

Summary
Subtilase cytotoxin (SubAB) is the prototype of a new family of AB5 cytotoxins produced by Shiga toxigenic Escherichia coli. Its cytotoxic activity is due to its capacity to enter cells and specifically cleave the essential endoplasmic reticulum (ER) chaperone BiP (GRP78). In the present study, we have examined its capacity to trigger the three ER stress-signalling pathways in Vero cells. Activation of PKR-like ER kinase was demonstrated by phosphorylation of eIF2α, which occurred within 30 min of toxin treatment, and correlated with inhibition of global protein synthesis. Activation of inositol-requiring enzyme 1 was demonstrated by splicing of X-box-binding protein 1 mRNA, while activating transcription factor 6 activation was demonstrated by depletion of the 90 kDa uncleaved form, and appearance of the 50 kDa cleaved form. The rapidity with which ER stress-signalling responses are triggered by exposure of cells to SubAB is consistent with the hypothesis that cleavage by the toxin causes BiP to dissociate from the signalling molecules.
Introduction
Subtilase cytotoxin (SubAB) is the prototype of a new AB5 toxin family that was discovered in a strain of Shiga toxigenic Escherichia coli (STEC) responsible for an outbreak of haemolytic uraemic syndrome (HUS). HUS is a life-threatening complication of STEC disease, which is characterized by a triad of microangiopathic haemolytic anaemia, thrombocytopoenia and renal failure. These clinical manifestations have long been considered to be directly attributable to effects of Shiga toxin (Stx) on endothelial cells (Paton and Paton, 1998), although it is possible that other virulence factors may contribute. The new toxin was named ‘subtilase cytotoxin’ because its 35 kDa A subunit (SubA) has sequence similarity with a subtilase-like serine protease of Bacillus anthracis (Paton et al., 2004). Members of the subtilase family are found in a wide variety of microorganisms, but none had previously been shown to be cytotoxic, nor have any been shown to be associated with a B subunit (SubB) (Siezen and Leunissen, 1997). Mutagenesis of a critical active site Ser residue in SubA abolished toxicity, indicating that serine protease activity is central to the mechanism of action of the holotoxin (Paton et al., 2004). The SubB is related to putative exported proteins from Yersinia pestis and Salmonella Typhi, and mediates binding to the surface of target cells. SubAB is more toxic for Vero (African green monkey kidney) cells than Stx in vitro, and is lethal for mice (Paton et al., 2004). Remarkably, we have recently shown that intraperitoneal injection of mice with purified toxin results in pathological features resembling those of Stx-induced HUS (Wang et al., 2007). Although it is not yet known whether SubAB contributes to development of HUS following exposure to STEC, the production of two potentially lethal cytotoxins by a single pathogen raises interesting questions about the relative importance of each toxin to human disease pathogenesis, and whether they might act in synergism. Thus, understanding how SubAB affects host cells is important.
Recently, we have reported that the extreme cytotoxicity of SubAB for eukaryotic cells is due to a specific single-site cleavage of BiP (also known as GRP78) (Paton et al., 2006). BiP is an Hsp70 family chaperone located in the endoplasmic reticulum (ER), and comprises an N-terminal ATPase and a C-terminal protein-binding domain. Although like other chaperones, it mediates correct folding of nascent secretory proteins, BiP has additional important functions. These include maintaining the permeability barrier of the ER membrane by sealing the luminal end of the Sec61 translocon pore, as well as targeting of terminally misfolded proteins to the Sec61 apparatus for degradation by the proteasome (Gething, 1999; Hendershot, 2004). BiP also plays a crucial role as the ER stress-signalling master regulator, as well as exhibiting anti-apoptotic properties through interference with caspase activation (Kim and Arvan, 1998; Rao et al., 2004). SubAB cleaves BiP at a di-leucine motif in the hinge connecting the ATPase and protein-binding domains (Paton et al., 2006), and such cleavage has ultimately fatal consequences for the cell.
We have been examining the downstream consequences of BiP cleavage in order to elucidate the mechanism by which this novel toxin kills target cells. We hypothesized that one of these consequences would be triggering of a severe ER stress response. ER stress can be induced by diverse perturbations in ER function and its purpose is to implement a series of changes in cellular activity, such that the ER stress is alleviated, enabling the cell to restore ER homeostasis and recover (Boyce and Yuan, 2006). ER stress is typified by the unfolded protein response (UPR). UPR is characterized by: (i) transcriptional upregulation of ER chaperones including BiP and other proteins, creating an ER milieu in which protein folding capacity is optimized, (ii) activation of proteasome-dependent ER-associated degradation (ERAD) to remove unfolded proteins from the ER lumen and (iii) modulation of translation to slow down the traffic of nascent proteins into the ER compartment that require folding. However, if these responses fail to restore ER homeostasis, apoptosis may result (Boyce and Yuan, 2006).
In mammals, ER stress responses can be triggered by activation of three distinct ER membrane-spanning signalling molecules, namely PKR-like ER kinase (PERK), inositol-requiring enzyme 1 (IRE1) and activating transcription factor 6 (ATF6). The luminal domains of all three of these sentinel proteins are known to interact with BiP, and accumulation of unfolded proteins in the ER lumen is thought to recruit BiP away, allowing these sentinels to initiate signalling. By phosphorylation of its target eIF2α, activated PERK (pPERK) inhibits global protein synthesis, yet allows translation of mRNAs such as ATF4, a transcriptional activator for genes that ultimately assist in re-establishing ER homeostasis (Szegezdi et al., 2006). Release from BiP enables ATF6 to traffic to the Golgi, where it undergoes limited proteolysis, releasing a 50 kDa activated form, which translocates to the nucleus and binds to the ER stress response element, thus inducing genes encoding ER chaperones such as BiP, GRP94 and protein disulphide isomerase, as well as the transcription factors C/EBP homologous protein (CHOP) and X-box-binding protein 1 (XBP1). Activated IRE1 splices XBP1 mRNA into a form that can be translated into XBP1 protein, which then upregulates genes encoding ER chaperones and the HSP40 family member P58IPK. P58IPK provides a feedback loop by binding and inhibiting PERK, thereby relieving the eIF2α-mediated translational block. As SubAB cleaves BiP, we hypothesized that activation of all three stress sentinels would occur. In this study, we have examined the effects of SubAB on the PERK, IRE1 and ATF6 pathways.
Results
Activation of PERK by SubAB
Direct detection of activated (phosphorylated) PERK is problematic because of the absence of commercial antibodies capable of reliably and specifically detecting pPERK. Accordingly, PERK activation was assessed by following phosphorylation of its target, eIF2α, by Western blotting. In the first instance, Vero cell monolayers were treated with 1 μg ml−1 native SubAB, or SubAA272B, for various times from 0.5 to 6 h. SubAA272B has a single amino acid substitution in the active site Ser272 residue, which reduces cytotoxic activity by approximately 99.9% (Paton et al., 2004). In cells treated with SubAB, significant eIF2α phosphorylation was evident at 0.5 h; this was maximal at 2 h, and had returned to baseline by 6 h. The earliest time point at which SubAB-induced eIF2α phosphorylation could be detected was 15 min using a 1 μg ml−1 dose, but it could not be detected at 5 min (data not shown). In contrast, there was no detectable eIF2α-P in untreated control cells, and only trace levels were detected in cells treated with SubAA272B. Total eIF2α was unaffected by any of the treatments (Fig. 1).
Fig. 1.
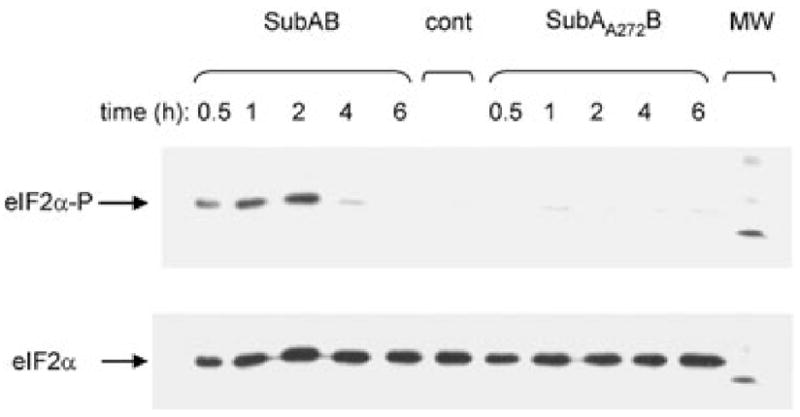
Time-course of eIF2α phosphorylation. Vero cell monolayers were treated with 1 μg ml−1 SubAB, or SubAA272B, for the indicated times. Proteins in cell extracts were separated by SDS-PAGE, electroblotted, probed with anti-phospho-eIF2α, and labelled species were detected using a HRP conjugate and ECL, as described in Experimental procedures. Filters were then stripped and re-probed using anti-eIF2α, to label total eIF2α. An untreated cell extract (cont) was also included. The visible band in the molecular marker (MW) track is 30 kDa.
Dose–response experiments (Fig. 2) showed that at 0.5 h, eIF2α-P levels were maximal at SubAB doses ≥ 100 ng ml−1. Responses were progressively lower at 10 and 1 ng ml−1. The small degree of eIF2α phosphorylation at 1 ng ml−1 was similar to that in cells treated with 1 μg ml−1 SubAA272B, which is consistent with the vestigial cytotoxic activity of the mutant toxin (and is similar to that observed in untreated cells). At this time point, global cellular protein synthesis, measured by [3H]-Leu incorporation, was significantly inhibited in cells treated with 10 ng ml−1, 100 ng ml−1 or 1 μg ml−1 SubAB (83.7%, 41.5% and 16.9% of that in control cells respectively; P < 0.002, P < 0.0001 and P < 0.0001 respectively). At these doses of SubAB, protein synthesis was further suppressed at 1 h (50.1%, 21.5% and 8.1% of that in control cells respectively; P < 0.0001 in all cases). The less dramatic inhibition of protein synthesis at 0.5 and 1 h in cells exposed to 10 ng ml−1 SubAB was consistent with the lower level of eIF2α-P, but was most pronounced at 2 h (30.2% of the control; P < 0.0001) when eIF2α phosphorylation may have peaked. At 2 and 4 h, significant inhibition of protein synthesis was also apparent at the lowest dose of SubAB tested (1 ng ml−1) (75.1% and 70.3% of that in control cells respectively; P < 0.0001 in both cases), and significant phosphorylation of eIF2α at this dose was evident from 1 h. However, from 2 h onwards, protein synthesis recovered in cells treated with the two highest doses of SubAB, in parallel with decreased levels of eIF2α-P. Indeed, for cells treated with 1 μg ml−1 SubAB, [3H]-Leu incorporation after 4 h exposure was not significantly different from that of control cells, which correlated with eIF2α-P returning to basal levels. Significant inhibition of protein synthesis was not observed in cells treated with 1 μg ml−1 SubAA272B at any time point (Fig. 2).
Fig. 2.
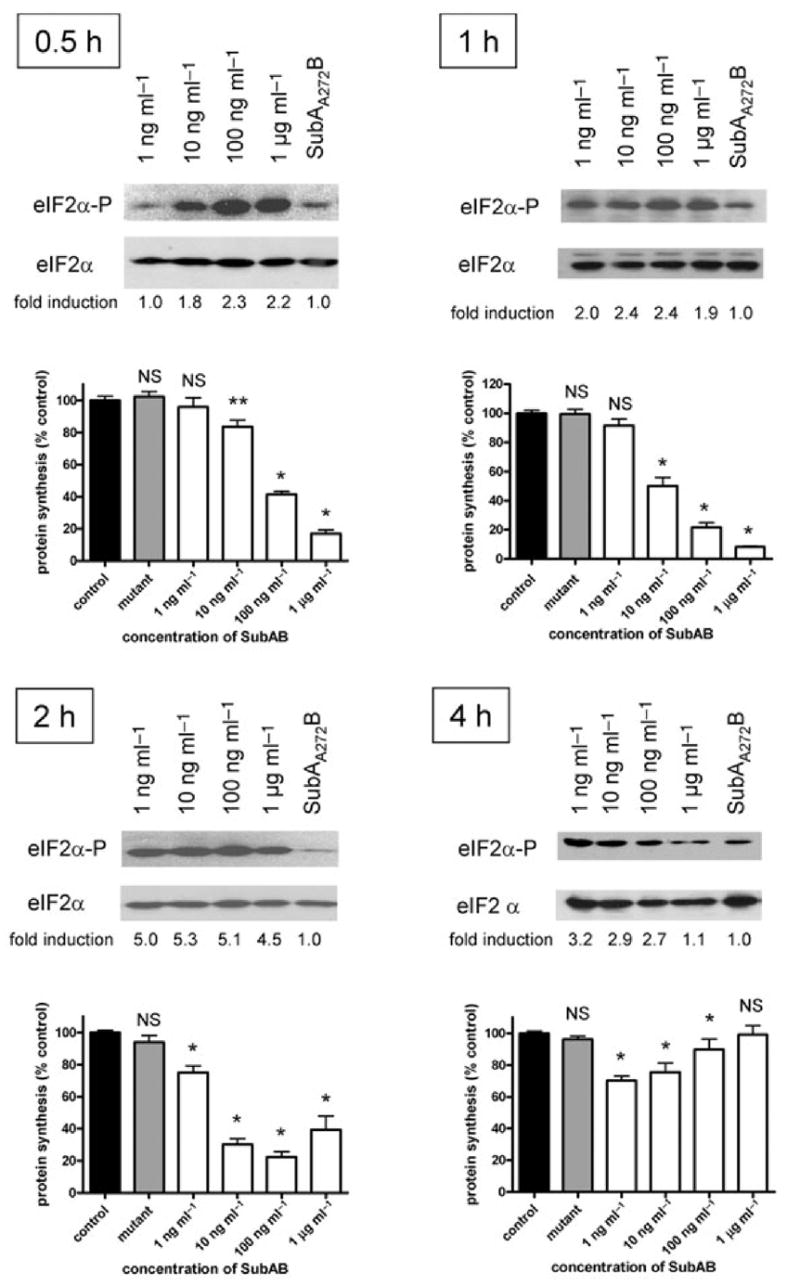
Effect of SubAB on eIF2α phosphorylation and protein synthesis. Vero cell monolayers were incubated with the indicated dose of SubAB, or 1 μg ml−1 SubAA272B for 0.5, 1, 2 or 4 h. Levels of eIF2α-P and total eIF2α were then determined by Western blotting, as for Fig. 1. Figures below each lane are fold induction of eIF2α-P following SubAB treatment, relative to treatment with SubAA272B (normalized to loading controls), determined by densitometry using ImageJ 1.38× software. Effects on global protein synthesis were also measured by assessment of [3H]-leucine incorporation into TCA-precipitable material during a 60 min pulse, as described in Experimental procedures. Data are the mean ± SEM from three independent experiments, expressed as a percentage of the untreated control for each time point. NS, not significant; *, P < 0.0001; **, P < 0.002 (two-tailed t-test).
To eliminate the possibility that an alternative pathway might be leading to eIF2α phosphorylation, we examined the effect of SubAB on eIF2α phosphorylation in wild type (wt) versus PERK −/− mouse embryonic fibroblasts (MEFs), which like Vero cells, are susceptible to the toxin, as judged by BiP cleavage (Fig. 3A). Cells were treated with 1 μg ml−1 native SubAB or control media at time points from 5 min to 4 h. In PERK −/− cells, only minimal phosphorylation over baseline values was detected. In contrast, phosphorylation of eIF2α in PERK wt cells was very similar in kinetics to those in Vero cells, with phosphorylation evident by 15 min, peaking at 1 h and then tapering off by 4 h (Fig. 3B). Protein synthesis inhibition was also measured by [3H]-Leu incorporation, as described above, in PERK −/− and PERK wt fibroblasts. Inhibition of protein synthesis was seen in SubAB-treated PERK wt cells, but not in SubAB-treated PERK −/− cells, when compared with untreated controls (Fig. 3C). These data show that PERK is necessary for eIF2α phosphorylation as well as inhibition of protein synthesis following SubAB treatment.
Fig. 3.
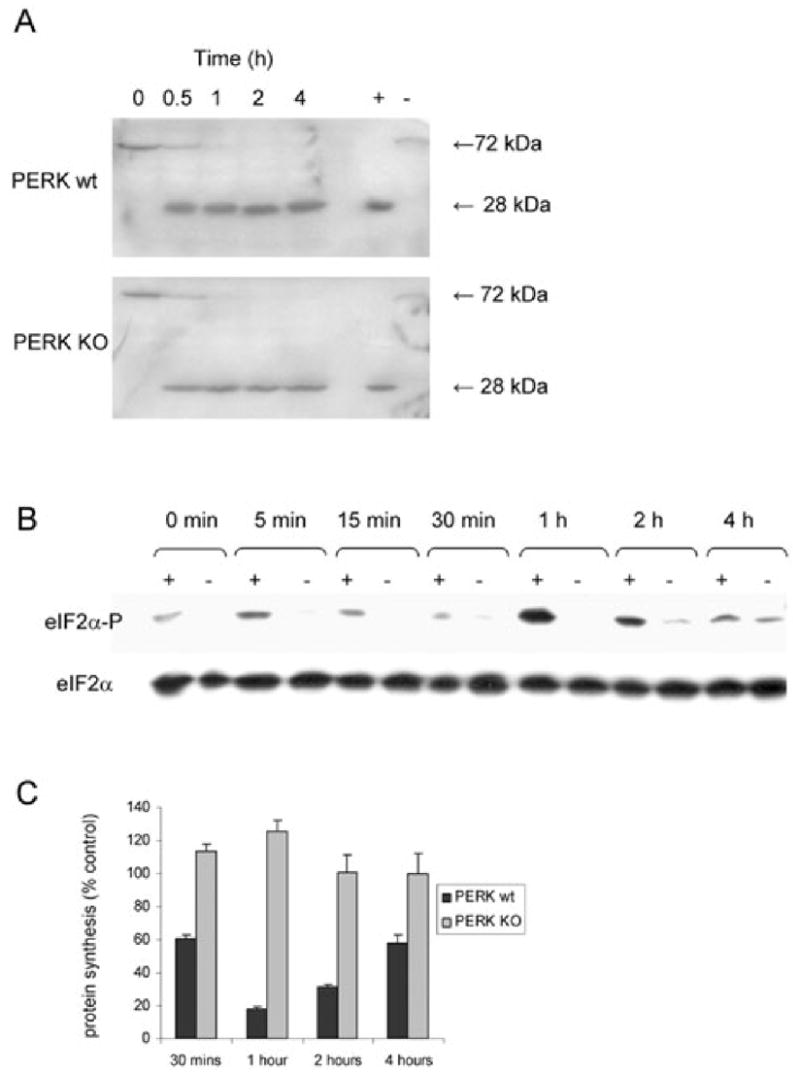
Effect of SubAB on BiP, eIF2α phosphorylation and protein synthesis in PERK −/− (KO) and PERK wt MEFs. MEF monolayers were incubated with 1 μg ml−1 SubAB for various times, as indicated.
A. Cleavage of BiP was assessed by Western blotting using anti-BiP C-20 (Santa Cruz), as described previously for Vero cells (Paton et al., 2006). The lanes marked + and − are Vero cells treated for 2 h with or without SubAB respectively.
B. Levels of eIF2α-P and total eIF2α were determined by Western blotting, as for Fig. 1; lanes marked + and − denote PERK wt and PERK KO cells respectively.
C. Global protein synthesis in PERK wt and PERK KO MEFs were measured as previously described in Fig. 2. Data are the mean ± SD 3H-Leu incorporation, expressed as a percentage of control cells, from a representative experiment conducted in triplicate.
Activation of IRE1 and induction of ER stress-associated genes
Activation of IRE1 was initially assessed by extracting RNA from Vero cells treated for 6 h with 100 ng ml−1 SubAB, 100 ng ml−1 SubAA272B or 0.2 μg ml−1 thapsigargin (a known ER stressor), and testing for splicing of XBP1 mRNA by reverse transcription (RT)-PCR. This assay generates amplicons of 398 and 424 bp for spliced or unspliced XBP1 mRNA variants respectively, with the presence of the former indicative of IRE1 activation (Shang, 2005). The assay also generates a higher amplicon species (450 bp), which is a hybrid of the two products (Shang, 2005). As shown in the upper panel of Fig. 4, splicing of XBP1 mRNA was virtually complete in cells treated with SubAB and thapsigargin, but there was negligible splicing in control cells and those treated with the mutant toxin. Additional RT-PCR analysis revealed qualitative increases in mRNA levels for BiP, CHOP, GRP94 and ATF4 in cells treated with either SubAB or thapsigargin, but not in those treated with SubAA272B. On the other hand, levels of the control transcript [glyceraldehyde-3-phosphate dehydrogenase (GAPDH)] were indistinguishable regardless of treatment (Fig. 4).
Fig. 4.
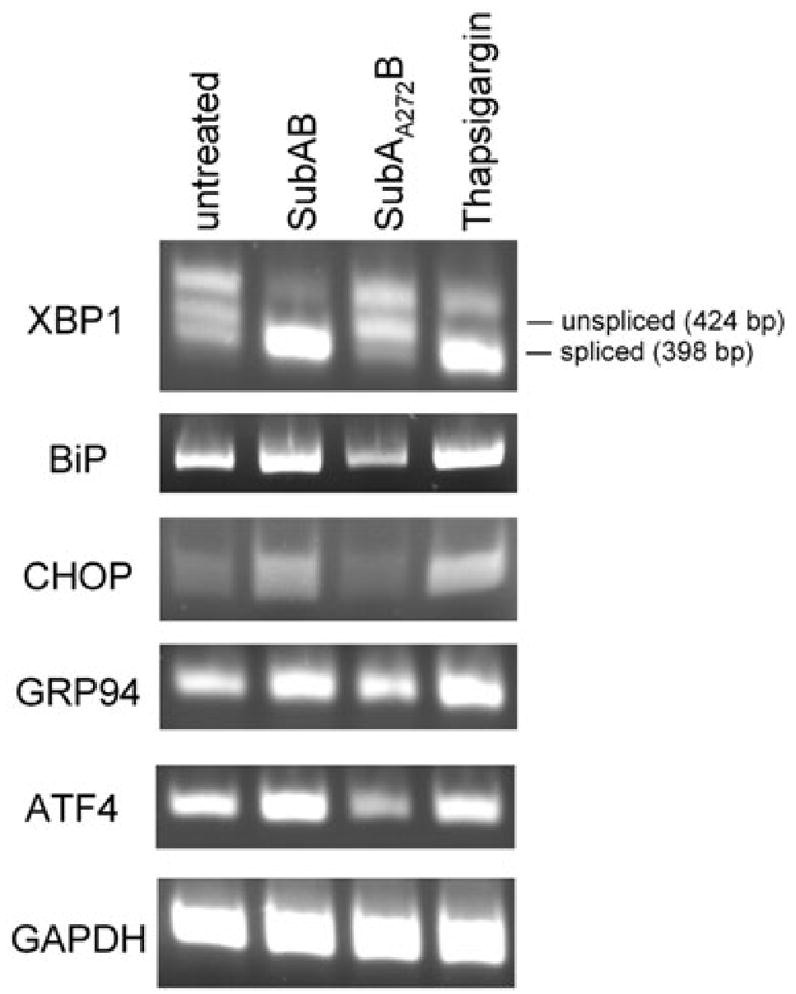
RT-PCR analysis. RNA extracts from Vero cells treated for 6 h with or without 100 ng ml−1 SubAB, 100 ng ml−1 SubAA272B or 0.2 μg ml−1 thapsigargin were analysed by RT-PCR using primers (Table 1) specific for XBP1, BiP, CHOP, GRP94, ATF4 and GAPDH (internal control), as described in Experimental procedures. The expected mobilities of spliced and unspliced XBP1 amplicons are indicated.
An extended time-course experiment over 30 h (Fig. 5) demonstrated that in the presence of 100 ng ml−1 SubAB, XBP1 mRNA splicing was nearly complete at 1 h, and was sustained for the entire period. BiP mRNA increased steadily from 2 h, reaching a peak at 24 h. Similarly, CHOP induction was evident at time points from 2 to 30 h. EDEM (ER degradation enhancing α-mannosidase-like protein) and GRP94 mRNA expression was most noticeable at 16 and 24 h. Increased ATF4 mRNA was evident at 1 h but, by 24–30 h, had returned to the level seen in untreated cells (Fig. 5). To investigate the kinetics of mRNA induction quantitatively, real-time PCR analysis of cDNA was employed using independent primer pairs (Fig. 6). These findings essentially confirmed those obtained by the qualitative RT-PCR analysis, although there were some minor variations. Statistically significant induction of BiP mRNA occurred by 6 h, reaching a plateau from 16 to 24 h. Similar kinetics were exhibited for GRP94, ATF4, EDEM and CHOP, with a plateau of maximal expression from 16 to 30 h, although lesser, but nevertheless statistically significant, increases in transcription were seen for ATF4 and CHOP at some earlier time points. GADD34, on the other hand, exhibited a steady increase in transcription over the entire time-course, with significant increases evident from 2 h onwards.
Fig. 5.
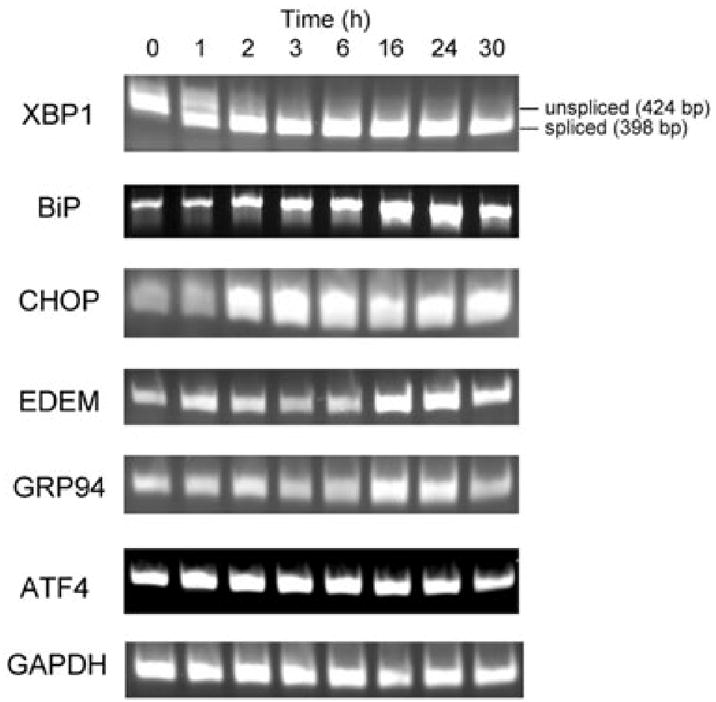
Time-course. RNA extracts from Vero cells treated with 100 ng ml−1 SubAB for 0–30 h were analysed by RT-PCR using primers (Table 1) specific for the indicated transcripts.
Fig. 6.
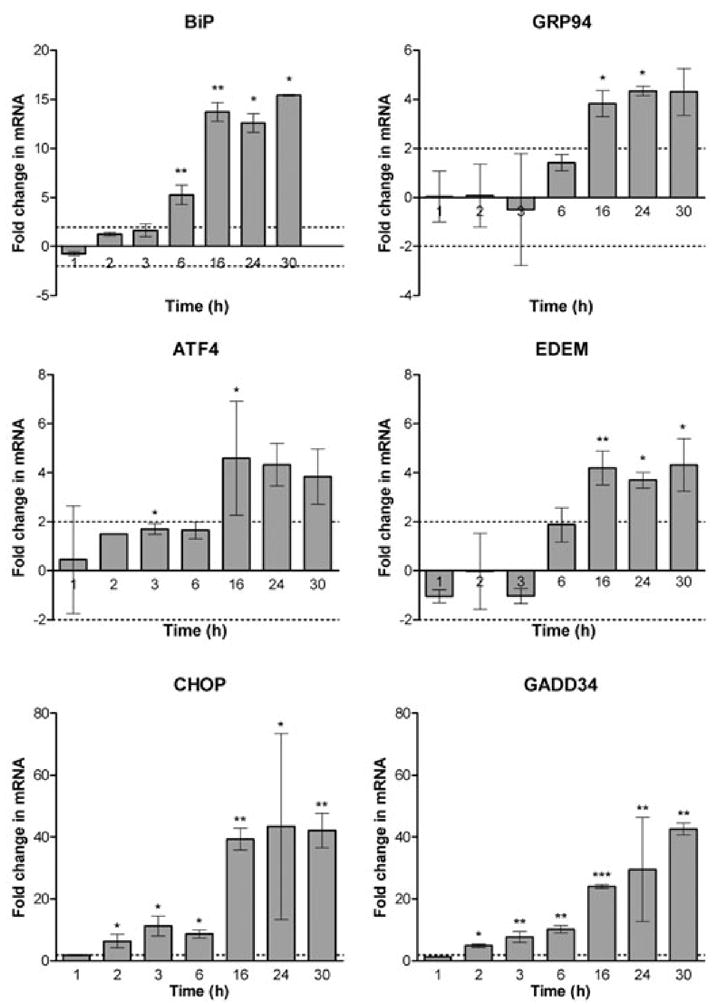
Quantitative real-time PCR analysis. RNA extracts from Vero cells treated with 100 ng ml−1 SubAB for 0–30 h were reverse-transcribed into cDNA and real-time PCR analysis was then performed using gene-specific primers (Table 2), as described in Experimental procedures. For each time point, the relative amounts of each transcript in SubAB-treated cells were compared with that in untreated cells, using the comparative cycle threshold (2ΔΔCt) method. Data represent the mean ± SD from two independent experiments, expressed as fold change in mRNA compared with the untreated control for each time point (*, P < 0.05; **, P < 0.01; ***, P < 0.001; Student’s unpaired two-tailed t-test).
Cell lysates from a similar time-course experiment were also subjected to Western blot analysis, and this showed a progressive increase in expression of GRP94 protein over the full 30 h period, whereas expression of CHOP was not seen until 6 h, peaked at 16 h and decreased thereafter (Fig. 7). Interpretation of BiP protein levels is complicated by the SubAB-mediated cleavage; the intact 72 kDa species was barely detectable after 1 h, but there was a steady increase in the levels of the 28 kDa cleavage product over 30 h. This is consistent with the progressive increases in BiP transcription seen in Figs 5 and 6, but newly translated BiP is being cleaved by the toxin in the ER lumen.
Fig. 7.
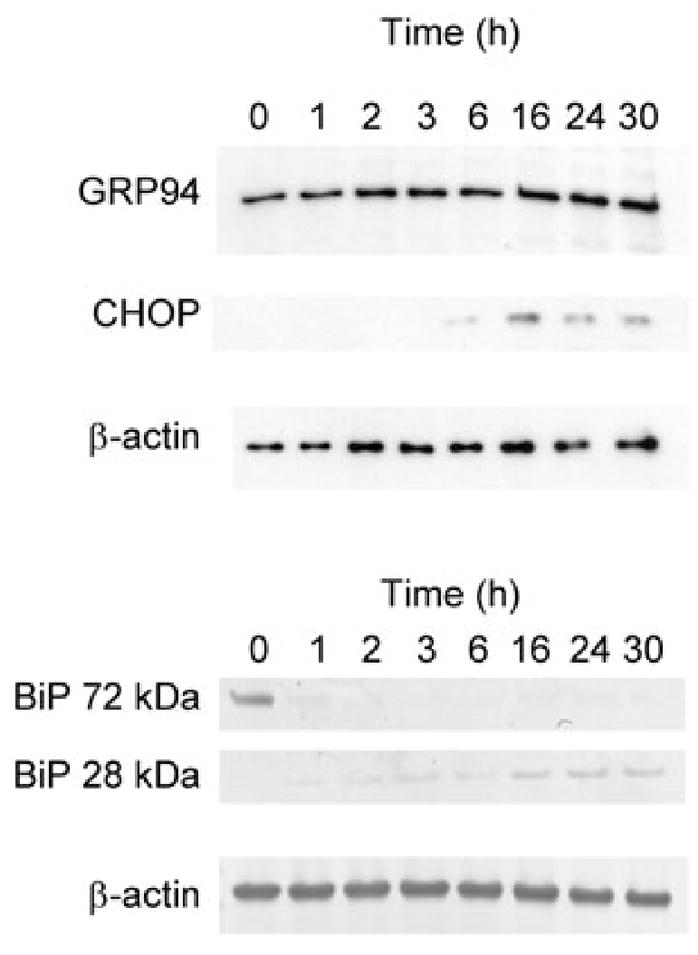
Time-course of GRP94, CHOP and BiP induction. Vero cells were treated with 100 ng ml−1 SubAB for 0–30 h, and lysates were analysed for GRP94, CHOP and BiP by Western blotting, as described in Experimental procedures. β-actin was used as an internal loading control. For BiP, separate panels showing intact (72 kDa) and cleaved (28 kDa) forms are presented.
Activation of ATF6 by SubAB
Direct Western blot analysis for ATF6 activation was not possible as a result of the non-availability of an antibody capable of detecting the intact (90 kDa) and the cleaved (50 kDa) forms of the protein. To overcome this, Vero cells were transiently transfected with plasmid p3×FLAG-CMV-7.1-ATF6 (Shen and Prywes, 2004), which directs coexpression of triple FLAG-tagged ATF6. After 24 h, cells were treated with or without SubAB, SubAA272B or thapsigargin; at various times thereafter, lysates were prepared and levels of intact ATF6 were examined by Western blotting using anti-FLAG (Fig. 8A). At 1 h, there were no obvious differences in 90 kDa ATF6 levels but, by 2 h, ATF6 levels were lower in transfected cells treated with SubAB. The reduction in 90 kDa ATF6 was more apparent in SubAB-treated cells after 4 h and, by 24 h, very little intact ATF6 remained. Thapsigargin treatment also reduced 90 kDa ATF6 levels at the 4 and 24 h time points. In contrast, the levels of 90 kDa ATF6 in cells treated with SubAA272B were similar to that in untreated transfected cells at all time points. Activation of the ATF6 pathway was also examined by detection of the 50 kDa cleaved form, although this was complicated by the presence of a closely migrating non-specific band present in untransfected cells that cross-reacted with the monoclonal anti-FLAG. Nevertheless, the 50 kDa cleaved form of ATF6 was clearly evident in cells treated with SubAB for 4 h (Fig. 8B); significant levels of 50 kDa ATF6 were not distinguishable from the non-specific background at other time points (result not shown), or in transfected cells treated with either mutant toxin or thapsigargin (Fig. 8B).
Fig. 8.
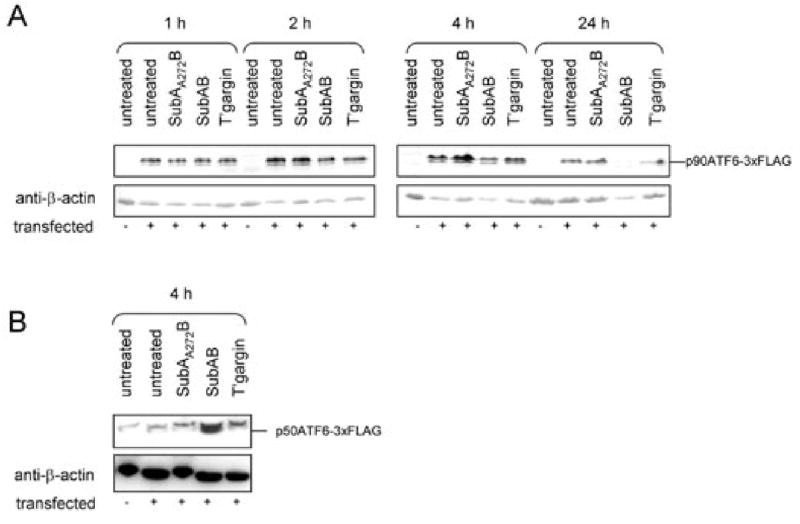
ATF6 activation. Vero cells transfected with p3×FLAG-CMV-7.1-ATF6 were treated with or without 100 ng ml−1 SubAB, 100 ng ml−1 SubAA272B or 0.2 μg ml−1 thapsigargin. At various times thereafter, lysates were prepared from these cells, as well as from untransfected Vero cells, and levels of (A) uncleaved tagged ATF6 (p90ATF6-3×FLAG), or (B) cleaved tagged ATF6 (p50ATF6-3×FLAG), were examined by Western blotting using anti-FLAG. After detection using alkaline phosphatase-conjugate and chromogenic substrate (A) or HRP and chemiluminescence (B), filters were stripped and re-probed with anti-β-actin as a loading control.
Induction of apoptosis by SubAB
The clear activation of all three ER stress-signalling pathways, and the ongoing degradation of newly translated BiP in SubAB-treated cells raises the likelihood that the mechanism of SubAB-induced cell death involves triggering apoptosis. This was investigated in Vero cells treated with 100 ng ml−1 SubAB using a DNA fragmentation assay. There was clear evidence of apoptosis at 30 h, but not at earlier time points. No DNA fragmentation was seen in cells treated with 100 ng ml−1 SubAA272B at 30 h (Fig. 9).
Fig. 9.
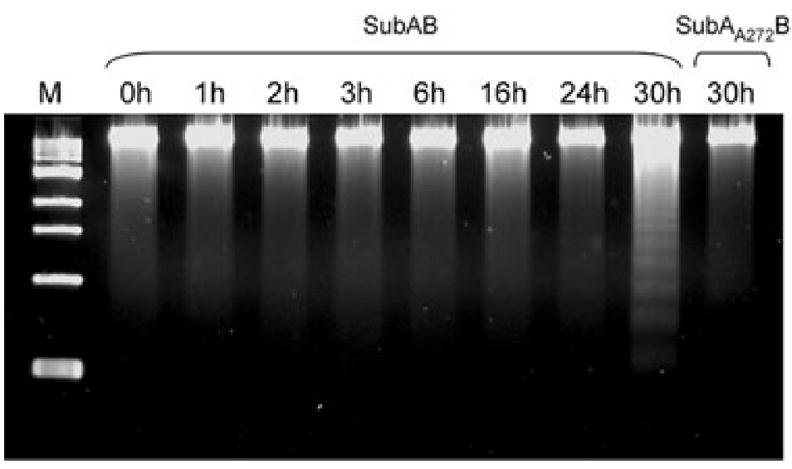
DNA fragmentation assay. Vero cells were treated with 100 ng ml−1 SubAB for the indicated times, or with SubAA272B for 30 h. DNA was then extracted and subjected to agarose gel electrophoresis, as described in Experimental procedures. M denotes molecular size markers.
Discussion
Our previous studies have established that the extreme cytotoxicity of SubAB is directly attributable to single site cleavage of the ER chaperone BiP at a di-leucine motif (L416-L417) in the hinge region connecting the ATPase and protein-binding domains of the molecule (Paton et al., 2006). As BiP is located in the ER lumen, SubAB added to the culture medium must first bind to receptors on the target cell surface, be internalized and then undergo clathrin-dependent retrograde trafficking via the Golgi to the ER, before it can make contact with its substrate (Chong et al., 2007). Nevertheless, BiP cleavage is rapid, and is evident within 15 min of exposure of Vero cells at high-toxin doses. Furthermore, virtually all of the BiP in the cell is degraded within 1–2 h, even at toxin doses lower than 10 ng ml−1 (Paton et al., 2006). However, effects on cell metabolism as measured by loss of ability of mitochondria to convert MTT [3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] to purple formazan crystals are not seen until 9–12 h following SubAB exposure (J. J. Wolfson and C. M. Thorpe, unpubl. data), and light microscopic evidence of a cytopathic effect on Vero cells does not become apparent for 36–48 h and takes at least 72 h to reach an end point. Accordingly, this study is a first step in a detailed investigation of the downstream consequences of SubAB-mediated BiP cleavage, and the chain of events that eventually, but inevitably, leads to cell death. We have shown unequivocally that BiP cleavage results in activation of all three ER stress-signalling pathways, as evidenced by eIF2α phosphorylation (for PERK), XBP1 mRNA splicing (for IRE1) and by depletion of the intact 90 kDa form of ATF6 and detection of its 50 kDa activated cleavage product. Typically, these three pathways are thought to be activated when accumulation of unfolded proteins in the ER lumen titrates BiP away from the respective membrane-spanning signalling molecule, collectively triggering a series of responses designed to repair the ailing endoplasmic reticulum, or if this cannot be accomplished, to promote apoptosis.
In this study, activation of an ER stress response was rapid, at least for the PERK and IRE1 pathways. Phosphorylation of eIF2α occurred within 30 min, and was associated with attenuation of global protein synthesis, whereas XBP1 mRNA splicing was observed at 60 min (the earliest time point tested). It seems unlikely that significant accumulation of unfolded proteins could have occurred in toxin-treated Vero cells in such a short time frame, and the rapidity of the responses suggests that cleavage of BiP by SubAB may cause it to immediately dissociate from these two signalling molecules, without the need for the accumulation of unfolded proteins in the ER lumen. However, ATF6 activation in response to SubAB treatment appeared to be markedly slower. Interestingly, Shen et al. (2005) have reported that BiP forms a stable interaction with ATF6, and that dissociation requires direct triggering mediated by an ER stress-responsive sequence in the luminal domain of ATF6. Thus, accumulation of unfolded proteins in SubAB-treated cells may be required before the ATF6 pathway is activated.
Further evidence of activation of ER stress-signalling pathways comes from our RT-PCR data showing increased mRNA levels for chaperones including GRP94 and BiP itself; steady increases in the total amount of GRP94 and BiP proteins were also evident by Western blotting. Such increases in chaperone expression are direct consequences of activation of the ATF6 and IRE1 pathways, and are designed to increase the protein-folding capacity of the ER (Szegezdi et al., 2006), although newly translated BiP continued to be degraded by SubAB, presumably in the ER lumen. Upregulation of EDEM mRNA also displayed similar time-course kinetics to BiP and GRP94. EDEM is induced by the IRE1/XBP1 pathway and increases the capacity of the cell for ERAD of terminally misfolded glycoproteins (Eriksson et al., 2004). The combination of induction of ATF4 mRNA and increased eIF2α phosphorylation likely results in translational re-programming, allowing preferential translation of ATF4 mRNA despite global repression of protein synthesis. Increase in levels of this transcriptional activator, along with spliced ATF6, promote the subsequent transcription of the transcription factor CHOP. Increased mRNA levels for CHOP were evident from 2 h, reaching a plateau at 16–30 h while, at the protein level, CHOP could only be detected from 6 to 30 h (having peaked at 16 h). These kinetics of CHOP induction are consistent with regulation at both the transcriptional and translational level. Such CHOP induction is indicative of prolonged ER stress and is likely to exert pro-apoptotic effects (Szegezdi et al., 2006). Indeed, clear evidence of apoptosis in SubAB-treated cells was seen at 30 h, as judged by DNA fragmentation. We also observed a steady increase in GADD34 mRNA in SubAB-treated Vero cells. Like CHOP, the GADD34 promoter has a binding site for ATF4, thus we would expect to see increased GADD34 mRNA after prolonged ER stress. GADD34 provides feedback inhibition of eIF2α phosphorylation, through its interaction with protein phosphatase 1, thereby contributing to restoration of global protein synthesis (Szegezdi et al., 2006). In SubAB-treated cells, eIF2α phosphorylation did in fact diminish and global protein synthesis was restored within 2 h in cells treated with the maximum toxin dose. Restoration of protein synthesis may also occur as a consequence of P58IPK induction via the IRE1/XBP1 pathway, which provides an alternative feedback loop on eIF2α phosphorylation (Szegezdi et al., 2006).
In summary, cleavage of BiP by SubAB into two domains results in activation of an early ER stress response, qualitatively similar to that observed with the UPR-inducing chemical agents, thapsigargin, dithiothreitol and tunicamycin. Although recent work has shown that subtle differences exist between the ER stress response induced by these chemical agents when examined side by side (DuRose et al., 2006), our data support the model in which disruption of BiP interactions with PERK, IRE1 and ATF6 is the key early event that results in downstream signalling. Although some of the expected later ER events occur, such as upregulation of mRNA for ER stress-responsive genes like GRP94, CHOP and BiP, one way in which SubAB-induced ER stress may differ from that observed with other agents is that BiP protein activity cannot be restored, as a result of continued SubAB activity. Thus, later ER stress events that are dependent on recovery of adequate BiP protein levels may be altered in SubAB-induced ER stress.
The exact contribution of SubAB to Stx-induced illness, if any, remains unknown. However, there are a number of different ways that SubAB-induced ER stress may play a role. One possibility is that like other ER stressors, SubAB-induced ER stress may act as a sensitizer to cytokine-mediated apoptosis (Hu et al., 2006). Another possibility is that SubAB and Stx may have synergistic activities on signal transduction pathways within co-intoxicated cells, potentiating inflammation and/or apoptosis. Also, there may be a more intimate interaction between SubAB and Stx, as BiP cleavage by SubAB may affect Stx trafficking (Yu and Haslam, 2005). This study represents the first step in understanding how SubAB-induced BiP cleavage may impact disease caused by STEC that carry this virulence factor.
Experimental procedures
Purification of toxin
SubAB and its non-toxic derivative SubAA272B were purified as described previously (Paton et al., 2004; Talbot et al., 2005).
Cell culture and toxin treatment
Vero (African Green Monkey kidney) cells were routinely grown in DMEM with 5% FCS, 100 U ml−1 penicillin G and 100 μg ml−1 streptomycin sulphate at 37°C. Cells were seeded into appropriately sized tissue culture plates, and confluent monolayers were exposed to either SubAB or SubAA272B at the indicated concentrations. Alternatively, cells were treated with 0.2 μg ml−1 thapsigargin. Where necessary, cells were transfected with plasmid p3×FLAG-CMV-7.1-ATF6 (Shen and Prywes, 2004) using Lipofectamine 2000 reagent (Invitrogen), as recommended by the supplier.
PERK wt and PERK −/− MEFs were the kind gift of Dr David Ron. These cells were grown in DMEM with 10% FCS, penicillin/streptomycin/glutamine, non-essential amino acids and β-mercaptoethanol at 37°C. The ability of SubAB to enter MEF cells was demonstrated by treating cells with SubAB and demonstrating BiP cleavage by Western blot with similar kinetics as that observed in Vero cells. Cells were grown and plated into appropriately sized tissue culture plates. For Western blotting experiments with anti-phospho-eIF2α, media were changed 2 h prior to toxin application. Sub-confluent monolayers were then exposed to SubAB at 1 μg ml−1.
Western blotting
For detection of eIF2α, following treatment and removal of culture media, cell culture plates were transferred to ice. Cells were washed twice with cold phosphate-buffered saline (PBS), then scraped into cold PBS to which 10 μg ml−1 leupeptin, 1 mM PMSF and 0.5 mM dithiothreitol were added. Cells were pelleted by centrifugation at 4°C, the supernatant was removed and pelleted cells were re-suspended in Triton lysis Buffer, consisting of 25 mM Hepes, 300 mM NaCl, 1.5 mM MgCl2, 2 mM EDTA, 0.05% Triton X-100, 0.1 mM Na3VO4, 20 mM β-glycerol phosphate, 10 μg ml−1 leupeptin, 1 mM PMSF and 0.5 mM dithiothreitol, pH 7.5. The re-suspended cells were gently rocked at 4°C for 20 min; cellular debris was then removed by centrifugation at 4°C and the supernatants were frozen at −80°C. The protein concentrations of the cell extracts were determined using Bio-Rad Protein Assay Reagent (Bio-Rad, Hercules, CA) according to manufacturer’s instructions. Alternatively, cells were lysed directly in sample buffer (50 mM Tris-HCl pH 6.8, 50 mM dithiothreitol, 1% SDS, 0.005% bromophenol blue and 10% glycerol) and boiled immediately for 5 min. Cell extracts were separated by SDS-PAGE, electroblotted onto nitrocellulose, blocked and incubated overnight with primary antibodies as indicated in the figure legends. Detection involved using the appropriate HRP-conjugated (KPL) or alkaline phosphatase-conjugated (Bio-Rad Laboratories) secondary antibodies with chemiluminescent substrate (Sigma) or NBT/X-phosphate substrate (Roche Molecular Diagnostics) respectively. The membranes were then stripped and re-probed with loading control antibodies as necessary. Anti-CHOP (#SC-11402), anti-BiP C-20 (#SC-1051) and anti-GRP94 (H-12) (#SC-11402) were obtained from Santa Cruz Biotechnology, USA. Anti-phospho-eIF2α (Ser51) (#9721) and anti-eIF2α (#9722) were from Cell Signalling, Beverly, Massachusetts, USA. Anti-β-actin (AC-74) (#A-5316) and anti-FLAG M2 (#F-3165) were from Sigma. Relative band intensities (normalized to loading controls) were determined using ImageJ 1.38× software.
Protein synthesis assays
Confluent Vero cells in 96-well plates were exposed to SubAB at concentrations ranging from 1 ng ml−1 to 1 μg ml−1, SubAA272B at 1 μg ml−1, or just culture medium alone (control) for 30 min to 4 h, as indicated. Incorporation of [3H]-leucine into TCA-precipitable material during a 60 min pulse was then assayed, as previously described (Thorpe et al., 1999). On every plate, the number of wells used for each toxin treatment ranged from 3 to 5, and the number of control wells ranged from 5 to 18. For each treatment, mean [3H]-leucine incorporation was expressed as a percentage of the mean incorporation for the untreated control cell monolayers (typically 20 000–40 000 d.p.m.). Experiments at each time point were performed three times on different days, and the data were pooled and expressed as mean percentage incorporation relative to the control cells ± SEM. Statistical analysis (two-tailed t-test) was carried out using Prism software.
RNA extraction, RT-PCR and real-time PCR
RNA was extracted from cell cultures using a RNeasy Mini Kit (QIAGEN, Hilden, Germany), as per the manufacturer’s instructions. RNasin RNase inhibitor (Promega, Madison, WI) was then added to the samples. Contaminating DNA was digested with RQ1 RNase-free DNase followed by DNase stop solution, according to the manufacturer’s instructions (Promega). The absence of DNA contamination in all RNA preparations was confirmed by RT-PCR analysis using primers specific for the gene encoding the housekeeping enzyme GAPDH (Table 1). The gene encoding GAPDH contains an intron such that the mRNA template directs amplification of a 239 bp product, whereas the chromosomal DNA template directs amplification of a 341 bp product. RT-PCR was performed using a one-step Access RT-PCR system (Promega) according to the manufacturer’s instructions. Each reaction was performed in a final volume of 20 μl containing 20 nmol of each oligonucleotide. The RT-PCR protocol included the following steps: 45 min of RT at 48°C followed by 2 min denaturation at 94°C and then 30–40 cycles of amplification at 94°C for 30 s, 50–60°C for 30 s and 72°C for 45 s (number of cycles and annealing temperature were optimized for each primer pair). Primer sequences are listed in Table 1. RT-PCR products were analysed by agarose gel electrophoresis, and images were captured using Quantity One software (Bio-Rad) after staining with ethidium bromide.
Table 1.
RT-PCR primers.
| Target | Primer sequences 5′-3′ | Reference |
|---|---|---|
| XBP1 | CTGGAAAGCAAGTGGTAGA
CTGGGTCCTTCTGGGTAG |
Shang (2005) |
| BiP | TTGCTTATGGCCTGGATAAGAGGG
TGTACCCTTGTCTTCAGCTGTCAC |
Shang (2005) |
| EDEM | TCATCCGAGTTCCAGAAAGCAGTC
TTGACATAGAGTGGAGGGTCTCCT |
Shang (2005) |
| ATF4 | AAGCCTAGGTCTCTTAGATGATTACC
CAACCTGGTCGGGTTTTGTTAAAC |
Leung et al. (2003) |
| CHOP | TGAGGAGAGAGTGTTCAAGAAG
TCCAGGAGGTGAAACATAGG |
Chan and Egan (2005) |
| GRP94 | TGAAAAGGCTGTGGTGTCTC
TCGTCTTGCTCTGTGTCTTC |
Park et al. (2004) |
| GAPDH | TCCTTGGAGGCCATGTGGGCCAT
TGATGACATCAAGAAGGTGGTGAAG |
Rogers et al. (2003) |
For quantitative PCR analysis, RNA (DNA-free) was reverse-transcribed into cDNA using random primers (Invitrogen Life Technologies) and Superscript III reverse transcriptase (Invitrogen Life Technologies). cDNA was then purified using the MinElute PCR Purification Kit (Qiagen) according to protocol outlined in the SenseAMP RNA Amplification Kit (Genisphere, http://www.genisphere.com/pdf/SenseAMP_Plus_12-15-06.pdf). Real-time PCR was performed using a different set of gene-specific primers (Table 2) and the Platinum SYBR Green qPCR SuperMix – UDG kit (Invitrogen Life Technologies) in a Rotor-gene RG-2000 cycler (Corbett Research, Mortlake, New South Wales, Australia). Amplification conditions included initial steps at 50°C for 2 min (uracil DNA glycosylase incubation) and 95°C for 2 min, then 35 repetitions of 95°C (15 s) and 60°C (30 s), followed by a melt curve analysis. The resulting cycle threshold values were recorded for each gene of interest (G.O.I.) and normalized with the house-keeping gene, GAPDH. The relative amounts of mRNA in untreated and SubAB-treated Vero cells (at each time point) were compared using the comparative cycle threshold (2ΔΔCt) method (Fig. 6). For each primer set used, a dilution series of cDNA was first established to determine the linear range for the primer pair and to verify that amplification efficiency was near 100%. Primers were designed to span at least one intron and reactions included a control where no reverse transcriptase was included in the cDNA synthesis reaction, to exclude amplification of genomic DNA. Data represent the mean ± SD from two independent experiments, expressed as a fold change in mRNA compared with the untreated control for each time point. * represents P < 0.05, ** represents P < 0.01 and *** represents P < 0.001 as determined by Student’s unpaired two-tailed t-test.
Table 2.
Quantitative real-time PCR primers.
| Target | Primer sequences 5′-3′ | Amplicon size (bp) | Reference |
|---|---|---|---|
| BiP | TGTGCAGCAGGACATCAAGT
AGTTCCAGCGTCTTTGGTTG |
109 | This study |
| EDEM | CTGGGTTGGAAAGCAGAGTG
TCTCCTTCATTGCAGGCTTC |
171 | This study |
| GADD34 | GGAGGAAGAGAATCAAGCCA
TGGGGTCGGAGCCTGAAGAT |
98 | Medigeshi et al. (2007) |
| ATF4 | CCAACAACAGCAAGGAGGAT
GTGTCATCCAACGTGGTCAG |
145 | This study |
| CHOP | AGCTGGAACCTGAGGAGAGA
TGGATCAGTCTGGAAAAGCA |
92 | Medigeshi et al. (2007) |
| GRP94 | GGCCAGTTTGGTGTCGGTTT
CGTTCCCCGTCCTAGAGTGTT |
159 | Wang et al. (2005) |
| GAPDH | TCCTTGGAGGCCATGTGGGCCAT
TGATGACATCAAGAAGGTGGTGAAG |
239 | Rogers et al. (2003) |
DNA fragmentation analysis
Post-treatment, Vero cells were washed with PBS and DNA was prepared using the Apoptotic DNA Ladder kit (Roche, Mannheim, Germany). Isolated DNA was treated with 100 μg ml−1 DNase-free RNase A (Roche) for 30 min and then equal amounts of DNA (2 μg) were loaded onto 1.5% agarose gels. Following electrophoresis, gels were stained with GelRed (Biotium, Hayward, CA) and photographed with the Gel Doc XR documentation system (Bio-Rad, Hercules, CA) using Quantity One software (Bio-Rad, Hercules, CA).
Acknowledgments
We are grateful to Ron Prywes for providing plasmid p3×FLAG-CMV-7.1-ATF6 and David Ron for providing PERK −/− and PERK wt fibroblasts. This research was supported by Program Grant 284214 from the National Health and Medical Research Council (NHMRC) of Australia (A.W.P. and J.C.P.), and the following Grants from the National Institutes of Health, USA: R01AI-068715 (A.W.P. and J.C.P.), R01AI-59509 (C.M.T.), T32AI-007329 (J.J.W. and D.M.J.) and P30DK-34928 (Center for Gastroenterology Research on Absorptive and Secretory Processes). J.C.P. is a NHMRC Australia Fellow.
References
- Boyce M, Yuan J. Cellular response to endoplasmic reticulum stress: a matter of life or death. Cell Death Differ. 2006;13:363–373. doi: 10.1038/sj.cdd.4401817. [DOI] [PubMed] [Google Scholar]
- Chan SW, Egan PA. Hepatitis C virus envelope proteins regulate CHOP via induction of the unfolded protein response. FASEB J. 2005;19:1510–1512. doi: 10.1096/fj.04-3455fje. [DOI] [PubMed] [Google Scholar]
- Chong DC, Paton JC, Thorpe CM, Paton AW. Clathrin-dependent trafficking of subtilase cytotoxin, a novel AB5 toxin that targets the ER chaperone BiP. Cell Microbiol. 2007;10:795–806. doi: 10.1111/j.1462-5822.2007.01085.x. [DOI] [PubMed] [Google Scholar]
- DuRose JB, Tam AB, Niwa M. Intrinsic capacities of molecular sensors of the unfolded protein response to sense alternate forms of endoplasmic reticulum stress. Mol Biol Cell. 2006;17:3095–3107. doi: 10.1091/mbc.E06-01-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson KK, Vago R, Calanca V, Galli C, Paganetti P, Molinari M. EDEM contributes to maintenance of protein folding efficiency and secretory capacity. J Biol Chem. 2004;279:44 600–44 605. doi: 10.1074/jbc.M407972200. [DOI] [PubMed] [Google Scholar]
- Gething MJ. Role and regulation of the ER chaperone BiP. Semin Cell Dev Biol. 1999;10:465–472. doi: 10.1006/scdb.1999.0318. [DOI] [PubMed] [Google Scholar]
- Hendershot LM. The ER function BiP is a master regulator of ER function. Mt Sinai J Med. 2004;71:289–297. [PubMed] [Google Scholar]
- Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol Cell Biol. 2006;26:3071–3084. doi: 10.1128/MCB.26.8.3071-3084.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim PS, Arvan P. Endocrinopathies in the family of endoplasmic reticulum (ER) storage diseases: disorders of protein trafficking and the role of ER molecular chaperones. Endocr Rev. 1998;19:173–202. doi: 10.1210/edrv.19.2.0327. [DOI] [PubMed] [Google Scholar]
- Leung YF, Tam PO, Lee WS, Lam DS, Yam HF, Fan BJ, et al. The dual role of dexamethasone on anti-inflammation and outflow resistance demonstrated in cultured human trabecular meshwork cells. Mol Vis. 2003;9:425–439. [PubMed] [Google Scholar]
- Medigeshi GR, Lancaster AM, Hirsch AJ, Briese T, Lipkin WI, DeFilippis V, et al. West Nile virus infection activates the unfolded protein 1 response leading to CHOP induction and apoptosis. J Virol. 2007;81:10849–10860. doi: 10.1128/JVI.01151-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HR, Tomida A, Sato S, Tsukumo Y, Yun J, Yamori T, et al. Effect on tumor cells of blocking survival response to glucose deprivation. J Natl Cancer Inst. 2004;96:1300–1310. doi: 10.1093/jnci/djh243. [DOI] [PubMed] [Google Scholar]
- Paton AW, Beddoe T, Thorpe CM, Whisstock JC, Wilce MC, Rossjohn J, et al. AB5 subtilase cytotoxin inactivates the endoplasmic reticulum chaperone BiP. Nature. 2006;443:548–552. doi: 10.1038/nature05124. [DOI] [PubMed] [Google Scholar]
- Paton JC, Paton AW. Pathogenesis and diagnosis of Shiga toxin-producing Escherichia coli infections. Clin Microbiol Rev. 1998;11:450–479. doi: 10.1128/cmr.11.3.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paton AW, Srimanote P, Talbot UM, Wang H, Paton JC. A new family of potent AB5 cytotoxins produced by Shiga toxigenic Escherichia coli. J Exp Med. 2004;200:35–46. doi: 10.1084/jem.20040392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao RV, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004;11:372–380. doi: 10.1038/sj.cdd.4401378. [DOI] [PubMed] [Google Scholar]
- Rogers TJ, Paton AW, McColl SR, Paton JC. Enhanced CXC chemokine responses of human colonic epithelial cells to locus of enterocyte effacement-negative Shiga toxigenic Escherichia coli. Infect Immun. 2003;71:5623–5632. doi: 10.1128/IAI.71.10.5623-5632.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J. Quantitative measurement of events in the mammalian unfolded protein response. Methods. 2005;35:390–394. doi: 10.1016/j.ymeth.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Shen J, Prywes R. Dependence of site-2 protease cleavage of ATF6 on prior site-1 protease digestion is determined by the size of the luminal domain of ATF6. J Biol Chem. 2004;279:43046–43051. doi: 10.1074/jbc.M408466200. [DOI] [PubMed] [Google Scholar]
- Shen J, Snapp EL, Lippincott-Schwartz J, Prywes R. Stable binding of ATF6 to BiP in the endoplasmic reticulum stress response. Mol Cell Biol. 2005;25:921–932. doi: 10.1128/MCB.25.3.921-932.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siezen RJ, Leunissen JA. Subtilases: the superfamily of subtilisin-like serine proteases. Protein Sci. 1997;6:501–523. doi: 10.1002/pro.5560060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot UM, Paton JC, Paton AW. Protective immunization of mice with an active-site mutant of subtilase cytotoxin of Shiga toxin-producing Escherichia coli. Infect Immun. 2005;73:4432–4436. doi: 10.1128/IAI.73.7.4432-4436.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorpe CM, Hurley BP, Lincicome LL, Jacewicz MS, Keusch GT, Acheson DW. Shiga toxins stimulate secretion of interleukin-8 from intestinal epithelial cells. Infect Immun. 1999;67:5985–5993. doi: 10.1128/iai.67.11.5985-5993.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, He Z, Zhang J, Wang Y, Wang T, Tong S, et al. Overexpression of endoplasmic reticulum molecular chaperone GRP94 and GRP78 in human lung cancer tissues and its significance. Cancer Detection Prevention. 2005;29:544–551. doi: 10.1016/j.cdp.2005.09.010. [DOI] [PubMed] [Google Scholar]
- Wang H, Paton JC, Paton AW. Pathological changes in mice induced by subtilase cytotoxin, an emerging Escherichia coli AB5 toxin that targets the endoplasmic reticulum. J Infect Dis. 2007;196:1093–1101. doi: 10.1086/521364. [DOI] [PubMed] [Google Scholar]
- Yu M, Haslam DB. Shiga toxin is transported from the endoplasmic reticulum following interaction with the luminal chaperone HEDJ/ERdj3. Infect Immun. 2005;73:2524–2532. doi: 10.1128/IAI.73.4.2524-2532.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]