

Abstract
Substitution of the Gla-domain of activated protein C (APC) with the Gla-domain of prothrombin (APC-PTGla) improves the anticoagulant activity of APC independent of protein S. Previous FRET studies showed that this substitution alters the active-site topography of this mutant, rendering it identical to the active-site of the APC-protein S complex. In this study, we characterized the functional properties and the active-site topography of another APC chimera containing the Gla-domain of factor X (APC-FXGla). We discovered that the anticoagulant activity of this mutant was similarly improved independent of protein S. The average distance of the closest approach (L) between the donor dye fluorescein attached to the active-site of APC derivatives and the acceptor dye octadecylrhodamine incorporated into PC/PS vesicles was determined to be 99 Å for APC and 84-86 Å for both APC-PTGla and APC-FXGla. Protein S minimally influenced the L values of the APC chimeras, however, it lowered this value to 87 Å for wild-type APC. Further studies revealed that neither chimera elicits a protective signaling response in the TNF-α-activated endothelial cells. These results suggest that unique structural features within the Gla-domain of APC enable the protease to interact with endothelial protein C receptor in the antiinflammatory pathway, while the same features also cause an inherently lower specific activity for APC in the anticoagulant pathway. This adaptation has made APC a cofactor dependent protease, requiring the cofactor function of protein S for its optimal anticoagulant function, which appears to involve the alteration of the active-site topography of APC above the membrane surface.
Keywords: FRET, APC, Prothrombin, Factor X, Gla-domain
1. Introduction
Activated protein C (APC) is a multi-domain vitamin K-dependent serine protease in plasma that is involved in the modulation of both anticoagulant and antiinflammatory pathways [1-5]. In the anticoagulant pathway, APC inactivates the procoagulant cofactors factor Va (FVa) and factor VIIIa (FVIIIa) by limited proteolysis [6]. The N-terminus γ-carboxyglutamic (Gla) domain of APC plays a key role in the anticoagulant function of the protease as it mediates the calcium dependent interaction of the protease with protein S on the membrane surface [7,8]. Protein S is an essential cofactor which promotes the anticoagulant function of APC in the proteolytic degradation of FVa and FVIIIa on the negatively charged phospholipid membrane [6,9]. APC inhibits thrombin generation by the rapid cleavage of FVa at Arg-506 and a slow membrane-dependent cleavage of the cofactor at the Arg-306 recognition site [10-12]. Protein S markedly promotes the activity of APC toward both cleavage sites, though it has been reported that it has a greater cofactor effect on the cleavage of the Arg-306 scissile bond [10,13]. Protein S has been demonstrated to bind the Gla-domain of APC, thereby enhancing its affinity for the physiological membrane surfaces and also relocating the active-site of APC to increase its specificity and efficiency toward its substrates [9,14]. In a previous study, the substitution of the Gla-domain of APC with the corresponding domain of prothrombin (APC-PTGla) had dramatically increased the anticoagulant activity of APC independent of protein S [15]. Subsequent fluorescence resonance energy transfer (FRET) studies have shown that protein S lowers the average distance of closest approach (L) between a fluorescein dye in the active-site of wild-type APC and octadecylrhodamine dyes at the membrane surface by ~10 Å (94 Å in the absence and 84 Å in the presence of protein S) [14,16]. Interestingly, an L value of ~89 Å for the membrane-bound APC-PTGla has been observed in both the presence and absence of protein S, suggesting that the cofactor function of protein S may involve an alteration of the topographical orientation of the active-site of the membrane-bound APC [16].
In addition to its key role in the anticoagulant pathway, the Gla-domain of APC also plays an essential role in the antiinflammatory function of the protease. The Gla-domain binds to endothelial protein C receptor (EPCR) to change the substrate specificity of APC, thereby eliciting cytoprotective signaling responses in vascular endothelial cells [9,17-19]. This property of APC is believed to contribute to its beneficial therapeutic effect in patients with severe sepsis [19, 20]. It has been demonstrated that the EPCR-bound APC can no longer function in the anticoagulant pathway [21]. Thus, identification of specific residues of the APC Gla-domain which are required for its either anticoagulant function or cytoprotective activities is of particular interest since it may facilitate engineering variants of APC which can exclusively and effectively function in either the anticoagulant pathway independent of protein S or in the antiinflammatory pathway. Although, the substitution of the Gla-domain of APC with other coagulation proteases either markedly improves [15] or does not negatively impact the anticoagulant function of APC in the whole plasma assay [22], nevertheless, the effect of the Gla-domain substitutions in the protective signaling activities of APC has not been investigated.
In this study, employing FRET and functional activity assays, we characterized the properties of a chimeric APC mutant in which the Gla-domain of the protease has been replaced with the same domain of factor X (APC-FXGla). Similar to APC-PTGla, both the anticoagulant activity and active-site topography of APC-FXGla was altered independent of protein S, thus mimicking the same characteristics of wild-type APC in the presence of protein S. Analysis of the activity of both APC derivatives in the TNF-α-induced apoptosis, permeability and neutrophil adhesion assays revealed that there is an absolute requirement for the Gla-domain of APC in eliciting protective signaling responses in endothelial cells.
2. Materials and methods
2.1. Construction, expression and purification of recombinant APC mutants
Expression, purification and preparation of wild-type protein C and its chimeric prothrombin Gla-domain (PTGla) or factor X Gla-domain (FXGla) derivatives in human embryonic kidney cells have been described previously [15,23]. The expressed proteins were purified by a combination of immunoaffinity and ion exchange chromatography using the HPC4 monoclonal antibody immobilized on Affi-gel 10 and FPLC Mono Q column, respectively, as described [24]. The protein C zymogens (~1 mg) were converted to APC by thrombin (50 μg) in 0.1 M NaCl, 0.02 M Tris-HCl, pH 7.4 (TBS) containing 5 mM EDTA for 2 h at 37 °C as described [25]. The APC derivatives were separated from thrombin and their concentrations were determined from the absorbance at 280 nm assuming a molecular mass of 56 kDa and an extinction coefficient (E1%1cm) of 14.5 and by an active-site titration assay using recombinant protein C inhibitor as described [25]. The purity of the APC derivatives was ensured by SDS-PAGE.
Human plasma protein S was purchased from Enzyme Research Laboratories (South Bend, IN). Human factors Xa (FXa) and Va (FVa), prothrombin and the fluorescein-labeled Phe-Pro-Arg-ck (Fl-FPR) were purchased from Haematologic Technologies Inc. (Essex Junction, VT) and octadecylrhodamine (OR) were from Invitrogen (Carlsbad, CA). Dioleoylphosphatidylcholine (PC) and dioleoylphosphatidylserine (PS) were purchased from Avanti Polar Lipids (Alabaster, AL). The chromogenic substrate Spectrozyme PCa (SpCa) was purchased from American Diagnostica (Greenwich, CT) and S2238 was from Kabi Pharmacia/Chromogenix (Franklin, OH).
2.2. Anticoagulant activity
The anticoagulant activity of APC derivatives were evaluated in both purified and plasma-based assay systems. In the purified system, the APC concentration dependence of FVa inactivation was measured by a three-stage assay as described [25]. Briefly, in the first stage, FVa (2.5 nM) was incubated with different concentrations of each APC derivative (1-5 nM) for 1-5 min on 20 μM phospholipid vesicles composed of 80% PC and 20 % PS in TBS containing 2.5 mM Ca2+, 1.0 mg/ml BSA, and 0.1% polyethylene glycol 8000. In the second stage, the remaining FVa activity was determined in a prothrombinase assay measuring the FVa-catalyzed prothrombin activation by FXa as described [25]. The remaining activity of FVa was determined from the decrease in the rate of thrombin generation as monitored by an amidolytic activity assay in the third stage using 200 μM S2238. A similar assay was used to evaluate the catalytic activity of APC derivatives toward FVa in the presence of a saturating concentration of protein S (220 nM). The anticoagulant activities in plasma were evaluated in an APTT assay using a STart 4 fibrinometer (Diagnostica/Stago, Asnieres, France). Briefly, 0.050 mL of TBS lacking or containing 0-20 nM final concentrations of each APC derivative was incubated with a mixture of 0.05 ml of normal pooled plasma plus 0.05 ml APTT reagent (Alexin) for 5 min before the initiation of clotting by the addition of 0.05 ml of 35 mM CaCl2 at 37 °C as described [25].
2.3. Permeability assay
The endothelial cell permeability was quantitated by the spectrophotometric measurement of the flux of Evans blue-bound albumin across functional EA.hy926 cell monolayer (Dr. C. Edgell, University of North Carolina at Chapel Hill, NC) using a modified 2-compartment chamber model as described previously [26]. Briefly, EA.hy926 cells were plated (5 × 104/well) in transwell of 3 μm pore size and 12-mm diameter for 4-6 days. The confluent monolayers were incubated with APC (0-100 nM) for 3 h followed by activation by thrombin (5 nM) for 10 min as described [26]. Inserts were washed with PBS, pH 7.4 before adding 0.5 mL Evans blue (0.67 mg/mL) (Sigma, St Louis, MO) diluted in growth medium containing 0.4% BSA. Fresh growth medium was added to the lower chamber, and the medium in the upper chamber was replaced with Evans blue/BSA. After 10 min the optical density at 650 nm was measured in the lower chamber. All experiments were performed in duplicate and repeated multiple times.
2.4. Apoptosis assay
The cytoprotective effect of different APC derivatives (0-100 nM) in EA.hy926 cells was evaluated in a TNF-α-induced (10 ng/mL for 4 hr) apoptosis assay as described [26]. The number of apoptotic cells was expressed as the percentage of TUNEL-positive cells of the total number of nuclei determined by Hoechst staining as described [26]. Results are expressed as mean ±SEM and all experiments were repeated three times. All APC assays were carried out in the presence of 4 units/mL hirudin.
2.5. Adhesion assay
Human neutrophil adherence to endothelial cells was evaluated by fluorescent labeling of neutrophils as described [26,27]. Briefly, peripheral blood neutrophils were labeled with 5 μM Vybrant DiD (Molecular Probes) for 20 min at 37 °C in phenol red-free RPMI containing 5% fetal bovine serum. Following twice washing, neutrophils (1.5 × 106/ml, 200 μl/well) were resuspended in adhesion medium (RPMI containing 2% fetal bovine serum and 20 mM Hepes) and added to confluent monolayers of EA.hy926 cells in 96-well plates which were treated with APC derivatives (10 nM for 24 h) followed by TNF-α (10 ng/mL for 4 h). The fluorescence of labeled cells was measured (total signal) using a fluorescence microplate reader (Molecular Device). After incubation for 60 min at 37 °C, non-adherent cells were removed by washing four times with pre-warmed RPMI and the fluorescent signals of adherent cells were measured by the same methods. The percentage of adherent leukocytes was calculated by the formula: % adherence = (adherent signal/total signal) × 100.
2.6. Phospholipid vesicle preparations
Phospholipid vesicles at a molar ratio of PC to PS of 4:1 containing or lacking different amounts of OR were prepared by the extrusion method as described [28]. Briefly, dried phospholipid mixtures were suspended at 1 mg/mL in 0.1 M NaCl, 0.05 M Hepes, pH 7.4, mixed for 10 min and passed twenty times through a 100 nm pore diameter polycarbonate membrane. The OR containing vesicles received various amounts of the dye in ethyl acetate prior to lyophilization and extrusion. The % yield of phospholipids recovery was determined by comparing the amount of choline from small samples before and after extrusion step by a colorimetric assay using Wako phospholipid B kit (Wako Chemicals USA, Inc., Richmond, VA). The % recovery for different preparations ranged from 85% to 90%. Phospholipid vesicles containing 100% PC vesicles were prepared by the same procedures for coating cuvettes. The concentration of OR in the acceptor-containing samples was determined from absorbance at 564 nm using a molar extinction coefficient of 95,400 M-1 cm-1 as described [14]. The acceptor density of OR (σ, in OR/Å2) was calculated using molecular weights 786.1 Da and 810.0 Da for PC and PS, respectively, assuming that acceptors were distributed randomly at the phospholipid surface, and assuming that each phospholipid molecule occupies 70 Å2 of the surface area as described [29].
2.7. Fluorescent labeling
All APC derivatives were incubated with 10-fold molar excess of Fl-FPR for 2 h at room temperature in the dark. The extent of active-site labeling was monitored by the loss of the enzymatic activity using SpPCa. Incubation was continued till more than 99.9% of the activity was inhibited. The free inhibitor or dye was separated from the labeled proteins by gel filtration on the PD-10 column followed by their extensive dialysis in 0.1 M NaCl and 0.05 M Hepes, pH 7.4 containing 5 mM Ca2+ at 4 °C in the dark. The extent of fluorescence labeling for all proteins was determined as described by Bock [30]. An extinction coefficient of 84,000 M-1 cm-1 at 498 nm was used to calculate the fluorescein concentration and the ratio ε280nm/ε498nm = 0.19 was used to correct for the contribution of the dye to 280-nm absorbance of the proteins as described [30]. The average number of dyes per protein was determined to range from 0.6-0.8 for all APC derivatives. The maximum excitation and emission wavelengths for all fluorescein labeled proteins were determined to be 493 nm and 521 nm, respectively.
2.8. Spectral measurements
All spectral measurements were performed using an Aminco-Bowman series 2 Spectrophotometer (Spectronic Unicam, Rochester, NY) as described [28]. The anisotropy of the fluorescein labeled proteins (25-50 nM) were measured both in the absence and presence of saturating concentrations of PC/PS vesicles (35 μM) and protein S (300 nM) using the excitation and emission wavelengths of 493 nm and 521 nm as described [28]. The anisotropy of OR labeled PC/PS vesicles containing limiting concentration of acceptor (σ = 0.7 × 10-4) was measured as described [28]. Absorbance measurements were made using a Beckman Coulter DU 800 spectrophotometer. The values for the quantum yield (Q), spectral overlap between donor and acceptor dyes (JDA), and the distance between donor and acceptor dyes at the 50% FRET efficiency (R0) were calculated as described [14]. The values of Q and JDA were separately measured for each fluorescein-labeled protein both in the absence and presence of PC/PS vesicles and Protein S.
2.9. Energy transfer measurements
Fluorescence energy transfer measurements were performed as described [14], except that the donor containing (cuvette D) and both donor (fluorescein) and acceptor (OR) containing cuvettes (cuvette DA) each received 50 nM fluorescein-labeled APC derivatives in 0.1 M NaCl, 0.05 M Hepes, pH 7.4, and 5 mM Ca2+ while blanks (cuvette B) and acceptor-containing cuvettes (cuvette A) received 50 nM of unlabeled APC derivatives in the same buffer. In the case of measurements in the presence of protein S, 50 nM of fluorescein-labeled APC derivatives were first incubated with a saturating concentration of OR containing PC/PS to obtain the maximum energy transfer in the DA sample followed by addition of protein S to a final concentration of 300 nM as described [14]. The net initial emission intensities were obtained by subtracting the initial intensities of A from DA (FDA-FA)o and B from D (FD-FB)o. Samples D and B were then titrated with PC/PS vesicles lacking the acceptor OR, while samples DA and A were titrated with PC/PS vesicles containing the acceptor. Similarly, the intensities of A and B were subtracted from DA and D, respectively and the values were then corrected for dilutions (less than 4% at the end of titration). Following 5 min incubation, emission intensities were measured. The ratio of the donor quantum yields (Q) in the D and DA samples, based on their fluorescence emission intensities (F), is given by equation 1 as described [14]:
| (1) |
at the end of each experiment, the fluorescent labeled proteins were released from the membrane surface by the addition of EDTA to 10 mM to ensure that energy transfer is reversible, however, in case of native APC the EDTA dissociation for APC from PC/PS vesicles was too slow thus DTT was used to reduce the disulphide bond between the two APC chains, thereby releasing the fluorescein-labeled heavy chain from the vesicle surface and thus reverting the fluorescence intensity of the donor as described [14]. For calculating the distance of the closest approach by equation 2 below, the QD/QDA value was calculated by dividing the value before EDTA by the value after the addition of EDTA. This normalization procedure corrects for the contribution of the acceptor inner filter effects and a potential membrane-binding-independent energy transfer as described [14,16].
2.10. Calculation of the distance of closest approach
Assuming that both donor and acceptor dyes are randomly and uniformly distributed (κ2 = 2/3), the distance of closest approach (L) between the plane of the donor dye attached to the active-site of the protein and the plane of the acceptor dyes at the surface of PC/PS vesicles can be determined using equation 2 as described [14,16]:
| (2) |
where π is 3.14, σ is the acceptor density at the membrane surface in OR/Å2 and Ro is the distance at which the singlet-singlet energy transfer from the donor dye to the acceptor dye is 50% efficient. The net QD/QDA values were plotted as a function of product between Ro2 and the acceptor density (σ) for at least 5-13 different energy transfer experiments to obtain the average value of L as described [14,16].
3. Results
3.1. Analysis of the anticoagulant activity of APC derivatives
Previous results have indicated that APC-PTGla exhibits markedly enhanced anticoagulant activity in both plasma-based clotting and FVa degradation assays independent of protein S [15]. Similar assays were employed to evaluate the anticoagulant activity of APC-FXGla. The results presented in Fig. 1 indicate that the substitution of the Gla-domain of APC with the same domain of factor X similarly improves the anticoagulant function of APC in both of these assays. In agreement with the literature [15], the FVa degradation assay indicated that protein S markedly improves the anti-FVa activity of APC in this assay (Fig. 1A). On the other hand, similar to APC-PTGla, the improvement in the activity of the APC-FXGla chimera was independent of protein S (Fig. 1A). The anticoagulant activity of APC-FXGla was more efficient than that of APC-PTGla both in FVa degradation and plasma-based APTT assays (Fig. 1). It has been hypothesized that the basis for the protein S-independent improvement in the activity of APC-PTGla may be due to its improved competitive effect on plasma proteins forming procoagulant activation complexes [15]. Nevertheless, the PC/PS concentration-dependence of FVa degradation in this study indicated that the APC derivatives have comparable binding affinities for PC/PS vesicles, thus yielding dissociation constants of ~1.2 μM for APC and ~0.9 μM for both of the Gla chimeras of APC. These results are in agreement with the previous study reporting similar binding affinities of ~0.5 μM for the interaction of both wild-type APC and APC-PTGla with PC/PS vesicles using direct binding approaches [15]. Thus, these results do not support the hypothesis that the Gla domains of the APC chimeras may have improved competitive effect on the formation of procoagulant complexes, but rather the results may suggest that, relative to procoagulant proteases, the Gla-domain of APC does not maintain the active-site of the protease at an optimal height and/or conformation to interact with its procoagulant substrates on the membrane surface, and that the cofactor function of protein S overcomes this inhibitory effect of the Gla-domain in the anticoagulant pathway.
Figure 1.
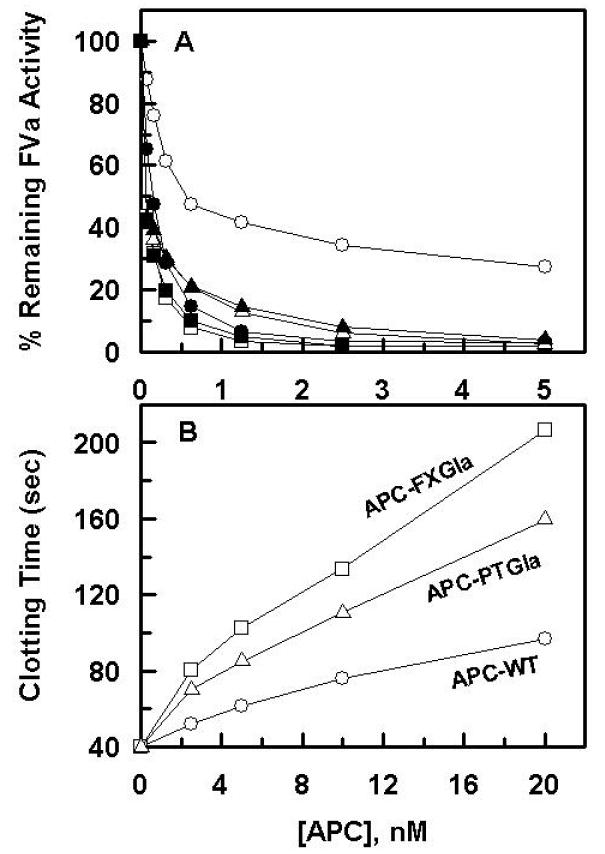
Factor Va degradation by the APC derivatives in the absence and presence of protein S. A. The APC concentration dependence of FVa (2.5 nM) degradation by wild-type APC (○,●), APC-FXGla (□,■) and APC-PTGla (△,▲) was carried out in the absence (open symbols) or presence of protein S (220 nM) (closed symbols) on PC/PS vesicles (20 μM) in TBS containing 0.5 mg/mL BSA, 0.1% PEG 8000 and 5 mM Ca2+. After 5 min incubation at room temperature, small aliquots of the inactivation reaction was transferred to a 96-well assay plate and the remaining cofactor activity of FVa was determined by a prothrombinase assay as described under “Materials and methods”. B. The anticoagulant activity of wild-type APC (○), APC-FXGla (□) and APC-PTGla (△) was measured in an APTT assay using normal human plasma as described under “Materials and Methods”.
3.2. Analysis of the topography of the active-sites of the APC-Gla domain chimeras
Previous FRET studies have indicated that the average distance of closest approach (L) between a fluorescein dye in the active-site of APC-PTGla and octadecylrhodamine (OR) dyes, randomly incorporated into PC/PS vesicles, is ~89 Å both in the absence and presence of protein S [16]. By comparison, L values of ~94 Å in the absence of protein S and ~84 Å in the presence of protein S for wild-type APC have been reported under the same experimental conditions [14,16]. Based on these results and the functional data showing a protein S-independent improvement in the anticoagulant activity of APC-PTGla, it has been hypothesized that protein S functions by altering the active-site topography of APC, thereby optimizing the recognition of FVa scissile bonds by APC above the membrane surface [14,16]. To determine whether a similar mechanism accounts for the protein S-independent improvement in the anticoagulant activity of APC-FXGla, the L values were determined for this mutant using essentially identical procedures. Thus, the change in the emission intensity of the donor fluorescein dye was monitored as a function of increasing concentration of PC/PS vesicles with or without the acceptor OR dyes both in the absence and presence of a saturating concentration of protein S. Similar to previous results [14,16], small changes in the fluorescence emission intensities of either wild-type APC or APC-FXGla were observed if PC/PS vesicles lacking OR were used for the titrations (data not shown). However, the fluorescence emission intensities of both wild-type APC and APC-FXGla were significantly decreased (10-15%) if PC/PS vesicles containing OR were used for the titrations. The EDTA or DTT reversible ratio of the donor quantum yields in the presence and absence of the acceptor (QDA/QD) was calculated according to equation 1. Plots of the data in the absence of protein S presented in Fig. 2 indicated that the change in the quantum yield of APC-FXGla is comparable to that of APC-PTGla and significantly larger than that of wild-type APC at all concentrations of PC/PS vesicles, suggesting that singlet-singlet energy transfer takes place with a higher efficiency in both of the APC chimeras. In the presence of protein S, plots of QDA/QD for both APC chimeras were essentially identical to those observed in the absence of protein S, however, they were altered for wild-type APC, thus exhibiting the same quantum yield observed with the mutants (data not presented). Since the efficiency of energy transfer depends on the density of OR (σ) at the membrane surface, the energy transfer was determined at several acceptor densities ranging from 1.5 - 5.6 × 10-4 and the average L values for each APC derivative in the absence and presence of protein S were calculated using equation 2, assuming a random orientation of transition dipoles for donor and acceptor dyes (κ2 = 2/3) as described previously [14]. The L values obtained from these measurements (Table 1) for APC both in the absence (99 ± 5 Å) and presence of protein S (87 ± 2 Å) are in close agreement with the corresponding values obtained for APC in a previous study (94.3 ± 4.0 Å in the absence and 84 ± 1 Å in the presence of protein S) [16]. Furthermore, the values obtained for APC-FXGla (86 ± 5 Å and 87 ± 2 Å in the absence and presence of protein S, respectively) were comparable to the same values obtained for APC-PTGla (84 ± 4 Å and 85 ± 2 Å in the absence and presence of protein S, respectively) in this study as well as in the previous study (88.7 ± 2.8 Å and 89 ± 1 Å in the absence and presence of protein S, respectively) [16], supporting the reliability of measurements. Since the magnitude of the energy transfer depends upon the density of the acceptor OR at the membrane surface [14,16], it was ensured that the plot of QD/QDA exhibited a linear dependence on the product between Ro2 and the acceptor density (σRo2) as demonstrated in Fig. 3.
Figure 2.
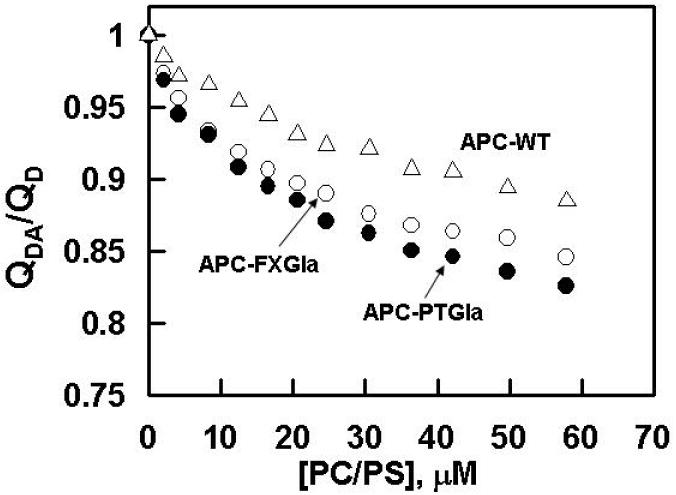
Dependence of energy transfer on PC/PS concentrations. The fluorescence labeled APC derivatives (50 nM each) in 0.1 M NaCl, 50 mM Hepes, 5 mM Ca2+, pH 7.4 were titrated with PC/PS vesicles containing or lacking OR at 25°C as described under “Materials and Methods”. The plots show the ratio of donor quantum yield (QDA/QD) in the presence or absence of OR for APC-WT (△), APC-FXGla (○) and APC-PTGla (●) which were calculated using equation 1 as described under “Material and Methods”. An acceptor density of 5.0 ×10-4 was used in all three experiments.
Table 1.
Distance of the fluorescein labeled active sites of APC derivatives from OR labeled PC/PS vesicles.
| APC | Ro (Å) | L (Å) | n | r (PC/PS) |
|---|---|---|---|---|
| APC-WT | 48± 1 | 99± 5 | 5 | 0.15 |
| APC- WT +protein S | 48± 1 | 87± 2 | 2 | 0.15 |
| APC-FXGla | 50± 1 | 86± 5 | 13 | 0.15 |
| APC-FXGla +protein S | 51± 1 | 87± 2 | 2 | 0.15 |
| APC-PTGla | 47± 1 | 84± 4 | 5 | 0.13 |
| APC-PTGla +protein S | 46± 1 | 85± 2 | 2 | 0.13 |
The relative distances were determined from the efficiency of energy transfer (FRET) from the donor fluorescein dye in the active-site of each APC derivative to the acceptor OR anchored to PC/PS vesicles with densities ranging from (1.5-5.6) × 10-4 OR/Å2 in 0.1 M NaCl and 0.05 M Hepes (pH 7.4) containing 5 mM Ca2+ using Equation 2 as described under “Material and methods”. In all experiments κ2 was assumed to be 2/3. The number of experiments used to calculate the average values are given as (n). The anisotropy (r) values for the fluorescein-labeled APC derivatives on PC/PS vesicles were measured at excitation and emission wavelengths of 493 nm and 521 nm, respectively. The anisotropy for the membrane bound OR at the limiting concentration of the acceptor (σ = 0.7 × 10-4) was determined to be 0.15.
Figure 3.
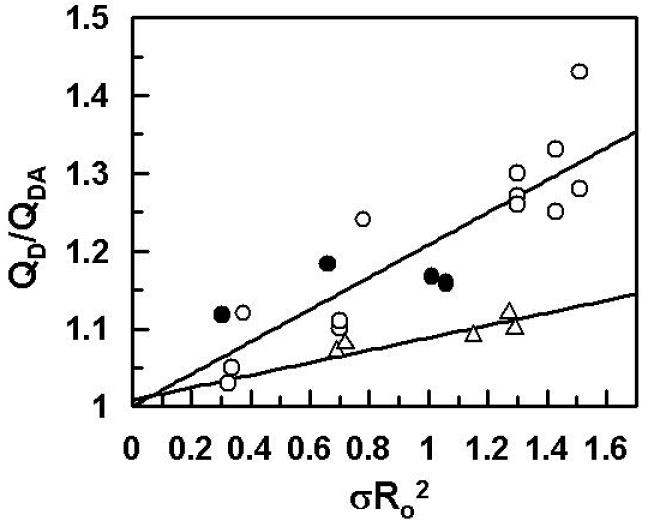
Linear dependence of FRET upon acceptor density for APC derivatives in the absence of protein S. The QD/QDA ratios, obtained using different densities of acceptor in the range of 1.5-5.6×10-4 OR/Å2, are plotted as the function of σRo2 for APC-WT (△), APC-FXGla (○) and APC-PTGla (●) as described under “Materials and Methods”.
To determine if protein S differentially changes the environment of the probe in the active-sites of the APC derivatives, the anisotropy values for the labeled proteins were determined in both the absence and presence of a saturating concentration protein S and PC/PS vesicles. The results indicated that neither PC/PS nor protein S influence the environment of the probe in the active-site pocket of APC derivatives, which is consistent with the literature [14,16]. The anisotropy values for all APC derivatives were similar ranging from 0.13-0.15 (Table 1), suggesting a nearly identical degree of rotational freedom for the fluorescein dye in the active-site pocket of proteins. The anisotropy of an OR containing PC/PS preparation with a density of 0.7 × 10-4 OR/Å2 was determined to be 0.15.
3.3. Analysis of the direct cellular effect of APC derivatives
Previous results have indicated that thrombin and proinflammatory cytokines enhance the permeability of endothelial cells and that APC exerts a potent protective effect by both EPCR and PAR-1 dependent mechanisms [19, 31]. The direct cellular effects of APC derivatives were evaluated in a permeability assay monitoring the flux of albumin across EA.hy926 cell monolayers in a dual chamber system as described [26]. As shown in Fig. 4A, thrombin increased the permeability of endothelial cells and wild-type APC effectively reversed the disruptive effect of thrombin. On the other hand, neither APC-PTGla nor APC-FXGla exhibited a protective effect even if their concentrations were increased to 100 nM, which is 10-fold higher than the concentration of APC required to obtain a maximal response (Fig. 4B). Similar results were obtained in two other cellular assays that are commonly used to investigate the cytoprotective properties of APC in in vitro systems. Thus, in both TNF-α-mediated endothelial cell apoptosis and neutrophil adhesion assays, wild-type APC prevented cell death (Fig. 4C) and inhibited the enhanced binding of neutrophils (Fig. 4D) to activated endothelial cells. However, neither Gla-domain chimeras of APC exhibited any activity in either one of these assays. These results suggest that the Gla-domain of APC is indispensable for its direct cellular effect in endothelial cells, suggesting that the Gla-domain substitution abolishes the ability of mutants to interact with EPCR. In support of this hypothesis, no binding for either APC-PTGla or APC-FXGla was observed in a solid phase binding-assay which yielded a KD of ~30 nM for wild-type APC interaction with soluble EPCR (data not presented). Nevertheless, both APC derivatives cleaved the exodomain of PAR-1 with identical efficiencies as determined in an alkaline phosphatase-based receptor cleavage assay [26], suggesting that inability of the Gla-domain mutants to signal in endothelial cells is exclusively related to their inability to interact with EPCR in endothelial cells (data not presented).
Figure 4.
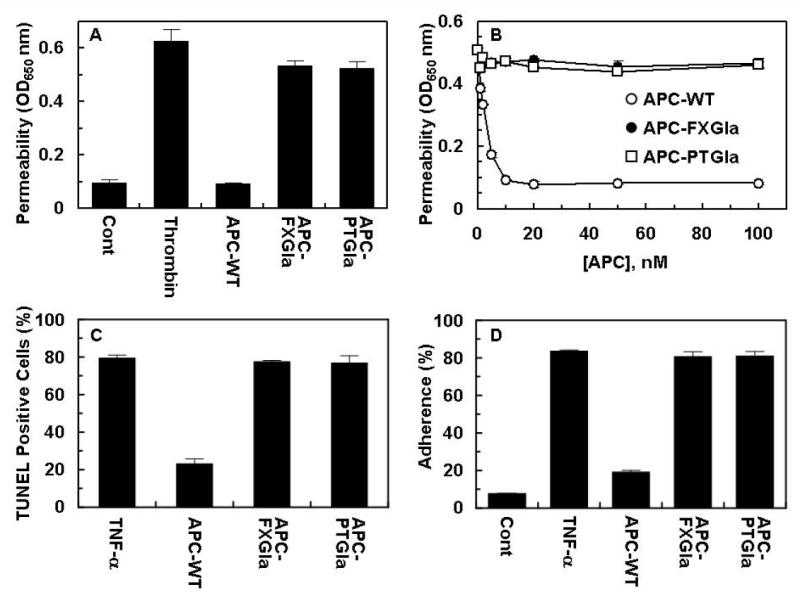
Comparisons of the protective effect of APC derivatives in the thrombin-induced permeability and TNF-α-induced apoptosis and neutrophil adhesion assays in EA.hy926 cells. A. The inhibition of thrombin-induced permeability by APC derivatives (10 nM each) was monitored from the flux of Evans blue-bound albumin across EA.hy926 cells as described under “Materials and Methods”. B. The concentration dependence of effect of APC derivatives in the thrombin induced permeability assay was carried as described above. C. Anti-apoptotic activity of APC derivatives were evaluated in confluent monolayers of EA.hy926 cells which were treated with APC derivatives (10 nM each) for 24 h prior to induction of apoptosis with TNF-α (10 ng/mL) for 4h. The number of apoptotic cells was expressed as the percentage of TUNEL-positive cells of the total number of nuclei as described under “Materials and Methods”. D. TNF-α (10 ng/mL)-mediated adherence of neutrophils to EA.hy926 monolayers was analyzed after treating monolayers with APC derivatives (10 nM each) as described under “Materials and Method”.
4. Discussion
The anticoagulant and antiinflammatory activities of APC require the interaction of the Gla-domain of the protease with protein S and endothelial protein C receptor (EPCR), respectively. In this study, we investigated three specific structure-function related questions in the APC anticoagulant and antiinflammatory pathways. First, using a different Gla-domain chimera, we investigated the hypothesis that a mechanism for the cofactor function of protein S in enhancing the anticoagulant activity of APC involves the alteration of the active-site topography of APC above the membrane surface as has been reported in the literature [14,16]. Secondly, we compared the anticoagulant properties of APC-FXGla with those of APC-PTGla to determine whether the Gla-domain of prothrombin is unique in its ability to enhance the anticoagulant function of APC independent of protein S or if this effect can also be recapitulated by the Gla-domain of other vitamin K-dependent proteins. Finally, we evaluated the direct cellular effects of the APC Gla-domain chimeras to understand the extent of the specificity requirement for this domain in interaction with and eliciting protective signaling responses in endothelial cells. As discussed below, our results provided important insight into the nature of each one of these questions.
Our results showed that protein S binding to APC leads to the lowering of the height of the active site of APC by ~12 Å above the membrane surface. The active-site of APC-PTGla was found to be closer to the membrane surface by a ~15 Å independent of protein S. Interestingly, a similar active-site height, that was ~13 Å closer to the membrane surface, was observed for APC-FXGla independent of protein S, providing further support for the hypothesis that protein S may improve the anticoagulant function of APC by altering the topography of the active-site of APC at the membrane surface [14,16]. Since the anticoagulant activity of this mutant was also improved independent of protein S, the results strongly suggest that the Gla-domain of APC contains unique structural features that maintain the active-site of APC in a topographical orientation that is not optimal for efficient recognition and degradation of its procoagulant substrates. Thus, APC has become a cofactor dependent protease, utilizing the cofactor function of protein S to compensate for its Gla-domain related inherently lower specific activity in the anticoagulant pathway. This is an adaptation that may have evolved to allow APC to interact with both protein S and EPCR in endothelial cells in order to function in the alternative anticoagulant and antiinflammatory pathways, respectively. It is known that APC cleavage of two P1-Arg scissile bonds, Arg-506 and Arg-306, on FVa is required for a complete inactivation of the cofactor at the membrane surface [4,11]. The cleavage of Arg-306 site is membrane dependent and the cofactor effect of protein S is primarily mediated through improving the APC cleavage of this site [10,13]. Thus, the Gla-domain substitution mimics the cofactor function of protein S by lowering the active-site of APC by 10-15 Å above the membrane surface to optimize its height for interaction with the Arg-306 cleavage site of the cofactor as has been hypothesized previously [16]. It should be noted that, as has been reported in the previous studies [14,16], this measurement assumes that the protein S effect is entirely translational and the κ2 value is equal to 2/3 and unaltered by protein S binding.
The observation that neither APC-PTGla nor APC-FXGla exhibited any cytoprotective activities in the cellular assays suggests that the Gla-domain of APC is highly specific for interaction with EPCR at the surface of vascular endothelial cells. It has been hypothesized that the cleavage of endothelial PAR-1 by APC, at least partially, may be responsible for the cytoprotective properties of the protease [19]. The PAR-1 cleavage efficiency of both Gla-domain chimeras were normal as tested by a cell-based PAR-1 cleavage assay monitoring the release of the soluble alkaline phosphatase fused to the N-terminus of the PAR-1 exodomain and anchored to the endothelial cell surface by the transmembrane domain of tissue factor as described [26] (data not presented). These results suggest that the lack of cellular signaling by either APC-PTGla or APC-FXGla is not due to their inability to cleave PAR-1, but rather to their inability to interact with EPCR on endothelial cells. Structural data have indicated that protein C residues responsible for specific interaction with EPCR are primarily located in the N-terminus of the ω-loop of the protein with Leu-8 playing a dominant role in the interaction [9]. It is interesting to note that Leu-8 and all other residues of the ω-loop have also been conserved on the same loop of factor VII, but not in prothrombin or factor X. Moreover, unlike the marked improvement in the anticoagulant activities of both APC-PTGla and APC-FXGla, the substitution of the Gla-domain of APC with the same domain of factor VII has resulted in a chimera with similar whole plasma anticoagulant activity [22]. Noting that factor VIIa can also bind to EPCR with similar high affinity [32], we speculate that the same features of the APC Gla-domain that endow high specificity for interaction with EPCR, are also responsible for maintaining the active-site of APC in the suboptimal height for interaction with the scissile bond(s) of FVa above the membrane surface. It should be noted that despite having a similar high affinity for interaction with EPCR, factor VIIa is not capable of eliciting PAR-1-dependent protective cellular responses in endothelial cells [32,33], suggesting that interactions outside of the ω-loop, which must be specific for protein C, are required for the EPCR-dependent protective signaling. Comparisons of the amino acid residues of the Gla-domain of APC with those of the procoagulant proteases described above indicate a remarkably high degree of conservation, with only few residues being variants. Results of several studies are consistent with the hypothesis that the variant residues of the N-terminal Gla-domain are critical for binding to EPCR and that those of the C-terminal end are involved in specific interactions with protein S at the membrane surface [9,15]. Among the residues of the C-terminal end of the Gla-domain, the surface residues at positions 23, 28, 32-36 are unique for protein C and may be responsible for the protein S cofactor activity by directly interacting with protein S [9]. Since most of these residues are charged in protein C, based on the molecular modeling studies [15], it has been postulated that electrostatic interactions in this region may change the angle between the Gla-domain and the reminder of the protein C molecule, thus explaining the non-optimal topographical orientation of the active-site of APC on the membrane surface in the absence of protein S.
Taken together, similar active-site topographies and functional properties for both APC-PTGla and APC-FXGla suggest that unique structural features within the Gla-domain are responsible for the specificity of APC interaction with EPCR in the antiinflammatory pathway, however, the same features are also the cause a lower specific activity for APC in the anticoagulant pathway. This adaptation has made APC a cofactor dependent protease requiring the cofactor function of protein S for its optimal anticoagulant function which appears to involve the alteration of the active-site topography of APC above the membrane surface. In light of the requirement for distinct residues in the Gla-domain of APC in each pathway, identification of these residues may lead to the design of recombinant APC derivatives with preferentially improved activity in either one of these pathways. Such variants may constitute useful therapeutic agents for treating thrombotic and inflammatory diseases under different conditions.
Acknowledgments
We would like to thank Audrey Rezaie for proofreading of this manuscript. The research discussed herein was supported by grants awarded by the National Heart, Lung, and Blood Institute of the National Institutes of Health (HL 62565 and HL 68571 to ARR).
Abbreviations
- FX
factor X
- FVa
activated factor V
- Gla
γ-carboxyglutamic acid
- APC
activated protein C
- APC-PTGla
a chimeric APC in which the Gla-domain and hydrophobic stack (residues 1-46) of the molecule has been replaced with the corresponding regions of prothrombin
- APC-FXGla
a chimeric APC in which the Gla-domain and hydrophobic stack (residues 1-46) of the molecule has been replaced with the corresponding regions of factor X
- EPCR
endothelial protein C receptor
- Fl
fluorescein
- OR
octadecylrhodamine
- FPR
(d-Phe)-Pro-Arg
- PC
dioleoylphosphatidylcholine
- PS
dioleoylphosphatidylserine
- PC/PS
phospholipid vesicles containing 80% PC and 20% PS
References
- 1.Esmon CT. Molecular events that control the protein C anticoagulant pathway. Thromb Haemost. 1993;70:1–5. [PubMed] [Google Scholar]
- 2.Esmon CT. Role of coagulation inhibitors in inflammation. Thromb Haemostas. 2001;86:51–56. [PubMed] [Google Scholar]
- 3.Shibata M, Kumar SR, Amar A, Fernandez JA, Hofman F, Griffin JH, Zlokovic BV. Anti-inflammatory, antithrombotic, and neuroprotective effects of activated protein C in a murine model of focal ischemic stroke. Circulation. 2001;103:1799–1805. doi: 10.1161/01.cir.103.13.1799. [DOI] [PubMed] [Google Scholar]
- 4.Dahlbäck B, Villoutreix BO. The anticoagulant protein C pathway. FEBS Letters. 2005;579:3310–3316. doi: 10.1016/j.febslet.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 5.Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science. 2002;296:1880–1882. doi: 10.1126/science.1071699. [DOI] [PubMed] [Google Scholar]
- 6.Walker FJ, Fay PJ. Regulation of blood coagulation by the protein C system. FASEB J. 1992;6:2561–2567. doi: 10.1096/fasebj.6.8.1317308. [DOI] [PubMed] [Google Scholar]
- 7.Stenflo J. Structure and function of protein C. Sem Thromb Hemost. 1984;10:109–121. doi: 10.1055/s-2007-1004413. [DOI] [PubMed] [Google Scholar]
- 8.Shen L, Shah AM, Dahlbäck B, Nelsestuen GL. Enhancement of human protein C function by site-directed mutagenesis of the γ-carboxyglutamic acid domain. J Biol Chem. 1998;273:31086–31091. doi: 10.1074/jbc.273.47.31086. [DOI] [PubMed] [Google Scholar]
- 9.Preston RJS, Ajzner E, Razzari C, Karageorgi S, Dua S, Dahlbäck B, Lane DA. Multifunctional specificity of the protein C/activated protein C Gla domain. J Biol Chem. 2006;281:28850–28857. doi: 10.1074/jbc.M604966200. [DOI] [PubMed] [Google Scholar]
- 10.Rosing J, Hoekema L, Nicolaes GAF, Thomassen MCLGD, Hemker HC, Varadi K, Schwarz HP, Tans G. Effects of protein S and factor Xa on peptide bond cleavages during inactivation of factor Va and factor VaR506Q by activated protein C. J Biol Chem. 1995;270:27852–27858. doi: 10.1074/jbc.270.46.27852. [DOI] [PubMed] [Google Scholar]
- 11.Kalafatis M, Rand MD, Mann KG. The mechanism of inactivation of human factor V and human factor Va by activated protein C. J Biol Chem. 1994;269:31869–31880. [PubMed] [Google Scholar]
- 12.Heeb MJ, Kojima Y, Greengard JS, Griffin JH. Activated protein C resistance: Molecular mechanisms based on studies using purified Gln506-factor V. Blood. 1995;85:3405–3411. [PubMed] [Google Scholar]
- 13.Norstrom EA, Steen M, Tran S, Dahlbäck B. Importance of protein S and phospholipid for activated protein C-mediated cleavage in factor Va. J Biol Chem. 2003;278:24904–24911. doi: 10.1074/jbc.M303829200. [DOI] [PubMed] [Google Scholar]
- 14.Yegneswaran S, Wood GM, Esmon CT, Johnson AE. Protein S alters the active site location of activated protein C above the membrane surface. J Biol Chem. 1997;272:25013–25021. doi: 10.1074/jbc.272.40.25013. [DOI] [PubMed] [Google Scholar]
- 15.Smirnov MD, Safa O, Regan LM, Mather T, Stearns-Kurosawa DJ, Kurosawa S, Rezaie AR, Esmon NL, Esmon CT. A chimeric protein C containing the prothrombin Gla domain exhibits increased anticoagulant activity and altered phospholipid specificity. J Biol Chem. 1998;273:9031–9040. doi: 10.1074/jbc.273.15.9031. [DOI] [PubMed] [Google Scholar]
- 16.Yegneswaran S, Smirnov MD, Safa O, Esmon NL, Esmon CT, Johnson AE. Relocating the active site of activated protein C eliminates the need for its protein S cofactor. J Biol Chem. 1999;274:5462–5468. doi: 10.1074/jbc.274.9.5462. [DOI] [PubMed] [Google Scholar]
- 17.Oganesyan V, Oganesyan N, Terzyan S, Qu D, Dauter Z, Esmon NL, Esmon CT. The crystal structure of the endothelial protein C receptor and a bound phospholipid. J Biol Chem. 2002;277:24851–24854. doi: 10.1074/jbc.C200163200. [DOI] [PubMed] [Google Scholar]
- 18.Castellino FJ. Human protein C and activated protein C: Components of the human anticoagulation system. Trends Cardiovasc Med. 1995;5:55–62. doi: 10.1016/1050-1738(94)00031-X. [DOI] [PubMed] [Google Scholar]
- 19.Mosnier LO, Griffin JH. The cytoprotective protein C pathway. Blood. 2007;109:3161–3172. doi: 10.1182/blood-2006-09-003004. [DOI] [PubMed] [Google Scholar]
- 20.Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW, Fisher CJJ. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Eng J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 21.Regan LM, Stearns-Kurosawa DJ, Kurosawa S, Mollica J, Fukudome K, Esmon CT. The endothelial protein C receptor. Inhibition of activated protein C anticoagulant function without modulation of reaction with proteinase inhibitor. J Biol Chem. 1996;271:17499–17503. doi: 10.1074/jbc.271.29.17499. [DOI] [PubMed] [Google Scholar]
- 22.Geng JP, Castellino FJ. Properties of recombinant chimeric protein in which the gamma-carboxyglutamic acid and helical stack domains of human anticoagulant protein C are replaced by those of human coagulation factor VII. Thromb Haemostas. 1997;77:926–933. [PubMed] [Google Scholar]
- 23.Iakhiaev AV, Rezaie AR, Idell S. Thrombomodulin-mediated catabolism of protein C by pleural mesothelial and vascular endothelial cells. Thromb Haemostas. 2007;98:627–634. [PubMed] [Google Scholar]
- 24.Manithody C, Yang L, Rezaie AR. Role of basic residues of the autolysis loop in the catalytic function of factor Xa. Biochemistry. 2002;41:6780–6788. doi: 10.1021/bi0255367. [DOI] [PubMed] [Google Scholar]
- 25.Yang L, Manithody C, Rezaie AR. Contribution of basic residues of the 70-80-loop to heparin binding and anticoagulant function of activated protein C. Biochemistry. 2002;41:6149–6157. doi: 10.1021/bi015899r. [DOI] [PubMed] [Google Scholar]
- 26.Bae J-S, Yang L, Manithody C, Rezaie AR. Engineering a disulfide bond to stabilize the calcium binding loop of activated protein C eliminates its anticoagulant but not protective signaling properties. J Biol Chem. 2007;282:9251–9259. doi: 10.1074/jbc.M610547200. [DOI] [PubMed] [Google Scholar]
- 27.Akeson AL, Woods CW. A fluorometric assay for the quantitation of cell adherence to endothelial cells. J Immunol Methods. 1993;163:181–185. doi: 10.1016/0022-1759(93)90121-m. [DOI] [PubMed] [Google Scholar]
- 28.Qureshi SH, Yang L, Yegneswaran S, Rezaie AR. FRET studies with factor X mutants provide insight into the topography of the membrane-bound factor X/Xa. Biochem J. 2007;407:427–433. doi: 10.1042/BJ20070735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang C, Mason JT. Geometric packing constraints in egg phosphatidylcholine vesicles. Proc Natl Acad Sci (USA) 1978;75:308–310. doi: 10.1073/pnas.75.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bock PE. Active site selective labeling of serine proteases with spectroscopic probes using thioester peptide chloromethyl ketones: Demonstration of thrombin labeling using Nα-[(acetylthio)acetyl]-D-Phe-Pro-Arg-CH2Cl. Biochemistry. 1988;27:6633–6639. doi: 10.1021/bi00417a063. [DOI] [PubMed] [Google Scholar]
- 31.Feistritzer C, Riewald M. Endothelial barrier protection by activated protein C through PAR1-dependent sphingosine 1-phosphate receptor-1 crossactivation. Blood. 2005;105:3178–3184. doi: 10.1182/blood-2004-10-3985. [DOI] [PubMed] [Google Scholar]
- 32.Ghosh S, Pendurthi UR, Esmon CT, Mohan Rao LV. Endothelial cell protein C receptor acts as a cellular receptor for factor VIIa on endothelium. J Biol Chem. 2007;282:11849–11857. doi: 10.1074/jbc.M609283200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bae J-S, Yang L, Manithody C, Rezaie AR. The ligand occupancy of endothelial protein C receptor switches the PAR-1-dependent signaling specificity of thrombin from a permeability-enhancing to a barrier-protective response in endothelial cells. Blood. 2007;110:3909–3916. doi: 10.1182/blood-2007-06-096651. [DOI] [PMC free article] [PubMed] [Google Scholar]