

Abstract
In cerebral adrenoleukodystrophy (cALD), an accumulation of very long chain fatty acids (VLCFA) stems from a defect of the peroxisomal ALD protein (ALDP) and results in the loss of myelin/oligodendrocytes, induction of inflammatory disease and mental deterioration. In brain white matter of cALD patients, we observed not only increased levels of VLCFA but also reduced levels of plasmenylethanolamine (PlsEtn) and increased levels of reactive oxygen species (ROS). The loss of PlsEtn was greatest in the plaque area and lesser but significant at histologically normal-looking areas of the cALD brain. The reduction in PlsEtn was related to oxidative stress, as supported by increased levels of reactive lipid aldehydes (4-hydroxynonenal and acrolein) and deleterious oxidized proteins (protein carbonyl) in all areas of the cALD brain. This inverse relationship between the levels of PlsEtn and ROS was further supported in an in vitro study using gene-silencing for dihydroxyacetone phosphate-acyl transferase (DHAP-AT), a key enzyme for PlsEtn biosynthesis. Levels of PlsEtn were also found decreased in vitro following gene-silencing for the ALDP/ALD related protein. Furthermore, low levels of PlsEtn were detected in brain white matter of ALDP knock out (KO) mice. A treatment of ALDP KO mice with lovastatin increased PlsEtn levels in the brain. Further, in an in vitro study, lovastatin treatment of rat C6 glial cells increased PlsEtn biosynthesis and reduced the cytokine-induced ROS accumulation. In summary, this study reports that altered metabolism of PlsEtn and ROS in cALD may be corrected by lovastatin treatment.
Keywords: adrenoleukodystrophy, plasmenylethanolamine, peroxisome, very long chain fatty acid, lovastatin, oxidative stress
Introduction
The most common peroxisomal disorder affecting males at early ages, adrenoleukodystrophy (ALD), results from deletion /mutation in the ABCD1 gene, leading to an absent or non-functioning adrenoleukodystrophy protein (ALDP) (Mosser et al. 1993). This defect causes an accumulation of very long chain fatty acids (VLCFA) in tissues and plasma via inhibition of peroxisomal β-oxidation. The accumulation of VLCFA is the hallmark of ALD disease. Two major clinical variants exist: cerebral ALD (cALD) and adrenomyeloneuropathy (AMN). Though caused by the same or a similar mutation or deletion, cALD is biochemically associated with redox alterations, inflammation and subsequent loss of myelin/oligodendrocytes. cALD is often fatal in childhood, whereas AMN patients live to adulthood with mild involvement of the peripheral nervous system (Moser et al. 1991; Powers 1995; Singh 1997, 2002; Moser et al. 2007). Unlike cALD, ALDP knock out (KO) mice do not show cerebral pathology. However, they present AMN like symptoms and show imbalance in antioxidant systems with increasing age (Powers et al. 2005; Fourcade et al. 2008). Therefore, a two hit hypothesis for cALD is proposed in which metabolic alterations cause destabilization of membrane structure and then a second event makes the membrane vulnerable to ROS and inflammation. We hypothesized that an imbalance in the endogenous antioxidant system due to intrinsic or extrinsic alterations leads to myelin membrane lipid oxidation, reactive oxygen species (ROS) formation, and immunogenic myelin fragmentation. These changes ultimately lead to manifestations of the inflammatory disease process. The major thrust of this investigation was to identify a trigger or causative factor which turns a metabolic disorder into a devastating inflammatory disease with profound demyelination and loss of oligodendrocytes.
ROS and inflammation are risk factors for several brain disorders. Both interfere with normal functioning of cellular organelles that harbor both ROS-producing and ROS-neutralizing systems. Locally-produced ROS, due to imbalances in its metabolizing systems, modify ROS-reactive lipid substrates. Among them, plasmenylethanolamine (PlsEtn; 1-alkyl-1’enyl-2-acyl-sn- glycero-3-phosphoethanolamine) and polyunsaturated fatty acids (PUFA) are major substrate targets of ROS. In the mammalian brain, PlsEtn is one of the major phospholipids, with the sn-1 position of glycerol bearing a long chain fatty alcohol linked through an ether bond having a cis-double bond between the first and second carbons (vinyl ether bond). The sn-2 position of glycerol is occupied mainly by PUFA, and the sn-3 position incorporates phosphoethanolamine moiety. The vinyl ether bond endows PlsEtn with a unique intramolecular antioxidant activity, which in turn protects PUFA present at the sn-2 position of the same molecule. Analysis of PlsEtn following acid hydrolysis from a normal brain shows three major alcohol components: 1-hexadecanol (16:0), 1- octadecanol (18:0) and 1- octadecenol (18:1 consists of cis- 9 and cis-7 species) (Poulos et al. 1991; Khan et al. 2005). Biosynthesis of the alkylether bond of PlsEtn, a rate-limiting reaction, requires the peroxisomal enzymes dihydroxyacetone-acyltransferase (DHAP-AT) and alkyldihydroxyacetonephosphate synthase (alkyl DHAP synthase) (Nagan et al. 1997; Liu et al. 2005). Alkyl alcohol, utilized by alkyl DHAP synthase for PlsEtn biosynthesis, is reported to be synthesized in peroxisomes using acetyl CoA generated by peroxisomal β-oxidation (Hayashi and Oohashi 1995; Hayashi and Hara 1997; Hayashi and Sato 1997). Because, 1-Alkylglycerol is formed exclusively in peroxisomes, the peroxisomal contribution to PlsEtn biosynthesis is reported to be determined by a ratio of the alkyl alcohol, represented as dimethlyacetal DMA, with the corresponding fatty acid following GC or GC/MS analysis (Bakovic et al. 2007). This ratio is used as the marker to measure PlsEtn levels in tissues and cells (Dacremont and Vincent 1995; Caruso 1996). The levels of PlsEtn in the brain are regulated by both biosynthetic enzyme system and hydrolytic activity of PlsEtn-specific phospholipase A2 (Farooqui and Horrocks 2004).
PlsEtn in white matter myelin contains mainly PUFA, including linoleic, arachidonic and docosahexaenoic acids (Norton and Cammer 1984; Norton 1984). A drastic reduction in PlsEtn leads to PUFA oxidation and compromise of membrane integrity and functions (Rodemer et al. 2003; Engelmann 2004). Furthermore, PlsEtn is suggested to have antioxidant activity (Zoeller et al. 1988), and its protection of biological structures such as neural membranes against free radical attack has been documented in vitro as well as in vivo (Zoeller et al. 2002; Hoffman-Kuczynski and Reo 2004, 2005; Kuczynski and Reo 2006). Evidence in favor of the antioxidant activity of PlsEtn was recently strengthened by gene-engineering of PlsEtn biosynthetic enzymes and by pharmacological manipulations of PlsEtn levels (Rodemer et al. 2003; Hoffman-Kuczynski and Reo 2004). Oxidative degradation of PlsEtn produced reactive aldehydes (Foglia et al. 1988) and caused oxidative stress in the human brain (Weisser et al. 1997). Excellent reviews of plasmalogens, peroxisomes and neurological disorders have been published (Farooqui et al. 2003; Mandel and Korman 2003).
Preservation of PlsEtn structure is significant for proper myelin function not only because PlsEtn is highly enriched in myelin but it acts also as an myelin endogenous antioxidant. The significance of PlsEtn in neural pathologies is strengthened by reports that aging and Alzheimer brains have reduced levels of PlsEtn (Ginsberg et al. 1995; Farooqui et al. 1997; Han et al. 2001; Helmy et al. 2003; Andre et al. 2006; Maeba et al. 2007). Circulating PlsEtn levels are significantly decreased in serum from diagnosed Alzheimer-type dementia at all stages, and the severity of this decrease correlated with dementia severity (Goodenowe et al. 2007). Drastically reduced PlsEtn levels are observed in peroxisome assembly diseases, including Zellweger syndrome (ZS) and rhizomelic chondrodysplasia punctata (RCDP) (Heymans et al. 1984; Hoefler et al. 1988; Aubourg et al. 1993; Schmitt et al. 1993). Therefore, peroxisomal dysfunction during a secondary insult may also contribute to reduced levels of PlsEtn in cALD‥
Neural membranes are considered a Pandora’s box of lipid mediators, many of which have powerful neurochemical effects and some of which are involved in signaling. Membrane lipid dysregulation in myelin/oligodendrocytes may not only cause dysmyelination/destabilization but also lead to membrane fragmentation, lipid peroxidation as well as produce chemotactic molecules for vascular immune cells (Latorre et al. 2003; Gorgas et al. 2006). Therefore, the objective of this investigation was to examine lipid alterations in different areas including the degenerative plaque and histologically normal looking areas of the cALD brain and to evaluate the peroxisomal contributions in these alterations. ALD disease is a peroxisomal defect with oxidative and inflammatory components in it. We focused this study on PlsEtn because key biosynthetic enzymes are localized in peroxisomes (Liu et al. 2005), and PlsEtn is known to modulate oxidative stress (Zoeller et al. 1988). Lovastatin treatment has been reported to increase PlsEtn levels in serum of hypercholesterolemic patients (Brosche et al. 1996) and to increase peroxisomal activity in keratinocytes (Williams et al. 1992). Moreover, lovastatin treatment is known not only to increase peroxisomal β-oxidation and decrease VLCFA in human skin fibroblasts from ALD patients (Singh et al. 1998a) but also to decrease VLCFA levels in plasma of ALD/AMN patients (Singh et al. 1998b; Pai et al. 2000). Therefore, the present study was directed to investigate the effects of lovastatin on the relationship between PlsEtn status and ROS levels as well as to examine the effects of lovastatin on the metabolism of PlsEtn in ALDP KO mice brain.
This study reports that reduction of PlsEtn levels due to ALDP dysfunction in cALD brain potentiated oxidative stress. The treatment of ALDP KO mice with lovastatin enhanced levels of PlsEtn in the brain white matter, providing support for the previously reported efficacy of lovastatin in reducing the VLCFA abnormality in ALD patients (Singh et al. 1998b; Singh et al. 1998a; Pai et al. 2000). These observations suggest that lovastatin therapy may be beneficial in correcting the overall metabolic defects observed in cALD.
Materials and methods
Human cALD brain and normal brain tissues
Frozen (−80°C) postmortem cALD brain (from four different patients) and age matched frozen control brain tissues (cerebrum section slices) were obtained from the Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore, MD, USA. Another frozen cALD brain tissue harvested within 2 h of death (age 9 years) at our hospital at Medical University of South Carolina (MUSC) was used in the study as described earlier (Paintlia et al. 2003). All five cALD brains had elevated levels of VLCFA measured as C26:0/C22:0 compared to age-matched control brains. The different areas of white matter (plaque, plaque shadow, around the plaque shadow, normal looking area away from plaque) from each cALD brain were identified and sliced by an experienced pathologist (Dr. Avtar K. Singh, M.D., Department of Pediatrics and Department of Pathology and Laboratory Medicine, MUSC, Charleston, SC). Three different white matter samples of each area from each cALD brain and three white matter brain samples from each normal control brain were dissected on a cold plate and weighed after removal of meninges, blood vessels and grey matter. Samples from grey matter were also analyzed and compared with white matter. Grey matter from the cALD brain showed no significant alterations in the levels of PlsEtn as well as VLCFA compared to grey matter from normal control brain.
Preparation of mouse primary astrocytes
C57BL6 breeding pairs were purchased from Jackson Laboratory (Ban Habor, ME) and maintained at the institution’s animal facility. Animal procedure was approved by the MUSC Animal Review Committee and received humane care in compliance with MUSC’s experimental guidelines and the National Research Council’s criteria for humane care (Guide for care and use of Laboratory Animals).
Primary astrocyte-enriched cultures were prepared from the whole cortex of 1-day-old C57BL/6 mice as described earlier (Pahan et al. 1998). Briefly, the cortex was rapidly dissected in ice-cold calcium/magnesium-free Hanks’ balanced salt solution (pH 7.4) as described previously (Won et al. 2001). The tissue was then minced, incubated in Hanks’ balanced salt solution containing trypsin (2 mg/ml) for 20 min, and washed twice with culture medium (Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum and 10 µg/ml gentamicin) and then disrupted by triturating through a Pasteur pipette, following which cells were seeded in 75-cm2 culture flasks (Falcon, Franklin, NJ, USA). After incubation at 37 °C in 5% CO2 for 1day, the medium was completely changed. The cultures received half exchanges with fresh medium twice a week. After 10 days, the cells were shaken for at least 30 min on an orbital shaker to remove the microglia and flasks were incubated for 1 day when they were shaken again for 8h to remove the oligodendrocytes. The remaining population was used for astrocyte cultures. All the cultured cells were maintained at 37 °C in 5% CO2.
Rat C6 glial cell culture
Rat C6 glial cells were grown to confluence in DMEM/F-12 containing 10% fetal bovine serum (FBS). They were allowed to grow in the same medium without serum for 48 h and then used for the experiments. Viability of the cells was assayed by trypan blue dye exclusion, and CNPase activity was measured to ascertain that C6 cells showed oligodendrocyte properties under serum free condition (Khan et al. 1998).
Lipid extraction and analysis from brain tissue
Total lipid from brain tissue was extracted as described earlier (Wilson and Sargent 1993; Khan et al. 1998). Lipids were resolved and purified as described earlier (Khan et al. 2000)
Preparation and analysis of dimethylacetal (DMA) and fatty acid methyl ester (FAME) by gas chromatography (GC)
FAME/DMA from the brain-extracted lipids and C6 cultured cells were prepared according to the procedure of Lepage and Roy (Lepage and Roy 1986) with modification as described (Khan et al. 2005). pentadecanoic (C15:0; 5 µg), heptadeca dimethyl acetal (C17:0 DMA, 5 µg) and heptacosanoic acid (C27:0; 2 µg) were used as internal standard. The purified samples of FAME/DMA were analyzed on a fused silica capillary column 25 M 007 Series methyl silicone, 0.25-mm internal diameter from Quadrex Corp (Woodbridge, CT) in a gas chromatograph either GC-15A or GC-17A connected with a flame ionization detector from Shimadzu Corporation. The peaks of individual FAME and DMA were identified by comparison of their relative retention times with those of known standards, and they were later verified by the addition of standards to prepared samples. The individual DMA and FAME were measured as absolute quantities by comparison with internal standard and also as area percent as described earlier (Pai et al. 2000).
Quantitation of phospholipids and VLCFA
Levels of the phospholipids including PlsEtn, diacylethanolamine (PtdEtn), phosphatidylcholine (PC) and phosphatidylethanolamine (PE or PlsEtn+PtdEtn) were measured by HPTLC and quantitated by densitometry. PlsEtn and PtdEtn were quantitated after purification and acid hydrolysis of PE fraction and quantitated by densitometry using PE (having both PlsEtn and PtdEtn) as standard (Ganser et al. 1988; Khan et al. 2000). PlsEtn was also quantitated by measuring long chain alcohol as DMA by GC and expressed as absolute concentration, percent change or as percent area of FAME+DMA. PlsEtn levels were also expressed as a ratio of DMA derivatives to corresponding fatty acid (FAME) as described elsewhere (Dacremont and Vincent 1995; Khan et al. 2005). The total amount of PlsEtn is reflected in the ratio of 16:0DMA, 18:0DMA and 18:1DMA. The measurement of VLCFA (C26:0) is represented as ratio of C26:0/C22:0 or as mass of 26:0 (nmol/ mg protein)as described earlier (Khan et al. 1998; Pai et al. 2000; Khan et al. 2005).
Studies on mouse
Animal procedure was approved by the MUSC Animal Review Committee, and all animals received humane care in compliance with the MUSC's experimental guidelines and the National Research Council's criteria (Guide for the Care and Use of Laboratory Animals).
ALDP KO (C57BL6) and control (normal) (C57BL6) breeding pairs were purchased from Jackson Laboratory (Ban Habor, ME) and maintained at the institution’s animal facility. The genotypes of all newborns from ALDP KO were determined by a PCR-based method as described (Forss-Petter et al. 1997) using the primers suggested in data sheets from Jackson Laboratory (http://jaxmice.jax.org). These primers were- 5′-CACAGCCTCTCTCCTTAAGACC-3′ (oIMR1120), 5′-TCGTTGTCTAGGCAACTGG-3′ (oIMR1121) and 5′-CTTCTATCGCCTTCTTGACG-3′ (oIMR1122). Two primers detect disrupted allele ALDP KO. Presence of the wild type allele was revealed in the same reaction by using a third primer (oIMR1121). PCR consisted of a 10 min heating step at 95 °C, followed by 35 cycles of denaturation at 95 °C for 1 min, annealing at 54 °C for 1 min, and extension at 72 °C for 1 min. The PCR products were then subjected to agarose gel electrophoresis. Lovastatin (Calbiochem, San Diego, CA, USA) therapy was performed on 40–45 day-old male mice. It was administered ip (0.5 mg/kg body weight in 0.1% Triton X-100 and PBS) daily for 3 weeks. Control ALDP KO and control (normal) mice were injected with 0.1% Triton X-100 and PBS. White matter from brain tissue was harvested from controls (normal and ALDP KO) and lovastatin-treated mice and analyzed for lipids, including PlsEtn, VLCFA and long chain fatty acids.
Biosynthesis of PlsEtn in cultured C6 cells
Biosynthesis rate of PlsEtn and PtdEtn was determined using [1,2-14C] ethanolamine; specific activity 110 mCi/mmol from American Radiolabeled Chemicals Inc, St. Louis, MO, USA. Cells were treated overnight (15 hr) with or without lovastatin. Lovastatin was prepared in DMSO, and DMSO was used as vehicle. After washing, the cells were pulsed with the labeled ethanolamine (0.1 µCi) for 2 h and/or 24 h. Viability of cells under experimental conditions was assayed by trypan blue dye exclusion as described (Khan et al. 1998). Incorporation of labeled ethanolamine was measured in total lipid extract as described (Bichenkov and Ellingson 2000). In brief, the lipids were separated on HPTLC plate as described earlier (Khan et al. 2000), and the area containing PE was scraped from the plate, extracted for lipids and reloaded on the plate along with standard PE (a mixture of PlsEtn and PtdEtn). After drying the plate, it was exposed to HCl vapor for hydrolysis of vinyl ether bond for 10 min and resolved in a solvent system described earlier (Khan et al. 2000). After exposing to iodine vapor, the spots of PtdEtn and hydrolyzed PlsEtn were analyzed for radioactivity using a liquid scintillation counter Beckman LS 3801 system.
Measurement of ROS in cultured C6 cells
ROS were determined using the membrane permeable fluorescent dye 6-carboxy 2′, 7′-dichlorodihydrofluorescein diacetate (DCFH2-DA) in serum-free medium. The cultured cells with or without treatment were incubated with 5 µM DCF dye in PBS for 2 hr at 37°C. The change in fluorescence was determined at excitation 485 nm and emission 530 nm using a Soft Max Pro spectrofluorometer (Molecular Devices, Sunnyvale, CA) as described (Khan et al. 2005).
Gene-silencing by SiRNA for ALDP and ALDRP in primary astrocytes
The Silencer siRNA (Ambion, Austin, TX, USA) was used for Abcd1 and /or Abcd2 silencing in primary mouse astrocytes. Briefly, mice astrocytes cultured in DMEM with 10% serum and in the presence of antibiotic were transfected with siRNA for Abcd1 and/or Abcd2 using siPORT NeoFX transfection agent (Ambion, Austin, TX, USA). Three siRNA for Abcd1 and Abcd2 (Ambion, Austin, TX, USA) each were used (Abcd1 siRNA 1, ID 162218; 5’-CCUCUACAACCUAAUUUAUtt-3’ 5’-AUAAAUUAGGUUGUAGAGGtg-3’ siRNA 2, ID 60153; 5’-GGUAUUUGAAGAUGUCAAAtt-3’ 5’-UUUGACAUCUUCAAAUACCtg-3’ siRNA 3, ID 60064; 5’-GGAAAUUGCCUUCUACGGGtt-3’ 5’-CCCGUAGAAGGCAAUUUCCtc-3’ Abcd2 SiRNA1, ID 188185; 5’-GGCUUUAGCUUACCAGAUGtt-3’ 5’-CAUCUGGUAAGCUAAAGCCtt-3’ siRNA 2, ID 214996; 5’-GGUAAAUGUCUAGAAAUGGtt-3’ 5’-CCAUUUCUAGACAUUUACCtg-3’ siRNA 3, ID 214997, 5’-GCUGUAGAGAUCAAUAGAGtt-3’ 5’-CUCUAUUGAUCUCUACAGCtc-3’). The siRNAs were mixed and diluted in OPTI-MEM1 medium to final concentration of 30nM/well. siRNA/transfection agent was dispensed into culture plates as directed by manufacturer. A positive control using GAPDH siRNA (Ambion, Austin, TX, USA) and negative control having sequence similarity to no known human, mice or rat gene were included. Cells were maintained in DMEM with reduced serum (2%). Silencing was observed with western blot and mRNA quantification. For protein analysis of the transfected cells, two wells per plate were lysed and used for protein measurements and protein levels (Western blot). Cells were maintained for 6 days in DMEM with 2% FBS before harvesting for the analysis.
Gene-silencing by SiRNA for DHAP-AT and ALDP+ALDRP in cultured C6 cells
Cultured C6 cells were grown to 50–60% confluency in DMEM/F-12 (1:1) with 10% serum and in the presence of antibiotic. The cells were transfected with DHAP-AT (Gnpat) SiRNA (30nM concentration) or Abcd1 (ALDP; 30nM concentration) or Abcd2 (ALDRP; 30nM concentration) or ALDP+ALDRP SiRNA (30 nM concentration of each) obtained from Ambion (Austin, TX, USA). The sequences of the specific siRNAs were as described: (DHAT-AT) SiRNA sequences ID# 190113 (Sense 5’-GCCAAGUGAUAUCAAGUCCtt-3’ Antisense 5’-GGACUUGAUAUCACUUGGCtt-3’, ID# 190114 (Sense 5’-CGUACUUUUGGAGGAAAUAtt-3’ Antisense 5’-UAUUUCCUCCAAAAGUACGtc-3’, ID# 55122 (Sense 5’-GGUAGACAGUAAAUAUGUGtt-3’ Antisense 5’-CACAUAUUUACUGUCUACCtt-3’).
Abcd1 SiRNA sequences ID# 162218 (Sense 5’-CCUCUACAACCUAAUUUAUtt-3’ Antisense 5’-AUAAAUUAGGUUGUAGAGGtg-3’), ID# 60153 (Sense 5’-GGUAUUUGAAGAUGUCAAAtt-3’ Antisense 5’-UUUGACAUCUUCAAAUACCtg-3’); ID# 60064 (Sense 5’-GGAAAUUGCCUUCUACGGGtt-3’ Antisense 5’-CCCGUAGAAGGCAAUUUCctc-3’).
Abcd2 SiRNA sequences ID# 195407 (Sense 5’-CCCUCUAUCCCAUCCUUGGtt-3’ Antisense 5’-CCAAGGAUGGGAUAGAGGGtt-3’), ID# 195408 (Sense 5’-GCUACUAGAACUUCGGAAAtt-3’ Antisense 5’-UUUCCGAAGUUCUAGUAGCtg-3’), ID#195409 (Sense 5’-CCUUACCAAACCGAUUUUAtt-3’ Antisense 5’-UAAAAUCGGUUUGGUAAGGtt-3’). Cells were maintained in DMEM/F-12 in absence of serum, and silencing was examined by Western blot at 72 h using antibody against DHAP-AT (Khan et al. 2005) or ALDP (Contreras et al. 1996) or ALDRP (custom made from ANASPEC against the mouse 20 residues c-terminal sequence; 722 CKILGEDSVLKTIQTPEKTS 741) specific antibodies. The cells were maintained up to 96 h before harvesting for the analysis of PlsEtn and VLCFA. Negative control SiRNA is a scrambled (Scr) sequence obtained from Ambion Inc (Austin, TX, USA) and bears no homology to human, mouse or rat genomes. Treatment with cytokines (Cyt), TNF-α (50ng/ml) + IL-1β (50ng/ml), was carried out at 72 h after treatment with SiRNA or Scr, and the cells were analyzed at 96 h as described above.
Expression of 4-HNE and acrolein by immunohistochemistry of brain section from cALD and control (normal) brains
Expressions of immunoreactive 4-HNE and acrolein were detected by immunohistochemical analysis using specific antibodies. Paraffin embedded sections from the formalin fixed brain tissues were stained for 4-HNE (Rabbit Polyclonal from A. G. Scientific, Inc., San Diego, CA, USA) and acrolein (Rabbit Polyclonal, Novus Biological Inc., Littleton, CO, USA) as described earlier (Paintlia et al. 2003). In brief, the brain tissue sections were deparaffinized, sequentially rehydrated in graded alcohol, and then immersed in phosphate-buffered saline (PBS, pH 7.4). Slides were then microwaved for 2 min in antigen unmasking solution, cooled and washed 3 times for 2 min in PBS. Sections were immersed for 25 min in 3% hydrogen peroxide in distilled water to eliminate endogenous peroxidase activity, then blocked in immunohistochemical grade 1% bovine serum albumin in PBS for 1 h and diluted goat serum for 30 min to reduce non-specific staining. Sections were incubated overnight with 4-HNE and acrolein antibody and then rinsed 3 times for 5 min in PBS containing 0.1% Tween-20. Secondary HRP labeled anti-rabbit or anti-goat or anti-mouse IgG was incubated on slides for 30 min. The HRP signal was amplified with TSA Biotin system NEL 700 (NEN Life Products, Boston, MA), and the slides were washed with PBS 3X 5 min each time and incubated with streptavidin TR (1:500) for 1 h, then again washed with PBS 3X5 min each, mounted in Permount cover slips. All sections were analyzed using an Olympus microscope, and images were captured using a digital video camera controlled by Adobe Photoshop 7.0 (Adobe Systems, CA, USA).
Immunoblot analysis and protein oxidation detection
Tissue extracts were subjected to SDS-PAGE and immunoblotting using rabbit polyclonal DHAP-AT antibody (custom-made) or β-actin as previously described (Haq et al. 2006). The protein oxidation was determined using Chemicon’s OxyBlot oxidized protein Detection Kit, protocol provided. Briefly, protein lysates were allowed to react with 2, 4-dinitrophenylhydrazine (DNP) reagent to convert oxo group to hydrazone. Lysates were subjected to SDS-PAGE and transferred to blotting membrane, blots were incubated with primary antibody to attach with hydrazone moiety of the proteins. Oxidized protein molecular weight markers provided in the kit were used as standard. Detection sensitivity is 10 femtomoles of hydrazone residues on a single protein.
Statistical evaluation
Statistical analysis was performed using software Graphpad Prism 3.0 (San Diego, CA, USA). Unless otherwise stated, all the values were expressed as mean ± SD of n determinations or as mentioned. The results were examined by unpaired t-test. Multiple comparisons were performed using ANOVA followed by Bonferroni test as appropriate. p value less than 0.05 was considered significant.
Results
Loss of PlsEtn in cALD brain
Different areas (marked as 1, 2, 3, 4 on X-axis of Fig 1) of brain white matter from cALD patient and white matter from age-matched control subjects (normal control brain) were dissected and identified (1, plaque; 2, plaque shadow; 3, around plaque shadow; 4, normal looking area away from the plaque) by a pathologist. Care was taken that white matter used in this study was not contaminated with grey matter. These samples were analyzed for VLCFA (26:0) along with all the three long chain alcohol species of PlsEtn as DMA using capillary GC. Masses of PC and PE, PlsEtn and PtdEtn were quantitated in the same samples using HPTLC. The ratio of 26:0/22:0 (Fig 1A) and the levels of 26:0 expressed as nmol/mg protein (Fig 1B) were significantly higher in plaque/plaque shadow areas and at least half to one fold elevated in histologically normal looking area (Fig 1A–B) compared to normal control, documenting a direct relationship between degree of VLCFA increase and the pathology of cALD brain. We also found an inverse relationship between the levels of VLCFA and PlsEtn in the same samples. Two (16:0DMA and 18:1DMA) of the three species of long chain alcohols incorporated in PlsEtn were significantly lower in a sequential manner (minimum in the plaque area and maximum in the normal looking area) measured as tissue concentration of DMA (Fig 1D) and the ratio of DMA vs. corresponding fatty acid (Fig 1C). Lack of reduced levels of the third PlsEtn species (18:0DMA) indicates a species specific loss rather than a generalized loss of PlsEtn in the cALD brain. Since PlsEtn is a component of the PE (PlsEtn+PtdEtn) class of phospholipids in brain white matter, we measured the mass of PE (Fig 1E) along with both of its fractions (PlsEtn + PtdEtn) (Fig 1F). In the same sample, we also measured the mass of PC to compare it with PE (Fig 1E). While the levels of PE reduced in accordance with the loss of DMA/PlsEtn, the PC levels did not change with the different areas of cALD brain. Unlike white matter, grey matter had no significantly reduced levels of PE (data not shown). Mass of both PlsEtn and PtdEtn decreased in cALD compared to normal brain (Fig 1F). However, the loss of PlsEtn compared to PtdEtn was observed in a sequential manner, with a greater decrease in the plaque (area 1) and lesser in the normal looking brain (area 4) (Fig 1F). Reduced levels of PlsEtn were also observed in cALD white matter by Wilson and Sargent (Wilson and Sargent 1993). These results indicate a loss of PlsEtn rather than a generalized loss of total phospholipids in the white matter of cALD brain.
Figure 1. Levels of very long chain fatty acids (VLCFA) and phospholipids (phosphatidylcholine, PC; phosphatidylethanolamine, PE; plasmenylethanolamine, PlsEtn; diacylethanolamine, PtdEtn) in different areas of cALD brain and in normal control brain.

Different areas [1 (plaque), 2 (plaque shadow), 3 (around the plaque shadow) and 4 (normal looking area away from the plaque)] of white matter from the same cALD and from white matter of age-matched control (normal) brains are analyzed. Lipids were extracted and analyzed for the mass of phospholipids on HPTLC. Dimethylacetal (DMA) and fatty acid methyl ester (FAME) were prepared using the total extracted lipids (Khan et al. 2000). Analysis was performed after the addition of C15:0, C17:0 DMA and C27:0 as internal standard. A) Levels of VLCFA measured as a ratio of 26:0/22:0. B) Mass of 26:0 measured as nmol/mg protein. C) Levels of PlsEtn presented as ratio of DMA vs. corresponding fatty acid. D) Mass of PlsEtn measured DMA (nmol/mg protein). E) Mass of PC and PE (nmol/mg protein). F) Mass of PlsEtn and PtdEtn (noml/mg protein). Data are presented as mean ± SD (n=5) from a triplicate analysis of each sample. ***P<0.001 vs. Control (Normal), *p<0.05 vs. Control (Normal).
SiRNA silencing of Abcd1 and Abcd2 genes for ALDP and ADRP expressions respectively increased the levels of VLCFA and decreased the levels of PlsEtn (DMA) in mouse primary astrocytes and in cultured C6 cells
We observed significantly reduced levels of DMA measured as % change compared to untreated control when expression of ALDP+ALDRP was blocked by SiRNA silencing of Abcd1 and Abcd2 genes together in mouse primary astrocytes (Fig 2C) and in cultured C6 cells (Fig 2D). The silencing, as expected, also increased VLCFA measured as % change of the levels of 26:0 in mouse primary astrocytes (Fig 2B) and in C6 cells (data not shown). Fig 2A shows that the silencing of Abcd1 and Abcd2 resulted in reduced expression of ALDP and ALDRP, respectively, in mouse primary astrocytes. However, the silencing of both Abcd1 and Abcd2 together resulted in increased VLCFA levels and decreased DMA levels. The observations may be due to gene redundancy of Abcd1 and Abcd2 to each other in cultured cells. These results support an inverse relationship between levels of VLCFA and PlsEtn due to the peroxisomal defect of ALDP/ALDRP. Because the silencing significantly reduced the levels of DMA in both mouse primary astrocytes and cultured C6 cells and the fact that C6 cells are readily available in our laboratory, we used C6 cells in subsequent experiments.
Figure 2. Effect of Abcd1, Abcd2 and Abcd1+Abcd2 silencing by SiRNA on expression of ALDP and ALDRP, levels of VLCFA and levels of PlsEtn (DMA) in mouse primary astrocytes and in cultured C6 cells.
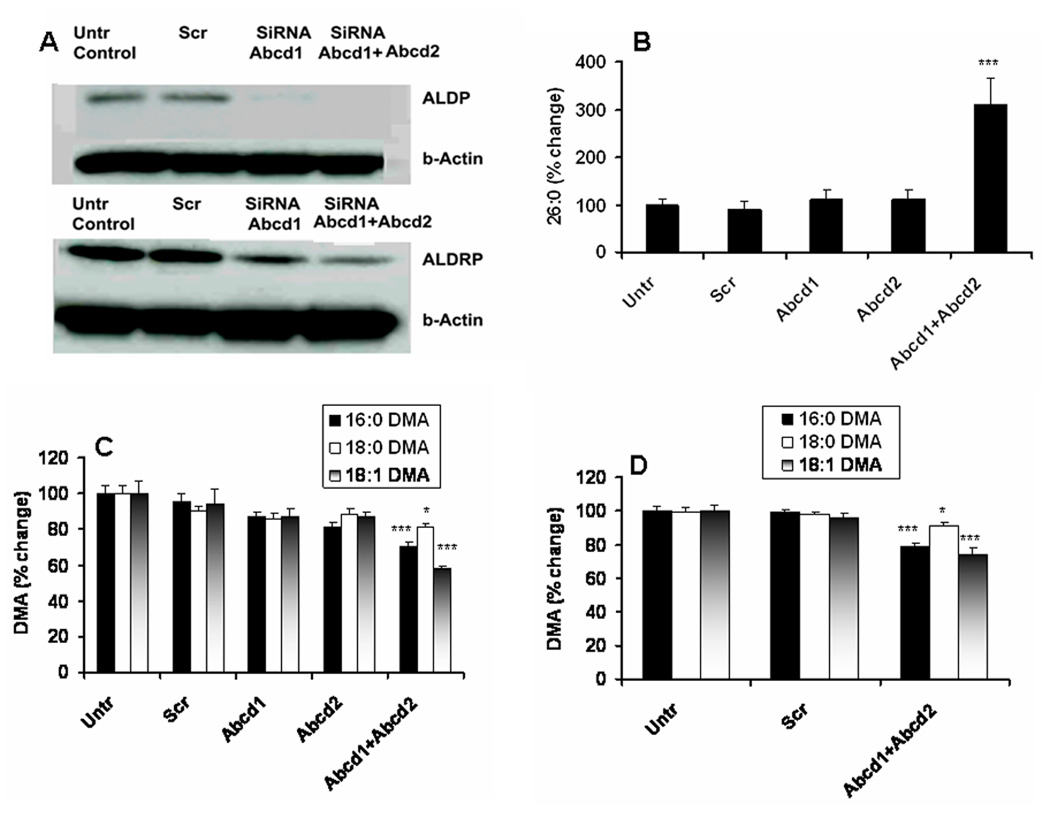
Procedure of SiRNA of Abcd1 or Abcd2 or scrambled (Scr) treatment in mouse primary astrocytes is described in Materials and Methods. Expression of ALDP and ALDRP was measured by Western blot (A). VLCFA was quantitated as % change in the levels of 26:0 compared to untreated (Untr) control cells (B). PlsEtn was measured as % change in the levels of DMA (all the three species) compared to untreated (Untr) control cells (C). DMA in cultured C6 cells (treated as described in Materials and Methods) was also measured as % change like in primary astrocytes (D). Results from individual silencing of Abcd1 or Abcd2 in C6 cells are not shown as the levels of DMA as well as VLCFA like astrocytes remained unchanged. Data are expressed as mean ± SD (n=7). *P<0.05 vs. Untr and Scr, ***P<0.001 vs. Untr and Scr
SiRNA silencing of Gnpat gene for DHAP-AT enzyme expression decreased the levels of PlsEtn (DMA) and increased the levels of ROS in cultured C6 cells
To investigate whether a decrease in the levels of PlsEtn results in increased levels of ROS, we reduced the levels of PlsEtn (measured as DMA) in C6 cells by silencing the Gnpat gene responsible for DHAP-AT expression, the rate limiting peroxisomal enzyme for the biosynthesis of PlsEtn. C6 cells under serum free condition, expressing an oligodendrocyte-like property and showing an increased levels of VLCFA by cytokine treatment (Khan et al. 1998), were treated with SiRNA for Gnpat or with scrambled SiRNA as negative control. SiRNA treatment reduced the expression of DHAP-AT as shown by Western blot (Fig 3A). This reduced expression of DHAP-AT resulted in reduced levels of PlsEtn measured as levels of DMA (Fig 3B) and as a ratio of DMA vs. corresponding fatty acid (Fig 3C). The silencing also increased the levels of ROS (Fig 3D). Treatment with the scrambler did not significantly alter either the expression of DHAP-AT protein, DMA or ROS (Fig 3A–D). Treatment of cells with proinflammatory cytokines or toxic lipids is known to decrease the levels of DMA and increase the levels of ROS (Khan et al. 2005). Therefore, we examined the effects of cytokines on cells under silenced Gnpat gene. The silencing rendered cultured C6 cells more sensitive to cytokines. It depleted not only PlsEtn measured as levels of DMA (Fig 3E) and ratio of DMA/acid (Fig 3F) to higher degree but also greatly increased the levels of ROS (Fig 3G), supporting further the relationship between inflammation, levels of PlsEtn and levels of cellular ROS.
Figure 3. Effect of Gnpat (DHAP-AT) silencing by SiRNA on DHAP-AT expression, levels of PlsEtn (DMA) and ROS, and sensitivity to cytokine in cultured C6 cells.
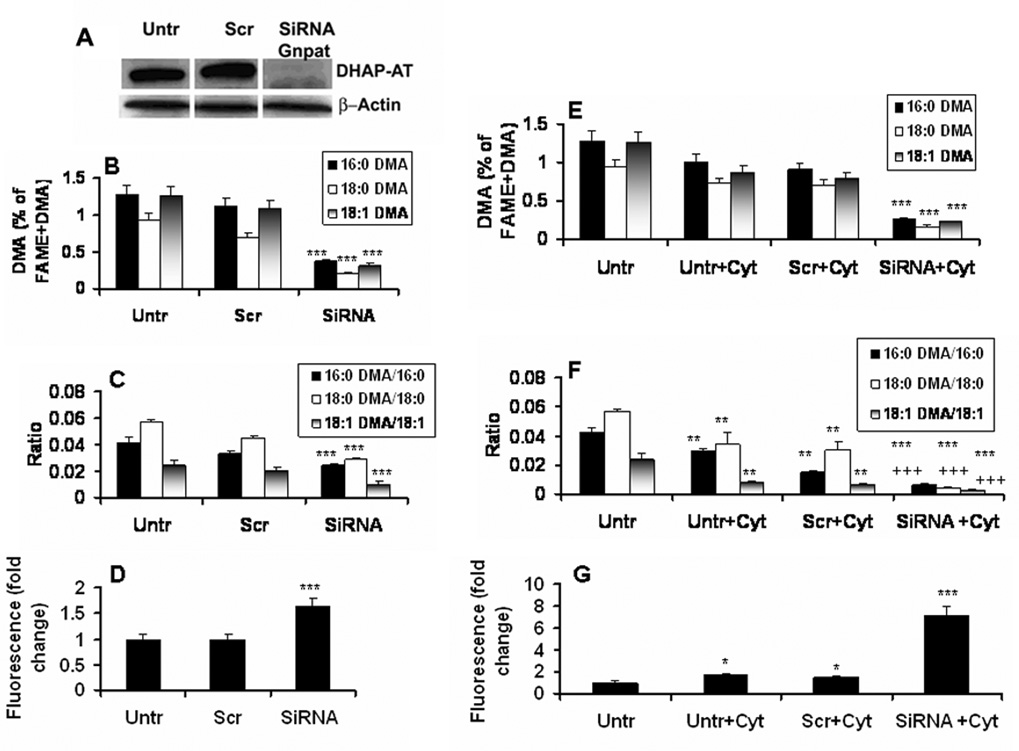
Procedure of SiRNA of Gnpat or scrambled (Scr) treatment and sensitization with Cyt (TNF-α, 50 ng/ml+IL-1β, 50 ng/ml) in cultured C6 cells is described in Materials and Methods. Expression of DHAP-AT was measured by Western blot using the specific antibody (A). PlsEtn species (all the three alcohols) were measured as % change of DMA (Fig B,E) as well as ratio of DMA/acid (Fig C,F). Levels of ROS were quantitated as fold change using DCF dye (D and G). Data are expressed as mean ± SD (n=3 for DHAPT expression and n=7 for DMA and ROS). *P<0.05 vs. untreated (Untr) and Scr, **P<0.001 vs. untreated (Untr) and Scr, ***P<0.001 vs. untreated (Untr) and Scr, +++p<0.001 vs. Untr, Untr+Cyt, Scr+Cyt.
Reactive lipid aldehydes and oxidized proteins in cALD brain
The mechanisms of the VLCFA accumulation-induced decrease in PlsEtn may involve increased cellular oxidative stress in the cALD brain. Therefore, we investigated the levels of the deleterious lipid peroxidation products, reactive aldehydes (4-HNE and acrnolein). These aldehydes modify and inactivate not only protein by covalent bond formation but also initiate and propagate PUFA peroxidation, hence continuously generating free radicals until exhaustion of the substrates. We observed increased levels of both 4-HNE and acrolein in the plaque area as well as in histologically normal looking areas of the cALD brain (Fig 4A). Acrolein is especially notorious to oxidize and convert proteins to deleterious protein carbonyls, thereby possibly compromising their structure/functions. We also observed increased expression of protein carbonyls in all areas of cALD brains (two patients), including histologically normal looking areas, whereas normal control brains did not show the expression of protein carbonyls (Fig 4B). Increased levels of 4-HNE in the juvenile ALD brain has already been shown by Powers et al (Powers et al. 2005). However, our results are unique in showing the levels of acrolein and protein carbonyls and also that these deleterious oxidative markers are expressed even in those areas otherwise histologically normal with lesser accumulated VLCFA compared to plaque and/or plaque shadow areas as shown in Fig 1A–B.
Figure 4. Photomicrograph of immunoreactive 4-HNE and acrolein and immunoblot of oxidized protein in two different brain samples from cALD and normal control.

A) Immunohistochemical staining was carried out using specific antibodies against 4-HNE and acrolein. One section each from two different brains (#1 and #2) is presented (a and g are from control brain #1; b and h are from control brain #2; c,e,i,k are from cALD brain #1; d,f,j,l are from cALD brain #2). Acrolein staining was found more prominent in all areas from cALD brains. Control normal brains did not show any significant staining for 4-HNE and acrolein. (Magnification 400X). B) Immunoblot analyses of brain extracts from two different normal control brains (1 and 2) and of three different areas (3, 5 normal looking; 4, 7 plaque shadow; 5, 8 plaque) from two different cALD brains were carried out according to manufacturer’s instructions described in the OxyBlot Oxidized Protein Detection Kit. Protein (30 µg) along with standard (Std) provided with the Kit was fractionated on immunoblot, and oxidized proteins were detected using antibody to DNP. β-actin was used as loading control.
Levels of PlsEtn and VLCFA in brain of ALDP knock out mice and their modulation by lovastatin treatment
To investigate whether lovastatin treatment increases the levels of PlsEtn in brain, ALDP KO mice were treated intraperitoneally for 3 weeks with a dose of 0.5 mg/kg of lovastatin suspended in triton X-100 as described earlier in studies with experimental autoimmune encephalomyelitis (EAE) animal models (Stanislaus et al. 1999). Similar to the normal looking area from cALD patients (Fig. 1C, 1D), white matter from the knock out mice brain had significantly reduced levels (p<0.05 compared to control normal) of two species (16:0 DMA and 18:1 DMA) of PlsEtn measured as a ratio of DMA/acid (Fig 5A) and mass of DMA (Fig 5B), whereas 18:0 DMA species of PlsEtn remained unaltered (Fig 5A–B). Treatment with lovastatin significantly (p<0.001 vs. ALDP-KO) increased the levels of all three species of PlsEtn (Fig 5A–B). Furthermore, the lovastatin treatment of ALD KO mice also reduced the levels of VLCFA measured both as a ratio of 26:0/22:0 (Fig 5C) and as an absolute amount of 26:0 (Fig 5D). Increased levels of PlsEtn and decreased levels of VLCFA by lovastatin treatment in brain of ALDP KO mice indicate the potential for the lovastatin treatment in cALD.
Figure 5. Effect of lovastatin treatment on the levels of PlsEtn (DMA) and VLCFA in brain from ALDP knock out mice.

Lovastatin (0.5 mg/kg body weight in 0.1% Triton X-100 and PBS) treatment was carried out for 3 weeks. Lipids from white matter were extracted, and the levels of PlsEtn as ratio of DMA/acid (A) as well as mass of DMA (B) and VLCFA as ratio of 26:0/22:0 (C) and mass of 26:0 (D) were measured. Results are expressed as mean ± SD from 7 different mice of duplicate determination. *p#0.05 vs. Normal, **p#0.01 vs. Normal and ALDP KO, ***p#0.001 vs. Normal and ALDP KO.
Increased biosynthesis of PlsEtn and decreased levels of cytokine-induced ROS after treatment with lovastatin in cultured C6 cells
To investigate whether lovastatin treatment increases PlsEtn biosynthesis or increases its levels by some other activity including antioxidation, cultured C6 cells under serum free condition (Khan et al. 1998) were treated with different concentrations of lovastatin. The biosynthesis of both PlsEtn and PtdEtn was measured using a chase for 2h with 14C-labeled ethanolamine as described in Material and Methods. Incorporation of 14C-labeled ethanolamine in PlsEtn was significantly increased (p<0.05 vs. untreated) in lovastatin-treated (1.0 and 2.0 µM) cells (Fig 6A). However, similar changes were not observed in PtdEtn (Fig 6A). These results indicate that lovastatin treatment significantly increased PlsEtn biosynthesis. However, the mechanisms of increased PlsEtn biosynthesis by lovastatin are not clear. Furthermore, lovastatin’s effect on the biosynthesis of PlsEtn was not significant at a 24 hr of pulse of the cell (data not shown). We further examined the effect of lovastatin under similar experimental conditions in the presence and absence of cytokines on ROS levels using DCF dye. Lovastatin treatment significantly decreased (p<0.05 compared with cytokine-treated) ROS levels (Fig 6B), indicating that an increase in PlsEtn directly correlates with a decrease in the levels of ROS.
Figure 6. Effect of lovastatin on incorporation of [1, 2-14C] ethanolamine in PlsEtn and PtdEtn as an index of biosynthesis of PlsEtn and PtdEtn and on the levels of ROS in cultured C6 cells.
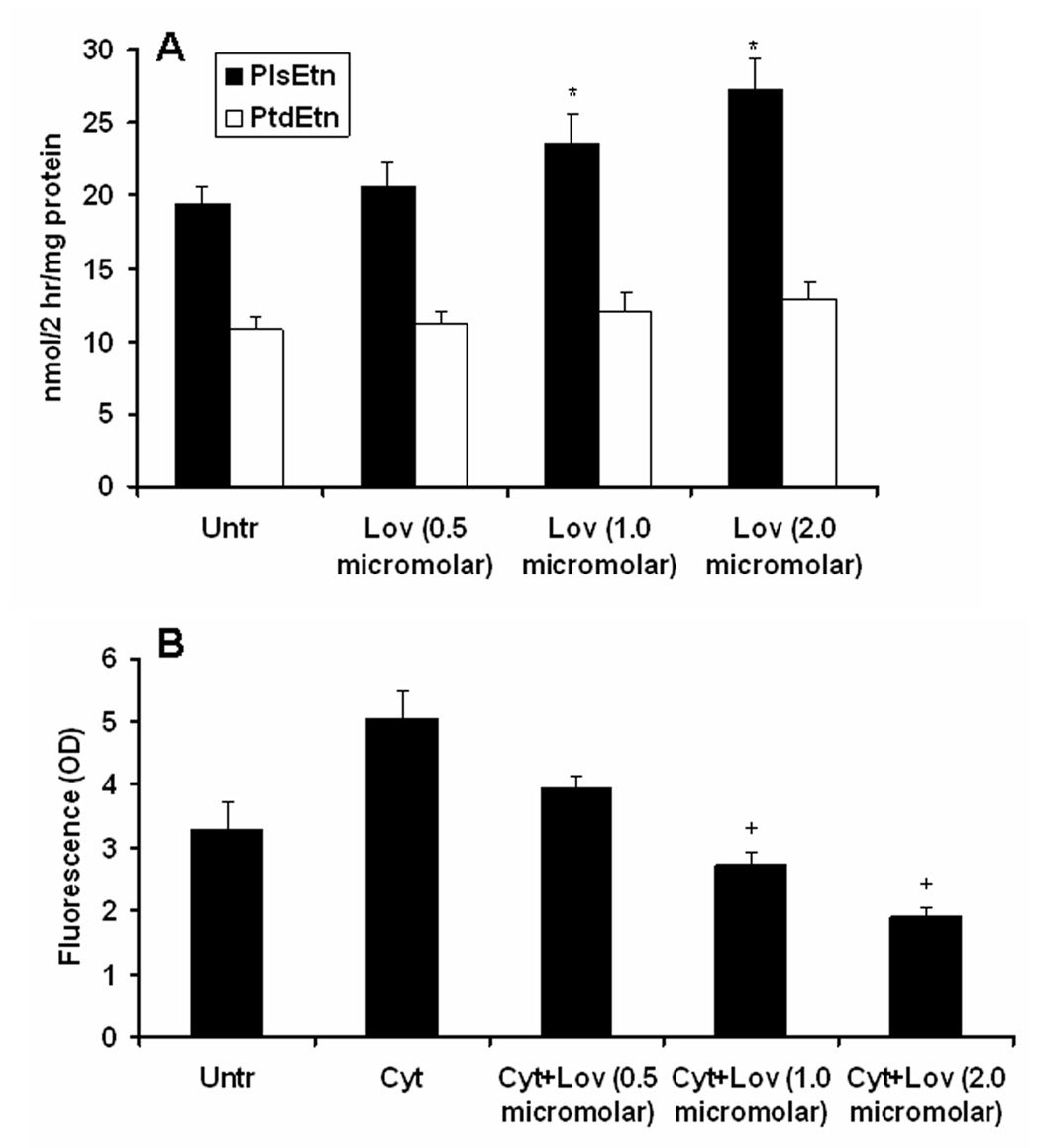
Cultured C6 cells were treated with different concentrations of lovastatin (in DMSO) overnight (15 hr) followed by a chase for 2 hr with the labeled ethanolamine. The cells were harvested, and lipids were resolved on HPTLC. PE was purified and hydrolyzed by HCl. Both the hydrolyzed and the non-hydrolyzed PE were resolved on HPTLC. Incorporation of radioactivity was measured in PE (PlsEtn+PtdEtn), PtdEtn and hydrolyzed PlsEtn fractions. Results are presented as nmol/2hr/mg protein (A). Levels of ROS were measured using DCF dye as described earlier (Khan et al. 2005) after cytokine (Cyt)-treatment (TNF-α, 50 ng/ml+IL-1β, 50 ng/ml) for 24 hr following lovastatin treatment. Results are expressed as arbitrary unit (OD) (B). Data are expressed as mean ± SD for 3 different experiments. *p<0.05 vs. Untr, +p<0.05 vs. Cyt.
Discussion
Our results demonstrated reduced levels of PlsEtn together with increased levels of VLCFA in white matter from cALD brain. In our in vitro model, this reduction in PlsEtn was directly related to the dysfunctions of ALDP/ALDRP. Reduced PlsEtn resulted in ROS accumulation that might cause the second hit we hypothesized in cALD, turning a metabolic disorder to a fatal neuroinflammatory disease process. When PlsEtn biosynthesis was increased by lovastatin, both ROS and VLCFA levels were decreased, revealing the potential of lovastatin in cALD treatment.
In cALD, it is understood that pathognomonic accumulation of VLCFA destabilizes myelin/oligodendrocyte membranes while a second hit triggers the loss/reduction of myelin-producing cells via oxidative injury. However, the nature and the origin of the events responsible for this pathologic transition have been enigmatic. The studies described here using different areas of brain from cALD patients and brain from ALDP KO mice document that loss of PlsEtn and alterations in cellular redox are early events that might be involved in the transition from metabolic to neurologic disease in X-ALD patients. Furthermore, this investigation finds loss of only two of the three long chain alcohol species of PlsEtn in cALD brain tissue as shown (Fig 1C, 1D, 1F). Even histologically normal looking areas from the cALD brain (area 4 in Fig 1A–F) with relatively little change in levels of VLCFA (Fig 1A–B) showed significant reductions in PlsEtn mass when compared with age-matched normal control brains (Fig 1D and 1F). Following silencing of Abcd1/Abcd2 in mouse primary astrocytes (Fig 2C) and in C6 cells (Fig 2D), the observed loss of PlsEtn further supports the relationship of loss of ALDP/ALDRP and consequent inhibition of peroxisomal β-oxidation of VLCFA with a decrease in the levels of PlsEtn (Fig 2C–D). Long chain alcohols incorporated in plasmalogens are documented to be synthesized, in part, by acetyl CoA generated during β-oxidation of VLCFA in liver peroxisomes (Hayashi and Oohashi 1995). Therefore, inhibition of VLCFA oxidation may result in reduced plasmalogen biosynthesis. Furthermore, the reduction of only two species of PlsEtn (16:0 DMA and 18:1 DMA) without a change in the third species (18:0 DMA) (Fig 1C–D) indicated that these selective losses are not due only to myelin loss in cALD. These observations directed our efforts to examine the status and role/function of PlsEtn in the pathobiology of cALD and to modulate its level in glial cells and in the brain of an animal model of ALD. Previous studies from this laboratory have reported that lovastatin treatment not only decreased VLCFA in ALD patients (Singh et al. 1998b; Pai et al. 2000) and in human skin fibroblast cell line (Singh et al. 1998a) but also ameliorated neuroinflammatory disease of EAE, an animal model of multiple sclerosis (Stanislaus et al. 1999) and also in spinal cord injury animal model (Bichenkov and Ellingson 2000; Pannu et al. 2005; Pannu et al. 2007). Moreover, lovastatin treatment has been reported to increase PlsEtn levels in patients with hypercholesterolemia (Brosche et al. 1996) and to up regulate peroxisomal activity in keratinocytes (Williams et al. 1992). Therefore, we investigated the use of lovastatin to modulate the levels of PlsEtn in ALDP KO mouse brain. These studies may also help to elucidate whether the lovastatin-mediated regulation of PlsEtn metabolism is peroxisome dependent mechanisms.
Abnormal myelin metabolism caused by excessive accumulation of VLCFA leads to ROS formation, possibly via activation of NADPH oxidase (Dhaunsi et al. 2005), resulting to peroxidation of PUFA, primarily embedded at the sn-2 position in PlsEtn. Consequent lipid peroxidation causes free radical generation and myelin lipid fragmentation. Therefore, the fragmented oxidized lipid-derived reactive aldehydes (4-HNE and acrolein) (Fig 4A) and consequent protein carbonyls (Fig 4B) are highly deleterious, initiating immunogenic reactions and inviting inflammation. Acrolein, a neurotoxin and the smallest reactive lipid aldehyde, has a half life of 7–10 days and is much more reactive than 4-HNE. It is a potent protein carbonyl-forming agent (Uchida 2003). Protein carbonyls interrupt normal cellular functions and may become immunogenic. Antigens derived from lipid peroxidation have been shown to contribute to the development of the immune responses associated with alcoholic liver disease (Albano 2002; Mottaran et al. 2002). Chronic fatigue syndrome is also characterized by an IgM-related immune response directed against disrupted lipid membrane components and lipid peroxidation products (Maes et al. 2006). The reactive aldehydes released from oxidized PlsEtn or PUFA react with nucleophiles, especially glutathione, and form Schiff base adducts which may have a deleterious effect on neural cell membranes (Kehrer and Biswal 2000; Stadelmann-Ingrand et al. 2004).
Compared to other brain cell types, oligodendrocytes and myelin membranes are more susceptible to oxidative stress, possibly because of their high content of iron and reduced levels of glutathione (Thorburne and Juurlink 1996; Wilson 1997; Dringen 2000). For these reasons and the fact that reduced PlsEtn resulted in excessive ROS formation in cultured C6 cells (Fig 3D and 3G), the focus of this study is on modulation of PlsEtn metabolism by therapeutics to reduce oxidative burden, normalize abnormal myelin metabolism and induce remyelination. Recent studies from our laboratory reported that lovastatin, by itself (Paintlia et al. 2005) and in combination with an activator of AMP-activated protein kinase, induces proliferation of OPC (oligodendrocyte precursor cells) and remyelination (Paintlia et al. 2006). This led us to examine the effect of lovastatin treatment on PlsEtn metabolism. The treatment increased PlsEtn biosynthesis in a dose-dependent manner in cultured C6 cells (Fig 6A). Secondly, ROS levels were decreased well in accordance with increased PlsEtn biosynthesis (Fig 6B), supporting that PlsEtn can effectively reduce oxidative stress. Plasma LDL from ALD patients has been shown to have altered oxidative parameters, which were normalized by simvastatin treatment (Di Biase et al. 2000). We hypothesize that this correction may be related to increased biosynthesis of PlsEtn by simvastatin treatment.
Next, we tested the effects of lovastatin treatment on the brain of ALDP KO mice (Fig 5). These mice are asymptomatic in spite of accumulation of mild levels of VLCFA (Forss-Petter et al. 1997; Lu et al. 1997; Kemp et al. 1998) and some oxidative stress observed by increased expression of MnSOD (Powers et al. 2005). Reactive lipid aldehyde adduct (malondialdehyde-lysine) are also observed in the spinal cords of these mice (Fourcade et al. 2008). The study of ALDP KO mice provided an opportunity to examine alterations in the metabolism of myelin lipids, including PlsEtn and VLCFA, and also to investigate whether lovastatin treatment can correct the observed lipid dysregulation. It is interesting to note that ALDP KO mice had mild reductions in only those PlsEtn species (16:0 DMA and 18:1 DMA) observed in human cALD brain. Presently, the reason for the lack of alteration in 18:0 DMA in both humans and mice is not understood. In spite of the low degree of change in PlsEtn, the changes in lipid peroxidation or glutathione reductions in ALDP KO mice brain were not significant (data not shown). A milder reduction in the mass of PlsEtn (Fig 5B) and remarkable lower mass of VLCFA (26:0) (Fig 5D) in the brain of ALDP KO mice compared to human cALD brain from normal looking area (Fig 1D and 1B respectively) may explain the absence of cerebral pathology in ALDP KO mice. Treatment of the KO mice with lovastatin for three weeks resulted in reduced VLCFA levels (Fig 5C–D) and increased levels PlsEtn (Fig 5A–B). Unlike the effect of lovastatin treatment on the levels of PlsEtn (Fig 5A–B), the levels of VLCFA were reduced only mildly (Fig 5C–D). Some laboratories have not observed decreased levels of VLCFA following treatment of ALDP KO mice with statins (Cartier et al. 2000; Yamada et al. 2000; Weinhofer et al. 2005). These differing results may be due to method of preparation of statin solution and route/mode of administration. Nevertheless, our studies with KO mice demonstrate that lovastatin treatment modified the metabolism of both PlsEtn and VLCFA in the brain, indicating that lovastatin therapy may also normalize alterations in PlsEtn and VLCFA in the brains of ALD patients.
In summary, the studies described here report reductions of PlsEtn in cALD brain and in cultured primary astrocytes or C6 cells due to silencing of Abcd1/Abcd2, documenting a relationship between the function of ALDP/ALDRP and the metabolism of PlsEtn. Secondly, the greater degree of reduction of PlsEtn in cALD brain as compared to the brain of ALDP KO mice indicates that the degree of reduction of PlsEtn may relate with the severity of the disease. As compared to ALDP KO mice brain, the cALD brain had an abundance of ROS-derived reactive lipid aldehydes and deleterious protein carbonyls, which may be immunogenic, recruiting inflammation and inducing loss of cells. Therapeutically, the studies with the lovastatin treatment showed potential to attenuate not only the accumulation of VLCFA but also to normalize the levels of ROS and PlsEtn, indicating the efficacy of statin therapy for X-ALD disease.
Acknowledgements
These studies were supported by grants (NS-22576, NS-34741, NS-37766 and NS-40810) from the NIH and (SCIRF 0406 and SCIRF 0506) from State of South Carolina Spinal Cord Injury Research Fund Board. This work was also supported by the NIH, Grants C06 RR018823 and No C06 RR015455 from the Extramural Research Facilities Program of the National Center for Research Resources. We thank Dr. Avtar K. Singh, M.D. (a pathologist) from the Ralph H Johnson VA Medical Center for dissecting and identifying different areas of human brain white matter. We would like to thank Ms. Joyce Bryan for procurement of animals and chemicals used in this study. We also acknowledge Yeong-Bin Im for her help in lipid analysis. The writing assistance of Dr Tom G Smith, MUSC Writing Center is valuable and acknowledged.
Abbreviations used
- ALDP
adrenoleukodystrophy protein
- ALDRP
adrenoleukodystrophy related protein
- AMN
adrenomyeloneuropathy
- cALD
cerebral adrenoleukodystrophy
- Cyt
cytokines
- DHAP-AT
dihydroxyacetone phosphate-acyl transferase
- DMA
dimethylacetal
- EAE
experimental autoimmune encephalomyelitis
- FAME
fatty acid methyl ester
- GC
gas chromatography
- 4-HNE
4-hydroxynonenal
- HPTLC
high performance thin layer chromatography
- KO
knock out
- Lov
lovastatin
- OPC
oligodendrocyte precursor cells
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- PlsEtn
plasmenylethanolamine
- PtdEtn
diacylethanolamine
- ROS
reactive oxygen species
- Scr
scrambled
- VLCFA
very long chain fatty acid
References
- Albano E. Free radical mechanisms in immune reactions associated with alcoholic liver disease. Free Radic Biol Med. 2002;32:110–114. doi: 10.1016/s0891-5849(01)00773-0. [DOI] [PubMed] [Google Scholar]
- Andre A, Juaneda P, Sebedio JL, Chardigny JM. Plasmalogen metabolism-related enzymes in rat brain during aging: influence of n-3 fatty acid intake. Biochimie. 2006;88:103–111. doi: 10.1016/j.biochi.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Aubourg P, Kremser K, Roland MO, Rocchiccioli F, Singh I. Pseudo infantile Refsum's disease: catalase-deficient peroxisomal particles with partial deficiency of plasmalogen synthesis and oxidation of fatty acids. Pediatr Res. 1993;34:270–276. doi: 10.1203/00006450-199309000-00006. [DOI] [PubMed] [Google Scholar]
- Bakovic M, Fullerton MD, Michel V. Metabolic and molecular aspects of ethanolamine phospholipid biosynthesis: the role of CTP:phosphoethanolamine cytidylyltransferase (Pcyt2) Biochem Cell Biol. 2007;85:283–300. doi: 10.1139/o07-006. [DOI] [PubMed] [Google Scholar]
- Bichenkov E, Ellingson JS. Effects of transient ethanol exposure on the incorporation of [(3)H]ethanolamine into plasmalogen in the differentiating CG-4 oligodendrocyte cell line. Biochem Pharmacol. 2000;60:1703–1711. doi: 10.1016/s0006-2952(00)00473-1. [DOI] [PubMed] [Google Scholar]
- Brosche T, Kral C, Summa JD, Platt D. Effective lovastatin therapy in elderly hypercholesterolemic patients - an antioxidative impact? Arch Gerontol Geriatr. 1996;22:207–221. doi: 10.1016/0167-4943(95)00694-x. [DOI] [PubMed] [Google Scholar]
- Cartier N, Guidoux S, Rocchiccioli F, Aubourg P. Simvastatin does not normalize very long chain fatty acids in adrenoleukodystrophy mice. FEBS Lett. 2000;478:205–208. doi: 10.1016/s0014-5793(00)01852-4. [DOI] [PubMed] [Google Scholar]
- Caruso U. Simple analysis of plasmalogens in erythrocytes using gas chromatography/mass spectrometry with selected-ion monitoring acquisition. Rapid Commun Mass Spectrom. 1996;10:1283–1285. doi: 10.1002/(SICI)1097-0231(19960731)10:10<1283::AID-RCM298>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Contreras M, Sengupta TK, Sheikh F, Aubourg P, Singh I. Topology of ATP-binding domain of adrenoleukodystrophy gene product in peroxisomes. Arch Biochem Biophys. 1996;334:369–379. doi: 10.1006/abbi.1996.0467. [DOI] [PubMed] [Google Scholar]
- Dacremont G, Vincent G. Assay of plasmalogens and polyunsaturated fatty acids (PUFA) in erythrocytes and fibroblasts. J Inherit Metab Dis. 1995;18 Suppl 1:84–89. doi: 10.1007/BF00711431. [DOI] [PubMed] [Google Scholar]
- Dhaunsi GS, Kaur J, Alsaeid K, Turner RB, Bitar MS. Very long chain fatty acids activate NADPH oxidase in human dermal fibroblasts. Cell Biochem Funct. 2005;23:65–68. doi: 10.1002/cbf.1173. [DOI] [PubMed] [Google Scholar]
- Di Biase A, Salvati S, Vari R, Avellino C, Sforza F, Cappa M, Masella R. Susceptibility to oxidation of plasma low-density lipoprotein in X-linked adrenoleukodystrophy: effects of simvastatin treatment. Mol Genet Metab. 2000;71:651–655. doi: 10.1006/mgme.2000.3100. [DOI] [PubMed] [Google Scholar]
- Dringen R. Metabolism and functions of glutathione in brain. Prog Neurobiol. 2000;62:649–671. doi: 10.1016/s0301-0082(99)00060-x. [DOI] [PubMed] [Google Scholar]
- Engelmann B. Plasmalogens: targets for oxidants and major lipophilic antioxidants. Biochem Soc Trans. 2004;32:147–150. doi: 10.1042/bst0320147. [DOI] [PubMed] [Google Scholar]
- Farooqui AA, Horrocks LA. Brain phospholipases A2: a perspective on the history. Prostaglandins Leukot Essent Fatty Acids. 2004;71:161–169. doi: 10.1016/j.plefa.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Farooqui AA, Rapoport SI, Horrocks LA. Membrane phospholipid alterations in Alzheimer's disease: deficiency of ethanolamine plasmalogens. Neurochem Res. 1997;22:523–527. doi: 10.1023/a:1027380331807. [DOI] [PubMed] [Google Scholar]
- Farooqui AA, Ong WY, Horrocks LA. Plasmalogens, docosahexaenoic acid and neurological disorders. Adv Exp Med Biol. 2003;544:335–354. doi: 10.1007/978-1-4419-9072-3_45. [DOI] [PubMed] [Google Scholar]
- Foglia TA, Nungesser E, Marmer WN. Oxidation of 1-O-(alk-1-enyl)-2,3-di-Oacylglycerols: models for plasmalogen oxidation. Lipids. 1988;23:430–434. doi: 10.1007/BF02535515. [DOI] [PubMed] [Google Scholar]
- Forss-Petter S, Werner H, Berger J, Lassmann H, Molzer B, Schwab MH, Bernheimer H, Zimmermann F, Nave KA. Targeted inactivation of the X-linked adrenoleukodystrophy gene in mice. J Neurosci Res. 1997;50:829–843. doi: 10.1002/(SICI)1097-4547(19971201)50:5<829::AID-JNR19>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Fourcade S, Lopez-Erauskin J, Galino J, Duval C, Naudi A, Jove M, Kemp S, Villarroya F, Ferrer I, Pamplona R, Portero-Otin M, Pujol A. Early oxidative damage underlying neurodegeneration in X-adrenoleukodystrophy. Hum Mol Genet. 2008 doi: 10.1093/hmg/ddn085. [DOI] [PubMed] [Google Scholar]
- Ganser AL, Kerner AL, Brown BJ, Davisson MT, Kirschner DA. A survey of neurological mutant mice. I. Lipid composition of myelinated tissue in known myelin mutants. Dev Neurosci. 1988;10:99–122. doi: 10.1159/000111961. [DOI] [PubMed] [Google Scholar]
- Ginsberg L, Rafique S, Xuereb JH, Rapoport SI, Gershfeld NL. Disease and anatomic specificity of ethanolamine plasmalogen deficiency in Alzheimer's disease brain. Brain Res. 1995;698:223–226. doi: 10.1016/0006-8993(95)00931-f. [DOI] [PubMed] [Google Scholar]
- Goodenowe DB, Cook LL, Liu J, Lu Y, Jayasinghe DA, Ahiahonu PW, Heath D, Yamazaki Y, Flax J, Krenitsky KF, Sparks DL, Lerner A, Friedland RP, Kudo T, Kamino K, Morihara T, Takeda M, Wood PL. Peripheral ethanolamine plasmalogen deficiency: a logical causative factor in Alzheimer's disease and dementia. J Lipid Res. 2007;48:2485–2498. doi: 10.1194/jlr.P700023-JLR200. [DOI] [PubMed] [Google Scholar]
- Gorgas K, Teigler A, Komljenovic D, Just WW. The ether lipid-deficient mouse: tracking down plasmalogen functions. Biochim Biophys Acta. 2006;1763:1511–1526. doi: 10.1016/j.bbamcr.2006.08.038. [DOI] [PubMed] [Google Scholar]
- Han X, Holtzman DM, McKeel DW., Jr Plasmalogen deficiency in early Alzheimer's disease subjects and in animal models: molecular characterization using electrospray ionization mass spectrometry. J Neurochem. 2001;77:1168–1180. doi: 10.1046/j.1471-4159.2001.00332.x. [DOI] [PubMed] [Google Scholar]
- Haq E, Contreras MA, Giri S, Singh I, Singh AK. Dysfunction of peroxisomes in twitcher mice brain: a possible mechanism of psychosine-induced disease. Biochem Biophys Res Commun. 2006;343:229–238. doi: 10.1016/j.bbrc.2006.02.131. [DOI] [PubMed] [Google Scholar]
- Hayashi H, Oohashi M. Incorporation of acetyl-CoA generated from peroxisomal beta-oxidation into ethanolamine plasmalogen of rat liver. Biochim Biophys Acta. 1995;1254:319–325. doi: 10.1016/0005-2760(94)00194-4. [DOI] [PubMed] [Google Scholar]
- Hayashi H, Sato A. Fatty alcohol synthesis accompanied with chain elongation in liver peroxisomes. Biochim Biophys Acta. 1997;1346:38–44. doi: 10.1016/s0005-2760(97)00020-9. [DOI] [PubMed] [Google Scholar]
- Hayashi H, Hara M. 1-Alkenyl group of ethanolamine plasmalogen derives mainly from de novo-synthesized fatty alcohol within peroxisomes, but not extraperoxisomal fatty alcohol or fatty acid. J Biochem. 1997;121:978–983. doi: 10.1093/oxfordjournals.jbchem.a021683. [DOI] [PubMed] [Google Scholar]
- Helmy FM, Hack MH, Juracka A. Age-related changes of the endogenous cardiolipin and plasmalogens of guinea pig kidney and their in vitro hydrolysis by endogenous phospholipases: a thin layer chromatographic analysis in conjunction with densitometric measurement. Cell Biochem Funct. 2003;21:337–344. doi: 10.1002/cbf.1035. [DOI] [PubMed] [Google Scholar]
- Heymans HS, vd Bosch H, Schutgens RB, Tegelaers WH, Walther JU, Muller-Hocker J, Borst P. Deficiency of plasmalogens in the cerebro-hepato-renal (Zellweger) syndrome. Eur J Pediatr. 1984;142:10–15. doi: 10.1007/BF00442582. [DOI] [PubMed] [Google Scholar]
- Hoefler G, Hoefler S, Watkins PA, Chen WW, Moser A, Baldwin V, McGillivary B, Charrow J, Friedman JM, Rutledge L, et al. Biochemical abnormalities in rhizomelic chondrodysplasia punctata. J Pediatr. 1988;112:726–733. doi: 10.1016/s0022-3476(88)80689-9. [DOI] [PubMed] [Google Scholar]
- Hoffman-Kuczynski B, Reo NV. Studies of myo-inositol and plasmalogen metabolism in rat brain. Neurochem Res. 2004;29:843–855. doi: 10.1023/b:nere.0000018859.23881.f9. [DOI] [PubMed] [Google Scholar]
- Hoffman-Kuczynski B, Reo NV. Administration of myo-inositol plus ethanolamine elevates phosphatidylethanolamine plasmalogen in the rat cerebellum. Neurochem Res. 2005;30:47–60. doi: 10.1007/s11064-004-9685-4. [DOI] [PubMed] [Google Scholar]
- Kehrer JP, Biswal SS. The molecular effects of acrolein. Toxicol Sci. 2000;57:6–15. doi: 10.1093/toxsci/57.1.6. [DOI] [PubMed] [Google Scholar]
- Kemp S, Wei HM, Lu JF, Braiterman LT, McGuinness MC, Moser AB, Watkins PA, Smith KD. Gene redundancy and pharmacological gene therapy: implications for X-linked adrenoleukodystrophy. Nat Med. 1998;4:1261–1268. doi: 10.1038/3242. [DOI] [PubMed] [Google Scholar]
- Khan M, Contreras M, Singh I. Endotoxin-induced alterations of lipid and fatty acid compositions in rat liver peroxisomes. J Endotoxin Res. 2000;6:41–50. doi: 10.1177/09680519000060010601. [DOI] [PubMed] [Google Scholar]
- Khan M, Pahan K, Singh AK, Singh I. Cytokine-induced accumulation of very long-chain fatty acids in rat C6 glial cells: implication for X-adrenoleukodystrophy. J Neurochem. 1998;71:78–87. doi: 10.1046/j.1471-4159.1998.71010078.x. [DOI] [PubMed] [Google Scholar]
- Khan M, Haq E, Giri S, Singh I, Singh AK. Peroxisomal participation in psychosine-mediated toxicity: implications for Krabbe's disease. J Neurosci Res. 2005;80:845–854. doi: 10.1002/jnr.20529. [DOI] [PubMed] [Google Scholar]
- Kuczynski B, Reo NV. Evidence that plasmalogen is protective against oxidative stress in the rat brain. Neurochem Res. 2006;31:639–656. doi: 10.1007/s11064-006-9061-7. [DOI] [PubMed] [Google Scholar]
- Latorre E, Collado MP, Fernandez I, Aragones MD, Catalan RE. Signaling events mediating activation of brain ethanolamine plasmalogen hydrolysis by ceramide. Eur J Biochem. 2003;270:36–46. doi: 10.1046/j.1432-1033.2003.03356.x. [DOI] [PubMed] [Google Scholar]
- Lepage G, Roy CC. Direct transesterification of all classes of lipids in a one-step reaction. J Lipid Res. 1986;27:114–120. [PubMed] [Google Scholar]
- Liu D, Nagan N, Just WW, Rodemer C, Thai TP, Zoeller RA. Role of dihydroxyacetonephosphate acyltransferase in the biosynthesis of plasmalogens and nonether glycerolipids. J Lipid Res. 2005;46:727–735. doi: 10.1194/jlr.M400364-JLR200. [DOI] [PubMed] [Google Scholar]
- Lu JF, Lawler AM, Watkins PA, Powers JM, Moser AB, Moser HW, Smith KD. A mouse model for X-linked adrenoleukodystrophy. Proc Natl Acad Sci U S A. 1997;94:9366–9371. doi: 10.1073/pnas.94.17.9366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeba R, Maeda T, Kinoshita M, Takao K, Takenaka H, Kusano J, Yoshimura N, Takeoka Y, Yasuda D, Okazaki T, Teramoto T. Plasmalogens in human serum positively correlate with high- density lipoprotein and decrease with aging. J Atheroscler Thromb. 2007;14:12–18. doi: 10.5551/jat.14.12. [DOI] [PubMed] [Google Scholar]
- Maes M, Mihaylova I, Leunis JC. Chronic fatigue syndrome is accompanied by an IgM-related immune response directed against neopitopes formed by oxidative or nitrosative damage to lipids and proteins. Neuro Endocrinol Lett. 2006;27:615–621. [PubMed] [Google Scholar]
- Mandel H, Korman SH. Phenotypic variability (heterogeneity) of peroxisomal disorders. Adv Exp Med Biol. 2003;544:9–30. doi: 10.1007/978-1-4419-9072-3_2. [DOI] [PubMed] [Google Scholar]
- Moser HW, Bergin A, Cornblath D. Peroxisomal disorders. Biochem Cell Biol. 1991;69:463–474. doi: 10.1139/o91-070. [DOI] [PubMed] [Google Scholar]
- Moser HW, Mahmood A, Raymond GV. X-linked adrenoleukodystrophy. Nat Clin Pract Neurol. 2007;3:140–151. doi: 10.1038/ncpneuro0421. [DOI] [PubMed] [Google Scholar]
- Mosser J, Douar AM, Sarde CO, Kioschis P, Feil R, Moser H, Poustka AM, Mandel JL, Aubourg P. Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature. 1993;361:726–730. doi: 10.1038/361726a0. [DOI] [PubMed] [Google Scholar]
- Mottaran E, Stewart SF, Rolla R, Vay D, Cipriani V, Moretti M, Vidali M, Sartori M, Rigamonti C, Day CP, Albano E. Lipid peroxidation contributes to immune reactions associated with alcoholic liver disease. Free Radic Biol Med. 2002;32:38–45. doi: 10.1016/s0891-5849(01)00757-2. [DOI] [PubMed] [Google Scholar]
- Nagan N, Hajra AK, Das AK, Moser HW, Moser A, Lazarow P, Purdue PE, Zoeller RA. A fibroblast cell line defective in alkyl-dihydroxyacetone phosphate synthase: a novel defect in plasmalogen biosynthesis. Proc Natl Acad Sci U S A. 1997;94:4475–4480. doi: 10.1073/pnas.94.9.4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton W, Cammer W. Isolation and characterization of myelin. New York: Plenum Press; 1984. pp. 147–195. [Google Scholar]
- Norton WT. Recent advances in myelin biochemistry. Ann N Y Acad Sci. 1984;436:5–10. doi: 10.1111/j.1749-6632.1984.tb14772.x. [DOI] [PubMed] [Google Scholar]
- Pahan K, Sheikh FG, Khan M, Namboodiri AM, Singh I. Sphingomyelinase and ceramide stimulate the expression of inducible nitric-oxide synthase in rat primary astrocytes. J Biol Chem. 1998;273:2591–2600. doi: 10.1074/jbc.273.5.2591. [DOI] [PubMed] [Google Scholar]
- Pai GS, Khan M, Barbosa E, Key LL, Craver JR, Cure JK, Betros R, Singh I. Lovastatin therapy for X-linked adrenoleukodystrophy: clinical and biochemical observations on 12 patients. Mol Genet Metab. 2000;69:312–322. doi: 10.1006/mgme.2000.2977. [DOI] [PubMed] [Google Scholar]
- Paintlia AS, Paintlia MK, Singh I, Singh AK. Immunomodulatory effect of combination therapy with lovastatin and 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside alleviates neurodegeneration in experimental autoimmune encephalomyelitis. Am J Pathol. 2006;169:1012–1025. doi: 10.2353/ajpath.2006.051309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paintlia AS, Gilg AG, Khan M, Singh AK, Barbosa E, Singh I. Correlation of very long chain fatty acid accumulation and inflammatory disease progression in childhood X-ALD: implications for potential therapies. Neurobiol Dis. 2003;14:425–439. doi: 10.1016/j.nbd.2003.08.013. [DOI] [PubMed] [Google Scholar]
- Paintlia AS, Paintlia MK, Khan M, Vollmer T, Singh AK, Singh I. HMG-CoA reductase inhibitor augments survival and differentiation of oligodendrocyte progenitors in animal model of multiple sclerosis. Faseb J. 2005;19:1407–1421. doi: 10.1096/fj.05-3861com. [DOI] [PubMed] [Google Scholar]
- Pannu R, Barbosa E, Singh AK, Singh I. Attenuation of acute inflammatory response by atorvastatin after spinal cord injury in rats. J Neurosci Res. 2005;79:340–350. doi: 10.1002/jnr.20345. [DOI] [PubMed] [Google Scholar]
- Pannu R, Christie DK, Barbosa E, Singh I, Singh AK. Post-trauma Lipitor treatment prevents endothelial dysfunction, facilitates neuroprotection, and promotes locomotor recovery following spinal cord injury. J Neurochem. 2007;101:182–200. doi: 10.1111/j.1471-4159.2006.04354.x. [DOI] [PubMed] [Google Scholar]
- Poulos A, Bankier A, Beckman K, Johnson D, Robertson EF, Sharp P, Sheffield L, Singh H, Usher S, Wise G. Glyceryl ethers in peroxisomal disease. Clin Genet. 1991;39:13–25. doi: 10.1111/j.1399-0004.1991.tb02980.x. [DOI] [PubMed] [Google Scholar]
- Powers JM. The pathology of peroxisomal disorders with pathogenetic considerations. J Neuropathol Exp Neurol. 1995;54:710–719. [PubMed] [Google Scholar]
- Powers JM, Pei Z, Heinzer AK, Deering R, Moser AB, Moser HW, Watkins PA, Smith KD. Adreno-leukodystrophy: oxidative stress of mice and men. J Neuropathol Exp Neurol. 2005;64:1067–1079. doi: 10.1097/01.jnen.0000190064.28559.a4. [DOI] [PubMed] [Google Scholar]
- Rodemer C, Thai TP, Brugger B, Gorgas K, Just W. Targeted disruption of ether lipid synthesis in mice. Adv Exp Med Biol. 2003;544:355–368. doi: 10.1007/978-1-4419-9072-3_46. [DOI] [PubMed] [Google Scholar]
- Schmitt K, Molzer B, Stockler S, Tulzer G, Tulzer W. Zellweger syndrome, neonatal adrenoleukodystrophy or infantile Refsum's disease in a case with generalized peroxisome defect? Wien Klin Wochenschr. 1993;105:320–322. [PubMed] [Google Scholar]
- Singh I. Biochemistry of peroxisomes in health and disease. Mol Cell Biochem. 1997;167:1–29. doi: 10.1023/a:1006883229684. [DOI] [PubMed] [Google Scholar]
- Singh I. Peroxisomal fatty acid oxidation and cellular redox. Methods Enzymol. 2002;352:361–372. doi: 10.1016/s0076-6879(02)52033-4. [DOI] [PubMed] [Google Scholar]
- Singh I, Pahan K, Khan M. Lovastatin and sodium phenylacetate normalize the levels of very long chain fatty acids in skin fibroblasts of X- adrenoleukodystrophy. FEBS Lett. 1998a;426:342–346. doi: 10.1016/s0014-5793(98)00370-6. [DOI] [PubMed] [Google Scholar]
- Singh I, Khan M, Key L, Pai S. Lovastatin for X-linked adrenoleukodystrophy. N Engl J Med. 1998b;339:702–703. doi: 10.1056/NEJM199809033391012. [DOI] [PubMed] [Google Scholar]
- Stadelmann-Ingrand S, Pontcharraud R, Fauconneau B. Evidence for the reactivity of fatty aldehydes released from oxidized plasmalogens with phosphatidylethanolamine to form Schiff base adducts in rat brain homogenates. Chem Phys Lipids. 2004;131:93–105. doi: 10.1016/j.chemphyslip.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Stanislaus R, Pahan K, Singh AK, Singh I. Amelioration of experimental allergic encephalomyelitis in Lewis rats by lovastatin. Neurosci Lett. 1999;269:71–74. doi: 10.1016/s0304-3940(99)00414-0. [DOI] [PubMed] [Google Scholar]
- Thorburne SK, Juurlink BH. Low glutathione and high iron govern the susceptibility of oligodendroglial precursors to oxidative stress. J Neurochem. 1996;67:1014–1022. doi: 10.1046/j.1471-4159.1996.67031014.x. [DOI] [PubMed] [Google Scholar]
- Uchida K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog Lipid Res. 2003;42:318–343. doi: 10.1016/s0163-7827(03)00014-6. [DOI] [PubMed] [Google Scholar]
- Weinhofer I, Forss-Petter S, Kunze M, Zigman M, Berger J. X-linked adrenoleukodystrophy mice demonstrate abnormalities in cholesterol metabolism. FEBS Lett. 2005;579:5512–5516. doi: 10.1016/j.febslet.2005.09.014. [DOI] [PubMed] [Google Scholar]
- Weisser M, Vieth M, Stolte M, Riederer P, Pfeuffer R, Leblhuber F, Spiteller G. Dramatic increase of alpha-hydroxyaldehydes derived from plasmalogens in the aged human brain. Chem Phys Lipids. 1997;90:135–142. doi: 10.1016/s0009-3084(97)00089-3. [DOI] [PubMed] [Google Scholar]
- Williams ML, Menon GK, Hanley KP. HMG-CoA reductase inhibitors perturb fatty acid metabolism and induce peroxisomes in keratinocytes. J Lipid Res. 1992;33:193–208. [PubMed] [Google Scholar]
- Wilson JX. Antioxidant defense of the brain: a role for astrocytes. Can J Physiol Pharmacol. 1997;75:1149–1163. [PubMed] [Google Scholar]
- Wilson R, Sargent JR. Lipid and fatty acid composition of brain tissue from adrenoleukodystrophy patients. J Neurochem. 1993;61:290–297. doi: 10.1111/j.1471-4159.1993.tb03567.x. [DOI] [PubMed] [Google Scholar]
- Won JS, Choi MR, Suh HW. Stimulation of astrocyte-enriched culture with C2 ceramide increases proenkephalin mRNA: involvement of cAMP-response element binding protein and mitogen activated protein kinases. Brain Res. 2001;903:207–215. doi: 10.1016/s0006-8993(01)02452-0. [DOI] [PubMed] [Google Scholar]
- Yamada T, Shinnoh N, Taniwaki T, Ohyagi Y, Asahara H, Horiuchi , Kira J. Lovastatin does not correct the accumulation of very long-chain fatty acids in tissues of adrenoleukodystrophy protein-deficient mice. J Inherit Metab Dis. 2000;23:607–614. doi: 10.1023/a:1005634130286. [DOI] [PubMed] [Google Scholar]
- Zoeller RA, Morand OH, Raetz CR. A possible role for plasmalogens in protecting animal cells against photosensitized killing. J Biol Chem. 1988;263:11590–11596. [PubMed] [Google Scholar]
- Zoeller RA, Grazia TJ, LaCamera P, Park J, Gaposchkin DP, Farber HW. Increasing plasmalogen levels protects human endothelial cells during hypoxia. Am J Physiol Heart Circ Physiol. 2002;283:H671–H679. doi: 10.1152/ajpheart.00524.2001. [DOI] [PubMed] [Google Scholar]