

Abstract
The cellular DNA damage response (DDR) is initiated by the rapid recruitment of repair factors to the site of DNA damage to form a multiprotein repair complex. How the repair complex senses damaged DNA and then activates the DDR is not well understood. Here we demonstrate that prolonged binding of DNA repair factors to chromatin can elicit DDR in an ATM- and DNAPK- dependent fashion in the absence of DNA damage. Targeting of single repair factors to chromatin revealed a hierarchy of protein interactions within the repair complex and suggests amplification of the damage signal. We conclude that activation of DDR does not require DNA damage and stable association of repair factors with chromatin is likely a critical step in triggering, amplifying and maintaining the DDR signal.
The DNA damage response (DDR) cascade senses genome damage and activates several downstream pathways including cell cycle checkpoints and apoptotic programs (1–3). The earliest step in the response is the rapid targeting and accumulation of DNA repair factors near the damage site giving rise to nuclear repair foci (4). The repair cascade consists of the early DNA damage sensor complex MRN (MRE11/Rad50/NBS1), the transducer proteins MDC1 and 53BP1, followed by the PI3 kinases ATM/DNAPK/ATR which in turn phosphorylate the histone variant H2AX and downstream effectors including the Chk1 and Chk2 cell cycle kinases (5). The functional relevance of the increased local concentration of DNA repair factors at sites of DNA damage has not been clear, nor is it known precisely how DDR is activated on chromatin.
To address these questions, we stably targeted individual DNA repair components to chromatin in living cells and assessed their contribution to DDR. DNA repair factors were fused to the E. coli lac-repressor (lacR) and tagged with Cherry-Red fluorescent protein. The fusion proteins were introduced into a NIH-3T3 cell line which contains 256 repeats of the lac operator sequence (lacO) stably integrated on chromosome 3 (6). The fusion proteins were effectively recruited and retained at sites of DNA damage upon UV laser damage (fig. S1A). As expected, fusion proteins accumulated at the lacO array as distinct nuclear foci (Fig. 1A). When assessing the effect of bound fusion proteins, we found that immobilization of NBS1 or MRE11 alone activated DDR as indicated by phosphorylation of H2AX at the lacO site in 60–70% of cells (Fig. 1B; fig S1B). Notably, activation of DDR upon immobilization of NBS1 or MRE11 occurred in the absence of DNA damage. The presence of DNA lesions was excluded by ligation-mediated PCR (Fig. 1C), the absence of BrdU incorporation at the lac array, and lack of staining for the single strand-binding protein RPA (fig. S2A, B). Activation of DDR was not due to overexpression of fusion proteins since no γH2AX was detected in the presence of IPTG, which interferes with binding of fusion proteins to LacO (fig. S3A). Induction of γH2AX phosphorylation was rapid and replication-independent (fig. S3B, C). The response to immobilizing single repair factors to chromatin was not limited to H2AX since immobilization also efficiently induced phosphorylation of S343 in NBS1 and S1987 in ATM, two key hallmarks of general DDR activation (Fig. 1D). These observations suggest that stable binding of individual early components of the DNA repair machinery is sufficient to induce cellular DDR even in the absence of DNA lesions.
Fig. 1.
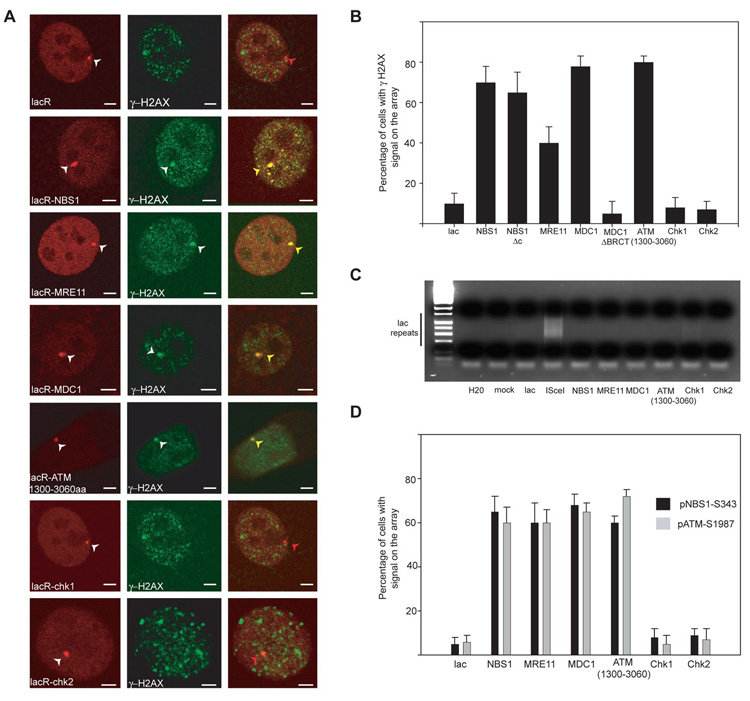
Immobilization of single repair factors on chromatin leads to DDR activation. (A) Immunofluorescence microscopy on NIH 2/4 cells transiently transfected for 16h with the indicated repair factor fused to Cherry-LacR (red). DDR activation is indicated by phosphorylation of H2AX (green). Scale Bar = 2µm. (B) Quantitative analysis of H2AX phosphorylation on the lacO array after transfection of the indicated repair factors for 16h. Values represent averages ± S.D (n = 40) from 4 independent experiments. (C) Absence of DNA lesions upon tethering. Ligation-mediated PCR on NIH2/4 cells transfected for 16h with the indicated repair factor fused to Cherry-LacR. The PCR product is a ladder due of the repetitive nature of the lac operator. Transfection of ISceI alone is used as a control. (D) Quantitative analysis of NBS1 (S343) or ATM (S1987) phosphorylation on the array after transfection of the indicated repair factor for 24 h. Values represent averages ± S.D (n = 60) from 2 independent experiments.
To extend these observations we tested the ability of downstream factors to induce DDR. Stable binding of MDC1 to chromatin led to activation of DDR with similar efficiency as NBS1 (Fig. 1A, B, D). This observation is consistent with the notion that H2AX phosphorylation and MDC1 foci formation are mutually inter-dependent (7–9). MDC1 ΔBRCT, which lacks the two C-terminal BRCT domains involved in its recruitment to γH2AX (9), did not elicit DDR (Fig. 1B), suggesting that downstream effects of MDC1 rely on the BRCT domain.
The downstream ATM kinase is a key player in triggering DDR (10). It is thought to exist as an inactive dimer in the absence of DNA damage, which becomes activated via autophosphorylation upon genomic insult (11). We find that immobilization of a major fragment of ATM (1300aa–3060aa), which includes the kinase domain, is sufficient to activate DDR as judged by phosphorylation of H2AX, NBS1 and ATM (Fig. 1A, B, D), suggesting that damaged DNA is not an absolute requirement for ATM activation. Unlike ATM, immobilization of the downstream effector kinases Chk1 and Chk2, which normally do not form DNA repair foci, did not lead to phosphorylation of H2AX (Fig. 1A, B, D). The correlation between accumulation at sites of DNA damage and ability to induce DDR when immobilized suggests that prolonged physical association of repair factors at sites of DNA damage might be a contributing factor to eliciting DDR in vivo.
Upon stable binding of NBS1, MRE11 or ATM1300–3060 to chromatin, phosphorylation of H2AX was largely blocked in the presence of the ATM inhibitor KU55933 (12) or the PI3 Kinase inhibitor caffeine, but not an inhibitor of DNAPK (Fig. 2A, B). H2AX phosphorylation by immobilized MDC1- lacR was dependent on both ATM and DNAPK kinase activities (Fig. 2B), but not on the DNAPK interacting partner Ku80 (fig. S4). Knockdown of ATR by RNAi had no effect on the extent of H2AX phosphorylation after immobilization of NBS1, MRE11 or MDC1 (fig. S5A, B). These results demonstrate that tethering-mediated DDR occurs via the ATM- and DNAPK-, but not ATR-, pathways.
Fig. 2.
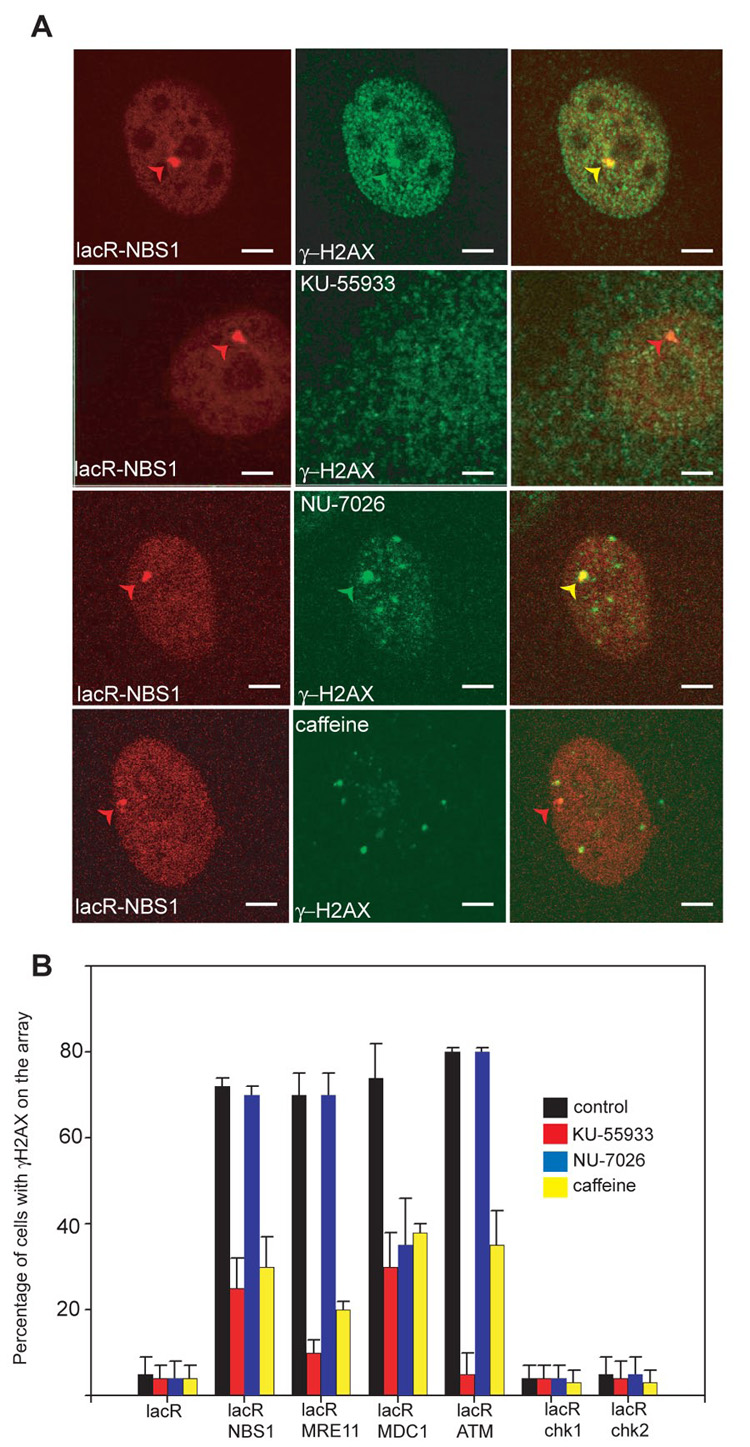
Activation of DDR depends on ATM and DNAPK. (A) Detection of γH2AX by indirect immunofluorescence in NIH 2/4 cells transiently transfected for 24h with lacR-NBS1- CherryRed (red) and treated with the indicated kinase inhibitor for 24h. γH2AX (green). Scale Bar = 2µm. (B) Quantitative analysis of H2AX phosphorylation on the lacO array after immobilization of the indicated repair factors in the presence of signaling inhibitors. Values represent averages ± S.D (n = 50) from 4 independent experiments.
We next used our experimental system to systematically determine the interdependencies of recruitment amongst repair factors in vivo. To this end, we probed the composition of repair foci formed upon stable binding of each individual factor. Immobilization of NBS1 led to recruitment of its direct interaction partner MRE11, but also of the downstream adaptors MDC1 and 53BP1 (Fig. 3A, B). Similarly, immobilization of MRE11 led to accumulation of NBS1, MDC1 and 53BP1 (Fig. 3B). Although MDC1 acts downstream of the early MRN sensor complex, immobilized MDC1 was able to efficiently recruit the upstream MRN components NBS1 and MRE11 and, to a lesser extent, 53BP1 (7, 13, 14), suggesting a feedback loop between MDC1 and the MRN complex, possibly as a means to amplify the damage response. This observation is consistent with the findings that MDC1 interacts with the MRN components and that downregulation of MDC1 in human cells fails to efficiently accumulate the MRN complex in irradiation-induced foci (15, 16). MDC1ΔBRCT, which does not induce DDR upon tethering, is still able to recruit NBS1 and MRE11. This observation is in agreement with the possibility that the BRCT domain may protect γH2AX from rapid dephosphorylation by phosphatases (9). LacR-ATM1300–3060 was unable to recruit any of the upstream factors with the exception of MDC1 (Fig. 3B). This effect is likely due to the ATM-induced phosphorylation of H2AX to which MDC1 binds with high affinity (Fig. 3B) (17). As expected, immobilized Chk1 and Chk2 did not recruit any upstream factors, further supporting their roles as loosely associated downstream components of the DDR pathway (Fig. 3B) (18). The differences in recruitment ability of ATM compared to NBS1, MRE11 and MDC1 suggests that ATM belongs to a distinct level of hierarchy within the repair complex.
Fig. 3.
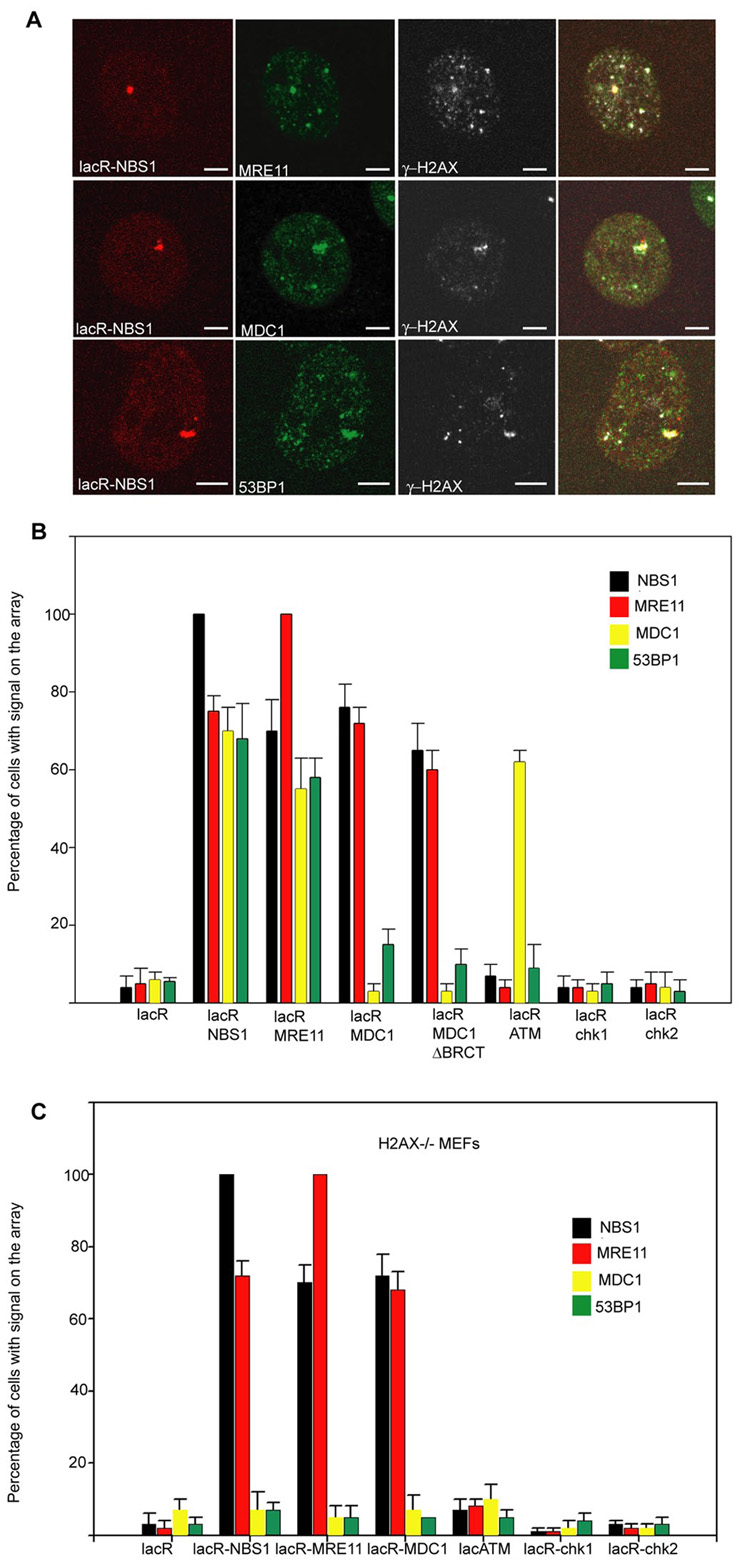
Cross-recruitment of repair factors. (A) Immunofluorescence microscopy on NIH 2/4 cells transiently transfected for 24h with lacR-NBS1 (red) with the indicated antibodies (green). Scale Bar = 2µm. (B) Quantitation of repair factor accumulation on the array in NIH 2/4 cells after immobilization of the indicated repair factors. Values represent averages ± S.D (n = 40) from 3 independent experiments. The antibody used to detect MDC1 recognizes only the mouse isoform and not human MDC1 present in the LacR fusion protein. (C) Quantitation of repair factor accumulation on the array on H2AX−/− MEFs containing the lacO array after immobilization of the indicated repair factors. Values represent averages ± S.D (n = 50) from 3 independent experiments.
To discriminate whether a given factor is recruited through protein-protein interactions with the immobilized protein or via phosphorylated H2AX, we assayed the composition of repair foci after binding individual proteins to chromatin in H2AX−/− MEFs containing the lacO array (6). Loss of H2AX had no effect on recruitment of NBS1 and MRE11 by either MRN-component or by MDC1 (Fig. 3C). However, upon immobilization of NBS1 or MRE11 the accumulation of the downstream factors MDC1 and 53BP1 was strongly impaired in the absence of H2AX (Fig. 3C). Recruitment of MDC1 by ATM1300–3060 was similarly decreased, suggesting that phosphorylation of H2AX is an important step in recruiting and maintaining these factors at sites of damage (17, 19).
To finally test whether individual repair factors are sufficient to induce a physiological DDR we assessed the effect of immobilization on cell cycle progression (Fig. 4). Upon targeting of NBS1, MRE11, MDC1 or ATM, but not Chk1 or Chk2, to chromatin, cells accumulated in G2 phase as determined by staining of pericentromeric heterochromatin with phosphoS10-H3 antibody (Fig. 4A) (20). Cell cycle delay was confirmed by increased phosphorylation of Rb at Ser 807/811 (fig. S6). Furthermore, the cell cycle delay was sensitive to the presence of chk2 and required ATM activity, suggesting a requirement for the checkpoint kinase chk2 (Fig. 4A). H2AX−/− cells were resistant to G2/M delays upon immobilization of repair factors (Fig. 4C). This observation is in line with the finding that cells lacking H2AX manifest a G2/M check point defect after exposure to low doses of irradiation (21).
Fig. 4.
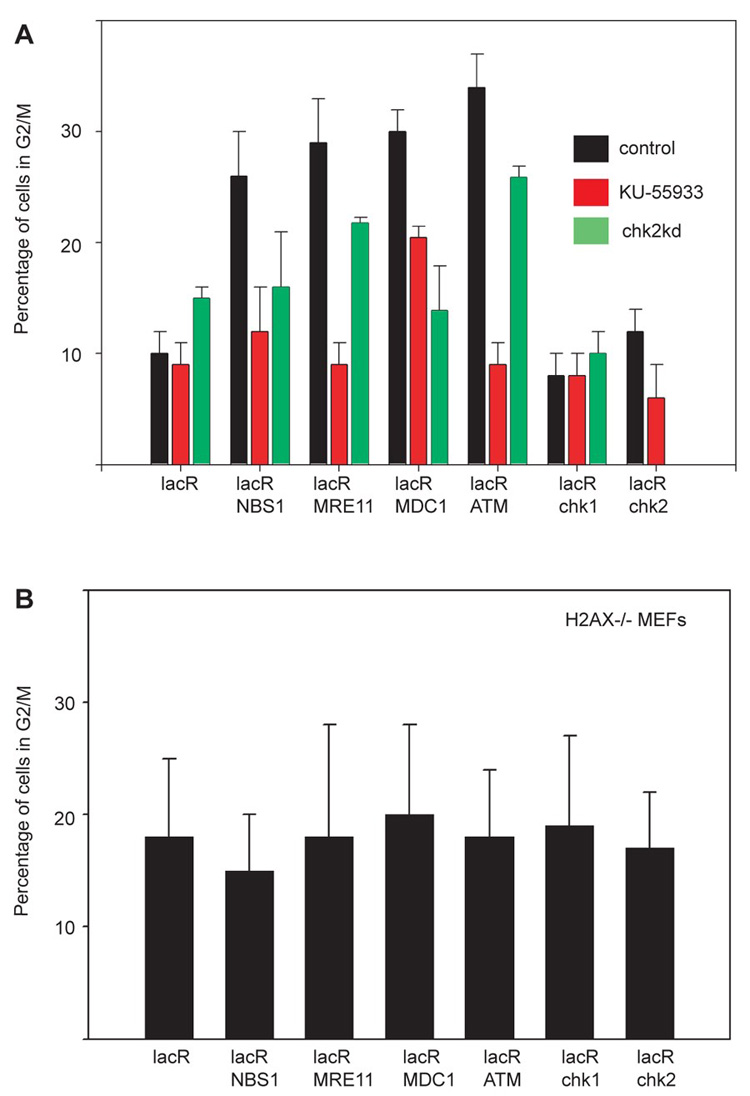
Targeting of single repair factors to chromatin induces G2 delay. (A) Quantitation of cells with phosphoH3 in Ser10 on pericentromeric chromatin on NIH 2/4 cells after immobilization of the indicated repair factors in wt cells, cells treated with ATM inhibitor or cells transfected with siRNA against chk2 . The siRNA pool eliminated also the expression of the fusion protein (absence of green bar in LacR-Chk2). Values represent averages ± S.D (n = 150) from at least two independent experiments. (B) Quantitation of cells with phosphoH3 in Ser10 on pericentromeric chromatin on H2AX−/− MEFs after immobilization of the indicated repair factors. The baseline level of phosphoS10H3 is slightly elevated in MEFs compared to NIH 2/4 cells. Values represent averages ± S.D (n = 150) from at least two independent experiments
We report here that activation of cellular DNA damage response pathways does not require DNA damage but can be triggered by stable association of single repair factors with chromatin. Our observations suggest that the physical interaction of DNA repair factors with chromatin is a key step in activation of the DDR signaling cascade, and that the observed build up at DNA damage foci probably contributes significantly to establishing the cellular response to damaged DNA (4). Our observation that immobilized downstream factors can recruit upstream components indicates that activation of a full DDR involves amplification via formation of multiple repair complexes and perpetuation of γH2AX phosphorylation. A critical role for signal amplification on DNA is also suggested by the findings that in the absence of γH2AX or MDC1, several repair factors, including NBS1 and 53BP1, are recruited to sites of DSBs, but do not accumulate and are not efficiently retained (16, 19). Our observation of phosphorylation of several key components of DDR including H2AX, NBS1 and ATM, and the appearance of cell cycle delays upon tethering indicate that the observed cellular response mimics to a large extent the physiological DDR. Given the apparent importance of the physical interaction of DNA repair factors with chromatin it will now be essential to uncover the precise role of higher order chromatin structure and chromatin remodeling complexes in triggering DDR.
Footnotes
Supporting Online Material www.sciencemag.org/cgi/content/full/1159051/DC1, Materials and Methods, Figs. S1 to S7, References
References and Notes
- 1.Lukas J, Bartek J. Cell. 2004;118:666. doi: 10.1016/j.cell.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 2.Shiloh Y. Curr Opin Genet Dev. 2001;11:71. doi: 10.1016/s0959-437x(00)00159-3. [DOI] [PubMed] [Google Scholar]
- 3.Jackson SP. Carcinogenesis. 2002;23:687. doi: 10.1093/carcin/23.5.687. [DOI] [PubMed] [Google Scholar]
- 4.Bekker-Jensen S, et al. J Cell Biol. 2006;173:195. doi: 10.1083/jcb.200510130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bartek J, Lukas J. Cancer Cell. 2003;3:421. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- 6.Soutoglou E, et al. Nat Cell Biol. 2007;9:675. doi: 10.1038/ncb1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lou Z, et al. Mol Cell. 2006;21:187. doi: 10.1016/j.molcel.2005.11.025. [DOI] [PubMed] [Google Scholar]
- 8.Peng A, Chen PL. J Biol Chem. 2003;278:8873. doi: 10.1074/jbc.C300001200. [DOI] [PubMed] [Google Scholar]
- 9.Stucki M, et al. Cell. 2005;123:1213. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 10.Shiloh Y. Cell Cycle. 2003;2:116. doi: 10.4161/cc.2.2.342. [DOI] [PubMed] [Google Scholar]
- 11.Bakkenist CJ, Kastan MB. Nature. 2003;421:499. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 12.Hickson I, et al. Cancer Res. 2004;64:9152. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 13.Bekker-Jensen S, Lukas C, Melander F, Bartek J, Lukas J. J Cell Biol. 2005;170:201. doi: 10.1083/jcb.200503043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. Nature. 2003;421:961. doi: 10.1038/nature01446. [DOI] [PubMed] [Google Scholar]
- 15.Goldberg M, et al. Nature. 2003;421:952. doi: 10.1038/nature01445. [DOI] [PubMed] [Google Scholar]
- 16.Lukas C, et al. Embo J. 2004;23:2674. doi: 10.1038/sj.emboj.7600269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stucki M, Jackson SP. DNA Repair (Amst) 2006;5:534. doi: 10.1016/j.dnarep.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 18.Lukas C, Falck J, Bartkova J, Bartek J, Lukas J. Nat Cell Biol. 2003;5:255. doi: 10.1038/ncb945. [DOI] [PubMed] [Google Scholar]
- 19.Celeste A, et al. Nat Cell Biol. 2003;5:675. doi: 10.1038/ncb1004. [DOI] [PubMed] [Google Scholar]
- 20.Monier K, Mouradian S, Sullivan KF. J Cell Sci. 2007;120:101. doi: 10.1242/jcs.03326. [DOI] [PubMed] [Google Scholar]
- 21.Fernandez-Capetillo O, et al. Nat Cell Biol. 2002;4:993. doi: 10.1038/ncb884. [DOI] [PubMed] [Google Scholar]
- 22.We thank A. Nussenzweig, J. Lukas and Y. Shiloh for reagents and comments; A. Belmont , T.Paull and M. Kastan for reagents; Sara Snyder for technical assistance; M. Kruhlak for help with microscopy. Imaging was performed at the National Cancer Institute (NCI) Fluorescence Imaging Facility. E.S. was supported by a fellowship from the Human Frontiers Science Program (HFSP). This research was supported by the Intramural Research Program of the National Institutes of Health (NIH), NCI, and Center for Cancer Research.