

Abstract
Background
Many inhaled anesthetics inhibit voltage-gated sodium channels at clinically relevant concentrations, and suppression of neurotransmitter release by these agents results, at least partly, from decreased presynaptic sodium channel activity. Volatile aromatic anesthetics can inhibit N-methyl-D-aspartate (NMDA) receptor function and enhance γ-amino butyric acid A (GABAA) receptor function, but these effects depend strongly on the chemical properties of the aromatic ompounds. The present study tested whether diverse aromatic anesthetics consistently inhibit sodium channel function.
Methods
We studied the effect of eight aromatic anesthetics on Nav1.2 sodium channels with β1 subunits, using whole-cell, two-electrode voltage-clamp techniques in Xenopus oocytes.
Results
All aromatic anesthetics inhibited INa (sodium currents) at a holding potential which produce half-maximal current (V1/2) (partial depolarization); inhibition was modest with 1,3,5-trifluorobenzene (8 ± 2%), pentafluorobenzene (13 ± 2%), and hexafluorobenzene (13 ± 2%), but greater with benzene (37 ± 2%), fluorobenzene (39 ± 2%), 1,2-difluorobenzene (48 ± 2%), 1,4-difluorobenzene (31 ± 3%), and 1,2,4-trifluorobenzene (33 ± 1%). Such dichotomous effects were noted by others for NMDA and GABAA receptors. Parallel, but much smaller inhibition, was found for INa at a holding potential which produced near maximal current (−90 mV) (VH-90), and hexafluorobenzene caused small (6 ± 1%) potentiation of this current. These changes in sodium channel function were correlated with effectiveness for inhibiting NMDA receptors, with lipid solubility of the compounds, with molecular volume, and with cation-π interactions.
Conclusion
Aromatic compounds vary in their actions on the kinetics of sodium channel gating and this may underlie their variable inhibition. The range of inhibition produced by MAC concentrations of inhaled anesthetics indicates that sodium channel inhibition may underlie the action of some of these anesthetics but not others.
Introduction
Voltage-gated sodium channels have an important role in the action potential initiation and propagation in excitable cells of nerve and muscle. They are established targets of local anesthetics, but have been considered to be resistant to general anesthetics.1 However, recent reports suggest that clinical concentrations of volatile anesthetics significantly suppress sodium channel function in neurons,2,3 and in recombinant mammalian sodium channels.4–6 Moreover, blocking of presynaptic sodium channels provides a mechanism for suppression of neurotransmitter release by volatile anesthetics.7–10
N-methyl-D-aspartate (NMDA) and γ-aminobutyric acid A (GABAA) receptors are targets for general anesthetics.11–14 Although not used as clinical anesthetics, volatile aromatic compounds have industrial applications, are drugs of abuse, and have anesthetic effects.15 They inhibit NMDA receptor function,16 and potentiate GABAA receptor function,17 doing so in a reciprocal manner: the aromatic anesthetics which inhibit NMDA receptors best enhance GABAA receptor function the least. The importance of aromatic structure to an effect on sodium channels is unknown and such knowledge might be important to an understanding of potential targets and mechanisms of volatile anesthetics.
Nine distinct pore-forming α subunits of sodium channels have been identified.18,19 Each α subunit has a different pattern of development and localization and demonstrates subtle differences in electrophysiological characteristics.20 We examined the effect of several structurally distinct volatile aromatics on Nav1.2, a sodium channel expressed primarily in the central nervous system, including the spinal cord, a major site of inhaled anesthetic action.21 We used the oocyte expression system to explore the mechanism of effect by volatile anesthetics on Nav1.2.
Methods
The Animal Care and Use Committees of the University of Texas approved this study.
Adult female Xenopus laevis frogs were obtained from Xenopus Express, Inc. (Plant City, FL). All aromatic anesthetics were purchased from Sigma-Aldrich (St. Louis, MO). We thank the following researchers for providing the subunit clones: cDNA for rat Nav1.2 αisubunit (Dr. W. A. Catterall, University of Washington, Seatle, WA), cDNA for human β1isubunit (Dr. A.L. George, Vanderbilt University).
Linearized cDNA templates with ClaI (Nav1.2 αisubunit), EcoRI (β1isubunit) were transcribed using T7 (Nav1.2 αisubunit), T6 (β1isubunit) RNApolymerase of mMESSAGE mMACHINE kit (Ambion, Ausin, TX). Preparation of Xenopus laevis oocytes and microinjection of the cRNA were performed as described previously.5 Stage IV–VI oocytes were manually isolated from an ovary. Oocytes were treated with collagenase (0.5 mg/ml) for 10 min and Nav1.2 αisubunit cRNAwas co-injected with the βjisubunit cRNAat a ratio of 1:10 (total volume was 20 ~ 40 ng/50 nl). Injected oocytes were incubated in modified Barth’s solution (88 mM NaCl, 1 mM KCl, 2.4 mM NaHCO3, 10 mM HEPES, 0.82 mM MgSO4, 0.33 mM Ca(NO3)2, 0.91 mM CaCl2, adjusted to pH 7.5) supplemented with 10,000 units of penicillin, 50 mg of gentamicin, 90 mg of theophylline, and 220 mg of sodium pyruvate per liter (incubation medium) at 19 °C.
Two to six days after injection, electrophysiological recordings were performed at room temperature (23 °C) using the whole-cell, two-electrode, voltage-clamp technique. Oocytes were placed in a 100 µl recording chamber and impaled with recording electrodes prepared with borosilcate glass using a puller (P-97; Sutter Instruments Company, Novato, CA) and filled with 3M KCl / 0.5% low melting point agarose and with resistances between 0.3 and 0.5 MΩ. The oocytes were perfused at 2 ml/min with Frog Ringer solution containing NaCl 115 mM, KCl 2.5 mM, HEPES 10 mM, CaCl2 1.8 mM, pH 7.2 using a peristaltic pump (Cole-Palmer Instrument Co., Chicago, IL). The whole-cell voltage clamp was achieved through these two electrodes using a Warner Instruments model OC-725C (Hamden, CT). Currents were recorded and analyzed using pCLAMP software (Axon Instruments, Foster City, CA). The sodium current amplitudes were typically 2 – 15 µA. Transients and leak currents were subtracted using the P/N procedure.
The voltage dependence of activation was determined by 50-ms depolarizing pulses from a holding potential of −90 mV (VH-90) or from a holding potential causing half-maximal current (V1/2) (approximately from −50 mV to −60 mV) to 60 mV in 10-mV increments. V1/2 value was determined individually for each oocyte in each experiment. Normalized activation data were fitted to a Boltzmann equation: G/Gmax = 1/(1 + exp(V1/2 − V)/k), where G is the voltage-dependent sodium conductance, Gmax is the maximal sodium conductance, G/Gmax is the normalized fractional conductance, V1/2 is the potential at which activation is half maximal, k is the slope factor. G values for each oocyte were calculated using: G = I/(Vt − Vr), where I is the peak sodium current, Vt is the test potential, and Vr is the reversal potential. Vr for each oocyte was estimated by extrapolating the linear ascending segment of the current voltage relationship (I−V) curve to the voltage axis. To measure steady-state inactivation, currents were elicited by a 50-ms test pulse to −20 mV after 200-ms prepulses ranging from −140 mV to 0 mV in 10-mV increments from a holding potential of −90 mV. Steady-state inactivation curves were fitted to a Boltzmann equation: I/Imax = 1/(1 + exp(V1/2 − V)/k), where Imax is the maximal sodium current, I/Imax is the normalized current, V1/2 is the voltage of half-maximal inactivation, and k is the slope factor.
The aromatic anesthetics were examined at the concentrations that are equivalent to 1MAC or 2MAC in rats.16,22 (MAC is the minimum anesthetic concentration needed to produce immobility in response to noxious stimulation in 50% of rats; it is equivalent to the EC50 for producing surgical immobility). All aromatic anesthetics were directly dissolved in the Frog Ringer, and were infused for 3 min to produce equilibrium. The loss of concentration from vial to bath was approximately 60–80% for all aromatic anesthetics, as determined by gas chromatography. Bath/vial concentrations of the four compounds that were tested were 1.1/2.5 mM (benzene), 0.40/1.4 mM (1,3,5-trifluorobenzene), 0.65/2.1 mM (pentafluorobenzene), and 0.12/0.58 mM (hexafluorobenzene).
Data Analysis
All values are presented as the mean ± SEM. The n values refer to the number of oocytes studied. Each experiment was performed with oocytes from at least two frogs. Statistical analyses were performed using a one-way analysis of variance (ANOVA) for multiple comparisons and a t test using GraphPad Prism software (GraphPad Software Inc., San Diego, CA).
Results
Effects of aromatic anesthetics on the peak Na+ inward currents elicited from two different holding potentials
All eight aromatic anesthetics suppressed INa at the V1/2 holding potential, and the magnitudes of these effects varied widely (Fig. 1B,D, Fig. 2). At V1/2, these anesthetics reduced INa by 37 ± 2% (benzene), 39 ± 2% (fluorobenzene), 48 ± 2% (1,2-difluorobenzene), 31 ± 3% (1,4-difluorobenzene), 33 ± 1% (1,2,4-trifluorobenzene), 8 ± 2% (1,3,5-trifluorobenzene), 13 ± 2% (pentafluorobenzene), and 13 ± 2% (hexafluorobenzene) (Fig. 2). Effects on INa at VH-90 were smaller than at V1/2 holding potential, and the magnitudes of the effects varied widely, with inhibitions of 8 ± 1% (benzene), 10 ± 1% (fluorobenzene), 15 ± 2% (1,2-difluorobenzene), 6 ± 0.5% (1,4-difluorobenzene), and 3 ± 0.7% (1,2,4-trifluorobenzene) (Fig. 1A, Fig. 2). 1,3,5-trifluorobenzene did not affect these currents. In contrast, pentafluorobenzene and hexafluorobenzene potentiated INa at VH-90 by 2 ± 1%, 6 ± 1%, respectively (Fig. 1C, Fig. 2). Thus, there are two effects of aromatic anesthetics on sodium channels: All produced inhibitory effects at V1/2 holding potential, albeit by widely varying amounts. At VH-90, benzene, fluorobenzene, 1,2-difluorobenzene, 1,4-difluorobenzene and 1,2,4-trifluorobenzene produced inhibition but pentafluorobenzene and hexafluorobenzene caused slight potentiation.
Figure 1.

Effects of benzene and hexafluorobenzene on sodium currents at different holding potentials in oocytes expressing Nav1.2 with β1. (A) The effect of benzene at −90 mV (VH-90) holding potential. Left panel; traces of sodium currents evoked by a 50-ms depolarizing pulse to −20 mV from a holding potential of VH-90 in the absence and presence of benzene. Right panel; time course figure of benzene effect at VH-90 holding potential on sodium currents. Currents were elicited by a 50-ms depolarizing pulses to −20 mV applied every 10 s from −90 mV. Normalized current is shown. Filled circles represent control and washout, open circles represent currents during 3 min benzene treatment. (B) The effect of benzene at a holding potential which induced half maximal current (V1/2). Left panel; traces of sodium currents evoked by a 50-ms depolarizing pulse to −20 mV from V1/2 holding potentials in the absence and presence of benzene. Right panel; time course of benzene action at V1/2 holding potential. Currents were elicited by a 50-ms depolarizing pulses to −20 mV applied every 10 s from V1/2 holding potential. (C) Effect of hexafluorobenzene at VH-90 holding potential. (D) Effect of hexafluorobenzene at V1/2 holding potential. Concentrations of anesthetics were equivalent to 1 MAC.
Figure 2.
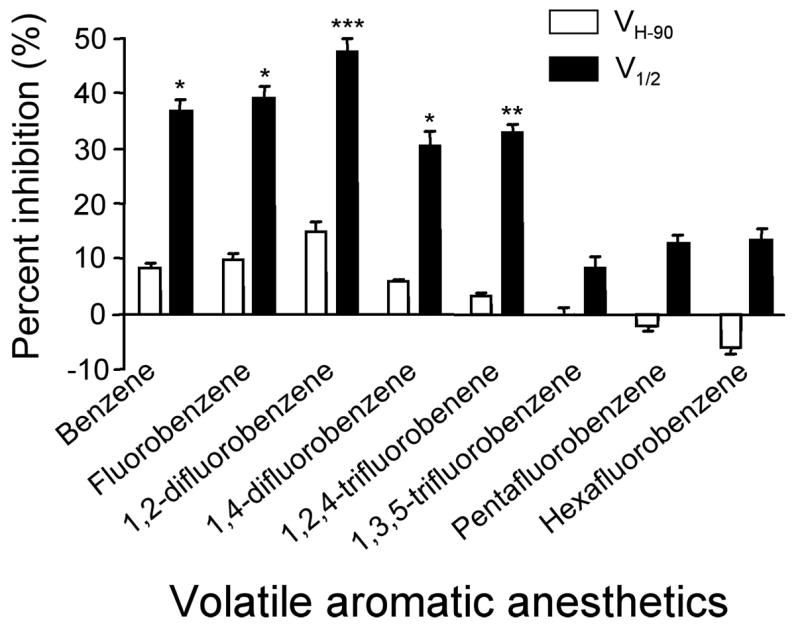
Effects of aromatic anesthetics on sodium currents at different holding potentials. Percent inhibitions were calculated from time course figure. Open columns represent the effect at VH-90 holding potential. Closed columns indicate the effect at V1/2 holding potential. Concentrations of anesthetics were equivalent to 1 MAC. V1/2 value was −53.5 ± 0.4 mV. Data were obtained from 6 oocytes and are mean ± S.E.M. (n = 6). *, p < 0.05; **, p < 0.01; ***, p < 0.001 (one-way ANOVA).
Effects of aromatic anesthetics on activation of sodium currents
Because of the different effects of aromatic anesthetics noted above, we examined actions of these two types compounds on gating of sodium channels. We selected benzene and 1,2-difluorobenzene as compounds with potent inhibitory effects at V1/2 holding potential plus smaller inhibitory effects at VH-90, and hexafluorobenzene as the compound with a much smaller inhibitory effect at V1/2 holding potential and slight potentiating effect at VH-90. The voltage dependence of activation was determined by 50-ms depolarizing pulses from a holding potential of −90 mV to 60 mV in 10-mV increments or from a holding potential of V1/2 to 60 mV in 10-mV increments. The activation curves were derived from the I−V curve (see Methods). Benzene and 1,2-difluorobenzene (2 MAC) reduced the peak INa at VH-90 and V1/2 (Fig. 3A, B). However, hexafluorobenzene (2 MAC) potentiated the peak INa at VH-90, especially in the depolarizing region where channel opening begins (around −40 ~ −30 mV) and suppressed the sodium currents at V1/2 more weakly than benzene or 1,2-difluorobenzene (Fig. 3C). Benzene and 1,2-difluorobenzene did not affect voltage dependence of activation at VH-90 or V1/2 (Fig. 4A, B, Table 1), but hexafluorobenzene shifted the midpoint of steady-state activation, V1/2 in hyperpolarizing direction by 4.9 and 2.2 mV at VH-90 and V1/2 (Fig. 4C, Table 1).
Figure 3.
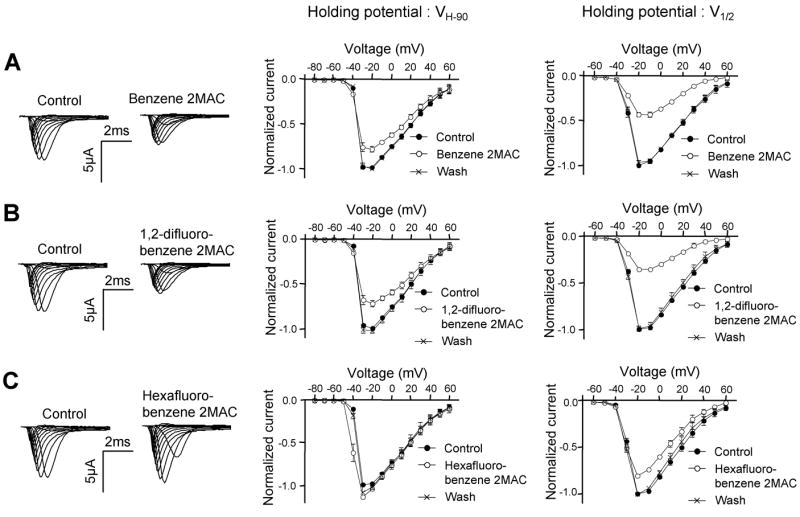
Effects of benzene (A), 1,2-difluorobenzene (B), and hexafluorobenzene (C) on I−V relations of sodium currents at VH-90 and V1/2 holding potentials in oocytes expressing Nav1.2 with β1 subunits. Left panel; representative INa traces in absence and presence of aromatic anesthetics at a VH-90 holding potential. Currents were elicited by 50-ms depolarizing steps between −80 and 60 mV in 10-mV increments from holding potentials of −90mV. Center panel; the effects of aromatic anesthetics on I−V curves elicited from a VH-90 holding potential (closed circles, control; open circles, aromatic anesthetics; cross, washout). The peak currents were normalized to the maximal currents which were observed at −20 or −30 mV. Right panel; the effects of aromatic anesthetics on I−V curves elicited from a V1/2 holding potential. Currents were elicited by 50-ms depolarizing steps between −60 and 60 mV in 10-mV increments from a V1/2 holding potential. Maximal currents were observed at −20 mV. Concentrations of anesthetics were equivalent to 2 MAC. V1/2 value was −53.3 ± 0.4 mV. Data were obtained from 5 oocytes and shown as mean ± S.E.M. (n = 5).
Figure 4.

Effects of benzene (A), 1,2-difluorobenzene (B), and hexafluorobenzene (C) on channel activation in oocytes expressing Nav1.2 with β1 from a VH-90 holding potential (upper) or V1/2 holding potential (down). Closed circles represent control, open circles indicate the effect of aromatic anesthetics, and cross indicate washout. Data shown as mean ± S.E.M. (n = 5). Activation curves were fitted to a Boltzmann equation and V1/2 are shown in Table 1. Concentrations of anesthetics were equivalent to 2 MAC.
Table 1.
Effects of Benzene, 1,2-fluorobenzene, and Hexafluorobenzene on Activation and Inactivation in Oocytes Expressing Nav1.2 with β1
| Activation | Holding VH-90 |
Holding V1/2 |
||||
|---|---|---|---|---|---|---|
| Control | Anesthetics | Shift | Control | Anesthetics | Shift | |
| V1/2(mV) | ||||||
|
| ||||||
| Benzene | −34.3 ± 0.5 | −34.6 ± 0.4 | −0.3 | −27.3 ± 0.5 | −26.9 ± 0.4 | +0.4 |
| 1,2-difluorobenzene | −33.5 ± 0.3 | −33.9 ± 0.6 | −0.4 | −27.0 ± 0.3 | −27.3 ± 0.6 | −0.3 |
| Hexafluorobenzene | −34.4 ± 0.4 | −39.3 ± 0.9** | −4.9 | −27.4 ± 0.5 | −29.6 ± 0.2** | −2.2 |
|
| ||||||
| Inactivation | Control | Anesthetics | Shift | |||
|
| ||||||
| V1/2(mV) | ||||||
|
| ||||||
| Benzene | −55.1 ± 0.4 | −62.5 ± 0.5*** | −7.4 | |||
| 1,2-trifluorobenzene | −55.8 ± 0.3 | −64.7 ± 0.7*** | −8.9 | |||
| Hexafluorobenzene | −56.6 ± 0.4 | −61.2 ± 0.4*** | −4.6 | |||
, p < 0.01;
p < 0.001, compared with control by one-way analysis of variance (mean ± S.E.M.; n = 5–7).
Effects of aromatic anesthetics on inactivation of sodium currents
The effects of benzene, 1,2-difluorobenzene and hexafluorobenzene on steady-state inactivation were also investigated. Benzene and 1,2-difluorobenzene shifted V1/2 in the hyperpolarizing direction significantly by 7.4 and 8.9 mV, respectively and also reduced sodium currents in the hyperpolarizing potential range (Fig. 5, Table 1). Hexafluorobenzene also significantly shifted V1/2 in the hyperpolarizing direction by 4.6 mV although the shift was smaller than that by benzene and 1,2-difluorobenzene, whereas hexafluorobenzene potentiated sodium current in the hyperpolarizing potential range slightly (Fig. 5, Table 1). The effects of benzene, 1,2-difluorobenzene and hexafluorobenzene in the hyperpolarizing range were consistent with the effects of these compounds on INa at VH-90, and the effects on these compounds on activation curve at VH-90.
Figure 5.

Effects of benzene (A), 1,2-difluorobenzene (B), and hexafluorobenzene (C) on inactivation in oocytes expressing Nav1.2 with β1. Left panel; representative INa traces in absence and presence of aromatic anesthetics. Currents were elicited by a 50-ms test pulse to −20 mV after 200-ms prepulses ranging from −140 to 0 mV in 10-mV increments from a holding potential of VH-90. Right panel; the effects of aromatic anesthetics on inactivation curves (closed circles, control; open circles, aromatic anesthetics; cross, washout). Data shown as mean ± S.E.M. (n = 7). Inactivation curves were fitted to a Boltzmann equation and the V1/2 values are shown in Table 1. Concentrations of anesthetics were equivalent to 2 MAC.
Discussion
We found that structurally distinct aromatic anesthetics differently affected INa at VH-90 and V1/2 holding potentials. At V1/2 holding potential, 1 MAC benzene, fluorobenzene, 1,2-difluorobenzene, 1,4-difluorobenzene, 1,2,4-trifluorobenzene suppressed INa by 30–50%. These inhibitory effects parallel those of other inhaled anesthetics. For example, clinically relevant concentrations (0.25–0.4 mM) of isoflurane suppress INa by 30–40% in several preparations: Chinese hamster ovary cells expressing Nav1.2,4 isolated rat neurohypophysial nerve terminals,3 and an oocyte system expressing Nav1.2, Nav1.4, or Nav1.6.5
Solt et al. separated these aromatic anesthetics into two groups based on the inhibition of NMDA receptors produced by 1 MAC.16 Less fluorinated compounds with the limited fluorination – benzene, fluorobenzene and 1,2-difluorobenzene –inhibited strongly (Group 1), and compounds with greater fluorination –1,4-difluorobenzene, 1,2,4-trifluorobenzene, 1,3,5-trifluorobenzene, pentafluorobenzene, hexafluorobenzene – inhibited weakly (Group 2). Solt et al. also studied the effect of these compounds on GABAA receptors,17 and found that aromatic anesthetics which inhibit NMDA receptor currents most (Group 1) potentiate GABAA receptor currents least, and anesthetics that inhibit NMDA receptor currents least (Group 2) potentiate GABAA receptor currents most. These findings suggest that distinct molecular properties determine the action of anesthetics on NMDA and GABAA receptors.
We found that benzene, fluorobenzene, and 1,2-difluorobenzene (Group 1 compounds), strongly inhibited sodium currents at V1/2, whereas 1,3,5-trifluorobenzene, pentafluorobenezene, and hexafluorobenzene (Group 2 compounds), inhibited weakly. But unlike the NMDA results, we found that 1,4-difluorobenzene and 1,2,4-trifluorobenzene (Group 2 compounds) inhibited sodium currents (Fig. 6A). Our previous study of isoflurane5 found similar sodium channel inhibition as for 1,4-difluorobenzene and 1,2,4-trifluorobenzene, and these three compounds are effective on both sodium channels and GABAA receptors (Fig. 6B).
Figure 6.

(A) Relationship between inhibitory potencies on sodium channels at VH-90 holding potential (open circle) or V1/2 holding potential (closed circle) and that on NMDA receptors of aromatic anesthetics at 1MAC. (B) Relationship between inhibitory potencies on sodium channels at VH-90 holding potential (open circle) or V1/2 holding potential (closed circle) and potencies of enhancement of aromatic anesthetics on GABAA receptors. NMDA receptor inhibition are from Solt et al16, and GABAA receptor potentiation are from Kelly et al.17 Data shown as mean ± S.E.M. (n = 6). Correlation between inhibitory potencies on sodium channels of aromatic anesthetics at VH-90 holding potential (open circle) or V1/2 holding potential (closed circle) and their abilities to oil/gas partition coefficient (C), molecular volumes (D), or cation-π interactions (E). The lines were derived from linear regression analysis. Oil/gas partition coefficients of aromatic anesthetics are from Fang et al,22 molecular volume and cation-π binding energy of aromatic anesthetics are from Raines et al.,23 In C, r2 = 0.88 and p = 0.0005 (V1/2), r2 = 0.87 and p = 0.0006 (VH-90); in D, r2 = 0.55 and p = 0.0353 (V1/2), r2 = 0.68 and p = 0.0113 (VH-90); in E, r2 = 0.52 and p = 0.0445 (V1/2), r2 = 0.67 and p = 0.0135 (VH-90).
Raines et al. showed that cation-π interactions predict the actions of aromatic anesthetics on NMDA receptors.23 In order to explore the physical properties important for actions of these compounds on sodium channels, we correlated drug effects with several parameters (Fig. 6C,D, and E). Sodium channel inhibition was correlated with cation-π interactions, but this relationship was weaker than seen for cation-π interactions and NMDA receptor inhibition by Raines et al. 23 Inhibition of sodium channels was strongly correlated with oil/gas partition coefficients. These results suggest that aromatic anesthetics interact with different channels by distinct mechanisms. We recently reported that n-alcohols inhibit sodium channel function and their potency increase with carbon number up to a cut-off at nonanol,24 suggesting that hydrophobic and steric interactions determine the potencies of n-alcohols. Hydrophobic and steric interactions might also be important for sodium channel inhibition by aromatic anesthetics, with a smaller contribution from cation-π interactions. Some fluorinated compounds are either not as potent anesthetics as would be predicted from their lipid solubility or do not produce anesthesia (nonimmobilizers).25 The nonimmobilizing fluorinated cyclobutanes do not inhibit sodium channels,26 indicating that factors other than lipid solubility are also important for channel inhibition by some compounds.
Analysis of gating parameters revealed differences in the actions of benzene, 1,2-fluorobenzene and hexafluorobenzene. Other volatile anesthetics generally enhance inactivation with no effect on activation.5,6 We also found this to be true for benzene and 1,2-difluorobenzene. However, hexafluorobenzene enhanced peak INa at VH-90, shifted activation in the hyperpolarizing direction and increased sodium currents in the hyperpolarizing range of inactivation. These changes indicate that hexafluorobenzene shifts channel gating equilibrium toward the open channel state. No other anesthetics have been reported to have an activating effect on sodium channels. This action of hexafluorobenzene on channel gating might offset the effects on the inactivated state and explain the small inhibition at V1/2 holding potential relative to the other compounds. Pentafluorobenzene, another Group 2 compound, may shift GABAA receptor gating toward the open channel state because pentafluorobenzene decreases the GABA EC50 and activates GABAA receptors directly.17 This observation suggests some commonalities in the action of aromatic compounds on sodium channels and GABAA receptors.
In conclusion, we find that volatile aromatic anesthetics inhibit Nav1.2 sodium channel function but the magnitude of the inhibition varies widely among the compounds. Differences in the actions of these compounds on sodium channel gating are likely responsible for the range of inhibitory effects. Although numerous inhaled anesthetics and n-alcohols inhibit sodium channel function, some inhibit weakly and others strongly (Group 1 versus Group 2) at concentrations corresponding to MAC. We applied aromatic compounds in the present study because they reciprocally affect GABAA and NMDA receptors; we tested whether the sodium channel might provide more consistent responses. However, neither the aromatics nor clinical agents (e.g., halothane, isoflurane) provide consistent inhibition of sodium channels. This indicates that sodium channel inhibition may be important for some anesthetics but not others as a mediator of the capacity of inhaled anesthetics to produce immobility in the face of noxious stimulation.
Acknowledgments
We thank Dr. Michael Laster for determining the concentrations of anesthetics used in this study.
Supported by National Institutes of Health Grants GM47818 and AA06399 (to R.A.H.).
Footnotes
Dr. Eger is a paid consultant to Baxter Healthcare Corp.
Implications Statement
Aromatic anesthetics can block NMDA receptors and enhance GABAA receptors responses. The present report finds various effects of these compounds on sodium channels. Although volatile aromatic anesthetics are not used clinically, exploring their effects on ion channels may aid in understanding mechanisms of clinical inhaled anesthetic actions.
References
- 1.Franks NP, Lieb WR. Molecular and cellular mechanisms of general anaesthesia. Nature. 1994;367:607–14. doi: 10.1038/367607a0. [DOI] [PubMed] [Google Scholar]
- 2.Ratnakumari L, Hemmings HC., Jr Inhibition of presynaptic sodium channels by halothane. Anesthesiology. 1998;88:1043–54. doi: 10.1097/00000542-199804000-00025. [DOI] [PubMed] [Google Scholar]
- 3.Ouyang W, Wang G, Hemmings HC., Jr Isoflurane and propofol inhibit voltage-gated sodium channels in isolated rat neurohypophysial nerve terminals. Mol Pharmacol. 2003;64:373–81. doi: 10.1124/mol.64.2.373. [DOI] [PubMed] [Google Scholar]
- 4.Rehberg B, Xiao YH, Duch DS. Central nervous system sodium channels are significantly suppressed at clinical concentrations of volatile anesthetics. Anesthesiology. 1996;84:1223–33. doi: 10.1097/00000542-199605000-00025. [DOI] [PubMed] [Google Scholar]
- 5.Shiraishi M, Harris RA. Effects of alcohols and anesthetics on recombinant voltage-gated Na+ channels. J Pharmacol Exp Ther. 2004;309:987–94. doi: 10.1124/jpet.103.064063. [DOI] [PubMed] [Google Scholar]
- 6.Ouyang W, Hemmings HC., Jr Isoform-selective effects of isoflurane on voltage-gated Na+ channels. Anesthesiology. 2007;107:91–8. doi: 10.1097/01.anes.0000268390.28362.4a. [DOI] [PubMed] [Google Scholar]
- 7.Schlame M, Hemmings HC., Jr Inhibition by volatile anesthetics of endogenous glutamate release from synaptosomes by a presynaptic mechanism. Anesthesiology. 1995;82:1406–16. doi: 10.1097/00000542-199506000-00012. [DOI] [PubMed] [Google Scholar]
- 8.Lingamaneni R, Birch ML, Hemmings HC., Jr Widespread inhibition of sodium channel-dependent glutamate release from isolated nerve terminals by isoflurane and propofol. Anesthesiology. 2001;95:1460–6. doi: 10.1097/00000542-200112000-00027. [DOI] [PubMed] [Google Scholar]
- 9.Westphalen RI, Hemmings HC., Jr Selective depression by general anesthetics of glutamate versus GABA release from isolated cortical nerve terminals. J Pharmacol Exp Ther. 2003;304:1188–96. doi: 10.1124/jpet.102.044685. [DOI] [PubMed] [Google Scholar]
- 10.Wu XS, Sun JY, Evers AS, Crowder M, Wu LG. Isoflurane inhibits transmitter release and the presynaptic action potential. Anesthesiology. 2004;100:663–70. doi: 10.1097/00000542-200403000-00029. [DOI] [PubMed] [Google Scholar]
- 11.Franks NP, Dickinson R, de Sousa SL, Hall AC, Lieb WR. How does xenon produce anaesthesia? Nature. 1998;396:324. doi: 10.1038/24525. [DOI] [PubMed] [Google Scholar]
- 12.Martin DC, Plagenhoef M, Abraham J, Dennison RL, Aronstam RS. Volatile anesthetics and glutamate activation of N-methyl-D-aspartate receptors. Biochem Pharmacol. 1995;49:809–17. doi: 10.1016/0006-2952(94)00519-r. [DOI] [PubMed] [Google Scholar]
- 13.Mihic SJ, Ye Q, Wick MJ, Koltchine VV, Krasowski MD, Finn SE, Mascia MP, Valenzuela CF, Hanson KK, Greenblatt EP, Harris RA, Harrison NL. Sites of alcohol and volatile anaesthetic action on GABA(A) and glycine receptors. Nature. 1997;389:385–9. doi: 10.1038/38738. [DOI] [PubMed] [Google Scholar]
- 14.Franks NP. Molecular targets underlying general anaesthesia. Br J Pharmacol. 2006;147(suppl 1):S72–81. doi: 10.1038/sj.bjp.0706441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neal MJ, Robson JM. The analgesic and anaesthetic actions of tetrafluorobenzene. Br J Pharmacol Chemother. 1966;26:482–93. doi: 10.1111/j.1476-5381.1966.tb01929.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Solt K, Eger EI, 2nd, Raines DE. Differential modulation of human N-methyl-D-aspartate receptors by structurally diverse general anesthetics. Anesth Analg. 2006;102:1407–11. doi: 10.1213/01.ane.0000204252.07406.9f. [DOI] [PubMed] [Google Scholar]
- 17.Kelly EW, Solt K, Raines DE. Volatile aromatic anesthetics variably impact human gamma-aminobutyric acid type A receptor function. Anesth Analg. 2007;105:1287–92. doi: 10.1213/01.ane.0000282829.21797.97. [DOI] [PubMed] [Google Scholar]
- 18.Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- 19.Catterall WA, Goldin AL, Waxman SG. International Union of Pharmacology. XLVII Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev. 2005;57:397–409. doi: 10.1124/pr.57.4.4. [DOI] [PubMed] [Google Scholar]
- 20.Goldin AL. Resurgence of sodium channel research. Annu Rev Physiol. 2001;63:871–94. doi: 10.1146/annurev.physiol.63.1.871. [DOI] [PubMed] [Google Scholar]
- 21.Antognini JF, Schwartz K. Exaggerated anesthetic requirements in the preferentially anesthetized brain. Anesthesiology. 1993;79:1244–9. doi: 10.1097/00000542-199312000-00015. [DOI] [PubMed] [Google Scholar]
- 22.Fang Z, Sonner J, Laster MJ, Ionescu P, Kandel L, Koblin DD, Eger EI, 2nd, Halsey MJ. Anesthetic and convulsant properties of aromatic compounds and cycloalkanes: implications for mechanisms of narcosis. Anesth Analg. 1996;83:1097–104. doi: 10.1097/00000539-199611000-00035. [DOI] [PubMed] [Google Scholar]
- 23.Raines DE, Gioia F, Claycomb RJ, Stevens RJ. The N-methyl-D-aspartate receptor inhibitory potencies of aromatic inhaled drugs of abuse: evidence for modulation by cation-pi interactions. J Pharmacol Exp Ther. 2004;311:14–21. doi: 10.1124/jpet.104.069930. [DOI] [PubMed] [Google Scholar]
- 24.Horishita T, Harris RA. n-Alcohols inhibit voltage-gated Na+ channels expressed in Xenopus oocytes. J Pharmacol Exp Ther. 2008 doi: 10.1124/jpet.108.138370. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koblin DD, Chortkoff BS, Laster MJ, Eger EI, 2nd, Halsey MJ, Ionescu P. Polyhalogenated and perfluorinated compounds that disobey the Meyer-Overton hypothesis. Anesth Analg. 1994;79:1043–8. doi: 10.1213/00000539-199412000-00004. [DOI] [PubMed] [Google Scholar]
- 26.Ratnakumari L, Vysotskaya TN, Duch DS, Hemmings HC., Jr Differential effects of anesthetic and nonanesthetic cyclobutanes on neuronal voltage-gated sodium channels. Anesthesiology. 2000;92:529–41. doi: 10.1097/00000542-200002000-00037. [DOI] [PubMed] [Google Scholar]