

Abstract
Previous studies have shown that ketone bodies (KB) exert antioxidant effects in experimental models of neurological disease. In the present study, we explored the effects of the KB acetoacetate (ACA) and β-hydroxybutyrate (BHB) on impairment of hippocampal long-term potentiation (LTP) in rats by hydrogen peroxide (H2O2) using electrophysiological, fluorescence imaging and enzyme assay techniques. We found that: (1) a combination of ACA and BHB (1 mM each) prevented impairment of LTP by H2O2 (200 μM); (2) KB significantly lowered intracellular levels of reactive oxygen species (ROS) — measured with the fluorescent indicator carboxy-H2DCFDA — in CA1 pyramidal neurons exposed to H2O2; (3) the effect of KB on LTP was replicated by the protein phosphatase 2A (PP2A) inhibitor fostriecin; (4) KB prevented impairment of LTP by the PP2A activator C6 ceramide; (5) fostriecin did not prevent the increase in ROS levels in CA1 pyramidal neurons exposed to H2O2, and C6 ceramide did not increase ROS levels; (6) PP2A activity was enhanced by both H2O2and rotenone – a mitochondrial complex I inhibitor that increases endogenous superoxide production; and (7) KB inhibited PP2A activity in protein extracts from brain tissue treated with either H2O2 or ceramide. We propose that oxidative impairment of hippocampal LTP is associated with PP2A activation, and that KB prevent this impairment in part by inducing PP2A inhibition through an antioxidant mechanism.
Keywords: ketones, oxidative stress, plasticity, long-term potentiation, protein phosphatase 2A
INTRODUCTION
Oxidative stress, a condition in which cellular defense mechanisms are overwhelmed by reactive oxygen species (ROS), has been repeatedly implicated in the pathogenesis of chronic neurodegenerative disorders such as Alzheimer’s and Parkinson’s diseases, and of acute brain injuries caused by stroke and head trauma (Keller et al., 2005; Mariani et al., 2005; Moreira et al., 2006; Reddy, 2006). Antioxidant therapies have therefore been the focus of intense research. Recent studies have shown that the ketone bodies (KB) acetoacetate (ACA) and β-hydroxybutyrate (BHB) protect neurons against excitotoxic injury by decreasing mitochondrial ROS production, but the functional significance of this antioxidant effect has not yet been investigated (Noh et al., 2006; Maalouf et al., 2007).
The hippocampus is a critical region for several forms of learning and memory, but it is highly susceptible to oxidative stress, and is hence a common site of injury in many neurological diseases (Sano and Kirino, 1990; Mahieux, 2003). Importantly, high concentrations of hydrogen peroxide (H2O2) have been shown to impair hippocampal long-term potentiation (LTP), a very useful measure of neuronal function and integrity in vitro (Kamsler and Segal, 2003, 2004). The mechanisms underlying the inhibitory effects of H2O2on LTP are incompletely understood, but serine/threonine phosphatases have previously been implicated (Winder and Sweatt, 2001; Kamsler and Segal, 2003, 2004). Specifically, hippocampal LTP has been associated with decreased activity of the protein serine/threonine phosphatase 2A (PP2A). In addition, the ketogenic diet, a high-fat and low-carbohydrate anticonvulsant diet that induces ketonemia to low millimolar concentrations, down-regulates PP2A activity and expression (Fukunaga et al., 2000; Noh et al., 2004). Consequently, we tested the hypothesis that KB prevent oxidative impairment of LTP by inhibiting PP2A.
We have previously shown that a combination of ACA and BHB in low millimolar concentrations provides optimal protection against excitotoxic and oxidative injury (Kim et al., 2007; Maalouf et al., 2007). ACA and BHB administered individually were neuroprotective as well but required higher concentrations. In the present study, we chose to continue using a combination of ketone bodies in low millimolar concentrations as this effective treatment paradigm more accurately reproduces the metabolic environment induced by calorie restriction or the ketogenic diet, two potential neuroprotective interventions (Haymond et al., 1982; Lamers et al., 1995; Thavendiranathan et al., 2000). Our results, based on electrophysiological, fluorescence imaging and direct biochemical measurements, indicate that H2O2-induced inhibition of LTP is associated with increased PP2A activity and that ACA and BHB (1 mM each) may prevent these changes by decreasing PP2A activity in an antioxidant manner.
Materials and methods
All protocols were approved by the Institutional Animal Care and Use Committee. Fostriecin and N-Hexanoyl-D-erythro-Sphingosine (C6 Ceramide) were purchased from EMD Biosciences (San Diego, CA). All other chemicals were purchased from Sigma (St. Louis, MO).
Slice preparation
Rats aged postnatal days 18-26 (P18-26) were anesthetized with isoflurane (Baxter; Deerfield, IL). After decapitation, the brain was rapidly removed and submerged in ice-cold, oxygenated artificial cerebrospinal fluid (aCSF in mM: NaCl 124, KCl 3, KH2PO41.25, MgSO42.5, CaCl2 3.4, NaHCO3 26, glucose 10, pH = 7.4). Horizontal slices (300–400 μm thick) were prepared with a vibratome (The Vibratome Company; St. Louis, MO). The hippocampal region and entorhinal cortex were dissected out and incubated in aCSF at room temperature for 1 hour before further experimentation.
Electrophysiology
All electrophysiological and fluorescence imaging experiments were conducted at room temperature. Hippocampal slices (400 μm) were transferred to a submerged recording chamber (RC-22; Warner Instruments; Hamden, CT) placed under an Axioskop FS 2 microscope (Carl Zeiss Microimaging, Inc.; Thornton, NY, USA). A MCE-100 bipolar concentric electrode (David Kopf Instruments, Tujunga, CA) was used to stimulate Schaffer collaterals, and a borosilicate recording electrode (1.5 mm external diameter, 0.75 mm internal diameter; WPI; Sarasota, FL) filled with 2M NaCl (impedance, 2 MΩ) was placed in the stratum radiatum of the CA1 subfield. Field potentials were recorded with a Multiclamp 700B amplifier (Axon Instruments; Union City, CA) and digitized with a Digidata 1322A (Axon Instruments). Stimulation was delivered every 30 s through an A365 stimulus isolator (WPI; Sarasota, FL). Field potential slopes were calculated with Clampfit (Axon Instruments) and normalized to the mean slope during the first 5 min of recording. Stimulus intensities were adjusted to elicit field potential amplitudes that were half of the maximal amplitude. Long-term potentiation of Schaffer collaterals was achieved with a single high-frequency burst (100 Hz for 1 s) using an intensity twice as high as during baseline recording.
Fluorescence imaging
To measure hydrogen peroxide levels in CA1 stratum pyramidale, slices (300 μm) were incubated with the intracellular hydrogen peroxide indicator carboxy-2’,7’-dichlorodihydrofluorescein diacetate (carboxy-H2DCFDA) for 30 min at room temperature, and then transferred to the recording chamber. Fluorescent light was generated with an X-Cite 120 lamp (Photonics Solutions Inc, Canada) and passed through a Zeiss filter set 10 (excitation 450 to 490 nm, emission 515 to 565 nm; Carl Zeiss Microimaging, Inc.). Fluorescence of pyramidal neurons in CA1 was measured with Axiovision 4.3 (Carl Zeiss Microimaging, Inc.) and analyzed after normalization to an adjacent area in stratum radiatum.
Protein phosphatase 2A assay
Protein phosphatase 2A activity was assayed with the Promega Serine/Threonine Phosphatase Assay System, a non-radioactive method that determines amounts of free phosphates by measuring the absorbance of a molybdate : malachite green : phosphate complex (Promega Corporation; Madison, WI). Slices (400 μm) were homogenized in Tris-EDTA buffer (Tris 10 mM, EDTA 1 mM, sodium azide 0.02%; pH = 7.5) containing a mixture of protease inhibitors with broad specificity for the inhibition of serine, cysteine, aspartic proteases and aminopeptidases (1 μl of 4-2-aminoethyl-benzenesulfonyl fluoride, pepstatinA, E-64, bestatin, leupeptin, and aprotinin per 1 ml of homogenization buffer) as well as the protein phosphatase 1A inhibitor Inhibitor-2 (0.1 μM). The homogenized lysate was centrifuged a first time at 100,000 × g for 1 h at 4°C to remove particulate matter and then a second time in the Promega Spin Column at 600 × g for 5 min at 4°C to exclude endogenous phosphates. Final protein concentration was measured with the Pierce BCA Protein Assay Kit (Pierce, Rockford, IL). To measure PP2A activity, protein extracts (5 μg) were mixed with 10 μl of PP2A reaction buffer (250 mM imidazole, 1 mM EGTA, 0.1% β-mercaptoethanol, 0.5 mg/ml BSA), 5 μl of 1mM phosphopeptide and 100 μl of Tris-EDTA buffer and then transferred to a 96-well plate (1.2 area, flat bottom). After incubation at 37°C, the enzymatic reactions were stopped at 0, 20 and 40 min by adding the Molybdate Dye. To determine phosphate levels, the optical densities of the samples were measured with a fluorescence plate reader (Tecan SPECTRA Fluor; Durham, NC) at 590 nm following a 30 min incubation period at room temperature. Finally, the percent increase in phosphate concentration between the 20thand 40thminute interval was calculated to estimate PP2A activity.
Statistics
Treatment groups were compared using Kruskal-Wallis One Way Analyses of Variance on Ranks with Dunn’s post-hoc test. All analyses were performed with SigmaStat V2.03 (SPSS Inc, Chicago, IL).
Results
KB prevent H2O2-mediated impairment of long-term potentiation
In the presence of 200 μM H2O2(n = 6, from 5 rats), high-frequency burst stimulation of the Schaffer collaterals (100 Hz for 1 s) led to a statistically insignificant decrease (3 ± 2%; mean ± S.E.M.) in the field EPSP slope of the CA1 field potential, whereas in the control situation (n = 6, from 5 rats), the slope increased by 76 ± 2% one hour after tetanic stimulation (H2O2 application was initiated 5 min prior to tetanic stimulation and did not have an appreciable effect on the baseline characteristics of the field EPSP) (Fig. 1A). When ACA and BHB together (1 mM each) were added 20 min prior to H2O2 (n = 6, from 4 rats), a 110 ± 15% increase in the EPSP slope occurred after tetanic stimulation (Fig. 1A). The differences between control and 200 μM H2O2 alone and between 200 μM H2O2 alone and 200 μM H2O2 with 1 mM KB were statistically significant (p < 0.05). Data collected in the last 5 minutes of each recording (i.e., 55 to 60 min after tetanic stimulation) were used for statistical analyses. Pre-incubation with ketone bodies for only 5 min did not counteract the effects of H2O2 (3 ± 4% decrease; n = 5, from 3 rats; not significant; data not shown). Moreover, LTP was not affected by exposure to ketone bodies alone (89 ± 12% increase; n = 6, from 3 rats; not significant) or to 20 μM H2O2 (71 ± 15% increase; n = 5, from 5 rats; not significant) (Fig. 2).
Figure 1.
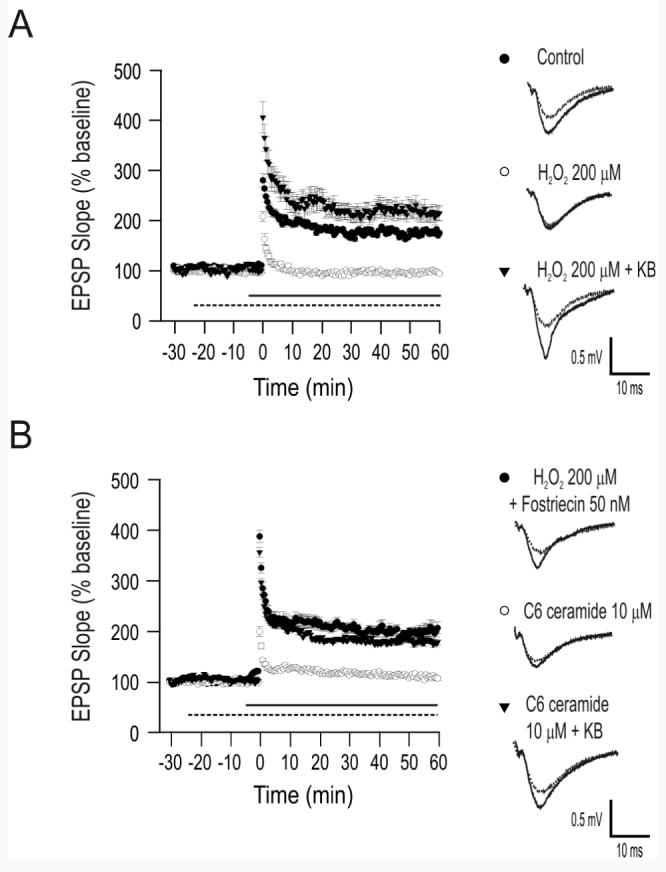
Ketone bodies (KB) prevent H2O2-mediated impairment of long-term potentiation (LTP) in CA1. (A) Long-term potentiation was significantly impaired by exposure to 200 μM H2O2 starting 5 min prior to high-frequency burst stimulation (straight horizontal line). Pre-treatment with a combination of the ketone bodies acetoacetate (ACA; 1 mM) and β-hydroxybutyrate (BHB; 1 mM) for 20 min prior to H2O2 administration (dotted horizontal line) resulted in the full expression of LTP. (B) The effects of KB were replicated by the protein phosphatase 2A (PP2A) inhibitor fostriecin (50 nM, applied 20 min prior to H2O2). Moreover, exposure to the PP2A activator ceramide (10 μM) 5 min prior to high-frequency burst stimulation (straight horizontal line) blocked LTP but, as with H2O2, pre-treatment with KB for 20 min (dotted horizontal line) prevented ceramide-induced impairment of LTP. Each set of traces in panels a and b reflect the mean ± S.E.M. of 5-6 recorded cells from 3-5 rats. Error bars, when not visible, were smaller than the symbols used.
Figure 2.

Summary of the effects of different pharmacological treatments on LTP, quantified by comparing the average field potential slope 1 h after induction to the average slope during the first 5 min of recording. The neuroprotective effects of KB were time-dependent. Furthermore, activation and inhibition of PP2A mimicked the effects of H2O2 and KB, respectively, thereby suggesting that PP2A mediates the effects of KB on LTP under conditions of oxidative stress. All differences were statistically significant (* indicates p < 0.05 relative to control, + indicates p < 0.05 relative to 200 μM H2O2, ## indicates p < 0.01 relative to 10 μM ceramide). Each vertical bar reflects the mean ± S.E.M. of 5-6 recorded cells from 3-5 rats.
Inhibition of PP2A replicates the protective effects of KB
Pre-incubation with 50 nM fostriecin, a specific PP2A inhibitor (Lewy et al., 2002), for 20 min prior to H2O2(200 μM) perfusion led to a 102 ± 13% increase in EPSP slope one hour after high-frequency burst stimulation (n = 5, from 3 rats; p < 0.05 relative to 200 μM alone; Fig. 1B). Treatment with 50 nM fostriecin alone prior to tetanic stimulation led to a 110 ± 2% increase in the EPSP slope (n = 4, from 2 rats) (Fig. 2). Moreover, inhibition of LTP by the PP2A activator C6 ceramide (N-Hexanoyl-D-erythro-Sphingosine) was prevented by KB (Fig. 1B). C6 ceramide is a short-chain, non-physiological form of ceramide that increases long-chain, endogenous ceramide synthesis in a series of enzymatic reactions that are dependent on ROS and that are inhibited by glutathione (Sultan et al., 2006; Won and Singh, 2006). Administration of 10 μM C6ceramide 10 min prior to high-frequency stimulation resulted in a statistically insignificant (i.e., 8 ± 15%) increase in the EPSP slope only (n = 5, from 4 rats; p < 0.05 relative to control) (Fig. 1B). Shorter pre-incubation times and lower concentrations did not produce any discernible effects on LTP, and C6ceramide did not affect the baseline characteristics of the field EPSP. The pre-incubation time in our experiments was consistent with previous findings indicating that C6ceramide conversion to endogenous ceramide requires at least 10 min (Sultan et al., 2006). When both ACA and BHB (1 mM each) were added 20 min prior to C6ceramide administration and continued throughout the experiment, the EPSP slope increased by 78 ± 6% (n = 6 from 4 rats; p < 0.01 relative to 10 μM ceramide alone). Summary data are presented in Fig. 2 (Analyses of Variance F score = 4.29).
KB decrease reactive oxygen species levels in CA1 pyramidal neurons exposed to H2O2
To determine the effects of exogenous H2O2 and KB on intracellular ROS levels, hippocampal slices were imaged during the first 30 minutes of each LTP experiment (from the first field potential recording to the high-frequency burst) with carboxy-H2DCFDA, a cell-permeant fluorescent indicator for H2O2 mainly but also possibly superoxide, nitric oxide (NO) and peroxynitrite (Hempel et al., 1999). Carboxy-H2DCFDA becomes fluorescent after its acetate groups are removed by intracellular esterases and after oxidation occurs within the cell. Therefore, in our model, carboxy-H2DCFDA fluorescence measurements reflect steady-state levels (i.e. both exogenously applied and endogenously produced H2O2).
ROS levels were measured in CA1 stratum pyramidale rather than in stratum radiatum, the site where LTP occurs. Although carboxy-H2DCFDA was designed to remain sequestered inside cells following activation by intracellular esterases, we have noticed that significant leakage does occur and can contaminate measurements of intracellular fluorescence. Cell bodies emit, however, a stronger and more easily delineated fluorescence signal than axons and dendrites. Consequently, intracellular ROS levels can be more accurately isolated in stratum pyramidale than in stratum radiatum where activated carboxy-H2DCFDA leaking from Schaffer collaterals and dendrites of CA1 pyramidal neurons cannot be distinguished from intracellular levels.
The neuroprotective effect of ketone bodies was associated with decreased fluorescence of carboxy-H2DCFDA following exposure to H2O2. CA1 neurons displayed a 46 ± 5% increase (over control values) in carboxy-H2DCFDA fluorescence following exposure to 200 μM H2O2for 5 min (n = 7, from 4 rats) (Fig. 3A). Pre-treatment with KB for 20 min resulted in a 4 ± 1% decrease (relative to control) of the signal following a 5 min exposure to H2O2 (n = 7 from 4 rats; p < 0.05 relative to 200 μM H2O2 alone; Fig. 3B). A similar antioxidant effect of ketone bodies was found at much higher concentrations of H2O2 (Kim et al., 2007). Application of 10 mM hydrogen peroxide for 5 min increased carboxy-H2DCFDA fluorescence by 67 ± 6% in the absence of ketone bodies (n = 5, from 2 rats) but only by 23 ± 3% in their presence (n = 5, from 3 rats; Fig. 3C). The difference was statistically significant (p < 0.05). The F score for the analysis of variance was 9.86.
Figure 3.
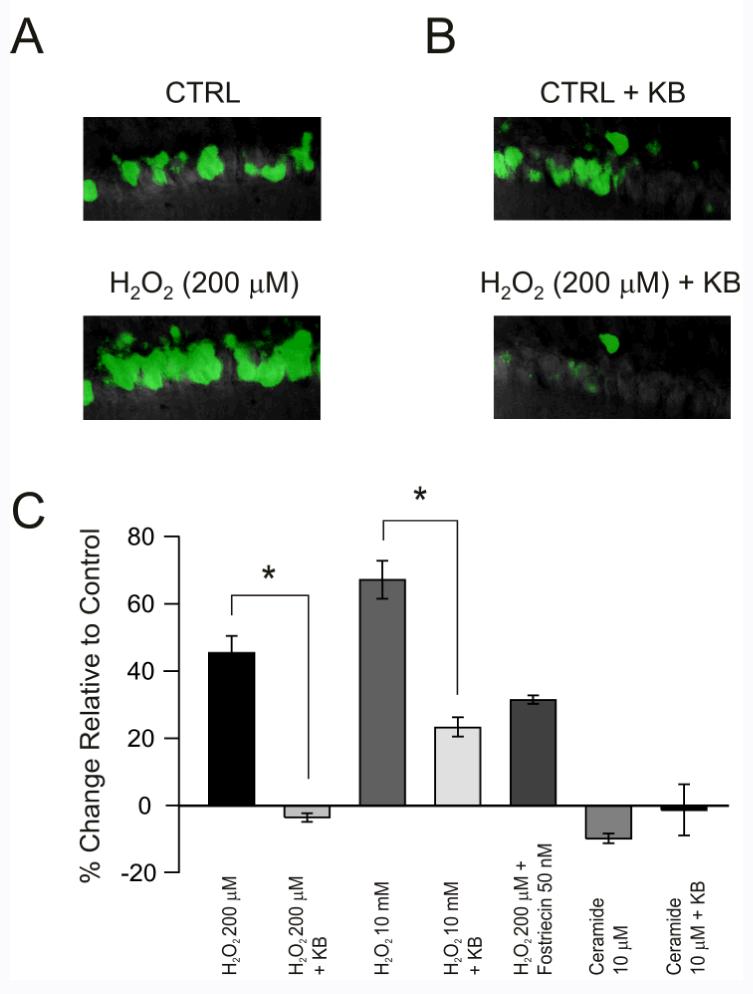
Intracellular levels of reactive oxygen (ROS) species in CA1 pyramidal neurons exposed to H2O2 are decreased by ketone bodies (KB), but are not affected by pharmacological manipulation of protein phosphatase 2A (PP2A). (A) Intracellular ROS levels, measured with the fluorescent indicator carboxy-H2DCFDA, increased following exposure to exogenous H2O2 (200 μM) for 5 min (top panel shows CA1 pyramidal neurons before H2O2 and bottom panel shows the same neurons after H2O2). (B) Pre-treatment with KB for 20 min significantly decreased intracellular ROS levels despite the presence of H2O2(top panel shows CA1 pyramidal neurons pre-treated with KB but before H2O2and bottom panel shows the same neurons after H2O2). (C) The antioxidant effects of KB were evident with much higher doses of H2O2 (10 mM). The PP2A inhibitor fostriecin did not significantly alter the effects of H2O2 and the PP2A activator ceramide did not affect intracellular ROS levels. Each vertical bar reflects the mean ± S.E.M. of 5-7 cells from 2-4 rats; *p < 0.05.
KB-induced reduction in ROS levels is independent of PP2A
In contrast to KB, pre-treatment of hippocampal slices with 50 nM fostriecin (n = 5, from 3 rats) for 20 min did not prevent the increase in carboxy-H2DCFDA fluorescence (31 ± 1%) caused by H2O2 exposure (200 μM for 5 min) (Fig. 3C). Consistently, stimulation of PP2A by C6ceramide (up to 100 μM) did not increase carboxy-H2DCFDA fluorescence above control levels (8 ± 1% decrease; not significant; Fig. 3C). Our data therefore suggested that PP2A activity was not associated with oxidative changes within the time-frame studied, despite the fact that ketone body effects on LTP were replicated by PP2A inhibition.
The protective effect of KB involves inhibition of PP2A
To further clarify the association between oxidative stress, KB and PP2A, we used an enzymatic assay to directly measure PP2A activity. Proteins were extracted from hippocampal slices exposed to various combinations of ROS, KB and PP2A activators and inhibitors. Following removal of endogenous phosphates, a phosphopeptide - specifically formulated to react with serine/threonine phosphatases - was added to the protein extracts and the amount of free phosphate generated by the reaction was measured. All procedures were performed in the absence of cations and in the presence of calcium chelators to minimize the activity of protein phosphatases 2B and 2C, and in the presence of a protein phosphatase 1 inhibitor.
Due to the lower sensitivity of the enzymatic assay relative to electrophysiological measurements, neurons were exposed to longer durations and higher concentrations of H2O2to ensure that the resultant phosphate levels were within the range detectable by the assay. For the same reasons, the use of entire slices, rather than specific micro-dissected regions of the hippocampus, was necessary to isolate adequate amounts of PP2A. The effective range of the assay is 100 to 4000 pmol of phosphate and, in our hands, levels below 200 pmol yielded highly variable results.
Exposure of hippocampal slices to H2O2for 20 min led to a dose-dependent increase in PP2A activity above control levels (41 ± 20% at 200 μM with n = 8, from 8 rats; 101 ± 6% at 2 mM with n = 15, from 15 rats) (Fig. 4). Exposure to 200 µM H2O2or incubations times shorter than 20 min did not considerably affect PP2A activity in this assay, most likely because phosphate levels were too low for detection by the assay (Fig. 4). Rotenone (10 μM for 20 min), a mitochondrial respiratory inhibitor that increases endogenous superoxide production at complex I (Sherer et al., 2003; Xu et al., 2003; Radad et al., 2006), produced the same outcome as exogenous H2O2 (190 ± 17% increase relative to control; n = 8, from 8 rats; p < 0.01 relative to control) (Fig. 4). Pre-incubation with a combination of acetoacetate and β-hydroxybutyrate (1 mM each; n = 11, from 11 rats) or fostriecin (50 nM; n = 7, from 7 rats) for 1 h inhibited the effects of 2 mM H2O2(13 ± 11% and 37 ± 7% increase above control levels, respectively) (Fig. 4). The inhibitory effects of KB and fostriecin were statistically significant (p < 0.05 relative to 2 mM H2O2 alone). A large increase in PP2A activity was also observed with 10 μM C6 ceramide for 20 min (142 ± 5%, n = 13, from 13 rats), further demonstrating the specificity of the assay for PP2A (Fig. 4). This increase was prevented by ketone bodies (77 ± 5% increase; n = 12, from 12 rats; p < 0.01 relative to 10 μM ceramide alone) (Fig. 4; analysis of variance F score = 3.20).
Figure 4.
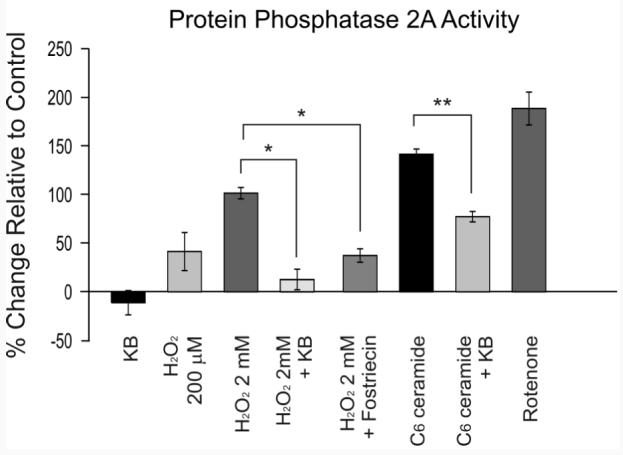
The neuroprotective effects of ketone bodies (KB) involve the inhibition of protein phosphatase 2A (PP2A). The results of a colorimetric assay measuring the amount of phosphate generated by the interaction of PP2A with a serine/threonine phosphopeptide clearly show that PP2A is activated by H2O2. Ketone bodies prevented PP2A activation by H2O2 and ceramide. Each vertical bar reflects the mean ± S.E.M. of 7-15 cells from 7-15 rats; *p < 0.05, **p < 0.01.
Discussion
The principal finding of this study is that ACA and BHB protect against H2O2-induced impairment of hippocampal LTP, possibly in part by inhibiting protein serine/threonine phosphatase 2A. Furthermore, activation of this enzyme appears to be critically involved in the disruption of LTP induction by oxidative stress. These results provide evidence that the neuroprotective effects of KB, which include reduced mitochondrial ROS formation and inhibition of mitochondrial permeability transition (mPT), not only decrease cellular loss but also improve neuronal function during oxidative stress (Maalouf et al., 2007; Kim et al., 2007).
The effects of ROS on long-term potentiation
The deleterious effects of ROS have been well documented using various experimental protocols. Under pathological conditions (e.g., following glutamate excitotoxicity), ROS production increases significantly, damaging nucleic acids, proteins and lipids, and triggers the opening of mPT pores, high conductance channels that form in mitochondrial membranes and cause swelling followed by apoptotic cell death (Mattson et al., 2003; Nicholls, 2004; Balaban et al., 2005; Bernardi and Forte, 2007). Whether ROS facilitates or inhibits hippocampal LTP remains less clear, however. Some studies have shown that H2O2 can impair LTP (Kamsler and Segal, 2003), while others have suggested that H2O2 can enhance LTP and that antioxidant treatments can inhibit LTP (Knapp and Klann, 2002; Serrano and Klann, 2004). These latter observations support the hypothesis that, under physiological conditions, ROS are mediators of synaptic plasticity. Consistent with this hypothesis, LTP was significantly attenuated by the administration of exogenous superoxide dismutase, which converts the superoxide radical to O2 and H2O2, and treatment of hippocampal slices with superoxide scavengers prevented the full expression of LTP in CA1 (Klann and Thiels, 1999).
The discordant effects of H2O2 on hippocampal LTP are most likely a function of H2O2 concentration and duration of exposure. High micromolar or millimolar concentrations of H2O2 that consistently produce oxidative stress (Hyslop et al., 1995; Lei et al., 1997), as was the case in the present study, inhibit LTP in CA1, whereas lower micromolar concentrations enhance LTP (Kamsler and Segal, 2003). There are other reasons why the results from previously published studies should be viewed cautiously (Klann and Thiels, 1999). First, enhanced superoxide dismutase activity might impair LTP because of a secondary increase in H2O2 concentration. This possibility is supported by the fact that catalase reversed the effects of superoxide dismutase. Second, the enhanced synaptic transmission might reflect abnormally increased neuronal excitability, an early sign of neuronal injury, rather than an increase in synaptic strength. Impairment of LTP might therefore represent an early manifestation of a pathological process.
Modulation of PP2A activity by ROS
Methodological differences could explain the contradictory effects of H2O2on PP2A, a type 2 serine/threonine phosphatase that, unlike other members of this enzyme family, can be activated in the absence of calcium or other divalent cations (Cohen, 1991). PP2A is composed of a catalytic subunit, a structural subunit and one of many possible regulatory subunits that determine substrate specificity and cellular localization (Janssens and Goris, 2001; Lechward et al., 2001). We found that PP2A was activated by both exogenous (i.e., H2O2) and endogenous (superoxide production by complex I following rotenone exposure) sources of oxidative stress. Interestingly, Whisler et al. (1995) showed that 100-200 μM H2O2 inhibited PP2A in Jurkat cells, a cell line derived from human T-cell leukemia. In contrast, we demonstrated that, in hippocampus, PP2A is stimulated by high concentrations of H2O2. Although the reasons behind this difference remain unclear, abnormal PP2A activity has been reported in chronic myelogenous leukemia (Neviani et al., 2005). Therefore, the neoplastic nature of Jurkat cells might confer differential responses to oxidative stress.
Considering previous studies that showed an association between decreased PP2A activity and facilitation of LTP (and inhibition of long-term depression) in the hippocampus (Fukunaga et al., 2000; Kang-Park et al., 2003), our findings suggest that H2O2 inhibits LTP by activating PP2A. Consistently, we observed that activation of PP2A with C6 ceramide inhibited LTP and that fostriecin, a PP2A inhibitor, reversed the inhibitory effects of H2O2 on LTP. Ceramide is endogenously synthesized from sphingomyelin, a ubiquitous membrane component, in a series of enzymatic reactions that are enhanced by ROS and inhibited by antioxidants such as glutathione (Won and Singh, 2006). C6 ceramide, an exogenous, short-chain, non-physiological form of ceramide causes endogenous ceramide production in a series of enzymatic reactions that are also stimulated by ROS (Sultan et al., 2006).
Kamsler and Segal (2003) described similar findings for the serine/threonine phosphatase 2B (PP2B, also termed calcineurin). Higher doses of hydrogen peroxide inhibited LTP and activated PP2B. In addition, both PP2A and PP2B have been implicated in cellular apoptosis. Consistently, the molecular targets of PP2A and PP2B are similar and include NMDA receptors, the Bcl-2 family and the mitochondrial permeability transition pore (Garcia et al., 2003; Mansuy and Shenolikar, 2006; Hara and Synder, 2007). It is presently not clear, however, if these two enzymes function in parallel or interact with each other to regulate synaptic plasticity and apoptosis.
The neuroprotective effects of KB
Following one day of fasting or exposure to the ketogenic diet, the levels of two ketone bodies, β-hydroxybutyrate and acetoacetate, increase significantly, reaching millimolar concentrations in the blood and moderately lower concentrations in the cerebrospinal fluid (Haymond et al. 1982; Lamers et al. 1995; Seymour et al. 1999; Thavendiranathan et al. 2000). The levels of other ketone bodies such as acetone increase as well but much more modestly (Laffel 1999). Therefore, to replicate the metabolic environment induced by fasting or by the ketogenic diet, we chose to study the neuroprotective effects of β-hydroxybutyrate and acetoacetate simultaneously at physiological (low millimolar) concentrations. In support of this protocol, previous findings have shown that a combination of β-hydroxybutyrate and acetoacetate in low millimolar concentrations (1 mM each) protects acutely isolated neurons against oxidative stress and glutamate excitotoxicity (Kim et al. 2007; Maalouf et al. 2007).
In the present study, KB decreased intracellular H2O2 during exposure to exogenous H2O2, inhibited PP2A activity and prevented oxidative impairment of LTP. The observed changes in ROS levels and PP2A activity were based, however, on measurements in cell bodies and in homogenates of whole hippocampal slices, respectively. These approaches were chosen to compensate for technical limitations but potentially limit the interpretation of the data. First, it was not possible for us to determine if KB decreased ROS levels specifically in Schaffer collaterals or in dendrites of CA1 neurons. Second, changes in PP2A activity may have occurred in the dentate gyrus or in entorhinal cortex rather than in CA1 and CA3. Nevertheless, although these data do not demonstrate the effects of KB directly on CA1 and CA3 neurons, we believe that they describe the general effects of KB on ROS levels and PP2A activity in neurons and that they support our electrophysiological findings.
In previous studies, we showed that, in isolated neocortical pyramidal neurons, KB decrease basal ROS production and block the increase in ROS production following oxidative injury (Kim et al., 2007; Maalouf et al., 2007). In the present study, we confirm that KB block ROS production following exposure to exogenous H2O2 and further show that KB inhibit PP2A. Given that, under our experimental conditions, PP2A modulation did not affect intracellular ROS levels, we propose that KB might inhibit PP2A through their antioxidant properties, thereby preventing oxidative impairment of LTP (Fig. 5).
Figure 5.

Illustration summarizes hypothetical mechanisms underlying the protective effects of ketone bodies (KB) on oxidative impairment of hippocampal long-term potentiation (LTP). Increased intracellular reactive oxygen species (ROS) levels caused by exposure to exogenous H2O2or rotenone, a complex I inhibitor that increases endogenous ROS formation, enhance endogenous ceramide synthesis from sphingomyelin. In turn, ceramide activates protein phosphatase 2A (PP2A), subsequently leading to inhibition of LTP. By decreasing intracellular ROS levels, KB inhibit the synthesis of ceramide and PP2A, and thereby prevent LTP inhibition.
Alternative interpretations of the results
Our results do not rule out the possibility that factors other than H2O2 and PP2A are involved. First, carboxy-H2DCFDA is possibly sensitive to oxidative and nitrogen species other than H2O2, namely superoxide, NO and peroxynitrite. Maalouf et al. (2007) have previously shown that KB decrease mitochondrial formation of superoxide, a possible mediator of LTP (Klann and Thiels, 1999). Moreover, Maalouf et al. (1998) showed that inhibition of NO synthase enhances synaptic plasticity. Given that changes in carboxy-H2DCFDA fluorescence following exposure to H2O2or KB might reflect changes in superoxide or NO levels, inhibition of LTP by exogenous H2O2 and the normal expression of LTP in the presence of KB could be secondary to changes in intracellular superoxide or NO levels. However, it is important to note that, although these observations raise the possibility of factors other than H2O2 modulating LTP in our model, they do not contradict the more general conclusion that KB reduce intracellular ROS levels.
Second, carboxy-H2DCFDA fluorescence also increases following direct activation by the antioxidant enzymes catalase and copper/zinc superoxide dismutase (Hempel et al., 1999). Increased activity of antioxidant enzymes most probably occurs as a compensatory mechanism to H2O2 administration and could therefore potentially explain the observed increase in carboxy-H2DCFDA fluorescence. Consistently, decreased ROS production following treatment with KB would lead to a parallel reduction of antioxidant enzyme activity and, consequently, carboxy-H2DCFDA fluorescence. More importantly however, this possibility supports again the general conclusion that KB reduce intracellular ROS levels.
Third, although the enzyme assay was optimized to measure the activity of PP2A, it does not distinguish among the various subtypes of this enzyme family that also includes protein phosphatase 4 (previously termed protein phosphatase X) and protein phosphatase 6 (Kloeker et al., 2003). The amino acid sequences and the pharmacological sensitivities of these related enzymes are highly similar. For instance, protein phosphatase 4, which is highly expressed in the brain, including the hippocampus, is inhibited by fostriecin (Kloeker et al., 1997; Cohen et al., 2005). Therefore, although our findings do not point to a specific PP2A subtype, they do in general implicate the protein phosphatase 2A family of enzymes in the impairment of LTP by oxidative stress.
Implications of the findings
KB decrease the mitochondrial production of ROS at complex I and delay the opening of the mitochondrial permeability transition (mPT) pore following exposure to exogenous H2O2 or to the thiol oxidant diamide (Kim et al., 2007; Maalouf et al., 2007). As it is known that PP2A activation facilitates the opening of the mPT pore (Garcia et al., 2003; Van Hoof and Goris, 2003), it would be expected that inhibition of PP2A might also be neuroprotective. Therefore, the present study suggests a further mechanistic link between KB and inhibition of mPT, which when activated, results in the release of cytochrome c into the cytoplasm and the initiation of the apoptotic cascade (Nicholls, 2004).
Our results further demonstrate that, in addition to inhibiting neuronal death and injury, KB treatment prevents the disruption of synaptic plasticity by oxidative stress. These effects are highly consistent with those of calorie restriction on long-term potentiation (Hori et al., 1992; Eckles-Smith et al., 2000; Okada et al., 2003). In fact, in vitro measures of neuronal health have revealed many similarities between the neuroprotective effects of calorie restriction, of the ketogenic diet and of KB following exposure to amyloid Aβ, mitochondrial toxins that can cause Parkinson’s disease or epileptogenic injuries (Kashiwaya et al., 2000; Mattson et al., 2003; Gasior et al., 2006). Whether ketone bodies can also improve cognitive function or reduce neurological deficits in behaving animals like calorie restriction and the ketogenic diet remains unknown however. To our knowledge, studies of KB in vivo have demonstrated their anticonvulsant properties (Rho et al., 2002) but have not looked at cognitive or other neurological measures. Nevertheless, given all the in vitro similarities between these three interventions, we strongly believe that KB, administered parenterally or as a dietary supplement, would be beneficial in neurological diseases and therefore deserve further investigation in the future.
Acknowledgements
This work was supported by NIH grant NS 044846 (JMR) and the Barrow Neurological Foundation.
Abbreviations
- LTP
long-term potentiation
- KB
ketone bodies
- PP2A
protein phosphatase 2A
- H2O2
hydrogen peroxide
- ROS
reactive oxygen species
- BHB
β-hydroxybutyrate
- ACA
acetoacetate
- (carboxy-H2DCFDA)
carboxy-2′,7′-dichlorodihydrofluorescein diacetate
- mPT
mitochondrial permeability transition.
Footnotes
Corresponding Author: Jong M. Rho, MD. Neurology Research, NRC 4thFl. Barrow Neurological Institute and St. Joseph’s Hospital & Medical Center 350 W. Thomas Road, Phoenix, AZ 85013 Tel. 602-406-3156 Fax. 602-406-5779 Email: jong.rho@chw.edu
References
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Bernardi P, Forte M. The mitochondrial permeability transition pore. Novartis Found Symp. 2007;287:157–164. [PubMed] [Google Scholar]
- Cohen P. Classification of protein-serine/threonine phosphatases: identification and quantitation in cell extracts. Methods Enzymol. 1991;201:389–398. doi: 10.1016/0076-6879(91)01035-z. [DOI] [PubMed] [Google Scholar]
- Cohen PT, Philp A, Vazquez-Martin C. Protein phosphatase 4 - from obscurity to vital functions. FEBS Lett. 2005;579:3278–3286. doi: 10.1016/j.febslet.2005.04.070. [DOI] [PubMed] [Google Scholar]
- Eckles-Smith K, Clayton D, Bickford P, Browning MD. Caloric restriction prevents age-related deficits in LTP and in NMDA receptor expression. Brain Res Mol Brain Res. 2000;78:154–162. doi: 10.1016/s0169-328x(00)00088-7. [DOI] [PubMed] [Google Scholar]
- Fukunaga K, Muller D, Ohmitsu M, Bako E, DePaoli-Roach AA, Miyamoto E. Decreased protein phosphatase 2A activity in hippocampal long-term potentiation. J Neurochem. 2000;74:807–817. doi: 10.1046/j.1471-4159.2000.740807.x. [DOI] [PubMed] [Google Scholar]
- Garcia A, Cayla X, Guergnon J, Dessauge F, Hospital V, Rebollo MP, Fleischer A, Rebollo A. Serine/threonine protein phosphatases PP1 and PP2A are key players in apoptosis. Biochimie. 2003;85:721–726. doi: 10.1016/j.biochi.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Gasior M, Rogawski MA, Hartman AL. Neuroprotective and disease-modifying effects of the ketogenic diet. Behav Pharmacol. 2006;17:431–439. doi: 10.1097/00008877-200609000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara MR, Snyder SH. Cell signaling and neuronal death. Annu Rev Pharmacol Toxicol. 2007;47:117–141. doi: 10.1146/annurev.pharmtox.47.120505.105311. [DOI] [PubMed] [Google Scholar]
- Haymond MW, Karl IE, Clarke WL, Pagliara AS, Santiago JV. Differences in circulating gluconeogenic substrates during short-term fasting in men, women, and children. Metabolism. 1982;31:33–42. [PubMed] [Google Scholar]
- Hempel SL, Buettner GR, O’Malley YQ, Wessels DA, Flaherty DM. Dihydrofluorescein diacetate is superior for detecting intracellular oxidants: comparison with 2′,7′-dichlorodihydrofluorescein diacetate, 5(and 6) - carboxy-2′,7′- dichlorodihydrofluorescein diacetate, and dihydrorhodamine 123. Free Radic Biol Med. 1999;27:146–159. doi: 10.1016/s0891-5849(99)00061-1. [DOI] [PubMed] [Google Scholar]
- Hori A, Hirotsu I, Davis PJ, Carpenter DO. Long-term potentiation is lost in aged rats but preserved by calorie restriction. Neuroreport. 1992;3:1085–1058. doi: 10.1097/00001756-199212000-00013. [DOI] [PubMed] [Google Scholar]
- Hyslop PA, Zhang Z, Pearson DV, Phebus LA. Measurement of striatal H2O2 by microdialysis following global forebrain ischemia and reperfusion in the rat: correlation with the cytotoxic potential of H2O2 in vitro. Brain Res. 1995;671:181–186. doi: 10.1016/0006-8993(94)01291-o. [DOI] [PubMed] [Google Scholar]
- Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J. 2001;353:417–439. doi: 10.1042/0264-6021:3530417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamsler A, Segal M. Hydrogen peroxide modulation of synaptic plasticity. J Neurosci. 2003;23:269–276. doi: 10.1523/JNEUROSCI.23-01-00269.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamsler A, Segal M. Hydrogen peroxide as a diffusible signal molecule in synaptic plasticity. Mol Neurobiol. 2004;29:167–178. doi: 10.1385/MN:29:2:167. [DOI] [PubMed] [Google Scholar]
- Kang-Park MH, Sarda MA, Jones KH, Moore SD, Shenolikar S, Clark S, Wilson WA. Protein phosphatases mediate depotentiation induced by high-intensity theta-burst stimulation. J Neurophysiol. 2003;89:684–690. doi: 10.1152/jn.01041.2001. [DOI] [PubMed] [Google Scholar]
- Kashiwaya Y, Takeshima T, Mori N, Nakashima K, Clarke K, Veech RL. D-beta-hydroxybutyrate protects neurons in models of Alzheimer’s and Parkinson’s disease. Proc Natl Acad Sci U S A. 2000;97:5440–5444. doi: 10.1073/pnas.97.10.5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller JN, Schmitt FA, Scheff SW, Ding Q, Chen Q, Butterfield DA, Markesbery WR. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology. 2005;64:1152–1156. doi: 10.1212/01.WNL.0000156156.13641.BA. [DOI] [PubMed] [Google Scholar]
- Kim DY, Davis LM, Maalouf M, Sullivan PG, van Brederode J, Rho JM. Ketone bodies protect against oxidative stress in neocortical neurons. J Neurochem. 2007;101:1316–1326. doi: 10.1111/j.1471-4159.2007.04483.x. [DOI] [PubMed] [Google Scholar]
- Klann E, Thiels E. Modulation of protein kinases and protein phosphatases by reactive oxygen species: implications for hippocampal synaptic plasticity. Prog Neuropsychopharmacol Biol Psychiatry. 1999;23:359–376. doi: 10.1016/s0278-5846(99)00002-0. [DOI] [PubMed] [Google Scholar]
- Kloeker S, Bryant JC, Strack S, Colbran RJ, Wadzinski BE. Carboxymethylation of nuclear protein serine/threonine phosphatase X. Biochem J. 1997;327:481–486. doi: 10.1042/bj3270481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloeker S, Reed R, McConnell JL, Chang D, Tran K, Westphal RS, Law BK, Colbran RJ, Kamoun M, Campbell KS, Wadzinski BE. Parallel purification of three catalytic subunits of the protein serine/threonine phosphatase 2A family (PP2A(C), PP4(C), and PP6(C)) and analysis of the interaction of PP2A(C) with alpha4 protein. Protein Expr Purif. 2003;31:19–33. doi: 10.1016/s1046-5928(03)00141-4. [DOI] [PubMed] [Google Scholar]
- Knapp LT, Klann E. Role of reactive oxygen species in hippocampal long-term potentiation: contributory or inhibitory? J Neurosci Res. 2002;70:1–7. doi: 10.1002/jnr.10371. [DOI] [PubMed] [Google Scholar]
- Laffel L. Ketone bodies: a review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab Res Rev. 1999;15:412–426. doi: 10.1002/(sici)1520-7560(199911/12)15:6<412::aid-dmrr72>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Lamers KJ, Gabreels FJ, Renier WO, Wevers RA, Doesburg WH. Fasting studies in cerebrospinal fluid and blood in children with epilepsy of unknown origin. Epilepsy Res. 1995;21:59–63. doi: 10.1016/0920-1211(95)00011-x. [DOI] [PubMed] [Google Scholar]
- Lechward K, Awotunde OS, Swiatek W, Muszynska G. Protein phosphatase 2A: variety of forms and diversity of functions. Acta Biochim Pol. 2001;48:921–933. [PubMed] [Google Scholar]
- Lei B, Adachi N, Arai T. The effect of hypothermia on H2O2 production during ischemia and reperfusion: a microdialysis study in the gerbil hippocampus. Neurosci Lett. 1997;222:91–94. doi: 10.1016/s0304-3940(97)13349-3. [DOI] [PubMed] [Google Scholar]
- Lewy DS, Gauss CM, Soenen DR, Boger DL. Fostriecin: chemistry and biology. Curr Med Chem. 2002;9:2005–2032. doi: 10.2174/0929867023368809. [DOI] [PubMed] [Google Scholar]
- Maalouf M, Dykes RW, Miasnikov AA. Effects of D-AP5 and NMDA microiontophoresis on associative learning in the barrel cortex of awake rats. Brain Res. 1998;793:149–168. doi: 10.1016/s0006-8993(98)00152-8. [DOI] [PubMed] [Google Scholar]
- Maalouf M, Sullivan PG, Davis L, Kim DY, Rho JM. Ketones inhibit mitochondrial production of reactive oxygen species following glutamate excitotoxicity by increasing NADH oxidation. Neurosci. 2007;145:256–64. doi: 10.1016/j.neuroscience.2006.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahieux F. Hippocampal sclerosis and dementia. Psychol Neuropsychiatr Vieil. 2003;1:179–186. [PubMed] [Google Scholar]
- Mansuy IM, Shenolikar S. Protein serine/threonine phosphatases in neuronal plasticity and disorders of learning and memory. Trends Neurosci. 2006;29:679–686. doi: 10.1016/j.tins.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Mariani E, Polidori MC, Cherubini A, Mecocci P. Oxidative stress in brain aging, neurodegenerative and vascular diseases: an overview. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;827:65–75. doi: 10.1016/j.jchromb.2005.04.023. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Duan W, Guo Z. Meal size and frequency affect neuronal plasticity and vulnerability to disease: cellular and molecular mechanisms. J Neurochem. 2003;84:417–431. doi: 10.1046/j.1471-4159.2003.01586.x. [DOI] [PubMed] [Google Scholar]
- Moreira PI, Honda K, Zhu X, Nunomura A, Casadesus G, Smith MA, Perry G. Brain and brawn: parallels in oxidative strength. Neurology. 2006;66:S97–101. doi: 10.1212/01.wnl.0000192307.15103.83. [DOI] [PubMed] [Google Scholar]
- Neviani P, Santhanam R, Trotta R, Notari M, Blaser BW, Liu S, Mao H, Chang JS, Galietta A, Uttam A, Roy DC, Valtieri M, Bruner-Klisovic R, Caligiuri MA, Bloomfield CD, Marcucci G, Perrotti D. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell. 2005;8:355–368. doi: 10.1016/j.ccr.2005.10.015. [DOI] [PubMed] [Google Scholar]
- Nicholls DG. Mitochondrial dysfunction and glutamate excitotoxicity studied in primary neuronal cultures. Curr Mol Med. 2004;4:149–177. doi: 10.2174/1566524043479239. [DOI] [PubMed] [Google Scholar]
- Noh HS, Lee HP, Kim DW, Kang SS, Cho GJ, Rho JM, Choi WS. A cDNA microarray analysis of gene expression profiles in rat hippocampus following a ketogenic diet. Brain Res Mol Brain Res. 2004;129:80–87. doi: 10.1016/j.molbrainres.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Noh HS, Hah YS, Nilufar R, Han J, Bong JH, Kang SS, Cho GJ, Choi WS. Acetoacetate protects neuronal cells from oxidative glutamate toxicity. J Neurosci Res. 2006;83:702–709. doi: 10.1002/jnr.20736. [DOI] [PubMed] [Google Scholar]
- Okada M, Nakanishi H, Amamoto T, Urae R, Ando S, Yazawa K, Fujiwara M. How does prolonged caloric restriction ameliorate age-related impairment of long-term potentiation in the hippocampus? Brain Res Mol Brain Res. 2003;111:175–181. doi: 10.1016/s0169-328x(03)00028-7. [DOI] [PubMed] [Google Scholar]
- Radad K, Rausch WD, Gille G. Rotenone induces cell death in primary dopaminergic culture by increasing ROS production and inhibiting mitochondrial respiration. Neurochem Int. 2006;49:379–386. doi: 10.1016/j.neuint.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Reddy PH. Mitochondrial oxidative damage in aging and Alzheimer’s disease: implications for mitochondrially targeted antioxidant therapeutics. J Biomed Biotechnol. 2006:31372. doi: 10.1155/JBB/2006/31372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rho JM, Anderson GD, Donevan SD, White HS. Acetoacetate, acetone, and dibenzylamine (a contaminant in l-(+)-beta-hydroxybutyrate) exhibit direct anticonvulsant actions in vivo. Epilepsia. 2002;43:358–361. doi: 10.1046/j.1528-1157.2002.47901.x. [DOI] [PubMed] [Google Scholar]
- Sano K, Kirino T. Ammon’s horn sclerosis: its pathogenesis and clinical significance. Tohoku J Exp Med. 1990;161(Suppl):273–295. [PubMed] [Google Scholar]
- Serrano F, Klann E. Reactive oxygen species and synaptic plasticity in the aging hippocampus. Ageing Res Rev. 2004;3:431–443. doi: 10.1016/j.arr.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, Miller GW, Yagi T, Matsuno-Yagi A, Greenamyre JT. Mechanism of toxicity in rotenone models of Parkinson’s disease. J Neurosci. 2003;23:10756–10764. doi: 10.1523/JNEUROSCI.23-34-10756.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultan I, Senkal CE, Ponnusamy S, Bielawski J, Szulc Z, Bielawska A, Hannun YA, Ogretmen B. Regulation of the sphingosine-recycling pathway for ceramide generation by oxidative stress, and its role in controlling c-Myc/Max function. Biochem J. 2006;393:513–521. doi: 10.1042/BJ20051083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thavendiranathan P, Chow C, Cunnane S, Burnham WM. The effect of the ‘classic’ ketogenic diet on animal seizure models. Brain Res. 2000;959:206–213. doi: 10.1016/s0006-8993(02)03744-7. [DOI] [PubMed] [Google Scholar]
- Van Hoof C, Goris J. Phosphatases in apoptosis: to be or not to be, PP2A is in the heart of the question. Biochim Biophys Acta. 2003;1640:97–104. doi: 10.1016/s0167-4889(03)00029-6. [DOI] [PubMed] [Google Scholar]
- Whisler RL, Goyette MA, Grants IS, Newhouse YG. Sublethal levels of oxidant stress stimulate multiple serine/threonine kinases and suppress protein phosphatases in Jurkat T cells. Arch Biochem Biophys. 1995;319:23–35. doi: 10.1006/abbi.1995.1263. [DOI] [PubMed] [Google Scholar]
- Winder DG, Sweatt JD. Roles of serine/threonine phosphatases in hippocampal synaptic plasticity. Nat Rev Neurosci. 2001;2:461–474. doi: 10.1038/35081514. [DOI] [PubMed] [Google Scholar]
- Won JS, Singh I. Sphingolipid signaling and redox regulation. Free Radic Biol Med. 2006;40:1875–1888. doi: 10.1016/j.freeradbiomed.2006.01.035. [DOI] [PubMed] [Google Scholar]
- Xu G, Perez-Pinzon MA, Sick TJ. Mitochondrial complex I inhibition produces selective damage to hippocampal subfield CA1 in organotypic slice cultures. Neurotox Res. 2003;5:529–538. doi: 10.1007/BF03033163. [DOI] [PubMed] [Google Scholar]