

Abstract
The serotonin (5-hydroxytryptamine; 5-HT)2C receptor is a G protein-coupled receptor (GPCR) exclusively expressed in CNS that has been implicated in numerous brain disorders, including anxio-depressive states. Like many GPCRs, 5-HT2C receptors physically interact with a variety of intracellular proteins in addition to G proteins. Here, we show that calmodulin (CaM) binds to a prototypic Ca2+-dependent “1-10” CaM-binding motif located in the proximal region of the 5-HT2C receptor C-terminus upon receptor activation by 5-HT. Mutation of this motif inhibited both β-arrestin recruitment by 5-HT2C receptor and receptor-operated extracellular signal-regulated kinase (ERK) 1,2 signaling in human embryonic kidney-293 cells, which was independent of G proteins and dependent on β-arrestins. A similar inhibition was observed in cells expressing a dominant-negative CaM or depleted of CaM by RNA interference. Expression of the CaM mutant also prevented receptor-mediated ERK1,2 phosphorylation in cultured cortical neurons and choroid plexus epithelial cells that endogenously express 5-HT2C receptors. Collectively, these findings demonstrate that physical interaction of CaM with recombinant and native 5-HT2C receptors is critical for G protein-independent, arrestin-dependent receptor signaling. This signaling pathway might be involved in neurogenesis induced by chronic treatment with 5-HT2C receptor agonists and their antidepressant-like activity.
INTRODUCTION
Serotonin (5-hydroxytryptamine; 5-HT)2C receptors still raise particular interest in view of their broad physiological role and implication in the actions of numerous psychoactive drugs (Giorgetti and Tecott, 2004; Millan, 2005, 2006). They play an essential role in the regulation of mood and alteration of their functional status has been involved in the etiology of anxio-depressive states. 5-HT2C receptors are themselves the target of various classes of antidepressants, including tricyclics, specific serotonin reuptake inhibitors, and “atypical” antidepressants such as mianserin, mirtazapine, and agomelatine, which behave as neutral antagonists (or inverse agonists) at 5-HT2C receptors (Millan, 2005; Chanrion et al., 2008). Antidepressant properties of 5-HT2C antagonists have largely been attributed to activation of dopaminergic and adrenergic pathways innervating corticolimbic structures, which exert a favorable influence upon mood and are tonically inhibited, via activation of GABAergic interneurons, by 5-HT2C receptors.
In spite of the inhibitory influence of 5-HT2C receptors on dopaminergic and adrenergic projections, 5-HT2C receptor agonists have shown unequivocal effectiveness in certain models of antidepressant activity (Moreau et al., 1996; Cryan and Lucki, 2000). One possible explanation for this paradoxical antidepressant action would be their positive influence on neurogenesis, a phenomenon possibly involved in therapeutic effects of antidepressant agents (Malberg et al., 2000; Santarelli et al., 2003). Activation of extracellular signal-regulated kinase (ERK)1,2 signaling pathway by 5-HT2C receptor agonists, which has recently been demonstrated in a Chinese hamster ovary cell line stably expressing 5-HT2C receptors (Werry et al., 2005), might represent a key signaling cascade underlying their growth-promoting activity and antidepressant effects. As a consequence, it is important to demonstrate whether 5-HT2C receptor agonists also activate ERK1,2 pathway in cells that endogenously express 5-HT2C receptors and to elucidate the mechanisms contributing to receptor-mediated ERK1,2 stimulation.
It has recently emerged that activation of ERK1,2 pathway by GPCR ligands is not exclusively mediated by G proteins but that it can involve protein complexes linking the receptor to ERK1,2 signaling. The β-arrestin–associated protein scaffold, which includes kinase members of the module that phosphorylates ERK1,2, has been identified as an essential component contributing to activation of ERK1,2 signaling pathway upon stimulation of various GPCRs (Lefkowitz and Shenoy, 2005). β-Arrestin–mediated activation of ERK1,2 seems to be intimately related to their ability to promote endocytosis of receptors in clathrin-coated pits.
The 5-HT2C receptor interacts with many intracellular proteins in addition to G proteins (mainly Gαq and Gα13) and β-arrestins (Bockaert et al., 2004, 2006). To date, the 5-HT2C receptor is certainly the GPCR for which interacting proteins have been the most extensively characterized by mean of proteomic approaches (Becamel et al., 2002, 2004). The majority of these interactions take place within the receptor C-terminus. For example, a set of postsynaptic density 95/disc-large/zona occludens proteins bind to the SSV motif on the distal C-terminal 5-HT2C receptor and have a key role in receptor desensitization and internalization after prolonged agonist exposure (Gavarini et al., 2006). The 5-HT2C receptor C-terminus also interacts with calmodulin (CaM) (Becamel et al., 2002), a ubiquitous modulator of Ca2+-dependent processes that has been found to interact with many other GPCRs, including the dopamine D2, metabotropic glutamate mGlu5 and mGlu7, μ-opioid, vasopressin V2, angiotensin II AT1A, and serotonin 5-HT1A and 5-HT2A receptors (Minakami et al., 1997; Nakajima et al., 1999; O'Connor et al., 1999; Thomas et al., 1999; Wang et al., 1999; Bofill-Cardona et al., 2000; Nickols et al., 2004; Turner et al., 2004; Turner and Raymond, 2005).
The roles that interaction with CaM plays in receptor function are remarkably diverse. Depending on the GPCR, CaM interaction with the receptor can affect its coupling with the G protein or its phosphorylation by protein kinase C (Nakajima et al., 1999; O'Connor et al., 1999; Wang et al., 1999; Bofill-Cardona et al., 2000; Turner et al., 2004; Turner and Raymond, 2005), suggesting a possible role in receptor desensitization. CaM can also directly contribute to receptor signaling (Della Rocca et al., 1999; O'Connor et al., 1999; Nickols et al., 2004). Notably, CaM has been involved in activation of ERK1,2 signaling pathway upon stimulation of certain GPCRs (Della Rocca et al., 1999; Belcheva et al., 2001). The mechanisms by which CaM influences GPCR-dependent ERK1,2 signaling remain to be clarified and may differ from one GPCR to another. For example, the role of CaM in 5-HT1A receptor-mediated ERK1,2 phosphorylation is critically dependent on its ability to promote receptor internalization (Della Rocca et al., 1999). Alternatively, CaM-dependent transactivation of the epidermal growth factor receptor has been involved in μ-opioid receptor-mediated ERK1,2 activation (Belcheva et al., 2001).
In this report, we have biochemically characterized the interaction between CaM and the 5-HT2C receptor C-terminal domain, and we provide evidence of an essential role of this interaction in 5-HT2C receptor-mediated ERK1,2 signaling. The data also reveal a previously unappreciated cooperativity between β-arrestin and CaM to transduce signals that may be shared by other GPCRs shown to interact with CaM.
MATERIALS AND METHODS
Chemicals, Plasmid Vectors, and Antibodies
All chemicals used in this study were obtained from Sigma-Aldrich (St. Louis, MO), unless otherwise indicated. The plasmid encoding the 5-HT2C receptor C terminus fused to glutathione transferase (GST) (pGEX2C) has been described previously (Becamel et al., 2002). Plasmids encoding truncated or mutated forms of the 5-HT2C receptor C-terminus (pGEX2CΔ21, pGEX2CΔ10, pGEX2CR376/377A, and pGEX2C22Stop; Figure 1) were generated from the pGEX2C plasmid by QuickChange mutagenesis (Stratagene, La Jolla, CA). The plasmid encoding cMyc-tagged human 5-HT2C receptor (nonedited version [INI], pRK5/cMyc-5-HT2CINI) has been described previously (Gavarini et al., 2006). The plasmid encoding nonedited cMyc-5-HT2CR376/377A receptor (mutated in the CaM binding motif, pRK5/cMyc-5-HT2CR376/377A) and the plasmid encoding fully edited (VGV) cMyc-5-HT2C receptor (pRK5/cMyc-5-HT2CVGV) were generated from the pRK5/cMyc-5-HT2CINI plasmid by QuickChange mutagenesis. The plasmid encoding the green fluorescent protein (GFP)-tagged 5-HT2C receptor (nonedited version, pEGFP/5-HT2C) was constructed by polymerase chain reaction (PCR) amplification of the 5-HT2C receptor sequence and in-frame ligation into the pEGFP-C1 plasmid (Clontech, Mountain View, CA). We verified that GFP-5-HT2C receptor exhibited similar pharmacological properties to those of Myc-5-HT2C receptor for modulation of Gαq-phospholipase C (PLC) β and ERK1,2 pathways. All constructs were verified by sequencing.
Figure 1.

Ca2+- and agonist-dependent interaction of calmodulin with the C-terminal domain of 5-HT2C receptor. (A) Solubilized protein extracts from mouse brain (1 mg of protein) or purified calmodulin (25 μg) was incubated with the 5-HT2C receptor C terminus fused to GST (GST-2C; 50 μg) in the presence of 1 mM CaCl2 or 0.5 mM EDTA. CaM retained by affinity was detected by immunoblotting using a monoclonal anti-CaM antibody. (B) The sequences of wild-type and mutated GST-tagged 5-HT2C receptor C-terminal fusion proteins are depicted. The box indicates the position of the putative 1–10 CaM binding motif identified using a search engine available on the web (http://calcium.uhnres.utoronto.ca/ctdb/pub_pages/search/index.htm). Brain protein extracts (1 mg) were incubated with the indicated GST fusion proteins (50 μg each) in the presence of 1 mM CaCl2, and affinity-purified CaM was detected as described in A. (C) Solubilized protein extracts of HEK-293 cells transfected with empty vectors (CTL) or cotransfected with GFP-CaM and either wild-type 5-HT2C or 5-HT2CR376/377A receptor were treated with either 5-HT (1 μM) or vehicle for 5 min. Solubilized protein extracts were immunoprecipitated with the polyclonal anti-GFP antibody. Immunoprecipitated proteins were analyzed by Western blotting using a monoclonal anti-GFP antibody and the polyclonal anti-5-HT2C receptor 522 antibody. Inputs represent 5% of the total protein amount used in immunoprecipitations. Data illustrated in A–C are representative of three independent experiments.
The cDNA encoding GFP-tagged CaM (pGFP-CaM) was obtained from Cell Signaling Technology (Danvers, MA). The cDNA encoding the Ca2+-insensitive CaM mutant (pJPA7/rCaM-DEF1234A) was generously provided by Dr. J. P. Adelman (Oregon Health and Science University, Portland, OR). The cDNAs encoding yellow fluorescent protein (YFP)-β-arrestin 2 (pcDNA-β-arr 2-YFP) and dominant-negative mutant of β-arrestin [pcDNA-DN-β-arr-(319-418), generated in the laboratory of Dr. J. L. Benovic] were generously provided by Dr. M. Bouvier (University of Montreal, Montreal, QC, Canada). The cDNA encoding dominant-negative Ras (RasN17) was generously provided by Dr. L. Journot (Institut de Génomique Fonctionnelle, Montpellier, France).
The mouse monoclonal anti-CaM, the rabbit polyclonal anti-ERK1,2 and anti-phospho-ERK1,2 (Thr202/Tyr204) antibodies were from Cell Signaling Technology; the rabbit polyclonal anti-GFP antibody was from Invitrogen (Carlsbad, CA), the mouse monoclonal anti-GFP antibody was from Roche Applied Science (Indianapolis, IN); and the mouse monoclonal anti-cMyc antibody (clone 9E-10) and the mouse monoclonal peroxidase-linked anti-FLAG antibody were from Sigma-Aldrich. The rabbit polyclonal 5-HT2C receptor antibody (522 antibody) has been described previously (Becamel et al., 2002). The rabbit polyclonal anti-Gαq, the rabbit polyclonal anti-Gα13, and the rat monoclonal anti-Ras antibody were from Santa Cruz Biotechnology (Santa Cruz, CA). The horseradish peroxidase-conjugated anti-rabbit and anti-mouse antibodies were from GE Healthcare (Chalfont St. Giles, Buckinghamshire, United Kingdom), and the Alexa Fluor 488-conjugated goat anti-mouse and Alexa Fluor 594-conjugated goat anti-rabbit antibodies from Invitrogen.
Cell Culture and Transfection
Human embryonic kidney (HEK)-293 cells, grown in DMEM (Invitrogen), supplemented with 10% dialyzed, heat-inactivated fetal calf serum and antibiotics, were transfected at 60–70% confluence either by electroporation for immunoblotting, coimmunoprecipitation, and calcium fluorescence measurement, as described previously (Chanrion et al., 2008), or by using Lipofectamine 2000 (Invitrogen) for Ca2+ imaging, immunocytochemistry, and experiments using siRNA, according to the manufacturer's instructions. The DNA ratio used for cotransfection was 1:2 (wild-type or mutant receptor cDNAs vs. cDNAs encoding either wild-type or dominant-negative forms of CaM or β-arrestin 2). Immunofluorescence experiments indicated that under these conditions >95% of cells expressing recombinant 5-HT2C receptors also expressed the cotransfected protein (data not shown).
Primary cultures of cortical neurons were prepared from the cerebral cortex of 16-d-old Swiss mouse embryos as described previously (Weiss et al., 1986). Neurons were grown in serum-free medium containing a 1:1 mixture of DMEM and F-12 nutrient supplemented with glucose (33 mM), glutamine (2 mM), NaHCO3 (13 mM), HEPES buffer (5 mM, pH 7.4), and a mixture of salt and hormones (transferrin, 100 μg/ml; insulin, 25 μg/ml; progesterone, 20 nM; putrescine, 60 nM; and Na2SeO3, 30 nM). Five days after seeding, neurons were transfected with the pEGFP/5-HT2C construct, in the presence or absence of the pJPA7/rCaM-DEF1234A plasmid, by using Lipofectamine 2000. Immunofluorescence experiments were performed 2 d after transfection. At this stage, cultures were shown to contain at least 95% of neurons (Weiss et al., 1986).
Primary cultures enriched in choroid plexus epithelial cells (CECs) were prepared from 6-d-old Swiss mice as described previously (Thouvenot et al., 2006). Briefly, choroid plexuses from the fourth ventricle were rapidly dissected, incubated with pronase (0.3 mg/ml; Sigma-Aldrich) and type I DNase (25 μg/ml; Sigma-Aldrich) in phosphate-buffered saline (PBS) containing 33 mM glucose (PBS-glucose) for 15 min at 37°C, and mechanically dissociated by eight forced passages through a 20-gauge needle. Dissociated cells were spun by centrifugation at 300 × g for 5 min, resuspended in culture medium, and enriched in CECs by their differential adhesion properties on plastic support. The culture medium consisted of DMEM/F-12 supplemented with glucose (33 mM), glutamine (2 mM), NaHCO3 (13 mM), HEPES buffer (5 mM; pH 7.4), penicillin-streptomycin (100 IU/ml and 100 μg/ml, respectively), and 15% Nu-serum (BD Biosciences, Franklin Lakes, NJ). After 10–12 d, cultures contained >80% of densely packed cells with a polygonal epithelial-like morphology and positively stained with an antibody against transthyretin, a specific marker of CECs in the CNS (Thouvenot et al., 2006). Cultured CECs were transfected after 7 d in culture by using Lipofectamine 2000, and the procedure used for HEK-293 cell transfection. Cotransfection of CECs with the pJPA7/rCaM-DEF1234A plasmid and a plasmid encoding GFP revealed a high transfection rate of the cultures (>80% of GFP-positive cells counted).
Small Interfering RNA (siRNA) Transfection
HEK-293 cells transiently transfected by electroporation with the pRK5/cMyc-5-HT2C plasmid were seeded in six-well dishes (100,000 cells/well) 24 h before transfection with siRNAs (Eurogentec, Seraing, Belgium) targeted against either CaM (hCalm3 gene) (positions 785–803) (5′-CUGAUGCAUUCAUAAGUUG-3′) or β-arrestin 1 (positions 439–459) (5′-AAAGCCUUCUGCGCGGAGAAU-3′) or β-arrestin 2 (positions 201–221) (5′-AAGGACCGCAAAGUGUUUGUG-3′) or Gαq/11 (positions 931–951) (5′-AAGATGTTCGT GGACCTGAAC-3′) or Gα13 (positions 96–114) (5′-GGAGATCGACAAATGCCTG-3′), or with control siRNA (5′-AAGUGGACCCUGUAGAUGGCG-3′), by using Lipofectamine 2000. The siRNAs targeting β-arrestins and Gα proteins have been validated previously (Birukova et al., 2004; Barnes et al., 2005; Kara et al., 2006). For calcium fluorescence experiments, cells in suspension were cotransfected at the same time with the pRK5/cMyc-5-HT2C plasmid and Gαq/11 siRNA by using Lipofectamine 2000 and then seeded in 96-well plates. All assays were performed 3 d after siRNA transfection.
GST Pull-Down Assay
Mice brains and HEK-293 cells were homogenized on ice with lysis buffer containing Tris-HCl (50 mM; pH 7.4), EDTA (0.5 mM), 3-[(3-cholamidopropyl)dimethylammonio]propanesulfonate (1.3%, wt/vol), and a protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN). Solubilized proteins (1 mg/condition) or purified CaM from bovine brain (25 μg/condition; Sigma-Aldrich) were incubated with GST fusion proteins (50 μg) immobilized onto glutathione-Sepharose 4B beads (40 μl; GE Healthcare) in the presence of either 0.5 mM EDTA or 1 mM CaCl2. Proteins retained by affinity were eluted off with SDS sample buffer (Tris-HCl [50 mM; pH 6.8], SDS [2%], glycerol (30%) DTT [100 mM], and bromphenol blue) and analyzed by immunoblotting.
Immunoprecipitation
Transfected HEK-293 cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (Sigma-Aldrich) supplemented with CaCl2 (1 mM). Cell lysates were centrifuged at 14,000 × g for 25 min, and solubilized proteins were incubated with the rabbit polyclonal anti-GFP antibody (4 μg) overnight at 4°C. Samples were incubated with 40 μl of protein A-Sepharose beads (GE Healthcare) for 1 h at 4°C, and immune complexes were collected by centrifugation, washed five times with RIPA buffer, eluted with SDS sample buffer, and analyzed by immunoblotting.
Immunoblotting
Proteins resolved by SDS-polyacrylamide gel electrophoresis were electrophoretically transferred onto nitrocellulose membranes. Membranes were blocked with 5% nonfat dry milk diluted in Tris-buffered saline-Tween (0.2%) and incubated with the primary antibodies (anti-GFP, 1:1000; anti-cMyc, 1:1000; anti-5-HT2C receptor, 1:500; anti-pERK1,2, 1:1000; anti-ERK1,2, 1:1000; anti-CaM, 1:1000; anti-Gαq, 1:1000; anti-Gα13, 1:1000; and anti-Ras, 1:1000 in blocking buffer) overnight at 4°C and with either anti-mouse or anti-rabbit horseradish peroxidase-conjugated secondary antibodies (1:5000) for 1 h at room temperature. Immunoreactivity was detected with an enhanced chemiluminescence method (ECL Plus detection reagent; GE Healthcare). Immunoreactive bands were quantified by densitometry using the NIH Image 1.62 software (National Institutes of Health, Bethesda, MD).
Calcium Fluorescence Measurement
HEK-293 cells grown in black-walled, clear-bottomed 96-well plates were washed with Locke's solution (140 mM NaCl, 1.2 mM KH2PO4, 5 mM KCl, 1.2 mM MgSO4, 10 mM HEPES, 1.8 mM CaCl2, and 10 mM glucose), supplemented with 2.5 mM probenecid and 0.5% bovine serum albumin, and loaded with 1 μM Fluo-4 acetoxymethyl ester (AM) (Invitrogen) for 1 h at 37°C. Cells were incubated with 50 μl of Locke's solution, and 5-HT was added in each well after 20 s of recording by using a fluorescence microplate reader (FlexStation II; Molecular Devices, Sunnyvale, CA). Fluorescence signals (excitation, 485 nm; emission, 525 nm) were then measured for 60 s at 2-s intervals. Kinetic parameters of 5-HT-evoked Ca2+ responses were determined using the Prism 3.0 software (GraphPad Software, San Diego, CA).
Calcium Imaging
HEK-293 cells grown in Lab-Tek II chamber slides Nalge Nunc International (Rochester, NY) were loaded with Fura-2 AM ester (Invitrogen) at a final concentration of 12.5 μM for 30 min at 37°C in Locke's solution. After loading, cells were rinsed twice and incubated for 30 min in dye-free Locke's buffer. Lab-Teks were then placed on the stage of an inverted IX70 Olympus microscope (Olympus, Tokyo, Japan) and continuously superfused with Locke's solution. Imaging of intracellular calcium changes in individual cells treated with agonist was performed by ratiometric imaging of Fura-2 fluorescence at 340- and 380-nm excitation by using the MetaFluor Imaging system (Molecular Devices). Fluorescence was excited by illumination via a 20× water immersion objective with rapid light wavelength switching provided by a DG4 filter wheel (Sutter Instrument, Novato, CA) and detected by a charge-coupled device camera under the control of MetaFluor software. Before agonist stimulation application, images were obtained for 30 s to establish a stable baseline Ca2+ measurement. Our standard protocol consisted of two sequential applications of agonist (5-HT; 1 μM), separated by 10-min washouts in presence or absence of either U73122 (5 μM) or EGTA (5 mM). Each Ca2+ trace illustrated is a mean response for a given field of cells (∼80 cells/field, three fields analyzed per experiment), representative of three experiments performed on different sets of cultured cells.
Analysis of 5-HT2C Receptor Cell Surface Expression by Enzyme-linked Immunosorbent Assay (ELISA)
HEK-293 cells grown in 96-well plates were fixed in 4% (wt/vol) paraformaldehyde in PBS for 20 min at room temperature. After two washes with glycine (0.1 M), cells were incubated in PBS containing 1% fetal calf serum for 30 min and then with a horseradish peroxidase-conjugated anti-FLAG monoclonal antibody (mAb; 1:5000) for 30 min. After five washes, the chromogenic substrate (SuperSignal ELISA Femto; Pierce Chemical, Rockford, IL) was added and immunoreactivity detected at 492 nm with a Wallac Victor2 luminescence counter (PerkinElmer Life and Analytical Sciences, Boston, MA). Control experiments were performed by omitting the primary antibody or by using cells transfected with empty vectors. Values were also normalized with respect to the total amount of protein. For each data point, four determinations were averaged and results were analyzed using analysis of variance (ANOVA) followed by Student–Newman–Keuls test.
Analysis of ERK1,2 Phosphorylation in Neurons by Immunocytochemistry
Cortical neurons grown on glass coverslips were fixed in methanol supplemented with tetramisole (5 mM) for 10 min at −20°C. They were washed three times with PBS containing tetramisole (5 mM) and permeabilized with 0.1% (wt/vol) Triton X-100 for 5 min. Neurons were then incubated in blocking buffer (10% goat serum in PBS containing 5 mM tetramisole) for 1 h at 37°C and overnight at 4°C with primary antibodies (monoclonal anti-GFP, 1:100; polyclonal anti-pERK1,2, 1:100) in blocking buffer. Cells were washed three times with blocking buffer and incubated for 1 h at room temperature with Alexa Fluor 488-conjugated goat anti-mouse or Alexa Fluor 594-conjugated goat anti-rabbit antibodies (2 μg/ml in blocking buffer). After three washes in PBS-tetramisole and a final wash in water, coverslips were mounted on glass slides in Mowiol 4.88 (Sigma-Aldrich). Fluorescent staining was observed with an Axiophot2 microscope (Carl Zeiss, Jena, Germany).
RESULTS
Ca2+- and Agonist-dependent Interaction of Calmodulin with the C-Terminal Domain of 5-HT2C Receptor
Confirming our previous results obtained using proteomics (Becamel et al., 2002), the entire C-terminus of the 5-HT2C receptor fused to GST and immobilized on glutathione-Sepharose beads specifically associated with CaM from mice brain cytosolic extract (Figure 1A). The receptor C-terminus similarly bound to purified bovine brain CaM, indicating a direct interaction between the receptor and CaM in vitro (Figure 1A). Furthermore, the interaction between CaM and the receptor 5-HT2C C-terminus was abolished by replacing Ca2+ (1 mM) with 0.5 mM EDTA in the binding medium (Figure 1A).
To determine the region of the 5-HT2C receptor C terminus important for CaM interaction, we first searched sequence motifs for Ca2+-dependent CaM recognition within the receptor's C-terminal domain. We used a search engine available on the web, which identifies such regions based on several criteria, including the presence of an amphipathic α-helix with hydrophobic/aromatic and basic residues at conserved positions (http://calcium.uhnres.utoronto.ca/ctdb/pub_pages/search/index.htm). We identified a putative CaM binding site localized at the juxtamembrane region of 5-HT2C receptor C-terminus (residues 370–379) and corresponding to “helix 8” present in many GPCRs (Figure 1B). This binding motif (LFNKIYRRAF) could be classified as a “1–10” motif. These motifs form amphipathic α-helices composed of basic residues and key hydrophobic residues (F, I, L, V, or W) localized at positions 1 and 10 (Yap et al., 2000). To confirm the involvement of this motif in CaM binding to the receptor, we designed several truncation mutants of the 5-HT2C receptor C-terminus and expressed them as GST fusion proteins (Figure 1B). Deleting the 21 N-terminal residues (residues 370–389, Δ21) of the 5-HT2C receptor C-terminal fusion protein or solely the 1-10 motif (residues 370–379, Δ10) prevented binding to CaM. In contrast, a truncated C-terminal mutant containing only the 22 N-terminal residues strongly associated with CaM in vitro (Figure 1B). Moreover, mutating two arginine residues within the 1–10 motif (R376/377A) that are critical for the amphipathic nature of the helix, almost abolished CaM interaction with the receptor C-terminal fusion protein (Figure 1B). We thus chose to introduce this modification into the entire receptor sequence to demonstrate the importance of the 1–10 motif for CaM binding to the receptor and its signaling properties within living cells. In absence of agonist, 5-HT2C receptor did not coimmunoprecipitate with CaM in homogenates from HEK-293 cells cotransfected with Myc-5-HT2C receptor and GFP-CaM (Figure 1C). In contrast, 5-HT2C receptor coimmunoprecipitated with GFP-CaM after acute exposure to 5-HT (1 μM; 5 min). Furthermore, GFP-CaM did not coimmunoprecipitate with 5-HT2C receptor mutated in the 1–10 motif (5-HT2CR376/377A), even after agonist exposure (Figure 1C). Collectively, these results indicate that 5-HT2C receptors associate with CaM in an agonist-dependent manner, a phenomenon corroborating the lack of coimmunoprecipitation of CaM with native 5-HT2C receptors in brain homogenates (data not shown).
Physical Interaction of Calmodulin with 5-HT2C Receptor C Terminus Is Essential for Receptor-Mediated ERK1,2 Signaling in HEK-293 Cells
5-HT (1 μM) induced a rapid and sustained increase in ERK1,2 phosphorylation state in HEK-293 cells transiently transfected with Myc-5-HT2C receptor (Figure 2A). Maximal response was detected at 2–5 min after the addition of 5-HT. Thereafter, the level of phosphorylated ERK1,2 progressively declined, but a robust increase in ERK1,2 phosphorylation was still detectable 3 h after the onset of agonist treatment (210 ± 30% of basal level corresponding to 37 ± 9% of maximal response). 5-HT-induced ERK1,2 activation was entirely inhibited by concomitant application of SB242,084 (1 μM), a specific 5-HT2C receptor neutral antagonist (Figure 2B). Furthermore, such a prolonged activation of ERK1,2 phosphorylation required permanent stimulation of 5-HT2C receptor by its ligand. Indeed, SB242,084 similarly prevented ERK1,2 phosphorylation induced by 60-min 5-HT exposure when added only for the last 10 min of the treatment (Figure 2B). 5-HT-induced ERK1,2 phosphorylation was concentration-dependent (pEC50 = 9.0 ± 0.19 and pEC50 = 8.6 ± 0.11 measured at 5 and 60 min, respectively; p = 0.14; Figure 2C).
Figure 2.
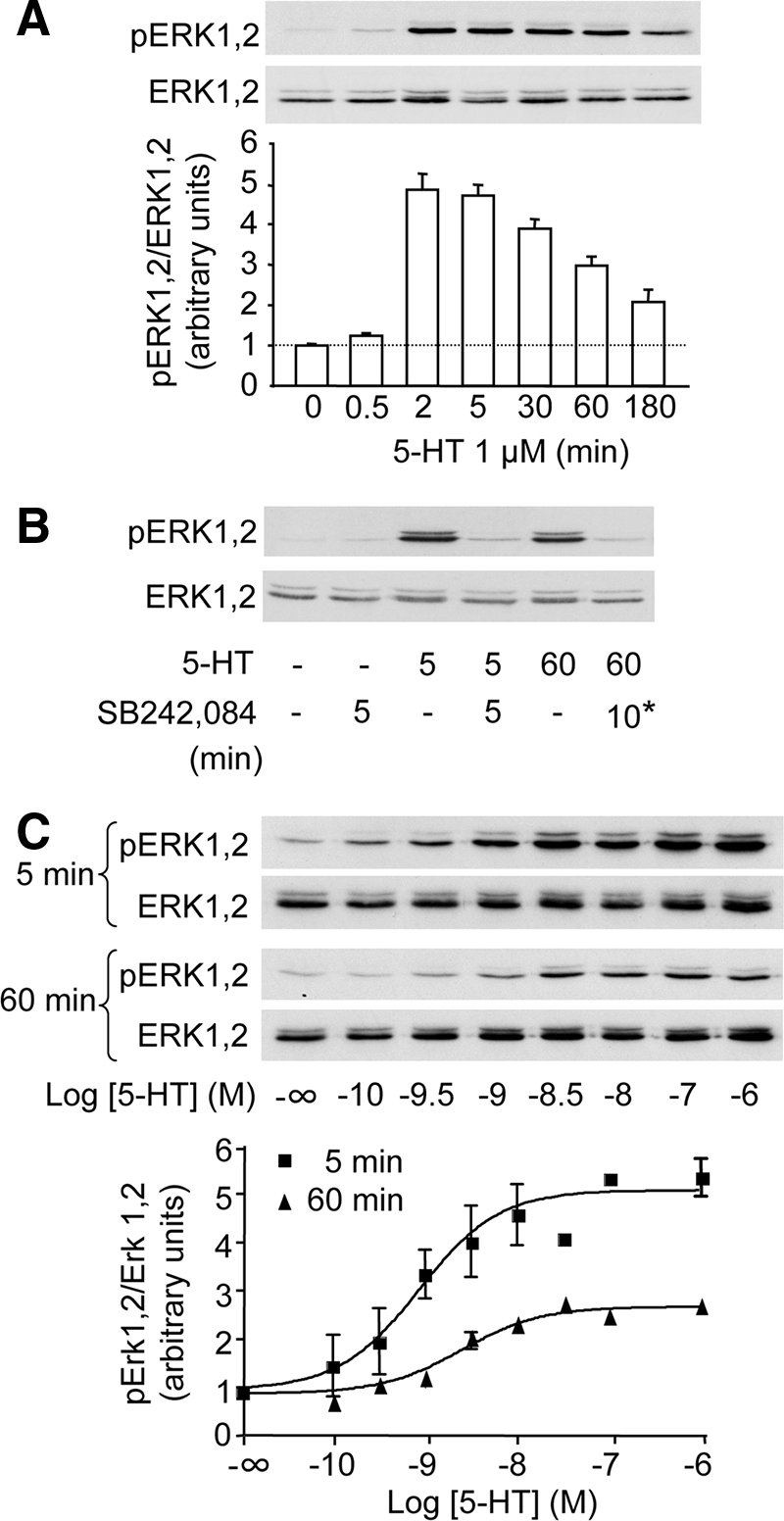
5-HT2C receptor activation induces a prolonged increase in ERK1,2 phosphorylation in HEK-293 cells. In A, B, and C, HEK-293 cells transfected with the 5-HT2C receptor were starved of serum for 18 h and then exposed to the indicated concentration of 5-HT for the indicated times. In B, SB242,084 (1 μM) was added either concomitantly with 5-HT for 5 min or for the last 10 min of treatment (*) in cultures exposed to 5-HT for 60 min. ERK1,2 phosphorylation was analyzed by sequential immunoblotting with a polyclonal antibody against phospho-Thr202/Tyr204 ERK1,2 and a polyclonal antibody recognizing ERK1,2 independently of their phosphorylation state. Representative immunoblots of three independent experiments performed on different sets of cultured cells are illustrated. Immunoreactive signals were quantified by densitometry, and data are expressed as a ratio of phosphorylated ERK1,2 (pERK1,2) to total ERK1,2. Each data point represents means ± SEM of values obtained in three experiments. In C, dose–response curves were fitted using Prism software.
Due to the lack of specificity of commonly used, membrane-permeable CaM inhibitors such as the naphthalene sulfonamide derivatives W7 and W13, trifluoperazine, and calmidazolium (Sengupta et al., 2007), we investigated the role of CaM in 5-HT2C receptor-mediated ERK1,2 signaling by transfecting cells with a Ca2+-insensitive CaM mutant, in which aspartate residues in the first Ca2+ ligand position of all four EF hand motifs of CaM have been mutated into alanine (CaM1,2,3,4) (Xia et al., 1998). CaM1,2,3,4 did not interact with the 5-HT2C receptor C-terminus in vitro, consistent with the Ca2+-dependent nature of CaM binding to 1–10 motif (Figure 3A, line 5), whereas wild-type GFP-CaM associated with the receptor C-terminus (Figure 3A, line 6). GFP-CaM was detected with a GFP antibody, due to the lack of sensitivity provided by the CaM antibody. A strong immunoreactive signal was observed with the CaM antibody in the fraction bound to the receptor C-terminus in presence of both CaM1,2,3,4 and wild-type GFP-CaM (Figure 3A, line 7). Thus, this signal likely corresponds to CaM1,2,3,4. This indicates that CaM1,2,3,4 binding to the 5-HT2C receptor C-terminus requires the presence of wild-type CaM and suggests that CaM might associate with the receptor as a dimer.
Figure 3.

5-HT2C receptor-operated ERK1,2 signaling is dependent on physical interaction of CaM with the receptor C-terminus. (A) The Ca2+-insensitive CaM mutant CaM1,2,3,4 binds to the 5-HT2C receptor C-terminus only in the presence of wild-type CaM. Homogenates from HEK-293 cells transfected with the indicated constructs were incubated with GST-fused 5-HT2C receptor C terminus. The amount of proteins used in pull-downs was adjusted to 1 mg in each condition with protein extracts from empty vector-transfected cells, which endogenously express negligible CaM amount, compared with GFP-CaM- or CaM1,2,3,4-transfected cells. GFP-CaM and CaM1,2,3,4 were analyzed by Western blotting using monoclonal antibodies against GFP and CaM, respectively, because GFP-CaM was not detected with the CaM antibody. The data illustrated are representative of three independent experiments. Inputs (lines 1–4) represent 5% of the total protein amount used for immunoprecipitations (lines 5–7). (B and C) HEK-293 cells were transfected with the indicated constructs and siRNAs. (B) Variations in cytosolic Ca2+ concentration upon exposure to increasing concentrations of 5-HT was analyzed by measuring fluo-4 fluorescence for 60 s. Values (intensities of 5-HT-evoked Ca2+ peak, normalized to maximal response measured in cells transfected with wild-type 5-HT2C receptor) are means ± SEM of quadruplicate determinations performed in a typical experiment. Two other experiments performed on different sets of cultured cells yielded similar results. (C) ERK1,2 phosphorylation was analyzed as indicated in the legend to Figure 2. Values, normalized to total ERK immunoreactivity, are means ± SEM of results obtained in three independent experiments. *p < 0.01 versus corresponding value in cells transfected with wild-type 5-HT2C receptor alone (ANOVA, followed by Student–Newman–Keuls test). (D) Endogenous CaM expression in HEK-293 cells transfected with control (CTL) or CaM siRNA (100 μg total protein per lane) was analyzed by immunoblotting. The data are representative of three independent experiments performed on different cultures.
Coexpressing CaM1,2,3,4 and 5-HT2C receptor in HEK-293 cells did not affect the amplitude of 5-HT–evoked Ca2+ increase nor the apparent affinity of 5-HT to induce Ca2+ mobilization, suggesting that CaM does not contribute to Gαq-dependent receptor signaling (Figure 3B). In contrast, coexpression of CaM1,2,3,4 with the receptor strongly decreased the magnitude of 5-HT-evoked ERK1,2 phosphorylation at all times of 5-HT exposure examined (Figure 3C). Further supporting the essential role of CaM in receptor-mediated ERK signaling, transfection of HEK-293 cells with siRNA directed against CaM, which strongly decreased CaM expression compared with control siRNA-transfected cells (Figure 3D), also markedly reduced ERK1,2 phosphorylation evoked by short (5-min) and prolonged (up to 180-min) 5-HT exposures (Figure 3C). Expression of wild-type GFP-CaM did not modify the amplitude of receptor-mediated Ca2+ signaling in HEK-293 cells (Figure 3B). It also did not enhance phosphorylation of ERK1,2 induced by 5-HT (Figure 3C), suggesting that the amount of endogenous CaM was not a limiting factor in 5-HT2C receptor-operated ERK signaling in HEK-293 cells.
The induction by 5-HT of ERK1,2 phosphorylation was also markedly reduced in HEK-293 cells transfected with 5-HT2CR376/377A receptor, compared with cells expressing wild-type 5-HT2C receptor (Figure 3C), indicating that physical interaction of calmodulin with 5-HT2C receptor C-terminus is essential for receptor-mediated ERK1,2 signaling. In contrast, the amplitude of Ca2+ response evoked by maximally effective concentrations of 5-HT was not affected by mutation of the receptor's CaM binding motif (Figure 3B). Nonetheless, the apparent affinity of 5-HT to increase cytosolic Ca2+ concentration was less elevated in cells expressing 5-HT2CR376/377A receptor than in cells expressing wild-type receptor (pEC50 = 8.0 ± 0.08 and 8.7 ± 0.10, respectively, p = 0.0054, as indicated by nonparametric two-tailed t test).
5-HT2C Receptor-mediated ERK1,2 Signaling Is Independent of Its Coupling to Heterotrimeric G Proteins and of Ras Engagement
Collectively, our data indicated that 5-HT2C receptor-mediated ERK1,2 signaling required Ca2+-dependent association of CaM to the receptor C-terminus. Treating cells with U73122 (5 μM), a PLCβ antagonist, did not affect the phosphorylation of ERK1,2 induced by 2-, 5- or 60-min 5-HT applications (Supplemental Figure 1 and Figure 4A), suggesting that binding of CaM to the receptor was not initiated by agonist-dependent stimulation of PLCβ and Ca2+ mobilization. Efficient inhibition of 5-HT-induced Ca2+ signaling in HEK-293 cells by U73122 was assessed by Fura-2 Ca2+ imaging. As shown in Figure 4B, two successive 5-HT challenges (1 min each) separated by a 10-min agonist washout, induced transient elevations of cytosolic Ca2+, and addition of U73122 during the washing period entirely inhibited Ca2+ elevation induced by the second 5-HT treatment. Consistent with the lack of inhibition of 5-HT-induced ERK1,2 phosphorylation by U73122, reducing Gαq protein expression by siRNA (Figure 4C) did not alter the level of phosphorylated ERK1,2 in cells treated for 2, 5, or 60 min with 5-HT (Supplemental Figure 1 and Figure 4A). It is worth noting that Gαq siRNA efficiently inhibited Gαq-dependent signaling in HEK-293 cells (i.e., 5-HT-induced Ca2+ elevation; Figure 4D). Further supporting a Gαq-PLCβ-independent mechanism, staurosporine (1 μM), a protein kinase C inhibitor, did not affect the level of ERK1,2 phosphorylation evoked by 5-HT (Figure 4A).
Figure 4.

5-HT2C receptor-mediated ERK1,2 signaling is independent of G proteins. (A) HEK-293 cells transfected with the plasmid encoding 5-HT2C receptor were pretreated or not (CTL) with either U73122 (5 μM; 10 min), EGTA (1 mM; 10 min), PTX (1 μg/ml; 24 h), or staurosporine (1 μM; 30 min). For experiments using siRNAs, cells were transfected with control (CTL siRNA), Gαq/11, or Gα13 siRNA, by using the transfection reagent Lipofectamine 24 h after receptor transfection. They were then exposed to 1 μM 5-HT for 5 or 60 min, and ERK1,2 phosphorylation was analyzed by immunoblotting as indicated in the legend to Figure 2. Data are means ± SEM of values obtained in three independent experiments. *p < 0.01 versus corresponding control values. (B) Variations in cytosolic Ca2+ levels in response to two successive 5-HT challenges (1 μM; 60 s each) separated by 10-min washout. U73122 (5 μM) or EGTA (5 mM) were added during the washout period. Traces represent average Ca2+ signals recorded in a field containing at least 80 cells and are representative of three experiments performed on different cultures (three fields analyzed per experiment). (C) Gαq and Gα13 expression in cells transfected with control, Gαq/11 or Gα13 siRNA was analyzed by immunoblotting. The data are representative of three independent experiments performed on different cultures. (D) Inhibition of 5-HT2C receptor-mediated Ca2+ signaling in cells transfected with Gαq/11 siRNA. Data represent maximal fluo-4 fluorescence signals in cells transfected with control or Gαq/11 siRNA and treated with vehicle or 1 μM 5-HT. They are means ± SEM of values obtained in three independent experiments performed in quadruplicate. *p < 0.001 versus 5-HT response measured in cells transfected with control siRNA.
Cell pretreatment with EGTA (1 mM; 10 min) induced a rapid decrease in basal cytosolic Ca2+ level and prevented further Ca2+ elevation upon 5-HT exposure, indicating efficient depletion of intracellular Ca2+ stores (Figure 4B). EGTA pretreatment also strongly inhibited the increase in ERK1,2 phosphorylation evoked by 5-HT exposure (Figure 4A). Collectively, these results indicate that basal cytosolic Ca2+ was prerequisite to CaM binding to the C-terminal domain of 5-HT2C receptor and to receptor-mediated ERK1,2 activation, whereas mobilization of intracellular Ca2+ mediated by the Gαq–PLC pathway was not required for 5-HT2C receptor-operated ERK1,2 signaling.
Although 5-HT2C receptor is reported to primarily couple to Gαq, it can also activate Gα13, a process involved in receptor-mediated activation of phospholipase D and rearrangement of the actin cytoskeleton (McGrew et al., 2002). Transfection of HEK-293 cells with siRNA directed against human Gα13, which almost abolished Gα13 expression compared with control siRNA-transfected cells (Figure 4C), did not attenuate induction of ERK1,2 phosphorylation by 5-HT (Supplemental Figure 1 and Figure 4A). Because 5-HT2C receptors were found to couple to several Gαi protein subunits in various expression systems (Cussac et al., 2002), we also explored possible involvement of Gαi/o proteins in receptor-mediated activation of ERK1,2 signaling pathway in HEK-293 cells. Pretreatment of cells with pertussis toxin (PTX; 1 μg/ml; 24 h), which uncouples GPCRs from Gαi/o protein subtypes, did not affect the increase in ERK1,2 phosphorylation induced by 2-, 5-, or 60-min 5-HT treatments (Supplemental Figure 1 and Figure 4A).
These findings suggested that 5-HT2C receptor-mediated activation of the ERK1,2 signaling pathway was independent of Gα protein subunits known to couple to the receptor. Nonetheless, in the aforementioned experiments, cells were transfected with unedited (INI) 5-HT2C receptor, which exhibits intracellular rather plasma membrane localization (Marion et al., 2004). Such an intracellular distribution might mask an important fraction of 5-HT2C receptor-mediated ERK signaling, including a G protein-dependent component. Consistent with this hypothesis, pretreatment of cells with the 5-HT2C receptor inverse agonist SB206,553 (100 nM; 18 h) followed by drug washout, which induces a redistribution of receptors to the plasma membrane (Chanrion et al., 2008), enhanced both 5-HT–stimulated CaM recruitment to the receptor and ERK1,2 phosphorylation (Supplemental Figure 2A and C). 5-HT-stimulated ERK signaling was likewise increased in cells expressing fully edited (VGV) 5-HT2C receptors (Supplemental Figure 2C), which do not exhibit constitutive activity and are mainly localized at the plasma membrane (Marion et al., 2004). Moreover, ERK1,2 phosphorylation was still unaffected by Gαq or Gα13 depletion by RNA interference in cells expressing 5-HT2CINI receptors and pretreated with SB206,553 (Supplemental Figure 2B and D). This indicates that 5-HT2C receptor-mediated ERK1,2 activation is independent of G proteins in cells expressing receptors mainly localized at the plasma membrane.
Activation of ERK1,2 signaling by treatments elevating cytosolic Ca2+ is often dependent on the Ras/Raf/mitogen-activated protein kinase kinase module. The activity of Ras proteins is under tight control of several exchange factors, such as Ras-GRF1, which is mainly activated by the Ca2+–CaM complex (Farnsworth et al., 1995). To determine whether 5-HT2C receptor-mediated ERK1,2 phosphorylation involved Ras activation, we coexpressed the dominant-negative Ras, RasN17, with 5-HT2C receptor in HEK-293 cells. RasN17 expression did not affect the level of phosphorylated ERK1,2 in HEK-293 cells exposed to 5-HT (Supplemental Figure 3), indicating that Ras was not involved in 5-HT2C receptor-operated ERK1,2 activation.
5-HT2C Receptor-mediated ERK1,2 Activation Is Dependent on β-Arrestins
Protein candidates for mediating G protein-independent 5-HT2C receptor signals are β-arrestins, which are well documented to bind several proteins of the module activating ERK1,2 (Lefkowitz and Shenoy, 2005). Transfection of HEK-293 with either β-arrestin 1 or β-arrestin 2 siRNA resulted in both a strong decrease in expression of the corresponding β-arrestin (Figure 5B) and a marked inhibition of 5-HT–evoked ERK1,2 phosphorylation (Figure 5A). Corroborating this result, induction by 5-HT of ERK1,2 phosphorylation was similarly decreased by expression of a dominant-negative mutant of β-arrestin known to affect clathrin-dependent GPCR internalization (319–418, DN β-arr; Figure 5A). Nonetheless, the DN β-arr effect did not reflect an inhibition of receptor endocytosis. Indeed, pretreatment of cells with concanavalin A (250 μg/ml), which induced a marked redistribution of receptor to the plasma membrane (Figure 5C), enhanced rather than inhibited ERK1,2 phosphorylation elicited by 5-HT (Figure 5A), reminiscent to that measured in SB206,553 pretreated cells (Supplemental Figure 2C).
Figure 5.

5-HT2C receptor-mediated ERK1,2 signaling is dependent on β-arrestins. (A) HEK-293 cells, transfected with the indicated constructs and siRNAs or treated with concanavalin A (Con A; 250 μg/ml; 2 h) were exposed to 1 μM 5-HT for 5 or 60 min. Phosphorylated ERK1,2 and total ERK1,2 were analyzed by immunoblotting as indicated in the legend to Figure 2. *p < 0.001 versus corresponding control value. (B) Endogenous β-arrestins 1 and 2 expression in HEK-293 cells transfected with control, β-arrestin 1 or β-arrestin 2 siRNA was analyzed by immunoblotting. The data are representative of three independent experiments performed on different cultures. (C) HEK-293 cells transfected with the 5-HT2C receptor were either cotransfected with DN-β-arr 2 or treated with Con A. The level of receptor expression at the cell surface was quantified by ELISA. Data, normalized to receptor level measured in cells transfected with wild-type 5-HT2C receptor and exposed to sham treatment (CTL), are means ± SEM of results obtained in four independent experiments performed in quadruplicate. *p < 0.001 versus CTL. (D) HEK-293 cells transfected with the indicated constructs, and siRNAs, were exposed to 5-HT (1 μM) for 5 min in the absence or presence of EGTA (5 mM, added for 10 min before the 5-HT challenge). Solubilized protein extracts were immunoprecipitated with either control immunoglobulin (CTL) or a monoclonal anti-GFP antibody. Immunoprecipitated proteins were analyzed by Western blotting using a polyclonal anti-GFP antibody and the polyclonal anti-5-HT2C receptor 522 antibody. Inputs represent 5% of the total protein amount used in immunoprecipitations. Data are representative of three independent experiments performed on different cultures.
CaM Binding to 5-HT2C Receptor C Terminus Promotes Recruitment of β-Arrestin 2 by the Receptor
Collectively, our observations indicate that CaM and β-arrestins act in concert to promote ERK1,2 signaling upon 5-HT2C receptor activation. Because β-arrestin 2 was found to be the predominant β-arrestin interacting with 5-HT2C receptors expressed in HEK-293 cells (Marion et al., 2004), we next explored the influence of CaM binding to the 5-HT2C receptor C-terminus on the recruitment of β-arrestin 2 by the receptor upon exposure to 5-HT. As shown in Figure 5D, Myc-5-HT2C receptors coimmunoprecipitated with YFP-tagged β-arrestin 2 in homogenates from HEK-293 cells treated for 5 min with 5-HT (1 μM). In contrast, association of 5-HT2C receptor with β-arrestin 2 was strongly decreased in cells expressing 5-HT2CR376/377A receptors. It was also reduced in cells coexpressing wild-type 5-HT2C receptor and CaM1,2,3,4 as well as in cells transfected with CaM siRNA (Figure 5D). In line with the inhibition by EGTA of CaM association with 5-HT2C receptors and of receptor-operated ERK1,2 signaling, a pretreatment of cells with EGTA (1 mM, 10 min) likewise prevented recruitment of β-arrestin 2 by the receptor upon 5-HT exposure (Figure 5D).
Role of Calmodulin in 5-HT2C Receptor-mediated ERK1,2 Activation in Cortical Neurons
To determine whether CaM similarly influences 5-HT2C receptor-mediated ERK1,2 phosphorylation in a more physiological cellular background, experiments were performed in primary cultures of mouse cortical neurons, which endogenously express 5-HT2C receptors (Chanrion et al., 2008). Cortical neurons exhibited no detectable increase in ERK1,2 phosphorylation upon 5-HT application, as assessed by immunoblotting, probably due to the low density of 5-HT2C receptors in these neurons. A similar result was obtained by immunocytochemical labeling of ERK1,2 phosphorylation in 5-HT–treated neurons. We thus analyzed ERK1,2 phosphorylation in cortical neurons transfected with GFP-5-HT2C receptor. Treatment of cultures with 1 μM 5-HT for 5 or 60 min increased phospho-ERK1,2 immunoreactivity in all neurons expressing Myc-5-HT2C receptors (n = 48 transfected neurons originated from five sets of cultured neurons counted in each experimental condition; Figure 6). Further supporting the involvement of 5-HT2C receptors, the 5-HT–evoked increase in ERK1,2 phosphorylation in neurons was prevented by application of SB242,084 (1 μM). No increase in phospho-ERK1,2 immunoreactivity was detected upon 5-HT exposure in neurons cotransfected with CaM1,2,3,4 (n = 36 neurons counted; Figure 6), corroborating our observations in HEK-293 cells.
Figure 6.

5-HT2C receptor-mediated ERK1,2 signaling in cortical neurons is dependent on calmodulin. Cortical neurons, grown for 5 d in serum-free medium, were transfected with GFP-tagged 5-HT2C receptor in absence or presence of CaM1,2,3,4. Forty-eight hours after transfection, they were preincubated or not with SB242,084 (1 μM; 10 min) and then exposed to 5-HT (1 μM) for 5 or 60 min. Phosphorylated ERK1,2 in transfected neurons were analyzed by immunofluorescence staining using a monoclonal anti-GFP and a polyclonal anti-phospho-ERK1,2 antibody. Representative fluorescence photomicrographs of five independent experiments performed on different sets of cultured neurons are illustrated (at least 36 5-HT2C receptor-transfected neurons counted in each experimental condition). Bar, 20 μm.
Role of Calmodulin in 5-HT2C Receptor-mediated ERK1,2 Activation in Choroid Plexus Epithelial Cells
We next used cultured epithelial cells from choroid plexus, a structure rich in 5-HT2C receptors and expressing a receptor density comparable to that measured in transfected HEK-293 cells (0.31 ± 0.07 pmol/mg protein; n = 3; assessed by [3H]mesulergine binding) (Chanrion et al., 2008), to determine the role of CaM in an endogenous system. In these cells, 5-HT (1 μM) produced a robust increase in ERK1,2 phosphorylation, reminiscent to that measured in HEK-293 cells (Figure 7). This effect was mediated by 5-HT2C receptors, because it was suppressed by SB242,084 pretreatment (1 μM; Figure 7). 5-HT did not increase ERK1,2 phosphorylation in choroid plexus epithelial cells overexpressing CaM1,2,3,4, underscoring the essential role of CaM in 5-HT2C receptor-operated ERK1,2 signaling in choroid plexus epithelial cells expressing native receptors.
Figure 7.

5-HT2C receptor-mediated ERK1,2 signaling in choroid plexus epithelial cells is dependent on calmodulin. Primary cultures of mice choroid plexuses enriched in epithelial cells were transfected or not with CaM1,2,3,4. Twenty-four hours after transfection, they were starved of serum for 2 h, pretreated or not with SB242,084 (1 μM; 10 min) and then exposed to sham treatment or 5-HT (1 μM, 5 min). Phosphorylated and total ERK1,2 were analyzed by immunoblotting as indicated in the legend to Figure 2. CaM1,2,3,4 expression was analyzed by immunoblotting using the monoclonal anti-CaM antibody. The data are means ± SEM of values obtained in three independent experiments. *p < 0.05 versus corresponding value in empty-vector–transfected cells.
DISCUSSION
Activation of the ERK1,2 signaling cascade by GPCRs is a complex process that sequentially involves several mechanisms, which are dependent and independent on activation of their cognate G protein (Lefkowitz and Shenoy, 2005; Reiter and Lefkowitz, 2006). The G protein-dependent response is generally rapid and transient, whereas the G protein-independent response, which is intimately related to the ability of β-arrestin to bind to the receptor and to promote receptor endocytosis in clathrin-coated pits, is characterized by slower onset but greater persistence. Our results indicate that activation of ERK1,2 signaling by 5-HT2C receptors differs from this general model in that it is entirely independent of the receptor coupling to heterotrimeric G proteins. Indeed, neither the early phase of receptor-dependent ERK1,2 activity nor the later phase was affected by depletion of cellular Gαq, the Gα subunit primarily coupled to 5-HT2C receptors. Further supporting a Gαq-independent process, 5-HT2C receptor-mediated ERK1,2 phosphorylation was not affected by the selective inhibition of PLCβ, the classical effector of Gαq. 5-HT2C receptor-mediated ERK1,2 signaling did not either involve Gα13 or Gαi/o, the two other Gα protein subunits known to be engaged by 5-HT2C receptors, as it was insensitive to Gα13 depletion and PTX pretreatment, respectively. To our knowledge, the 5-HT2C receptor is one of the first wild-type GPCRs capable of activating the ERK1,2 pathway exclusively via a G protein-independent mechanism in response to its natural ligand. Such a property has recently been suggested for both 5-HT4 and V2 vasopressin receptors expressed in HEK-293 cells (Barthet et al., 2007; Charest et al., 2007). Nonetheless, a Gαs protein-dependent component has been involved in ERK1,2 activation mediated by V2 receptors in another study (Ren et al., 2005).
Our results also indicate that 5-HT2C receptor-operated ERK1,2 signaling is critically dependent on the presence of β-arrestins. Although β-arrestin 2 was identified as the predominant β-arrestin isoform interacting with 5-HT2C receptors (Marion et al., 2004), depletion of either β-arrestin 1 or β-arrestin 2 by RNA interference strongly reduced 5-HT2C receptor-mediated ERK1,2 phosphorylation. These results are coherent with several reports indicating that both β-arrestin isoforms are required for G protein-independent ERK1,2 signaling mediated by various GPCRs such as β2-adrenergic and parathyroid hormone type 1 receptors (Gesty-Palmer et al., 2006; Shenoy et al., 2006). Nonetheless, activation of ERK1,2 by other GPCRs, including angiotensin II type 1a and V2 receptors, is critically dependent on β-arrestin 2, whereas β-arrestin 1 has a minor influence or an antagonistic action on ERK1,2 signaling (Ahn et al., 2004; Ren et al., 2005), indicating that specificity of β-arrestin isoforms for ERK signaling is dependent on the GPCR.
β-Arrestin–dependent ERK1,2 signaling has been intimately related to its ability to form a stable signaling complex with the internalized receptor that is sequestered in endocytic vesicles where ERK1,2 remains activated for prolonged periods. The increase in ERK1,2 phosphorylation induced by 5-HT2C receptor stimulation persisted for several hours, similar to that measured upon V2 receptor activation (Tohgo et al., 2003), and it was abolished in cells expressing a β-arrestin mutant defective in interactions with clathrin. Nonetheless, this effect might rather result from blockade of interaction between arrestins and their downstream partners than inhibition of receptor internalization. Indeed, 5-HT2C receptor-operated ERK1,2 signaling required permanent agonist receptor stimulation and was not affected by treatments that stabilize the receptor at the plasma membrane. Hence, persistent β-arrestin–dependent ERK1,2 signaling evoked by GPCR ligands might not be solely mediated by receptor sequestered within endocytic vesicles.
Collectively, our observations point out that β-arrestin–dependent ERK1,2 signaling can operate independently of G protein signaling. They corroborate previous reports showing that mutants of GPCRs that cannot couple with G proteins can efficiently stimulate ERK1,2 phosphorylation in a β-arrestin–dependent manner (Wei et al., 2003). Independence of G protein coupling was also suggested by the demonstration that GPCR ligands, which do not promote G protein coupling or behave as inverse agonists for G protein-dependent signaling, are capable of increasing ERK activity (Azzi et al., 2003; Wei et al., 2003). In this study, we have identified CaM as an essential protein contributing to both recruitment of β-arrestin 2 by 5-HT2C receptor and receptor-mediated ERK1,2 signaling independently of activation of heterotrimeric G proteins. Indeed, expression of either a dominant-negative CaM mutant or a 5-HT2C receptor unable to bind CaM or CaM depletion by RNA interference strongly decreased the amount of β-arrestin 2 bound to the receptor upon agonist treatment and almost prevented receptor-mediated ERK1,2 phosphorylation. A previous study based on several GPCR models including the 5-HT2C receptor has identified a critical β-arrestin binding determinant common to the rhodopsin family GPCRs formed by the proximal 10 residues of the second intracellular loop (Marion et al., 2006), whereas the C-terminal tail was proposed to govern the stability of the receptor–β-arrestin complex (Tohgo et al., 2003). Accordingly, CaM bound to the juxtamembrane region of the 5-HT2C receptor C-terminus might function to stabilize 5-HT2C receptor–β-arrestin interaction. Interestingly, putative CaM binding motifs have been identified in the same region of several GPCRs, including muscarinic, adrenergic, and serotonergic receptors (Turner and Raymond, 2005). Whether physical association of CaM with these receptors promotes β-arrestin recruitment and/or stabilizes receptor–β-arrestin complex is an important issue requiring experimental validation.
The precise architecture of the complex formed by 5-HT2C receptors, CaM and β-arrestin upon 5-HT stimulation also remains to be resolved. CaM was recently demonstrated to physically interact with β-arrestins (Wu et al., 2006; Xiao et al., 2007). The CaM binding site identified in β-arrestin sequences by Wu et al. shows significant overlap with the GPCR binding site. However, it is unlikely that CaM competes with 5-HT2C receptor to bind to this site, due to the relatively low affinity of the arrestin/CaM interaction (∼ 7 μM), compared with the nanomolar affinity of arrestin/receptor binding (Wu et al., 2006). In fact, search for CaM binding motifs in the β-arrestin sequences identified two motifs with a high probability score. One of them was located outside the GPCR binding site, permitting simultaneous interaction of β-arrestin with both 5-HT2C receptor (i2 loop) and CaM. Moreover, CaM can form noncovalent dimers (Lafitte et al., 1999). This is consistent with the in vitro binding properties of the Ca2+-insensitive CaM mutant CaM1,2,3,4 to the 5-HT2C receptor C-terminus: we only detected a strong association of CaM1,2,3,4 in presence of an excess of wild-type GFP-CaM, suggesting that CaM1,2,3,4 binds to the receptor via formation of a dimer with wild-type CaM. Therefore, we propose a model in which β-arrestin 2 might be connected to both i2 loop of 5-HT2C receptor and receptor C-terminus via a CaM dimer, which acts to stabilize the 5-HT2C receptor/β-arrestin complex (see Supplemental Figure 4).
In conclusion, this study identifies for the first time a G protein-independent signaling mechanism (i.e., activation of the ERK1,2 pathway) engaged by the 5-HT2C receptor, one of the major antidepressant targets. Importantly, this signaling pathway was not only established in transfected HEK-293 cells but also in authentic cellular contexts, including choroid plexus epithelial cells, which express the highest receptor density, and cortical neurons. We also demonstrate that CaM physically associated with the C-terminal domain of the 5-HT2C receptor acts in concert with β-arrestin to promote G protein-independent receptor-operated ERK1,2 signaling. This pathway might provide a molecular substrate for the increase in neurogenesis induced by chronic treatment with 5-HT2C agonists, a phenomenon possibly involved in their paradoxical antidepressant action (Millan, 2005).
Supplementary Material
ACKNOWLEDGMENTS
Cytosolic Ca2+ measurements were carried out using facilities of the Pharmacological Screening platform of the Institut de Génomique Fonctionnelle. We are grateful to Drs. Federica Bertaso and Aline Dumuis for critical reading of the manuscript. This study was supported by grants from Centre National de la Recherche Scientifique, Institut National de la Santé et de la Recherche Médicale, the French Minister of Research (contract ACI-JC 5075 and ANR Neurosciences-2005) and the Fondation pour la Recherche Médicale (FRM Team 2005). M. L. was a recipient of fellowships from the French Minister of Research and FRM.
Abbreviations used:
- CaM
calmodulin
- CEC
choroid plexus epithelial cell
- ERK
extracellular signal-regulated kinase
- GPCR
G protein-coupled receptor
- PLC
phospholipase C
- PTX
pertussis toxin.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E08-04-0422) on September 3, 2008.
REFERENCES
- Ahn S., Wei H., Garrison T. R., Lefkowitz R. J. Reciprocal regulation of angiotensin receptor-activated extracellular signal-regulated kinases by beta-arrestins 1 and 2. J. Biol. Chem. 2004;279:7807–7811. doi: 10.1074/jbc.C300443200. [DOI] [PubMed] [Google Scholar]
- Azzi M., Charest P. G., Angers S., Rousseau G., Kohout T., Bouvier M., Pineyro G. beta-Arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proc. Natl. Acad. Sci. USA. 2003;100:11406–11411. doi: 10.1073/pnas.1936664100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes W. G., Reiter E., Violin J. D., Ren X. R., Milligan G., Lefkowitz R. J. beta-Arrestin 1 and Galphaq/11 coordinately activate RhoA and stress fiber formation following receptor stimulation. J. Biol. Chem. 2005;280:8041–8050. doi: 10.1074/jbc.M412924200. [DOI] [PubMed] [Google Scholar]
- Barthet G., Framery B., Gaven F., Pellissier L., Reiter E., Claeysen S., Bockaert J., Dumuis A. 5-Hydroxytryptamine 4 receptor activation of the extracellular signal-regulated kinase pathway depends on Src activation but not on G protein or beta-arrestin signaling. Mol. Biol. Cell. 2007;18:1979–1991. doi: 10.1091/mbc.E06-12-1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becamel C., Alonso G., Galeotti N., Demey E., Jouin P., Ullmer C., Dumuis A., Bockaert J., Marin P. Synaptic multiprotein complexes associated with 5-HT(2C) receptors: a proteomic approach. EMBO J. 2002;21:2332–2342. doi: 10.1093/emboj/21.10.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becamel C., Gavarini S., Chanrion B., Alonso G., Galeotti N., Dumuis A., Bockaert J., Marin P. The serotonin 5-HT2A and 5-HT2C receptors interact with specific sets of PDZ proteins. J. Biol. Chem. 2004;279:20257–20266. doi: 10.1074/jbc.M312106200. [DOI] [PubMed] [Google Scholar]
- Belcheva M. M., Szucs M., Wang D., Sadee W., Coscia C. J. mu-Opioid receptor-mediated ERK activation involves calmodulin-dependent epidermal growth factor receptor transactivation. J. Biol. Chem. 2001;276:33847–33853. doi: 10.1074/jbc.M101535200. [DOI] [PubMed] [Google Scholar]
- Birukova A. A., Birukov K. G., Smurova K., Adyshev D., Kaibuchi K., Alieva I., Garcia J. G., Verin A. D. Novel role of microtubules in thrombin-induced endothelial barrier dysfunction. FASEB J. 2004;18:1879–1890. doi: 10.1096/fj.04-2328com. [DOI] [PubMed] [Google Scholar]
- Bockaert J., Claeysen S., Becamel C., Dumuis A., Marin P. Neuronal 5-HT metabotropic receptors: fine-tuning of their structure, signaling, and roles in synaptic modulation. Cell Tissue Res. 2006;326:553–572. doi: 10.1007/s00441-006-0286-1. [DOI] [PubMed] [Google Scholar]
- Bockaert J., Fagni L., Dumuis A., Marin P. GPCR interacting proteins (GIP) Pharmacol. Ther. 2004;103:203–221. doi: 10.1016/j.pharmthera.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Bofill-Cardona E., Kudlacek O., Yang Q., Ahorn H., Freissmuth M., Nanoff C. Binding of calmodulin to the D2-dopamine receptor reduces receptor signaling by arresting the G protein activation switch. J. Biol. Chem. 2000;275:32672–32680. doi: 10.1074/jbc.M002780200. [DOI] [PubMed] [Google Scholar]
- Chanrion B., la Cour C. M., Gavarini S., Seimandi M., Vincent L., Pujol J. F., Bockaert J., Marin P., Millan M. J. Inverse agonist and neutral antagonist actions of antidepressants at recombinant and native 5-hydroxytryptamine2C receptors: differential modulation of cell surface expression and signal transduction. Mol. Pharmacol. 2008;73:748–757. doi: 10.1124/mol.107.041574. [DOI] [PubMed] [Google Scholar]
- Charest P. G., Oligny-Longpre G., Bonin H., Azzi M., Bouvier M. The V2 vasopressin receptor stimulates ERK1/2 activity independently of heterotrimeric G protein signalling. Cell Signal. 2007;19:32–41. doi: 10.1016/j.cellsig.2006.05.020. [DOI] [PubMed] [Google Scholar]
- Cryan J. F., Lucki I. Antidepressant-like behavioral effects mediated by 5-hydroxytryptamine(2C) receptors. J. Pharmacol. Exp. Ther. 2000;295:1120–1126. [PubMed] [Google Scholar]
- Cussac D., Newman-Tancredi A., Duqueyroix D., Pasteau V., Millan M. J. Differential activation of Gq/11 and Gi(3) proteins at 5-hydroxytryptamine(2C) receptors revealed by antibody capture assays: influence of receptor reserve and relationship to agonist-directed trafficking. Mol. Pharmacol. 2002;62:578–589. doi: 10.1124/mol.62.3.578. [DOI] [PubMed] [Google Scholar]
- Della Rocca G. J., Mukhin Y. V., Garnovskaya M. N., Daaka Y., Clark G. J., Luttrell L. M., Lefkowitz R. J., Raymond J. R. Serotonin 5-HT1A receptor-mediated Erk activation requires calcium/calmodulin-dependent receptor endocytosis. J. Biol. Chem. 1999;274:4749–4753. doi: 10.1074/jbc.274.8.4749. [DOI] [PubMed] [Google Scholar]
- Farnsworth C. L., Freshney N. W., Rosen L. B., Ghosh A., Greenberg M. E., Feig L. A. Calcium activation of Ras mediated by neuronal exchange factor Ras-GRF. Nature. 1995;376:524–527. doi: 10.1038/376524a0. [DOI] [PubMed] [Google Scholar]
- Gavarini S., Becamel C., Altier C., Lory P., Poncet J., Wijnholds J., Bockaert J., Marin P. Opposite effects of PSD-95 and MPP3 PDZ proteins on serotonin 5-hydroxytryptamine2C receptor desensitization and membrane stability. Mol. Biol. Cell. 2006;17:4619–4631. doi: 10.1091/mbc.E06-03-0218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gesty-Palmer D., et al. Distinct beta-arrestin- and G protein-dependent pathways for parathyroid hormone receptor-stimulated ERK1/2 activation. J. Biol. Chem. 2006;281:10856–10864. doi: 10.1074/jbc.M513380200. [DOI] [PubMed] [Google Scholar]
- Giorgetti M., Tecott L. H. Contributions of 5-HT(2C) receptors to multiple actions of central serotonin systems. Eur. J. Pharmacol. 2004;488:1–9. doi: 10.1016/j.ejphar.2004.01.036. [DOI] [PubMed] [Google Scholar]
- Kara E., Crepieux P., Gauthier C., Martinat N., Piketty V., Guillou F., Reiter E. A phosphorylation cluster of five serine and threonine residues in the C-terminus of the follicle-stimulating hormone receptor is important for desensitization but not for beta-arrestin-mediated ERK activation. Mol. Endocrinol. 2006;20:3014–3026. doi: 10.1210/me.2006-0098. [DOI] [PubMed] [Google Scholar]
- Lafitte D., Heck A. J., Hill T. J., Jumel K., Harding S. E., Derrick P. J. Evidence of noncovalent dimerization of calmodulin. Eur. J. Biochem. 1999;261:337–344. doi: 10.1046/j.1432-1327.1999.00284.x. [DOI] [PubMed] [Google Scholar]
- Lefkowitz R. J., Shenoy S. K. Transduction of receptor signals by beta-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- Malberg J. E., Eisch A. J., Nestler E. J., Duman R. S. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J. Neurosci. 2000;20:9104–9110. doi: 10.1523/JNEUROSCI.20-24-09104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion S., Oakley R. H., Kim K. M., Caron M. G., Barak L. S. A beta-arrestin binding determinant common to the second intracellular loops of rhodopsin family G protein-coupled receptors. J. Biol. Chem. 2006;281:2932–2938. doi: 10.1074/jbc.M508074200. [DOI] [PubMed] [Google Scholar]
- Marion S., Weiner D. M., Caron M. G. RNA editing induces variation in desensitization and trafficking of 5-hydroxytryptamine 2c receptor isoforms. J. Biol. Chem. 2004;279:2945–2954. doi: 10.1074/jbc.M308742200. [DOI] [PubMed] [Google Scholar]
- McGrew L., Chang M. S., Sanders-Bush E. Phospholipase D activation by endogenous 5-hydroxytryptamine 2C receptors is mediated by Galpha13 and pertussis toxin-insensitive Gbetagamma subunits. Mol. Pharmacol. 2002;62:1339–1343. doi: 10.1124/mol.62.6.1339. [DOI] [PubMed] [Google Scholar]
- Millan M. J. Serotonin 5-HT2C receptors as a target for the treatment of depressive and anxious states: focus on novel therapeutic strategies. Therapie. 2005;60:441–460. doi: 10.2515/therapie:2005065. [DOI] [PubMed] [Google Scholar]
- Millan M. J. Multi-target strategies for the improved treatment of depressive states: conceptual foundations and neuronal substrates, drug discovery and therapeutic application. Pharmacol. Ther. 2006;110:135–370. doi: 10.1016/j.pharmthera.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Minakami R., Jinnai N., Sugiyama H. Phosphorylation and calmodulin binding of the metabotropic glutamate receptor subtype 5 (mGluR5) are antagonistic in vitro. J. Biol. Chem. 1997;272:20291–20298. doi: 10.1074/jbc.272.32.20291. [DOI] [PubMed] [Google Scholar]
- Moreau J. L., Bos M., Jenck F., Martin J. R., Mortas P., Wichmann J. 5HT2C receptor agonists exhibit antidepressant-like properties in the anhedonia model of depression in rats. Eur. Neuropsychopharmacol. 1996;6:169–175. doi: 10.1016/0924-977x(96)00015-6. [DOI] [PubMed] [Google Scholar]
- Nakajima Y., Yamamoto T., Nakayama T., Nakanishi S. A relationship between protein kinase C phosphorylation and calmodulin binding to the metabotropic glutamate receptor subtype 7. J. Biol. Chem. 1999;274:27573–27577. doi: 10.1074/jbc.274.39.27573. [DOI] [PubMed] [Google Scholar]
- Nickols H. H., Shah V. N., Chazin W. J., Limbird L. E. Calmodulin interacts with the V2 vasopressin receptor: elimination of binding to the C terminus also eliminates arginine vasopressin-stimulated elevation of intracellular calcium. J. Biol. Chem. 2004;279:46969–46980. doi: 10.1074/jbc.M407351200. [DOI] [PubMed] [Google Scholar]
- O'Connor V., El Far O., Bofill-Cardona E., Nanoff C., Freissmuth M., Karschin A., Airas J. M., Betz H., Boehm S. Calmodulin dependence of presynaptic metabotropic glutamate receptor signaling. Science. 1999;286:1180–1184. doi: 10.1126/science.286.5442.1180. [DOI] [PubMed] [Google Scholar]
- Reiter E., Lefkowitz R. J. GRKs and beta-arrestins: roles in receptor silencing, trafficking and signaling. Trends Endocrinol. Metab. 2006;17:159–165. doi: 10.1016/j.tem.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Ren X. R., Reiter E., Ahn S., Kim J., Chen W., Lefkowitz R. J. Different G protein-coupled receptor kinases govern G protein and beta-arrestin-mediated signaling of V2 vasopressin receptor. Proc. Natl. Acad. Sci. USA. 2005;102:1448–1453. doi: 10.1073/pnas.0409534102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santarelli L., et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science. 2003;301:805–809. doi: 10.1126/science.1083328. [DOI] [PubMed] [Google Scholar]
- Sengupta P., Ruano M. J., Tebar F., Golebiewska U., Zaitseva I., Enrich C., McLaughlin S., Villalobo A. Membrane-permeable calmodulin inhibitors (e.g. W-7/W-13) bind to membranes, changing the electrostatic surface potential: dual effect of W-13 on epidermal growth factor receptor activation. J. Biol. Chem. 2007;282:8474–8486. doi: 10.1074/jbc.M607211200. [DOI] [PubMed] [Google Scholar]
- Shenoy S. K., Drake M. T., Nelson C. D., Houtz D. A., Xiao K., Madabushi S., Reiter E., Premont R. T., Lichtarge O., Lefkowitz R. J. beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J. Biol. Chem. 2006;281:1261–1273. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- Thomas W. G., Pipolo L., Qian H. Identification of a Ca2+/calmodulin-binding domain within the carboxyl-terminus of the angiotensin II (AT1A) receptor. FEBS Lett. 1999;455:367–371. doi: 10.1016/s0014-5793(99)00904-7. [DOI] [PubMed] [Google Scholar]
- Thouvenot E., Lafon-Cazal M., Demettre E., Jouin P., Bockaert J., Marin P. The proteomic analysis of mouse choroid plexus secretome reveals a high protein secretion capacity of choroidal epithelial cells. Proteomics. 2006;6:5941–5952. doi: 10.1002/pmic.200600096. [DOI] [PubMed] [Google Scholar]
- Tohgo A., Choy E. W., Gesty-Palmer D., Pierce K. L., Laporte S., Oakley R. H., Caron M. G., Lefkowitz R. J., Luttrell L. M. The stability of the G protein-coupled receptor-beta-arrestin interaction determines the mechanism and functional consequence of ERK activation. J. Biol. Chem. 2003;278:6258–6267. doi: 10.1074/jbc.M212231200. [DOI] [PubMed] [Google Scholar]
- Turner J. H., Gelasco A. K., Raymond J. R. Calmodulin interacts with the third intracellular loop of the serotonin 5-hydroxytryptamine1A receptor at two distinct sites: putative role in receptor phosphorylation by protein kinase C. J. Biol. Chem. 2004;279:17027–17037. doi: 10.1074/jbc.M313919200. [DOI] [PubMed] [Google Scholar]
- Turner J. H., Raymond J. R. Interaction of calmodulin with the serotonin 5-hydroxytryptamine2A receptor. A putative regulator of G protein coupling and receptor phosphorylation by protein kinase C. J. Biol. Chem. 2005;280:30741–30750. doi: 10.1074/jbc.M501696200. [DOI] [PubMed] [Google Scholar]
- Wang D., Sadee W., Quillan J. M. Calmodulin binding to G protein-coupling domain of opioid receptors. J. Biol. Chem. 1999;274:22081–22088. doi: 10.1074/jbc.274.31.22081. [DOI] [PubMed] [Google Scholar]
- Wei H., Ahn S., Shenoy S. K., Karnik S. S., Hunyady L., Luttrell L. M., Lefkowitz R. J. Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc. Natl. Acad. Sci. USA. 2003;100:10782–10787. doi: 10.1073/pnas.1834556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss S., Pin J. P., Sebben M., Kemp D. E., Sladeczek F., Gabrion J., Bockaert J. Synaptogenesis of cultured striatal neurons in serum-free medium: a morphological and biochemical study. Proc. Natl. Acad. Sci. USA. 1986;83:2238–2242. doi: 10.1073/pnas.83.7.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werry T. D., Gregory K. J., Sexton P. M., Christopoulos A. Characterization of serotonin 5-HT2C receptor signaling to extracellular signal-regulated kinases 1 and 2. J. Neurochem. 2005;93:1603–1615. doi: 10.1111/j.1471-4159.2005.03161.x. [DOI] [PubMed] [Google Scholar]
- Wu N., Hanson S. M., Francis D. J., Vishnivetskiy S. A., Thibonnier M., Klug C. S., Shoham M., Gurevich V. V. Arrestin binding to calmodulin: a direct interaction between two ubiquitous signaling proteins. J. Mol. Biol. 2006;364:955–963. doi: 10.1016/j.jmb.2006.09.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X. M., Fakler B., Rivard A., Wayman G., Johnson-Pais T., Keen J. E., Ishii T., Hirschberg B., Bond C. T., Lutsenko S., Maylie J., Adelman J. P. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature. 1998;395:503–507. doi: 10.1038/26758. [DOI] [PubMed] [Google Scholar]
- Xiao K., McClatchy D. B., Shukla A. K., Zhao Y., Chen M., Shenoy S. K., Yates J. R., 3rd, Lefkowitz R. J. Functional specialization of beta-arrestin interactions revealed by proteomic analysis. Proc. Natl. Acad. Sci. USA. 2007;104:12011–12016. doi: 10.1073/pnas.0704849104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap K. L., Kim J., Truong K., Sherman M., Yuan T., Ikura M. Calmodulin target database. J. Struct. Funct. Genomics. 2000;1:8–14. doi: 10.1023/a:1011320027914. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.