

Abstract
We identified a unique family with autosomal dominant heart disease variably expressed as restrictive cardiomyopathy (RCM), hypertrophic cardiomyopathy (HCM), and dilated cardiomyopathy (DCM), and sought to identify the molecular defect that triggered divergent remodeling pathways. Polymorphic DNA markers for nine sarcomeric genes for DCM and/or HCM were tested for segregation with disease. Linkage to eight genes was excluded, but a cardiac troponin T (TNNT2) marker cosegregated with the disease phenotype. Sequencing of TNNT2 identified a heterozygous missense mutation resulting in an I79N substitution, inherited by all nine affected family members but by none of the six unaffected relatives. Mutation carriers were diagnosed with RCM (n = 2), non-obstructive HCM (n = 3), DCM (n = 2), mixed cardiomyopathy (n = 1), and mild concentric left ventricular hypertrophy (n = 1). Endomyocardial biopsy in the proband revealed non-specific fibrosis, myocyte hypertrophy, and no myofibrillar disarray. Restrictive Doppler filling patterns, atrial enlargement, and pulmonary hypertension were observed among family members regardless of cardiomyopathy subtype. Mutation of a sarcomeric protein gene can cause RCM, HCM, and DCM within the same family, underscoring the necessity of comprehensive morphological and physiological cardiac assessment in familial cardiomyopathy screening.
Keywords: dilated cardiomyopathy, hypertrophic cardiomyopathy, mutation, restrictive cardiomyopathy, sarcomere, TNNT2
The three basic types of cardiomyopathy - dilated cardiomyopathy (DCM), hypertrophic cardiomyopathy (HCM), and restrictive cardiomyopathy (RCM) - are classified by prototypic morphological and physiological features, yet clinically distinct cardiomyopathies in unrelated families may be triggered by different mutations within the same gene (1, 2). Conversely, phenotypic traits may be variably expressed within certain families and sometimes overlap (3, 4). While genetic studies have provided significant insight into the pathogenesis of DCM and HCM (5), less is known about the molecular basis of RCM. Clinical manifestations of RCM reflect an inherent impairment of ventricular relaxation leading to elevated end-diastolic pressure, reduced ventricular filling, and pulmonary hypertension (6). Echocardiographic features include bi-atrial enlargement, impaired or restricted ventricular filling, normal or decreased diastolic ventricular volume, and near normal ventricular systolic function and wall thickness (7, 8). Ventricular wall thickness, cavity size and function vary, however, depending on the duration of RCM (8). Indeed, mild septal hypertrophy in the presence of classic features of RCM does not preclude diagnosis of RCM (9). While RCM can occur as a variable manifestation of desmin-related myopathies (10), cardiac troponin I (11, 12), cardiac troponin T (13), and beta-myosin heavy chain (14-16), also implicated in the pathogenesis of HCM and DCM, are the only known genes for primary RCM. All but one of these reports (11), however, describe isolated cases of RCM either attributable to de novo mutations or with limited clinical and genetic on family members. Consequently, data are scarce on segregation and variable expression of RCM-associated mutations within multigeneration families. Here, we identified a large family with an autosomal dominant form of cardiomyopathy characterized by RCM in the proband and clinical features of RCM, HCM, and/or DCM among relatives. Postulating a monogenic basis for disease, we performed targeted genetic linkage analyses for nine sarcomeric protein genes. An amino acid substitution in troponin T (I79N), previously reported in patients with HCM (17), segregated with the disease phenotype. The functional consequences of this molecular defect have been previously studied in a transgenic murine model (18-21), which exhibits the restrictive physiology observed in human mutation carriers.
Methods
Subjects
Informed written consent was obtained from study participants under a protocol approved by the Institutional Review Board of the Mayo Clinic. The proband was referred to a medical geneticist who obtained a detailed family history and constructed an extended pedigree. Medical records were acquired from the proband’s relatives, the majority of whom had undergone clinically indicated cardiac evaluation for symptoms and/or recognized risk of familial myocardial disease. Two asymptomatic study participants were screened by echocardiography and electrocardiography under the auspices of the research study. Echocardiographic measurements of cardiac structure and function were interpreted by indexing for body surface area and/or age, utilizing established reference values of the Mayo Clinic echocardiographic laboratory. Rhythm and conduction abnormalities were similarly defined using established criteria. Genomic DNA was extracted from whole blood samples (Puregene; Gentra Systems, Inc., Minneapolis, MN) or paraffin-embedded post-mortem tissue obtained at autopsy (QIAamp® DNA Mini Kit; QIAGEN, Inc., Valencia, CA).
Genotyping and segregation analysis
We selected one to three polymorphic short tandem repeat DNA markers within or spanning each of nine genes for DCM and/or HCM: troponin T (TNNT2), titin (TTN), essential light chain of myosin (MYL3), myosin-binding protein C (MYBPC), regulatory light chain of myosin (MYL2), beta-myosin heavy chain (MYH7), cardiac actin (ACTC1), alpha-tropomyosin (TPM1), and troponin I (TNNI3) (primer sequences available on request). Markers were identified using the Map Viewer link on the National Center for Biotechnology Information web site (http://www.ncbi.nih.gov), amplified from genomic DNA by the polymerase chain reaction (PCR), and resolved on polyacrylamide gels as previously described (22). Scored genotypes were placed on the pedigree and inferred, when possible, for individuals from whom DNA samples were not available. Segregation of a specific genotype or haplotype with cardiomyopathy was considered suggestive of genetic linkage, whereas genes were considered excluded when there was lack of a shared allele or haplotype in one or more affected relatives.
DNA sequencing
Genotyping and segregation analyses implicated TNNT2 as a candidate gene, so mutational analyses were performed. Oligonucleotide primer pairs were designed for exon-specific PCR amplification of coding and splice junction regions for the 15 translated exons of TNNT2 (primer sequences available on request), using Oligo 6.51 Primer Analysis Software (National Biosciences, Plymouth, MN). PCR products were treated with the PCR Product Presequencing Kit (USB Corporation, Cleveland, OH) and sequenced by the dye-terminator method in a core facility, using an ABI PRISM 377 DNA Sequencer (Applied Biosystems, Foster City, CA). DNA sequence was viewed and analyzed using the Sequencher computer program (Gene Codes Corporation, Ann Arbor, MI).
Results
Clinical features of proband
The proband (individual II.11 in Tables 1 and 2 and Fig. 1a) presented with dyspnea on exertion, palpitations and exercise intolerance at 53 years of age. Echocardiography revealed massive biatrial enlargement (Fig. 2a) and markedly abnormal diastolic function, diagnostic for RCM. Left ventricular dimensions and systolic function were normal. At diagnosis, she had mild hypertrophy localized to the mid-septum (maximal thickness = 14 mm) with no systolic anterior motion of the anterior mitral valve leaflet or left ventricular outflow tract obstruction, both at rest and with exercise and Valsalva maneuver. Diastolic stress echocardiography revealed worsening of diastolic function with exercise.
Table 1.
Clinical findings in individual family membersa
| ID | I79N | Sex | Age at diagnosis (evaluation) (years) | Age at death (years) | ECG, holter | Exercise testing | Cardiac catheterization |
|---|---|---|---|---|---|---|---|
| I.1 | - | M | - | 71 (heart disease) | - | - | - |
| I.2 | - | F | - | 94 (stroke) | - | - | No CAD |
| II.2 | +/- | F | 50s | 64 (heart failure) | - | Normal | No CAD |
| II.4 | +/- | F | 68 (70-71) | 73 (heart failure) | AF/FL, SB (HR = 52), BFB, 3° AVB, LAE, ST-T | - | No CAD |
| II.6 | +/- | F | 66 (69-70) | - | NSVT, BAE, ST-T, LVH | ↓VO2max, dyspnea | No CAD |
| II.7 | +/- | M | 46 (68) | 69 (cancer) | AF/FL | - | No CAD |
| II.9 | +/- | M | 58 (61-69) | - | AF, RAD, LAE, ST-T, LVH | ↓VO2max, dyspnea | - |
| II.10 | -/- | M | (60-61) | - | Normal | - | - |
| II.11 | +/- | F | 53 (53-63) | - | AF, SB (HR = 49), RAD, 3°AVB, LBBB, LAE, ST-T | ↓VO2max, dyspnea, ↓HR response, ↓blood pressure, ↑diastolic failure | No CAD, LVEDP = 23 mmHg (37 with exercise), PASP = 28 mmHg (57 with exercise) |
| III.1 | -/- | F | (50-54) | - | Normal | - | - |
| III.2 | -/- | F | (49) | - | Normal | - | - |
| III.3 | +/- | F | 49 (40-49) | - | LAE, IRBBB, ST-T | - | - |
| III.4 | +/- | F | 40 (40-45) | - | SVT, LAE, LVH, ST-T | ↓VO2max, dyspnea, ↓HR response, ↑ diastolic failure | No CAD, LVEDP = 31, PASP = 44 mmHg |
| III.5 | -/- | F | (42-48) | - | Normal | - | - |
| III.6 | +/- | F | (48-51) | - | SB (HR = 55) | - | - |
| III.7 | -/- | F | (44) | - | Normal | - | - |
| III.8 | -/- | F | (48) | - | Normal | - | - |
+/-, mutation carrier; -/-, mutation absent; -, not available/applicable; AF, atrial fibrillation; AFL, atrial flutter; AVB, atrioventricular block; BAE, biatrial enlargement; BFB, bifasicular block; CAD, coronary artery disease; HR, heart rate; ID, pedigree position; LAE, left atrial enlargement; LBBB, left bundle branch block; LVEDP, left ventricular end-diastolic pressure; LVH, left ventricular hypertrophy; NSVT, four-beat run of ventricular tachycardia; PASP, pulmonary artery systolic pressure; RAD, right axis deviation; SB, sinus bradycardia; ST-T, non-specific ST-T wave changes; SVT, supraventricular tachycardia; VO2max, peak oxygen consumption.
Normal values: LVEDP <15 mmHg; PASP ≤30 mmHg.
Table 2.
Echocardiographic findings in individual family membersa
| ID | I79N | MST/MPWT (mm) | LVDD/LVSD (mm) | EF% | E/A | DT (ms) | E’ (cm/s) | E/E’ | Atrial size | Atrioventricular valve regurgitation | PASP (mmHg) | Clinical diagnosis |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| I.1 | - | - | - | - | - | - | - | - | - | - | - | - |
| I.2 | - | - | - | - | - | - | - | - | - | - | - | Enlarged heart |
| II.2 | +/- | - | - | 23 | - | - | - | - | - | - | - | DCM |
| II.4 | +/- | 10/9 | 62/55 | 21 | 2.5 | 100 | - | - | BAE +++ | TR+ MR+ | 52 | DCM, PAH, restrictive physiology |
| II.6 | +/- | 18/10 | 43/31 | 51 | 0.4 | - | - | 13 | LAE ++ | TR+ MR+ | 56 | HCM, PAH, restrictive physiology |
| II.7 | +/- | 16/12 | 58/45 | 43 | 1.7 | 270 | - | - | BAE + | TR+ MR+ | 51 | Mixed DCM/HCM, restrictive physiology, PAH |
| II.9 | +/- | 12/10 | 55/35 | 60 | 2.0 | 155 | 3 | 17 | BAE +++ | TR+ MR+ | 54 | RCM, PAH |
| II.10 | -/- | 13/10 | 52/34 | 62 | 1.0 | 260 | - | - | nl | None | 26 | Normal |
| II.11 | +/- | 14→11→11/9 | 53→49/33→42 | 60→40 | 1.2→2.7 | 180→130 | 3 | 27 | BAE +++ | MR+ TR++ | 55 | RCM, PAH/systolic dysfunction |
| III.1 | -/- | 11/9 | 47/30 | 63 | 1.1 | 198 | 6 | 12 | nl | None | 23 | Normal |
| III.2 | -/- | 9/9 | 44/28 | 64 | 1.1 | 148 | 9 | 9 | nl | None | 24 | Normal |
| III.3 | +/- | 10→16/6→9 | 48/32 | 67 | - | - | - | - | LAE ++ | MR+ | 40 | HCM, PAH, mild diastolic dysfunction |
| III.4 | +/- | 24→21/8→9 | 45/24 | 72 | 2.0→3.0 | 175→133 | 6 | 17 | LAE +++ | MR+ | 51 | HCM, PAH, marked diastolic dysfunction and restrictive physiology |
| III.5 | -/- | 7/8 | 46/28 | 67 | 1.5 | 199 | - | 5 | nl | None | 21 | Normal |
| III.6 | +/- | 12→14/10→10 | 49/28 | 71 | 0.7 | 224 | 4 | 15 | LAE ++ | TR+ | 24 | Mild concentric LVH, mild diastolic dysfunction and restrictive physiology |
| III.7 | -/- | 12/10 | 48/26 | 74 | 0.9 | 202 | 9 | 7 | nl | None | 21 | Normal |
| III.8 | -/- | 10/9 | 51/31 | 67 | 1.3 | 156 | 7 | 14 | nl | None | 20 | Normal |
-, not available; →, significant change in measurement on follow up; +, mild; ++, moderate; +++, severe; BAE, bi-atrial enlargement; DCM, dilated cardiomyopathy; DT, mitral E wave deceleration time; E’, medial mitral annulus tissue Doppler E velocity; E/A, mitral inflow E/A ratio; EF%, left ventricular ejection fraction; HCM, hypertrophic cardiomyopathy; LVH, left ventricular hypertrophy; LVDD/LVSD, left ventricular diastolic/systolic dimension; MPWT, maximal postwall thickness; MR, mitral valve regurgitation; MST, maximal septal thickness; nl, normal; PAH, pulmonary artery hypertension; RCM, restrictive cardiomyopathy; TR, tricuspid valve regurgitation.
Normal values: MST <13 mm; MPWT <13 mm; LVDD ≤45.3 (body surface area)1/3 - 0.03 (age) 2 7.2 × 1.12; LVSD ≤28.8(body surface area)1/3 - 0.03 (age) - 4.1 × 1.18; EF ≥50%; E/A = 0.9-1.5; DT = 132-140 ms; E’ ≥7 m/s; E/E’ <15. Abnormal values are in given in bold.
Fig. 1.
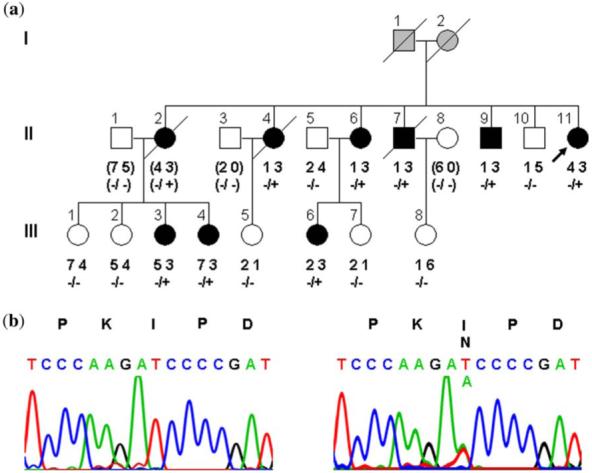
Pedigree structure of family with cardiomyopathy caused by TNNT2 mutation. (a) The proband (II.11) has five siblings and three nieces with cardiomyopathy, segregating as an autosomal dominant trait. Genotyping with a polymorphic dinucleotide repeat marker for TNNT2 showed inheritance of the ‘3’ allele by the nine affected family members with cardiac disease but not by the six relatives with normal echocardiograms. (b) Sequencing identified a heterozygous missense mutation in TNNT2, substituting an asparagine for an isoleucine at amino acid position 79 in cardiac troponin T2. Circles, females; squares, males; filled, cardiomyopathy; unfilled, normal cardiac phenotype; gray, unknown cardiac phenotype; diagonal line, deceased; +, Asn79 mutant allele; -, Ile79 wild-type allele; ( ), inferred genotype.
Fig. 2.

Morphological features of mutation carriers shown by 2D echocardiography. (a) Massive biatrial enlargement, normal ventricular size, depicting restrictive cardiomyopathy phenotype (II.11). (b) Dilated LV with severe biatrial enlargement, depicting dilated cardiomyopathy (DCM) phenotype and restrictive physiology (II.4). (c) Midseptal hypertrophy (arrow) with severe biatrial enlargement and mildly dilated LV, depicting mixed hypertrophic cardiomyopathy and DCM phenotype and restrictive physiology (II.7). RA, right atrium; RV, right ventricle; LA, left atrium; LV, left ventricle.
Exercise testing was notable for limitation of functional capacity with a peak oxygen consumption of 15 ml/kg/min. Blood pressure and heart rate responses to exercise were suboptimal. She had baseline non-specific ST-T wave changes and developed 1.5-2.0 mm ST segment depression in leads V4-V6, II, III, and AVF with exercise.
Cardiac catheterization (Fig. 3) excluded coronary artery disease but revealed low cardiac output (2.0 l/min) and elevated left and right ventricular end-diastolic pressures, pulmonary capillary wedge pressure, and pulmonary artery systolic pressure. Similar to findings on stress echocardiography, supine exercise during cardiac catheterization caused further elevation in biventricular end-diastolic filling pressures. Right ventricular endomyocardial biopsy showed non-specific interstitial fibrosis and moderate myocardial hypertrophy, with no myofibrillar disarray. The sample was negative for eosinophilic infiltrate, myocarditis, sarcoidosis, amyloidosis and hemochromatosis.
Fig. 3.

Physiological features of proband (II.11) shown by Doppler echocardiography and cardiac catheterization. Left panels show reduced mitral annulus E’, increased E/E’ ratio, increased mitral inflow E to A ratio, and reduced mitral inflow E wave deceleration time, typical echocardiographic features of restrictive cardiomyopathy (RCM) with elevated LV filling pressures. Right panels show elevated ventricular end-diastolic and pulmonary artery pressures and ‘square root sign’, characteristic findings in RCM. E’, medial mitral annulus tissue Doppler E velocity; E/A, mitral inflow E/A ratio; LV; left ventricle; LVEDP, left ventricular end-diastolic pressure; MPA, main pulmonary artery pressure; RVEDP, right ventricular end-diastolic pressure.
Electrocardiography revealed non-specific ST-T wave changes, left atrial enlargement and first-degree atrioventricular block. She subsequently developed sinus bradycardia and progression to complete heart block. Atrial flutter/fibrillation developed in association with progressive atrial enlargement. She ultimately underwent radiofrequency ablation of arrhythmogenic foci and pacemaker/cardioverter-defibrillator implantation.
Over a 10-year follow-up interval (ages 53-63 years), the patient developed increasing dyspnea, palpitations, presyncope, pedal edema, and exertional chest pain. She was treated with angiotensin-converting enzyme inhibitors, calcium channel blockers, beta-receptor blockers, diuretics, and warfarin for a left atrial thrombus. With treatment she experienced initial symptomatic improvement, but progressively decompensated with worsening diastolic function and recurrent symptoms of heart failure and low cardiac output. Left ventricular hypertrophy regressed (maximal septal thickness = 11 mm), while systolic function deteriorated (ejection fraction = 40%) associated with progressive atrial dilation. She is currently undergoing evaluation for cardiac transplantation.
Pedigree analysis
The proband’s family history was remarkable for diagnosis of cardiomyopathy in five siblings and three nieces (Tables 1 and 2). Pedigree analysis (Fig. 1a) implicated a single gene defect, inherited as either an autosomal or X-linked dominant trait. Age at diagnosis of disease varied from 40 to 68 years. Cardiomyopathic phenotypes of RCM, HCM, or DCM were variously noted in different family members. In addition to the proband, a brother (individual II.9) was diagnosed with RCM after presenting with dyspnea on exertion and atrial fibrillation. He had classic echocardiographic findings of normal ventricular wall thickness and chamber size, restrictive Doppler filling pattern, severe biatrial enlargement, and pulmonary hypertension.
By contrast, the proband’s two oldest siblings were diagnosed with idiopathic DCM following exclusion of coronary artery disease, although end-stage RCM or HCM could not be excluded due to lack of presymptomatic echocardiograms. Individual II.4 presented with atrial fibrillation and signs of heart failure at the age of 68 years. Cardiomegaly and decreased ejection fraction persisted on diuretic, angiotensin-converting enzyme inhibitor, and beta-receptor blocker therapy and complete heart block developed, prompting implantation of a cardioverter/defibrillator and dual-chamber pacemaker. Ventricular wall thickness was normal, but echocardiography revealed findings of restrictive physiology with severe atrial enlargement (Fig. 2b) and pulmonary hypertension. She died of progressive heart failure at 73 years of age, without documented ventricular arrhythmia on serial defibrillator/pacemaker interrogations over the preceding 3 years. Individual II.2 died of congestive heart failure at 64 years of age. Review of limited outside medical records confirmed the diagnosis of idiopathic DCM in her sixth decade of life, together with prominent right heart enlargement suggesting pulmonary artery hypertension.
Individuals II.6, III.3, and III.4 had HCM with asymmetrical septal hypertrophy. In contrast to the typical morphology of HCM, however, septal thickening was maximal at the mid-to-apical septum. Systolic anterior motion of the mitral valve and left ventricular outflow obstruction were absent, even on provocation. One individual (III.3) had mild pulmonary hypertension on an initial screening echocardiogram at 40 years of age, prior to development of HCM 9 years later. Her younger sibling (III.4) presented with symptomatic heart disease at 40 years of age, characterized by severe septal hypertrophy with markedly elevated ventricular filling pressures. All three had moderate to severe left atrial enlargement and pulmonary hypertension together with Doppler patterns indicative of variable degrees of restrictive physiology.
Two additional clinically affected family members had abnormal echocardiographic findings not easily classified as RCM, DCM, or HCM. One (II.7) had a history of asymptomatic left ventricular hypertrophy discovered on a routine electrocardiogram at 47 years of age. He presented for cardiac evaluation at 68 years of age due to dyspnea on exertion and concern about his family history. Echocardiography revealed mixed features of HCM and DCM (Fig. 2c) with asymmetric septal hypertrophy, left ventricular enlargement, reduced ejection fraction, and regional wall motion abnormalities in the absence of coronary artery disease. Like other family members, he also had restrictive physiology. He ultimately developed paroxysmal atrial fibrillation and symptoms of congestive heart failure and died from cancer. The other family member (III.6) had an initial screening echocardiogram at 48 years of age showing mild left atrial enlargement and Doppler indices indicative of restrictive left ventricular filling. She remained asymptomatic and on follow-up evaluation 3 years later had developed moderate left atrial enlargement and mild concentric left ventricular hypertrophy, in the absence of systemic hypertension.
Electrocardiography showed non-specific ST-T wave changes and atrial enlargement in the majority of affected family members. Progressive atrial dilation was associated with atrial fibrillation and flutter in four and non-sustained supraventricular tachycardia in one. Only one relative had a documented ventricular arrhythmia, limited to a four-beat run of ventricular tachycardia on Holter monitoring.
In summary, the phenotype in this family includes RCM, DCM, and HCM with mild to severe restrictive physiology developing early in the remodeling process. There was a high incidence of atrial tachyarrhythmia, but an absence of significant ventricular arrhythmias or sudden cardiac death.
Molecular genetic analysis
A candidate gene linkage approach was used to identify the gene responsible for cardiomyopathy in the proband and her family. Lack of cosegregation of gene-specific markers and cardiomyopathy excluded eight of nine candidate genes (data not shown). By contrast, a polymorphic dinucleotide repeat marker for TNNT2, located 208 kb 5′ of the start codon, was inherited by all nine affected family members and by none of the six relatives without cardiac disease (Fig. 1a). Once cosegregation between the TNNT2 marker and the disease phenotype was established, sequence analysis of this positional candidate gene was performed. A heterozygous missense mutation, c.236T>A, was identified in exon 8 (Fig. 1b), resulting in substitution of isoleucine (I) with asparagine (N) at amino acid position 79. Consistent with the linkage data, the mutation was inherited by all affected family members and none of the unaffected family members. This mutation disrupts the tropomyosin-binding domain important for calcium-dependent cardiac muscle contractile function (23).
Discussion
We identified a pathogenic mutation in TNNT2 in a large family with autosomal dominant cardiomyopathy. Mutations in TNNT2 were first described in patients with HCM in 1994 (17) and in patients with DCM in 2000 (24). In 2006, a de novo TNNT2 mutation was reported in an infant with RCM associated with mild to moderate left ventricular dysfunction and myocyte disarray (13). One of the reported HCM-associated mutations, I79N, is identical to the mutation identified in our family. Since the initial report, at least 30 distinct mutations in TNNT2 have been identified in patients with HCM. Several studies have shown that patients with troponin-associated HCM have less hypertrophy and a higher incidence of sudden cardiac death at a young age (25, 26). Specifically, the I79N mutation was associated with a malignant form of HCM characterized by a high incidence of sudden death and mild or absent cardiac hypertrophy (25). A similar lethal phenotype has been reported in DCM-associated mutations in TNNT2, albeit this correlation is not without exception (27, 28). The infant with an RCM-associated TNNT2 mutation presented with profound hemodynamic instability and ultimately underwent cardiac transplantation (13).
Despite variable morphologies, all affected members of our family exhibited restrictive physiology, and two mutation carriers were diagnosed with idiopathic RCM prior to enrollment in our study. Restrictive ventricular filling and diastolic dysfunction are recognized features of HCM caused by heritable sarcomeric protein defects (4); yet, only a few reports describe a primary phenotype of RCM in the absence of significant ventricular hypertrophy in members of families with a primary diagnosis of familial HCM (11, 12, 29). In a study of diastolic parameters in 104 patients with HCM, mean left atrial volume index in the subset with ‘large left atrial volume’ was 49.6 ± 12 ml/m2 (normal = 14-28 ml/m2) (30). However, none of the patients with a left ventricular outflow gradient <30 mmHg had left atrial volume index >21 ml/m2. Left atrial volume index was also proportional to the degree of hypertrophy in non-apical segments and mitral regurgitation. By contrast, seven of nine mutation carriers in our family had moderate to severe left atrial enlargement in the absence of left ventricular outflow tract obstruction, significant mitral regurgitation, or posterior left ventricular wall hypertrophy. On cardiac catheterization, RCM is characterized by elevated end-diastolic pressures, a ‘dip and plateau’ pattern in the right ventricle, pulmonary artery hypertension, elevated pulmonary capillary wedge pressure, and concordant changes in left and right ventricular pressure with respiration (ventricular concordance). Both patients carrying the I79N mutation who underwent hemodynamic catheterization (II.11 and III.4) showed these classic features.
Transition to a DCM phenotype has been reported in patients with HCM caused by certain TNNT2 mutations (31, 32). Overall, progression to a ‘burned out’ DCM phase is seen in 5-15% of patient with HCM, typically with regression of left ventricular outflow tract obstruction, wall thinning, and clinical heart failure (33). In contrast to mutation carriers in our family, however, risk for development of DCM has been linked to young age at diagnosis and increased left ventricular wall thickness.
In cardiac muscle, the thin filament troponin complex has three subunits: troponin T binds to tropomyosin, troponin C binds to calcium and troponin I inhibits actin and myosin cross-bridging. Muscle contraction is initiated by binding of calcium, released from sarcoplasmic reticulum, to troponin C. This results in a conformational change of the troponin complex attenuating the inhibitory effects of troponin I on actomyosin cross-bridge formation. Troponin T thus plays a pivotal role in stabilizing the tropomyosin-troponin complex and modulating calcium-mediated excitation-contraction coupling. The I79N substitution in TNNT2 is located in a conserved region of the protein (23). Transgenic mice with cardiac-targeted expression of human troponin T-I79N have played a key role in characterizing the molecular pathophysiology of this mutation (18-21). These mice exhibit no hypertrophy, yet fiber studies show enhanced calcium-activated force generation and ATPase activity, leading to rapid exhaustion of ATP stores and increased cost of force generation. Decrease in the rate of calcium dissociation from troponin C in diastole results in slower relaxation and increased baseline muscle tension, leading to elevation of end-diastolic pressure and RCM with diastolic heart failure. Diltiazem prevents isoproterenol stress-induced acute heart failure and sudden death, attributed to inhibition of L-type calcium current and decreased intracellular calcium during diastole (21). These findings may provide a rationale for mechanism-based calcium channel blocker therapy in human mutation carriers.
In summary, we identified a family with an I79N substitution in cardiac troponin T leading to variable cardiac remodeling and exhibiting phenotypic features of RCM, HCM, and DCM. While segregation analyses excluded a primary pathogenic role for eight other sarcomeric protein genes, we cannot exclude potential modifying effects of variants within these or other genes on cardiac phenotype. Notwithstanding, the physiological phenotype of mutation-carriers recapitulates the diastolic heart disease previously reported in transgenic mice expressing this mutation. Our findings further implicate genetic defects in the sarcomeric proteins in the pathogenesis of the three major types of cardiomyopathy. This study underscores the necessity of comprehensive morphological and physiological cardiac assessment when screening for presymptomatic familial cardiomyopathy.
Acknowledgements
The authors are grateful to family members for their participation in this study. This work was supported by a grant from the National Institutes of Health (HL071225) and the Marriott Heart Disease Research Program.
References
- 1.Olson TM, Michels VV, Thibodeau SN, Tai YS, Keating MT. Actin mutations in dilated cardiomyopathy, a heritable form of heart failure. Science. 1998;280:750–752. doi: 10.1126/science.280.5364.750. [DOI] [PubMed] [Google Scholar]
- 2.Olson TM, Doan TP, Kishimoto NY, Whitby FG, Ackerman MJ, Fananapazir L. Inherited and de novo mutations in the cardiac actin gene cause hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2000;32:1687–1694. doi: 10.1006/jmcc.2000.1204. [DOI] [PubMed] [Google Scholar]
- 3.Song L, DePalma SR, Kharlap M, et al. Novel locus for an inherited cardiomyopathy maps to chromosome 7. Circulation. 2006;113:2186–2192. doi: 10.1161/CIRCULATIONAHA.106.615658. [DOI] [PubMed] [Google Scholar]
- 4.Olson TM, Karst ML, Whitby FG, Driscoll DJ. Myosin light chain mutation causes autosomal recessive cardiomyopathy with mid-cavitary hypertrophy and restrictive physiology. Circulation. 2002;105:2337–2340. doi: 10.1161/01.cir.0000018444.47798.94. [DOI] [PubMed] [Google Scholar]
- 5.Ahmad F, Seidman JG, Seidman CE. The genetic basis for cardiac remodeling. Annu Rev Genomics Hum Genet. 2005;6:185–216. doi: 10.1146/annurev.genom.6.080604.162132. [DOI] [PubMed] [Google Scholar]
- 6.Kushwaha SS, Fallon JT, Fuster V. Restrictive cardiomyopathy. N Engl J Med. 1997;336:267–276. doi: 10.1056/NEJM199701233360407. [DOI] [PubMed] [Google Scholar]
- 7.Tam JW, Shaik N, Sutherland E. Echocardiographic assessment of patients with hypertrophic and restrictive cardiomyopathy: imaging and echocardiography. Curr Opin Cardiol. 2002;17:470–477. doi: 10.1097/00001573-200209000-00005. [DOI] [PubMed] [Google Scholar]
- 8.Wood MJ, Picard MH. Utility of echocardiography in the evaluation of individuals with cardiomyopathy. Heart. 2004;90:707–712. doi: 10.1136/hrt.2003.024778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richardson P, McKenna W, Bristow M, et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the definition and classification of cardiomyopathies. Circulation. 1996;93:841–842. doi: 10.1161/01.cir.93.5.841. [DOI] [PubMed] [Google Scholar]
- 10.Arbustini E, Pasotti M, Pilotto A, et al. Desmin accumulation restrictive cardiomyopathy and atrioventricular block associated with desmin gene defects. Eur J Heart Fail. 2006;8:477–483. doi: 10.1016/j.ejheart.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 11.Mogensen J, Kubo T, Duque M, et al. Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations. J Clin Invest. 2003;111:209–216. doi: 10.1172/JCI16336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kostareva A, Gudkova A, Sjöberg G, et al. Deletion in TNNI3 gene is associated with restrictive cardiomyopathy. Int J Cardiol. 2007 doi: 10.1016/j.ijcard.2007.07.108. doi:10.1016/j.ijcard.2007.07.108. [DOI] [PubMed] [Google Scholar]
- 13.Peddy SB, Vricella LA, Crosson JE, et al. Infantile restrictive cardiomyopathy resulting from a mutation in the cardiac troponin T gene. Pediatrics. 2006;117:1830–1833. doi: 10.1542/peds.2005-2301. [DOI] [PubMed] [Google Scholar]
- 14.Kubo T, Gimeno JR, Bahl A, et al. Prevalence, clinical significance, and genetic basis of hypertrophic cardiomyopathy with restrictive phenotype. J Am Coll Cardiol. 2007;49:2419–2426. doi: 10.1016/j.jacc.2007.02.061. [DOI] [PubMed] [Google Scholar]
- 15.Ware SM, Quinn ME, Ballard ET, Miller E, Uzark K, Spicer RL. Pediatric restrictive cardiomyopathy associated with a mutation in beta-myosin heavy chain. Clin Genet. 2008;73:165–170. doi: 10.1111/j.1399-0004.2007.00939.x. [DOI] [PubMed] [Google Scholar]
- 16.Karam S, Raboisson MJ, Ducreux C, et al. A de novo mutation of the beta cardiac myosin heavy chain gene in an infantile restrictive cardiomyopathy. Congenit Heart Dis. 2008;3:138–143. doi: 10.1111/j.1747-0803.2008.00165.x. [DOI] [PubMed] [Google Scholar]
- 17.Thierfelder L, Watkins H, MacRae C, et al. Alphatropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell. 1994;77:701–712. doi: 10.1016/0092-8674(94)90054-x. [DOI] [PubMed] [Google Scholar]
- 18.Rust EM, Albayya FP, Metzger JM. Identification of a contractile deficit in adult cardiac myocytes expressing hypertrophic cardiomyopathy-associated mutant troponin T proteins. J Clin Invest. 1999;103:1459–1467. doi: 10.1172/JCI6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller T, Snzczesna D, Housmans PR, et al. Abnormal contractile function in transgenic mice expressing a familial hypertrophic cardiomyopathy-linked troponin T (I79N) mutation. J Biol Chem. 2001;276:3743–3755. doi: 10.1074/jbc.M006746200. [DOI] [PubMed] [Google Scholar]
- 20.Harada K, Potter JD. Familial hypertrophic cardiomyopathy mutations from different functional regions of troponin T result in different effects on the pH and Ca2+ sensitivity of cardiac muscle contraction. J Biol Chem. 2004;279:14488–14495. doi: 10.1074/jbc.M309355200. [DOI] [PubMed] [Google Scholar]
- 21.Westermann D, Knollmann BC, Steendijk P, et al. Diltiazem treatment prevents diastolic heart failure in mice with familial hypertrophic cardiomyopathy. Eur J Heart Fail. 2006;8:115–121. doi: 10.1016/j.ejheart.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 22.Olson TM, Keating MT. Mapping a cardiomyopathy locus to chromosome 3p22-p25. J Clin Invest. 1996;97:528–532. doi: 10.1172/JCI118445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palm T, Graboski S, Hitchcock-DeGregori SE, Greenfield NJ. Disease-causing mutations in cardiac troponin T: identification of a critical tropomyosin-binding region. Biophys J. 2001;81:2827–2837. doi: 10.1016/S0006-3495(01)75924-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kamisago M, Sharma SD, DePalma SR, et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med. 2000;343:1688–1696. doi: 10.1056/NEJM200012073432304. [DOI] [PubMed] [Google Scholar]
- 25.Watkins H, McKenna W, Thierfelder L, et al. Mutations in the genes for cardiac troponin T and alpha-tropomyosin in hypertrophic cardiomyopathy. N Engl J Med. 1995;332:1058–1064. doi: 10.1056/NEJM199504203321603. [DOI] [PubMed] [Google Scholar]
- 26.Moolman JC, Corfield VA, Posen B, et al. Sudden death due to troponin T mutations. J Am Coll Cardiol. 1997;29:549–555. doi: 10.1016/s0735-1097(96)00530-x. [DOI] [PubMed] [Google Scholar]
- 27.Li D, Czernuszewicz GZ, Gonzalez O, et al. Novel cardiac troponin T mutation as a cause of familial dilated cardiomyopathy. Circulation. 2001;104:2188–2193. doi: 10.1161/hc4301.098285. [DOI] [PubMed] [Google Scholar]
- 28.Mogensen J, Murphy RT, Shaw T, et al. Severe disease expression of cardiac troponin C and T mutations in patients with idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 2004;44:2033–2040. doi: 10.1016/j.jacc.2004.08.027. [DOI] [PubMed] [Google Scholar]
- 29.Varnava A, Baboonian C, Davison F, et al. A new mutation of the cardiac troponin T gene causing familial hypertrophic cardiomyopathy without left ventricular hypertrophy. Heart. 1999;82:621–624. doi: 10.1136/hrt.82.5.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang H, Woo A, Monakier D, et al. Enlarged left atrial volume in hypertrophic cardiomyopathy: a marker for disease severity. J Am Soc Echocardiogr. 2005;18:1074–1082. doi: 10.1016/j.echo.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 31.Fujino N, Shimuzu M, Ino H, et al. Cardiac troponin T Arg92Trp mutation and progression from hypertrophic to dilated cardiomyopathy. Clin Cardiol. 2001;24:397–402. doi: 10.1002/clc.4960240510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fujino N, Shimuzu M, Ino H, et al. A novel mutation Lys273Glu in the cardiac troponin T gene shows high degree of penetrance and transition from hypertrophic to dilated cardiomyopathy. Am J Cardiol. 2002;89:29–33. doi: 10.1016/s0002-9149(01)02158-0. [DOI] [PubMed] [Google Scholar]
- 33.Maron BJ, Spirito P. Implications of left ventricular remodeling in hypertrophic cardiomyopathy. Am J Cardiol. 1998;81:1339–1344. doi: 10.1016/s0002-9149(98)00164-7. [DOI] [PubMed] [Google Scholar]