholar]
- Villafane R, Bechhofer DH, Narayanan CS, Dubnau D. Replication control genes of plasmid pE194. J Bacteriol. 1987;169:4822–4829. doi: 10.1128/jb.169.10.4822-4829.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegscheid B, Condon C, Hartmann RK. Type A and B RNase P RNAs are interchangeable in vivo despite substantial biophysical differences. EMBO Rep. 2006;7:411–417. doi: 10.1038/sj.embor.7400641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisblum B. Inducible resistance to macrolides, lincosamides and streptogramin type B antibiotics: the resistance phenotype, its biological diversity, and structural elements that regulate expression--a review. J Antimicrob Chemother. 1985;16(Suppl A):63–90. doi: 10.1093/jac/16.suppl_a.63. [DOI] [PubMed] [Google Scholar]
- Withey JH, Friedman DI. A salvage pathway for protein structures: tmRNA and trans-translation. Annu Rev Microbiol. 2003;57:101–123. doi: 10.1146/annurev.micro.57.030502.090945. [DOI] [PubMed] [Google Scholar]
- Yao S, Blaustein JB, Bechhofer DH. Processing of Bacillus subtilis small cytoplasmic RNA: evidence for an additional endonuclease cleavage site. Nucleic Acids Res. 2007;35:4464–4473. doi: 10.1093/nar/gkm460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusupova GZ, Yusupov MM, Cate JH, Noller HF. The path of messenger RNA through the ribosome. Cell. 2001;106:233–241. doi: 10.1016/s0092-8674(01)00435-4. [DOI] [PubMed] [Google Scholar]