

Abstract
CREB is a prototypic bZIP transcription factor and a master regulator of glucose metabolism, synaptic plasticity, cell growth, apoptosis, and tumorigenesis. Transducers of regulated CREB activity (TORCs) are essential transcriptional coactivators of CREB and an important point of regulation on which various signals converge. In this study, we report on the activation of TORC1 through MEKK1-mediated phosphorylation. MEKK1 potently activated TORC1, and this activation was independent of downstream effectors MEK1/MEK2, ERK2, JNK, p38, protein kinase A, and calcineurin. MEKK1 induced phosphorylation of TORC1 both in vivo and in vitro. Expression of the catalytic domain of MEKK1 alone in cultured mammalian cells sufficiently caused phosphorylation and subsequent activation of TORC1. MEKK1 physically interacted with TORC1 and stimulated its nuclear translocation. An activation domain responsive to MEKK1 stimulation was mapped to amino acids 431–650 of TORC1. As a physiological activator of CREB, interleukin 1α triggered MEKK1-dependent phosphorylation of TORC1 and its consequent recruitment to the cAMP response elements in the interleukin 8 promoter. Taken together, our findings suggest a new mechanism for regulated activation of TORC1 transcriptional coactivator and CREB signaling.
INTRODUCTION
cAMP response element-binding protein (CREB) is one of the most well-characterized transcription factors (Shaywitz and Greenberg, 1999). In response to a variety of stimuli including mitogens, neurotransmitters, and other agents that elevate intracellular cAMP or Ca2+ levels, CREB is activated rapidly through phosphorylation at S133 in the kinase inducible domain (KID), leading to up-regulated transcription of more than 4000 target genes that have a significant impact on many aspects of cell physiology and pathology (Mayr and Montminy, 2001).
In addition to induced phosphorylation at S133, CREB activity can also be regulated through a new family of transcriptional coactivators, termed transducers of regulated CREB activity (TORCs; Conkright et al., 2003; Iourgenko et al., 2003). Three members named TORC1, TORC2, and TORC3 have been found in this family. All three TORCs activate CREB-dependent transcription, but are differentially expressed in different tissues and cells (Conkright et al., 2003). TORCs represent a critical regulatory point in a complex network of signals (Bittinger et al., 2004; Sceraton et al., 2004; Koo et al., 2005; Shaw et al., 2005; Katoh et al., 2006). In one model for the regulation of TORC activity, tumor suppressor protein LKB1, which is frequently inactivated in the cancer-predisposing Peutz-Jeghers syndrome (Alessi et al., 2006), can phosphorylate and activate AMP-activated protein kinase (AMPK). The AMPK kinases then phosphorylate TORCs, leading to inhibition of O-glycosylation, binding with 14-3-3 proteins, sequestration in the cytoplasm, and prevention of transcriptional activation (Takemori et al., 2007a; Dentin et al., 2008). It is noteworthy that S171-phosphorylated TORC2 retained in the cytoplasm undergoes polyubiquitination at K628 followed by proteosome-dependent degradation (Dentin et al., 2007). In addition, other secondary messengers such as calcium and cAMP can also regulate TORC activity (Screaton et al., 2004; Ohmae et al., 2006; Amelio et al., 2007). While dephosphorylation of TORC2 at S171 and S275 is induced by a calcium-activated phosphatase known as calcineurin (Jansson et al., 2008), cAMP stimulates protein kinase A (PKA)-mediated phosphorylation of an AMPK-related kinase termed salk-inducible kinase 2 (SIK2) at S587, leading to its inhibition and subsequent activation of TORCs (Screaton et al., 2004).
Both TORC and p300 families of transcriptional coactivators are essential for optimal activity of CREB (Siu et al., 2006). TORCs and p300/CREB-binding protein (CBP) activate transcription through different mechanisms, but they also synergize with each other (Ravnskjaer et al., 2007). While phosphorylation of CREB at S133 promotes the recruitment of p300/CBP but not TORCs, TORCs enhance the association of CREB with TAFII130 independent of S133 phosphorylation (Conkright et al., 2003). On the other hand, TORC2 binds CBP/p300 directly and cooperates with them to confer specificity in the activation of CREB target genes (Ravnskjaer et al., 2007).
The physiological and pathological roles of TORC coactivators have been demonstrated in different contexts. First, TORC2 is a key regulator of cell metabolism. It is a sensor of glucose and modulates gluconeogenesis and insulin signaling (Canettieri et al., 2005; Koo et al., 2005; Dentin et al., 2007, 2008). TORCs are also transcriptional activators of peroxisome proliferator–activated receptor γ coactivator 1α (PGC1α), which regulates mitochondrial biogenesis and energy metabolism (Wu et al., 2006), and steroidogenic acute regulatory protein (StAR), a mitochondrial protein that transfers cholesterol (Takemori et al., 2007b). In addition, TORC2 coordinates glucose-dependent insulinotropic polypeptide (GIP)-induced expression of antiapoptotic BCL2 gene (Kim et al., 2008). Second, TORC2 regulates the normal program of germinal center gene activation and repression to promote B-cell development (Kuraishy et al., 2007). Third, TORC1 is required for late-phase long-term synaptic potentiation in the hippocampus (Zhou et al., 2006; Kovács et al., 2007). Finally, TORCs are critically involved in tumorigenesis (Siu and Jin, 2007). TORC1-MAML2 is a fusion oncoprotein found in malignant salivary gland tumor and promotes oncogenesis through activating CREB and its target genes (Coxon et al., 2005; Wu et al., 2005). TORCs are also essential coactivator of the Tax oncoprotein of human T-cell leukemia virus type 1 (HTLV-1) in the activation of viral long-terminal repeats (Koga et al., 2004; Siu et al., 2006), and this coactivation is inhibited by BCL3 (Hishiki et al., 2007). Suffice it to say, TORC coactivators are important regulators of CREB relevant to many biological systems and human diseases.
Different stimuli of CREB converge on just a few signaling pathways governed by calcium, cAMP, and mitogen-activated protein kinase (MAPK; Johannessen et al., 2004). MAPKs are evolutionarily conserved kinases that link extracellular signals ultimately to nuclear transcriptional machinery. MAPK modules generally comprise of three tiers in which a specific MAPK is regulated by a MAPK kinase (MAP2K), which in turn is activated by a MAPK kinase kinase (MAP3K; Turjanski et al., 2007). Mitogen-activated/extracellular signal–regulated kinase kinase 1 (MEKK1) is a MAP3K that regulates c-Jun N-terminal kinase (JNK) as well as other kinases including extracellular signal-regulated kinases (ERK1/2), p38, and IκB kinases (IKK; Cuevas et al., 2007). MEKK1 contains a large N-terminal regulatory domain and a C-terminal catalytic domain (Xu et al., 1996). The cleavage of MEKK1 at D874 releases the active kinase domain, resulting in constitutive activation of the kinase (Gibson et al., 1999). MEKK1 is a known regulator of several transcriptional activators and coactivators such as c-Myc, c-Jun, p53, CCAAT/enhancer binding protein β (C/EBPβ), silencing mediator for retinoid and thyroid hormone receptors (SMRT), and p300 (Fuchs et al., 1998; Hong and Privalsky, 2000; See et al., 2001; Alarcon-Vargas et al., 2002; Roy et al., 2002; Xia et al., 2007).
To investigate whether and how MAPK might impact on TORC activation, in this study we screened a panel of MAPK, MAP2K, and MAP3K and identified MEKK1 as a potent activator of TORC1-mediated transcription. The mechanism of MEKK1-induced activation of TORC1 was further investigated. We demonstrated the ability of MEKK1 to induce phosphorylation of TORC1, which does not occur at S167 equivalent to S171 of TORC2. In addition, we found that the expression of MEKK1 caused nuclear translocation of TORC1, followed by its recruitment to the cAMP response element (CRE) in the promoter region. Our work suggests a new signaling pathway for the activation of TORCs and CREB.
MATERIALS AND METHODS
Plasmids
Eukaryotic and bacterial expression plasmids for TORC1 have been described elsewhere (Siu et al., 2006). TORC1 S167A mutant was constructed by PCR method using primers 5′-GAGAAGGACCAATGCTGACTCCGCCCTGCA-3′ and 5′-TGCAGGGCGGAGTCAGCATTGGTCCTTCTC-3′. Expression plasmids for hemagglutinin (HA)-tagged MEKK1 and MEKK1 D1369A (Xu et al., 1996) were kindly provided by Dr. Melanie Cobb (The University of Texas Southwestern Medical Center, Dallas, TX). Expression plasmid for myc-tagged MEKK-C was constructed by subcloning MEKK-C gene in the pFC-MEKK plasmid (Stratagene, La Jolla, CA) into pCMV-Tag3B (Stratagene) via BamHI and XhoI restriction sites. Expression plasmids for ERK2, JNK, and p38 (Teramoto et al., 1996; Jin et al., 1997) were kindly provided by Dr. Silvio Gutkind (National Institute of Dental and Craniofacial Research, Bethesda, MD). Expression vectors for TORC1 and its truncated mutants fused to Gal4 DNA-binding domain (e.g., GalTORC1, GalM1, and GalM4) were derived from pM (Clontech, Mountain View, CA). Expression vectors for shGFP and shTORC1a, which target green fluorescent protein (GFP) and TORC1 transcripts, respectively, have been described elsewhere (Siu et al., 2006; Kok et al., 2007). An additional short hairpin RNA (shRNA) against TORC1 designated shTORC1b was also constructed, and it targets nucleotides 1738–1758 (5′-CAGUGGCAUCCCCAACAUCAU-3′) of TORC1 mRNA. An expression vector for A-CREB, a dominant inactive version of CREB (Ahn et al., 1998), was kindly provided by Dr. Charles Vinson (National Institute of Neurological Disorders and Stroke, Bethesda, MD). An expression plasmid for MEKK-KR, a dominant inactive mutant of MEKK-1 (Yan et al., 1994), was kindly provided by Dr. Dennis Templeton (University of Virginia, Charlottesville, VA).
Reporter Assay
HeLa and HEK293 cells were cultured in DMEM and transfected with Genejuice transfection reagent (EMD Chemicals, Gibbstown, NJ). Cells were harvested 48 h after transfection. Luciferase activity was determined as described (Ching et al., 2006; Siu et al., 2006) using the Dual-Luciferase reagents (Promega, Madison, WI). Transcriptional activity on canonical CRE or Gal4-binding elements was measured with plasmids pCRE-Luc (Stratagene) and pGal-Luc (Clontech), respectively. A plasmid containing −1481 to +44 nucleotides of the promoter region of interleukin 8 (IL-8) gene (Mukaida et al., 1990) was kindly provided by Dr. Naofumi Mukaida (Kanazawa University, Kanazawa, Japan). The IL-8 promoter was subcloned into pGL3-basic (Promega) via XhoI and HindIII restriction sites to generate pIL8-Luc. Plasmid pIL8ΔCRE-Luc in which the CRE is disrupted was constructed by PCR method using primers 5′-AATTTCCTCTCGATCAATGAAAAGATGAGGGT-3′ and 5′-ATCTTTTCATTGATCGAGAGGAAATTCCACGA-3′. Transfection efficiencies were normalized to a control plasmid (pSV-RLuc from Promega) expressing Renilla luciferase.
Western Blotting
HeLa and HEK293 cells were lysed in lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, and 1 mM sodium vanadate) supplemented with protease inhibitor (Roche, Indianapolis, IN). Samples were separated by SDS-PAGE and transferred onto Immobilon-P membranes (Millipore, Billerica, MA). Membranes were blocked with 5% nonfat milk and then probed with primary antibody and horseradish peroxidase–conjugated secondary antibody. Proteins were visualized by enhanced chemiluminescence (Amersham, Piscataway, NJ). For alkaline phosphatase (AP) treatment before SDS-PAGE and Western blotting, cell lysates were incubated with 10 U of calf intestinal AP (Invitrogen, Carlsbad, CA) at 30°C for 1 h.
In Vitro Kinase Assay
Phosphorylation of TORC1 by MEKK-C in vitro was assayed essentially as described (Jin et al., 1999). Escherichia coli–produced His-tagged TORC1 protein was affinity-purified through nickel-nitrilotriacetic acid resin (Qiagen, Valencia, CA). Myc-tagged MEKK-C protein was immunoprecipitated from cultured HeLa cells with rabbit anti-Myc (Santa Cruz Biotechnology, Santa Cruz, CA). Briefly, 1 μg of recombinant TORC1 was mixed with MEKK-C precipitate in kinase buffer (25 mM Tris-HCl, pH 7.5, 0.02 mM EGTA). The reaction was started with the addition of 10 μCi [γ-32P]ATP. After incubation at 30°C for 20 min, phosphorylation was terminated by the addition of sample buffer (60 mM Tris, 2% SDS, 6% glycerol, 1% β-mercaptoethanol, and 0.002% bromophenol blue). Samples were separated by SDS-PAGE and detected by autoradiography.
Coimmunoprecipitation
HEK293T cells grown in a 100-mm Petri dish were harvested in 0.5 ml lysis buffer (20 mM Tris-HCl pH 7.5, 100 mM NaCl, 0.1% NP-40, and 0.5 mM EDTA). V5-tagged TORC1 was immunoprecipitated from the cleared lysate by overnight incubation at 4°C with rabbit anti-V5 (Sigma-Aldrich, St. Louis, MO). The immunocomplex was collected by protein A agarose (Invitrogen), washed three times with lysis buffer, and resuspended with SDS-PAGE loading buffer.
Confocal Microscopy
Multicolor immunofluorescence imaging was performed on a Zeiss LSM510 confocal microscope as described (Carl Zeiss, Thornwood, NY; Chan et al., 2006; Ching et al., 2006). HeLa cells were fixed and permeabilized with ice-cold methanol:acetone (1:1) for 10 min. Cells were blocked with 3% bovine serum albumin (BSA) for 20 min. Cells were incubated with primary and conjugated secondary antibodies for 1 h individually in 0.3% BSA. Cells were stained with 0.5 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) before mounting.
RT-PCR
Semiquantitative analysis of IL-8 transcript by RT-PCR was performed as previously described (Choy et al., 2008a). Total RNA was purified by using Trizol reagent (Invitrogen). cDNA was synthesized by using ThermoScript RT-PCR System (Invitrogen). Primers for amplification of IL-8 mRNA were 5′-ATGACTTCCAAGCTGGCCGTGGCTC-3′ and 5′-TAATTTCTGTGTTGGCGCAGTGTGG-3′. Transcript of GAPDH was amplified with primers 5′-GCAGGGGGGAGCCAAAAGGG-3′ and 5′-TGCCAGCCCCAGCGTCAAAG-3′. Amplification of TORC1 mRNA was carried out with primers 5′-CCCACCTTCCCTGCA-3′ and 5′-CTGGCCAGGGTTCTC-3′. The number of PCR cycles was optimized to ensure that DNA amplification is within a quantitative linear range.
Quantitative Real-Time RT-PCR
Quantitative real-time RT-PCR was carried out as described (Choy et al., 2008a). Amounts of mRNA transcripts were measured by using Bio-Rad CFX96 real-time PCR detection system (Bio-Rad, Hercules, CA). SYBR Green PCR master mix (Applied Biosystems, Foster City, CA) was used to detect double-stranded DNA. Relative expression levels of TORC1 mRNA were calculated by normalizing to the level of GAPDH mRNA by using comparative threshold cycle (ct) method, in which fold difference = 2−(Δct of target gene−Δct of reference). Primers for amplification of TORC1 mRNA were 5′-GCCTCTGATGTGGTCTCTTCC-3′ and 5′-CCACGCAGC CAGAATCCTTTAG-3′. GAPDH transcript was amplified with primers 5′-AACGTGTCAGTGGTGGACCTG-3′ and 5′-AGTGGGTGTCGCTGTTGAAGT-3′.
Northern Blotting
Northern blot analysis of IL-8 mRNA was carried out as described (Choy et al., 2008b). Briefly, total RNA of HeLa cells was extracted by using RNeasy reagents (Qiagen). RNA was immobilized onto Zeta-Probe GT membrane (Bio-Rad) by UV cross-linking. IL-8 and GAPDH probes were generated with the same primers listed above for RT-PCR analysis. These gene-specific probes were labeled with [α-32P]dCTP (Applied Biosystems) using Ready-To-Go DNA Labeling beads (Amersham). Unincorporated nucleotides were removed by using illustra ProbeQuant G-50 microcolumns (GE Healthcare, Piscataway, NJ). Membrane was hybridized with labeled probes in NorthernMax prehybridization and hybridization buffer (Ambion, Austin, TX) at 42°C overnight. After hybridization, the blot was rinsed twice with 2× SSC containing 0.1% SDS at 42°C for 5 min and then washed twice with 0.1× SSC containing 0.1% SDS at 42°C for 15 min.
Chromatin Immunoprecipitation Assay
The association of TORC1 with IL-8 promoter in cultured cells was detected by the chromatin immunoprecipitation (ChIP) assay as described (Chun and Jin, 2003; Chin et al., 2005). Briefly, HeLa cells were cross-linked by 1% formaldehyde for 10 min at 37°C. The DNA–protein complex was immunoprecipitated, and the genomic DNA was purified by phenol-chloroform extraction. Promoter sequence spanning −121 to +62 nucleotides of the IL-8 gene was PCR-amplified by using primers 5′-GGGCCATCAGTTGCAAATCGT-3′ and 5′-TTCCTTCCGGTGGTTTCTTC-3′.
RESULTS
MEKK1 Activates TORC1-mediated Transcription
Although MAPKs are critically involved in the regulation of CREB activation, the underlying mechanisms are not well understood (Johannessen et al., 2004). To investigate whether and how MAPKs, MAP2Ks, and MAP3Ks might modulate the activation of TORCs, we screened a panel of 13 kinases (ERK1, ERK2, JNK, p38, ERK5, MEK1, MEK2, MEK4, MEK7, MEKK1, MEKK3, Raf, and ASK1) for stimulation of TORC1-dependent activation of CRE-driven expression of firefly luciferase reporter. Because MEKK1 exhibited the most significant stimulatory activity on TORC1 among the 13 kinases tested, we sought to characterize its activation of TORC1 in detail.
We first performed cotransfection experiments to assess MEKK1-induced activation of TORC1-dependent transcription. In HeLa cells where the level of endogenous TORCs was low (Conkright et al., 2003), the expression of MEKK1 alone, when compared with that of TORC1, did not significantly activate CRE-driven transcription (Figure 1; groups 1–4 compared with group 5). In contrast, when TORC1 and MEKK1 were coexpressed, MEKK1 induced moderate and dose-dependent enhancement of TORC1 activity on CRE (Figure 1A; groups 5–8). Because MEKK1 is a MAP3K, we next investigated the requirement of the kinase activity for the activation of TORC1 by using two forms of MEKK1. First, we used constitutively active MEKK-C that contains the catalytic domain only. More than 10-fold stimulation of TORC1 transcriptional activity by MEKK-C (Figure 1B; groups 5–8) suggested that the activation of TORC1 was likely triggered by an MEKK1-induced phosphorylation event. Similar findings were also obtained from HEK293 cells (Supplemental Figure S1; groups 5–8), indicating that MEKK1-dependent activation of TORC1 occurred in different types of cells. Second, we exploited MEKK1 D1369A, a kinase-dead mutant of MEKK1 (Xu et al., 1996). The inability of MEKK1 D1369A to stimulate TORC1-mediated transcription (Figure 1C, groups 5–8) lent further support to the notion that MEKK1-induced phosphorylation mediates the activation of TORC1.
Figure 1.

Activation of TORC1 transcriptional activity by MEKK1. HeLa cells were transfected with the indicated plasmids. All groups received reporter constructs pCRE-Luc and pSV-RLuc. TORC1 (A–C) or TORC1 S167A (D–F) expression plasmid was also introduced into cells in groups 5–8. The amounts of expression vector for full-length MEKK1 (A and D), MEKK-C (B and E), and MEKK1 D1369A (C and F) were progressively increased as indicated. Relative luciferase activity (RLA) represents firefly luciferase activity recovered form pCRE-Luc normalized to Renilla luciferase activity recovered from pSV-RLuc. Results represent three independent experiments; error bars, SDs.
The regulation of TORC1 through phosphorylation at S167 by AMPK-related kinases such as SIK2 has been suggested (Screaton et al., 2004). To determine the involvement of this phosphorylation in MEKK1-induced activation of TORC1, we constructed the S167A mutant of TORC1 and characterized it for activity on CRE and stimulation by MEKK1. Because the behaviors of TORC1 wild-type and TORC1 S167A in all three settings of the reporter assay were almost identical (Figure 1, D–F, compared with A–C), MEKK1-induced activation of TORC1 was independent of phosphorylation at S167.
TORC1-activated Transcription Is Not Mediated by ERK2, MEK1/2, JNK, p38, PKA, or Calcineurin
MEKK1 is a MAP3K that activates downstream MAPKs such as ERK2, JNK, and p38 (Turjanski et al., 2007). In our initial screening for activators of TORC1, none of these MAPKs was found to stimulate TORC1-induced activation of CRE-dependent transcription. To formally address the influence of MAPKs on TORC1 activity, we measured TORC1 transcriptional activity with progressively increasing doses of ERK2, JNK, and p38 (Figure 2, A–C; lanes 5–8). Although expression of ERK2 and JNK did not affect TORC1 activity significantly, TORC1 appeared to be inhibited in high concentrations of p38, which was likely caused by cytotoxicity.
Figure 2.

TORC1- and MEKK1-mediated transcriptional activation is not mediated through ERK2, JNK, p38, PKA, or calcineurin. HeLa cells were transfected with the indicated plasmids and RLA was measured as in Figure 1. Progressively increasing amounts of expression vector for full-length ERK2 (A), JNK (B), p38 (C), and MEKK-C (D) were used. The indicated inhibitors (U0126, SP600125, SB205380, H89, and CsA) were added from a stock solution in DMSO to the cell culture to a final concentration of 5 μM at 14 h before harvest.
To determine whether downstream kinases MEK1/2, JNK and p38 are involved in MEKK1-dependent activation of TORC1, we also used chemical inhibitors U0126, SP600125, and SB203580, which specifically block MEK1/2, JNK, and p38 activity, respectively (Clerk and Sugden, 1998; Favata et al., 1998; Bennett et al., 2001). To prevent nonspecific effect on the CRE, we tethered TORC1 to the promoter through Gal4 DNA-binding domain and stimulated with MEKK-C. In this setting, an approximately twofold activation of GalTORC1 by MEKK-C was observed (Figure 2D, groups 1–4). However, neither the basal activity of GalTORC1 nor the stimulation by MEKK-C was significantly affected by U0126, SP600125, or SB203580 (Figure 2D, groups 5–8, 9–12, and 13–16). Consistent with this, the addition of U0126, SP600125, or SB203580 did not influence TORC1-induced activation of CRE-dependent transcription (Supplemental Figure S2, A–D). Thus, MEKK1-induced activation of TORC1-dependent transcription was unlikely mediated through MEK1/2, ERK2, JNK, or p38.
Coactivator function of TORCs is regulated by PKA and calcineurin (Screaton et al., 2004; Jansson et al., 2008), which could also be activated directly or indirectly by MEKK1. To exclude the possibility that PKA and calcineurin might be involved in MEKK1-induced activation of TORC1, we also assessed the impact of PKA and calcineurin inhibitors on the activation of GalTORC1 by MEKK-C. H89 is a specific inhibitor of PKA (Chijiwa et al., 1990), whereas cyclosporin A (CsA) potently inhibits calcineurin (Liu et al., 1991). Notably, treatment of cells with H89 or CsA did not alter the activity profile of GalTORC1 in the presence or in the absence of MEKK-C (Figure 2D, groups 17–20 and 21–24). In addition, neither H89 nor CsA exerted a significant effect on the activation of CRE-dependent transcription by TORC1 (Supplemental Figure S2, E and F). Hence, PKA or calcineurin is not involved in MEKK1-induced activation of TORC1.
MEKK1 Induces S167-independent Phosphorylation of TORC1
Activation of TORC1 by MEKK1 prompted us to determine whether MEKK1 sufficiently induced phosphorylation of TORC1 in vivo and in vitro. We overexpressed TORC1 and MEKK1 in HeLa cells and monitored the status of TORC1 proteins by Western blotting. While a doublet TORC1 band was observed in the lysate of cells expressing TORC1 alone (Figure 3, A and B; lane 1), expression of MEKK1 or dominant active MEKK-C resulted in the accumulation of the slow-migrating TORC1 species (Figure 3, A and B; lane 2). To verify that the slow-migrating band represents hyperphosphorylated form of TORC1 (pTORC1), we treated the protein samples with AP before SDS-PAGE analysis. Disappearance of the slow-migrating band and emergence of the fast-migrating species in the presence of AP indicated that TORC1 was indeed phosphorylated upon expression of MEKK1 or MEKK-C (Figure 3, A and B, lane 3). In contrast, no alteration of the doublet TORC1 band pattern was appreciated when the kinase-deficient mutant MEKK1 D1369A was expressed (Figure 3C). Additionally, similar results were also obtained from HEK293 cells (Supplemental Figure S3). Thus, the kinase activity of MEKK1 was required for phosphorylation of TORC1.
Figure 3.

MEKK1-induced phosphorylation of TORC1. (A–C) TORC1 phosphorylation in HeLa cells expressing MEKK1, MEKK-C, and MEKK1 D1369A. Cells were transfected with expression plasmids for TORC1 and full-length MEKK1 (A), MEKK-C (B), or MEKK1 D1369A (C). Cell lysate in lane 3 was treated with alkaline phosphatase (AP). TORC1, MEKK1, and MEKK-C were detected with anti-V5, anti-HA, and anti-Myc antibodies, respectively. α-Tubulin was also probed to verify equal loading. pTORC1 represents hyperphosphorylated form of TORC1 protein. (D) MEKK-C–mediated phosphorylation of TORC1 in vitro. E. coli–produced His-tagged TORC1 (lanes 2 and 3) was incubated with MEKK-C recovered from HeLa cells (lanes 1 and 3). Phosphorylated TORC1 was analyzed by SDS-PAGE followed by autoradiography.
We next carried out in vitro kinase assay with recombinant TORC1 and MEKK1 proteins. Despite many attempts, MEKK1 proteins either purified from E. coli or purchased from different sources were found to be inactive in the phosphorylation of myelin basic protein (MBP). In light of this, we expressed Myc-tagged MEKK-C in HeLa cells and immunoprecipitated MEKK-C protein with a mAb against Myc. As shown in Figure 3D, incubation of recombinant TORC1 purified from E. coli with MEKK-C precipitated from HeLa cells led to phosphorylation of TORC1.
Because TORC1 is thought to be phosphorylated at S167 equivalent to S171 of TORC2 (Screaton et al., 2004), we expressed TORC1 S167A in HeLa and HEK293 cells and repeated the phosphorylation assays with MEKK1, MEKK-C, and MEKK1 D1369A (Figure 4 and Supplemental Figure S4). It is noteworthy that the patterns of pTORC1 S167A and TORC1 S167A proteins observed were very similar to those of pTORC1 and TORC1 (compare Figure 4 with Figure 3 and Supplemental Figure S4 to Supplemental Figure S3). Hence, MEKK1-induced phosphorylation of TORC1 did not occur at the S167 phosphorylation site.
Figure 4.

MEKK1-induced and S167-independent phosphorylation of TORC1. Phosphorylation of TORC1 S167A in HeLa cells expressing MEKK1, MEKK-C, and MEKK1 D1369A (A–C) was analyzed as in Figure 3.
MEKK1 Interacts with TORC1 and Relocalizes It to the Nucleus
The induction of TORC1 phosphorylation by MEKK1 in vivo and in vitro (Figures 3 and 4 and Supplemental Figures S3 and S4) led us to examine whether these two proteins might associate with each other. When we performed a coimmunoprecipitation assay in HeLa cells expressing TORC1 and MEKK1, a protein complex of TORC1 and MEKK1 was detected (Figure 5, lane 4). This association between TORC1 and MEKK1 was specific because coprecipitation did not occur when either TORC1 or MEKK1 alone was expressed (Figure 5, lanes 2 and 3).
Figure 5.

Interaction of MEKK1 with TORC1 in cultured cells. HeLa cells were transfected with expression plasmids for V5-tagged TORC1 (lanes 3 and 4) and HA-tagged MEKK1 (lanes 2 and 4). TORC1 was immunoprecipitated with anti-V5. The precipitates were analyzed by Western blotting with anti-V5 (A) and anti-HA (B) to detect TORC1 and MEKK1, respectively. The input lysates were also probed for MEKK1 (C). Similar results were also obtained from other cell lines such as HEK293T.
To verify the interaction of TORC1 and MEKK1, we went on to determine their subcellular localizations in HeLa cells. We noted that both MEKK1 and TORC1 localized to the cytoplasm (Figure 6, panels 1–4). Interestingly, while MEKK1 D1369A did not influence the distribution of TORC1 within the cell (Figure 6, panels 5–8), dominant active MEKK-C was predominantly found in the nucleus, and it relocalized TORC1 to intranuclear compartment (Figure 6, panels 9–12). These results were compatible with the notion that MEKK1-induced phosphorylation of TORC1 promotes nuclear translocation.
Figure 6.

Colocalization of MEKK1 with TORC1. HeLa cells were transfected with expression plasmids for V5-tagged TORC1 and HA-tagged MEKK1 (panels 1–4), HA-tagged MEKK1 D1369A (panels 5–8), or Myc-tagged MEKK-C (panels 9–12). TORC1 and MEKK proteins were detected by fluorescein- and tetramethylrhodamine isothiocyanate (TRITC)-conjugated secondary antibodies, respectively. Nuclei were visualized by DAPI staining. Transfected cells are highlighted with arrows. Bar, 30 μm.
MEKK1-induced Phosphorylation Is Mediated through a C-terminal Activation Domain in TORC1
Although the C-terminal 200 amino acids of TORC1 have been thought to be important for transcriptional activity (Conkright et al., 2003), it is not understood whether they can sufficiently mediate transcriptional activation. To address this issue, we constructed a series of truncated mutants of TORC1 (Figure 7A) and expressed them in HEK293T cells as Gal4 DNA-binding domain fusions. Results from luciferase reporter assays indicated that a C-terminal domain comprising amino acids 431–650 was sufficient for transcriptional activation (Figure 7B, group 6 compared with groups 1 and 2). Consistent with this, a C-terminal-truncated TORC1 mutant deprived of amino acids 441–650 exhibited a substantially reduced transcriptional activity on CRE (Supplemental Figure S5, group 5 compared with group 2). Interestingly, the transcriptional activity of GalM4 containing the C-terminal activation domain only could be further stimulated by MEKK-C (Figure 7C, groups 21–24 compared with groups 1–4 and 5–8). Thus, this C-terminal activation domain likely contains the site(s) that are modified by MEKK1-induced phosphorylation.
Figure 7.

Definition of an MEKK1-responsive activation domain in TORC1. (A) A diagram of truncated TORC1 mutants. (B) Transcriptional activity of GalTORC1 and mutants. HEK293T cells were transfected with reporter construct pGal-Luc paired with increasing amounts of empty pM vector (vec) or an expression vector for full-length TORC1 (T1) or one of the TORC1 mutants (M1–M4) fused to Gal4 DNA-binding domain. (C) Stimulation of GalM4 transcriptional activity by MEKK-C. HEK293T cells were transfected with progressively increasing amounts of MEKK-C plasmid. (D) MEKK-C–induced phosphorylation of GalM4. HEK293T cells were transfected with expression plasmids for GalM4 and MEKK-C. Cell lysate in lane 3 was treated with AP. GalM4 and MEKK-C were detected with anti-Gal4 (mouse monoclonal; clone RK5C1 from Santa Cruz) and anti-HA antibodies, respectively. pGal4M4 represents hyperphosphorylated form of GalM4 protein.
To verify the modification of GalM4 in response to MEKK1, we coexpressed GalM4 and MEKK-C in HEK293T cells. Indeed, slow-migrating GalM4 bands were observed in MEKK-C–expressing cells (Figure 7D, lane 2 compared with lane 1). Treatment with AP confirmed that the slow-migrating GalM4 species were hyperphosphorylated forms, whereas the single band seen in the absence of MEKK-C was derived from hypophosphorylated GalM4 (Figure 7D, lane 3 compared with lanes 2 and 1). Together, our results demonstrated that MEKK1-induced phosphorylation occurs within the C-terminal activation domain of TORC1.
Physiological Activation of IL-8 Promoter Is Mediated through MEKK1 and TORC1
Above we demonstrated MEKK1 activation of TORC1-dependent transcription on canonical CRE (Figures 1 and 2). This raised a major concern about the physiological relevance of our findings. To address this issue, we chose IL-8 promoter for further analysis because IL-8 was one of cAMP-responsive genes initially identified in the functional cloning of TORCs (Iourgenko et al., 2003). We first confirmed that the expression of luciferase reporter driven by IL-8 promoter was activated by TORC1 (Figure 8, A–C, group 5 compared with group 1). Furthermore, this activation was dependent on the CRE because disruption of CRE suppressed the response to TORC1 almost completely (Figure 8, D-–F, group 5 compared with group 1). The expression of MEKK1 or MEKK-C alone also activated the transcription from IL-8 promoter in a dose-dependent manner (Figure 8, A and B, group 2–4 compared with group 1). The activation by MEKK1 was substantially inhibited but not completely abolished when the CRE in the IL-8 promoter was disrupted (Figure 8, D and E, group 2–4 compared with group 1). Importantly, coexpression of MEKK1 or MEKK-C with TORC1 significantly enhanced transcriptional activity of TORC1 on IL-8 promoter (Figure 8, A–B, group 6–8 compared with group 5). The activity of MEKK1, MEKK-C, and TORC1 was substantially reduced on the CRE-deficient IL-8 promoter (Figure 8, D and E, group 6–8 compared with group 5). In addition, the kinase-dead mutant MEKK1 D1369A was unable to activate IL-8 promoter significantly (Figure 8, C and F). Collectively, our results suggested that MEKK1 could enhance the activity of TORC1 on CRE in the context of IL-8 promoter.
Figure 8.

Stimulation of TORC1 activity on IL-8 promoter by MEKK1. HeLa cells were cotransfected with reporter plasmid pIL-8-Luc (A–C, G, and H) or pIL-8ΔCRE-Luc (D–F) plus progressively increasing amounts of expression vector for MEKK1 (A and D), MEKK-C (B and E), MEKK1 D1369A (C and F), and GalM1 (GM1; G and H).
To establish the role of TORC1 in MEKK1-induced activation of IL-8 promoter, we made use of a dominant inactive form of TORC1. Previously, a truncated mutant of TORC1/2 comprising the N-terminal CREB-binding domain only has been shown to have dominant interfering activity on wild-type TORC1/2 (Bittinger et al., 2004; Xu et al., 2007). A similar construct was made and we noted that the N-terminal 152 residues of TORC1 fused to Gal4 DNA-binding domain (i.e., GalM1 or GM1) had a more potent interfering activity on TORC1 (Supplemental Figure S6, B compared with A). When this GalM1 mutant was expressed in HeLa cells, in which TORC1 is a major TORC coactivator (data not shown), the activation of IL-8 promoter by MEKK1 or MEKK-C was inhibited almost completely (Figure 8, G and H). Thus, endogenous TORC1 was critically involved in MEKK1-induced activation of IL-8 promoter.
IL-1α is a physiological inducer of IL-8 (Larsen et al., 1989) and is also an activator of MEKK1 (Xia et al., 2000). This prompted us to investigate whether IL-1α activates IL-8 production through TORC1 and MEKK1. However, the activation of IL-8 production by IL-1α is a rapid process that could not be accurately reflected by luciferase reporter assay. In addition, it would be desirable to measure the activation of endogenous IL-8 gene. Thus, we used semiquantitative RT-PCR to monitor the change of IL-8 transcript in response to treatment with IL-1α in HeLa cells (Figure 9A). Consistent with previous findings (Larsen et al., 1989), activation of IL-8 transcription by IL-1α peaked at 1 h after treatment and diminished at later time points (Figure 9A, lanes 3–5 compared with lane 2). Although expression of an irrelevant shRNA (shGFP) did not influence the induction of IL-8 transcript by IL-1α (Figure 9A, lane 7 compared with lanes 2 and 6), prior expression of shTORC1a and shTORC1b, two independent shRNAs that were specific and effective in suppressing the expression of TORC1 mRNA (Siu et al., 2006 and Supplemental Figure S7), led to a significant reduction in the steady-state amount of endogenous IL-8 transcript in HeLa cells (Figure 9A, lanes 8 and 9 compared with lanes 6 and 7). In contrast, neither shTORC1a nor shTORC1b had any influence on the expression of the irrelevant GAPDH transcript.
Figure 9.
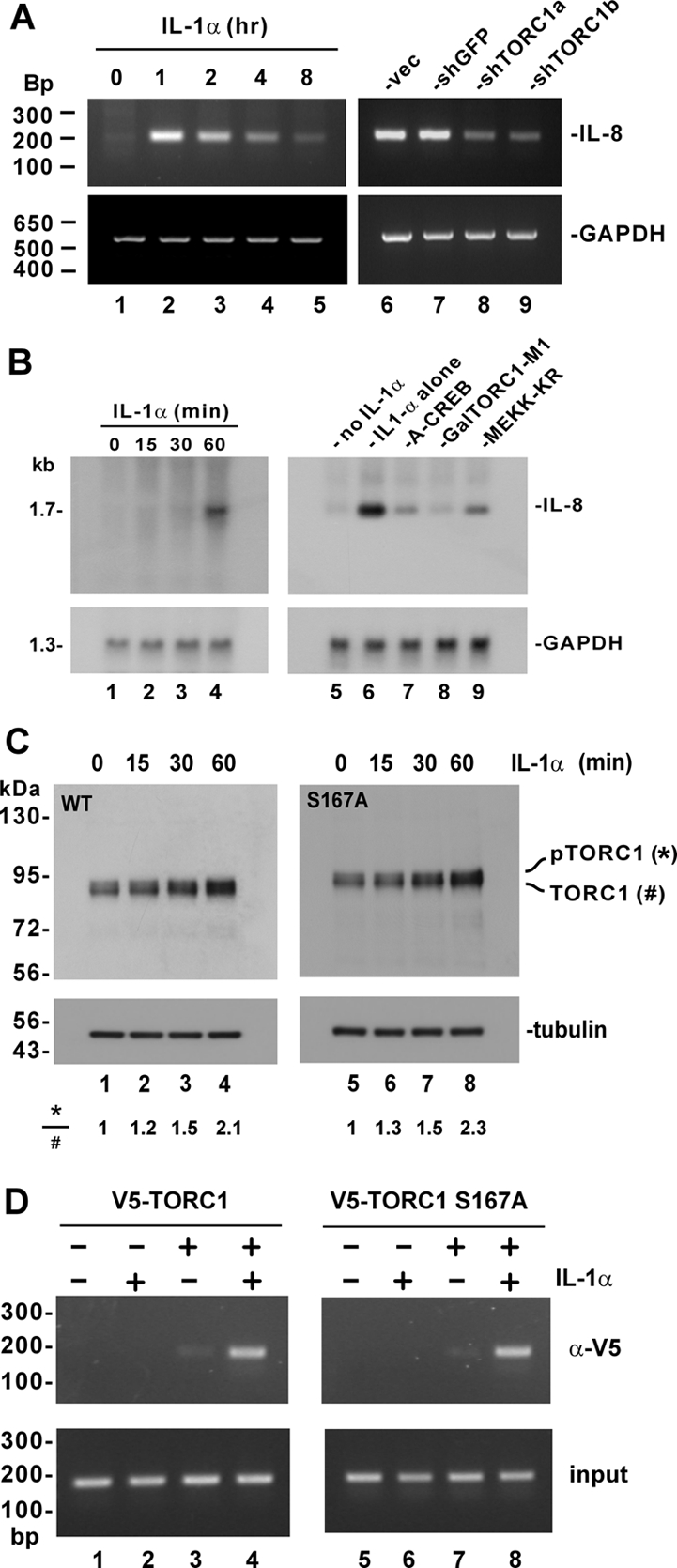
IL-1α–induced activation of IL-8 promoter through TORC1. (A) Requirement of TORC1 for IL-1α–induced production of IL-8. In lanes 1–5, HeLa cells were treated with 10 ng/ml IL-1α for the indicated duration of time. In lanes 6–9, cells were transfected with empty shRNA expression vector (vec) and expression plasmids for shGFP, shTORC1a, and shTORC1b, respectively, before treatment with 10 ng/ml IL-1α for 1 h. IL-8 and GAPDH transcripts were analyzed by semiquantitative RT-PCR. The ability of shTORC1a and shTORC1b to knockdown TORC1 expression and activity was confirmed by RT-PCR analysis of TORC1 transcript and by luciferase assay (Siu et al., 2006 and Supplemental Figure S7). (B) Requirement of CREB, TORC1, and MEKK1 for IL-1α induction of IL-8. In lanes 1–4, HeLa cells were treated with 10 ng/ml IL-1α for the indicated duration of time. In lanes 7–9, cells transfected, respectively, with expression vectors for A-CREB, GalTORC1-M1, and MEKK-KR were treated with 10 ng/ml IL-1α for 60 min. IL-8 and GAPDH transcripts were analyzed by Northern blotting. (C) IL-1α–induced phosphorylation of TORC1 is independent of S167. HeLa cells were transfected with expression plasmid for V5-tagged TORC1 (lanes 1–4) or TORC1 S167A (lanes 5–8). Cells were treated with 10 ng/ml IL-1α for 0–60 min as indicated at 48 h after transfection. Cell lysates were immunoblotted for TORC1 using anti-V5. Similar results were also obtained if cells were transfected with an expression plasmid for GalTORC1 driven by SV40 promoter and enhancer. (D) TORC1 recruitment to the IL-8 promoter. HeLa cells were transfected with expression plasmid for V5-tagged TORC1 (lanes 3 and 4) or TORC1 S167A (lanes 7 and 8). Cells were treated with 10 ng/ml IL-1α for 1 h before harvest (lanes 2, 4, 6, and 8). The DNA/TORC1 complex was immunoprecipitated using anti-V5 antibody and the IL-8 sequence was PCR-amplified.
Next we also performed Northern blotting to quantify the steady-state amounts of IL-8 mRNA. The dynamics of IL-1α–induced IL-8 transcription in HeLa cells was reassessed and a pronounced induction was observed at 60 min after treatment (Figure 9B, lanes 4 compared with lanes 1–3, and lane 6 compared with lane 5). When cells were pretransfected with expression plasmids for A-CREB, GalTORC1-M1, and MEKK-KR, which are well-characterized dominant inactive forms of CREB, TORC1, and MEKK1, respectively (Yan et al., 1994; Ahn et al., 1998; Ching et al., 2004; Siu et al., 2006; Xu et al., 2007), substantial reduction of IL-8 transcript was observed (Figure 9B, lanes 7 and 8 compared with lane 3). That is to say, loss of CREB, TORC1, or MEKK1 function as a result of the expression of dominant inactive mutant prevented the induction of IL-8 transcription by IL-1α. In other words, endogenous CREB, TORC1, and MEKK1 were required for IL-1α–induced activation of IL-8 promoter.
To shed light on the mechanism by which TORC1 mediates the stimulation of IL-8 promoter by IL-1α, we next investigated whether IL-1α might have an impact on MEKK1-induced phosphorylation of TORC1. Because the induction of IL-8 transcription by IL-1α peaked at 1 h after treatment (Figure 9, A and B), we monitored the dynamics of TORC1 proteins in IL-1α–treated HeLa cells within the first hour (Figure 9C). Indeed, an approximately twofold elevation of phosphorylated TORC1 was found at 60 min after treatment with IL-1α (Figure 9C, lane 4 compared with lanes 1–3). Importantly, phosphorylation of TORC1 S167A mutant could also be induced by IL-1α (Figure 9C, lanes 5–8), suggesting that the phosphorylation of TORC1 could be mediated by MEKK1. Because the CMV promoter used to drive TORC1 expression could also be stimulated by MEKK1 (Bruening et al., 1998), we also compared the levels of TORC1 transcript in untreated and IL-1α–treated transfected cells by semiquantitative and quantitative real-time RT-PCR. We did not notice a significant change within 1 h after treatment (Supplemental Figure S8). In addition, similar results were also obtained when the expression of TORC1 was driven by the SV40 promoter and enhancer (data not shown). Thus, IL-1α–induced activation of CMV promoter and the consequent elevation of TORC1 transcript were not a significant concern in our experimental setting.
Finally, we performed a ChIP assay to determine the recruitment of TORC1 to the IL-8 promoter (Figure 9D). We immunoprecipitated TORC1–DNA complex from HeLa cells and then PCR-amplified the CRE-containing sequence in the IL-8 promoter. A much more pronounced band of amplified DNA was observed only when HeLa cells were treated with IL-1α (Figure 9D, lane 4 compared with lane 3), suggesting that IL-1α induced the recruitment of TORC1 to IL-8 promoter. Because IL-1α was also found to stimulate the recruitment of TORC1 S167A, this process was independent of TORC1 phosphorylation at S167. Collectively, our findings are consistent with the model that IL-1α activates MEKK1 to induce TORC1 phosphorylation and its subsequent recruitment to IL-8 promoter.
DISCUSSION
In this study, we demonstrated a new MEKK1-mediated signaling pathway that regulates the activity of TORC1 coactivator in the activation of CRE-dependent transcription. We showed that MEKK1, but not its downstream kinases MEK1/2, ERK2, JNK, or p38, stimulated TORC1-mediated activation of CRE in a manner that required MEKK1-induced phosphorylation of TORC1, which did not occur at S167 (Figures 1–4). Activation of TORC1 by MEKK1 was not mediated through PKA or calcineurin (Figure 2). Instead, MEKK1 physically associated with TORC1 (Figure 5), and the kinase activity of MEKK1 sufficiently induced TORC1 phosphorylation in vitro and in vivo (Figures 3 and 4). Mechanistically, MEKK1-induced phosphorylation of TORC1 in the 220-residue C-terminal activation domain (Figure 7) promoted nuclear translocation of TORC1 and its subsequent recruitment to CRE promoters (Figures 6 and 9). Importantly, we provided the first evidence for the operation of this MEKK1-TORC1 signaling pathway in the physiological activation of IL-8 promoter by IL-1α (Figures 8 and 9). Although our findings were made with TORC1, similar results were also obtained in experiments conducted with TORC2 and TORC3 (data not shown). Thus, our work reveals a general mechanism for the regulation of TORC activity.
TORCs are essential coactivators of CREB and have important regulatory roles in glucose metabolism, neuronal function, and cell growth (Takemori et al., 2007a; Dentin et al., 2008). One well-characterized signaling pathway for regulation of TORC activity involves LKB-AMPK kinases and targets TORCs for posttranslational modifications such as O-linked glycosylation and polyubiquitination (Screaton et al., 2004; Dentin et al., 2007, 2008). The phosphorylation of TORC1 by AMPK is thought to take place at S167 equivalent to S171 of TORC2 (Screaton et al., 2004). However, our various experiments performed with TORC1 and TORC1 S167A yielded essentially the same results (Figures 1, 4, 8, and 9, and Supplemental Figures S2 and S4), suggesting that what we described in this work could be independent of AMPK-mediated phosphorylation. Although our findings do not exclude the possibility for cross-talk between the two pathways, existing evidence does suggest that they respond to different stimuli and lead to opposite consequences. In contrast to AMPK kinases that are regulated by LKB1, AMP, and calcium (Takemori et al., 2007a), MEKK1 is activated by IL-1α (Figure 9). More importantly, whereas AMPK-catalyzed phosphorylation of TORC1 at S167 caused sequestration of TORC1 in the cytoplasm and prevention of its transcriptional activity (Screaton et al., 2004), MEKK1-induced phosphorylation of TORC1 in the C-terminal activation domain of 220 amino acids promoted nuclear translocation and its recruitment to CRE promoter (Figures 6, 7, and 9).
While our work was under review, an independent study suggested a new mechanism for regulation of TORC2 activity by cAMP and glucose (Jansson et al., 2008). In their model, TORC2 is inhibited by MARK2-catalyzed phosphorylation at S275 and activated by calcineurin-dependent dephosphorylation at both S171 and S275. Although both their and our findings support the notion that S171 phosphorylation can insufficiently account for induced translocation and activation of TORC coactivators, it is noteworthy that the components and consequence of the two signaling pathways demonstrated in the two studies are still different. MARK2 is another AMPK-related kinase and MEKK1 is a MAP3K. Moreover, similar to phosphorylation at S171 by SIK2, phosphorylation of TORC2 at S275 by MARK2 sequesters the protein in the cytoplasm and therefore inhibits transcriptional activity. On the contrary, phosphorylation of TORC1 within the C-terminal region of 220 residues by MEKK1 leads to transcriptional activation (Figure 7). Hence, these two pathways that control intracellular activity of TORC coactivators through phosphorylation at S167/S261 and another site within the C-terminal activation domain likely operate in different contexts and serve different physiological roles. Particularly, because various cellular targets of CREB differ in their requirement for TORCs (Ravnskjaer et al., 2007; Xu et al., 2007), it is plausible that only a subset of CREB target genes are regulated by MEKK1.
Here, we identified IL-8 as one of the CREB targets responsive to MEKK1 stimulation (Figure 8). In addition, as a physiological stimulus of IL-8 production, IL-1α rapidly induced MEKK1-dependent phosphorylation of TORC1 and its subsequent recruitment to IL-8 promoter (Figure 9). IL-8 is a prototypic chemokine that plays pivotal roles in cytokine signaling, tumor progression, angiogenesis and tissue remodeling (Rossi and Zlotnik, 2000). Because of its importance and inducibility, transcriptional activation of IL-8 promoter has been extensively studied (Hoffmann et al., 2002). Nuclear factor κB (NF-κB) and AP1 are thought to be key regulators of IL-8 promoter (Yasumoto et al., 1992). Plausibly, the activation of IL-8 promoter by MEKK1 in the absence of TORC1 overexpression (Figure 8, A and B) was attributed at least in part to the stimulation of NF-κB and AP1. Although a functional CRE has been identified in IL-8 promoter and its activation by TORCs have been shown (Iourgenko et al., 2003), the physiological function of this CRE is not understood. In this regard, our findings that MEKK1 stimulates TORC1-dependent activation of IL-8 promoter and that IL-1α induces IL-8 transcription through phosphorylation and promoter recruitment of TORC1 reveal a new facet in the regulation of IL-8 gene expression. It will be of great interest to see whether other physiological and pathological inducers of IL-8 such as tumor necrosis factor α and Ras (Sparmann and Bar-Sagi, 2004) might also act through MEKK1 and TORCs. In addition, the cooperation of different activation pathways and transcription factors including NF-κB, AP1, and CREB in the activation of IL-8 promoter also merits further investigations. Likewise, identification and characterization of additional CREB target genes that are regulated through MEKK1 and TORCs are much warranted.
We were able to exclude the involvement of MEK1/2, ERK2, JNK, p38, PKA, calcineurin, and several other downstream kinases in MEKK1-induced activation of TORC1 (Figure 2). Our results implicate a more direct role for MEKK1 in S167- and S261-independent phosphorylation of TORC1 (Figures 1 and 4 and Supplemental Figure S4). However, we cannot completely rule out the possibility that another kinase tightly associated with MEKK1 in the immunoprecipitate might be responsible for the modification of TORC1. To establish direct phosphorylation of TORC1 by MEKK1, an in vitro kinase assay should be performed with functional recombinant MEKK1 protein free of contaminating kinases.
Although we presented evidence that MEKK1-induced phosphorylation of TORC1 occurred within the 220-residue activation domain at the C-terminus (amino acids 431–650; Figure 7), but not at S167 (Figures 1 and 4 and Supplemental Figure S4) or S261, our multiple attempts to determine the exact phosphorylation site(s) by using mass spectrometry and site-directed mutagenesis were unsuccessful. TORC1 is likely phosphorylated at multiple sites (Screaton et al., 2004; Jansson et al., 2008) and phosphorylation at any site is often substoichiometric. Hence, in future experiments it will be crucial to increase the yield of phosphorylated TORC1 upon stimulation with MEKK1 and to achieve a higher stoichiometry of MEKK1-induced phosphorylation.
Our results obtained from luciferase reporter assays (Figures 1 and 8 and Supplemental Figure S1) and confocal microscopy (Figure 6) consistently supported the notion that MEKK-C was more active than full-length MEKK1 in inducing TORC1 nuclear translocation and transcriptional activation. These data are also in agreement with the dominant active property of MEKK-C (Gibson et al., 1999) and the negative regulatory function of the N-terminal domain of MEKK1 (Xu et al., 1996; Lu et al., 2002; Witowsky and Johnson, 2003). However, we also noted that MEKK1 and MEKK-C exhibited comparable activity to induce phosphorylation of TORC1 as analyzed by electrophoretic mobility shift of TORC1 proteins (Figures 3 and 4, and Supplemental Figures S3 and S4). Generally, luciferase reporter assays are highly sensitive and produce high-resolution quantitative data for comparison. In contrast, the analysis of phosphorylation by electrophoretic mobility shift might be less accurate and less quantitative. In light of this, it is not too surprising that a higher activity of MEKK-C in phosphorylating TORC1 was not observed when hyperphosphorylated TORC1 was analyzed by electrophoresis (Figures 3 and 4, and Supplemental Figures S3 and S4).
Although our findings support the notion that MEKK1 induces nuclear translocation and promoter recruitment of TORC1 (Figures 6 and 8), exactly how MEKK1-induced phosphorylation of TORC1 might influence its nuclear import, nuclear export as well as transcriptional activity remains elusive. Moreover, whether and how MEKK1-induced phosphorylation might modulate other forms of post-translational modifications of TORC1 such as ubiquitination and O-lined glycosylation (Dentin et al., 2007, 2008) have not been formally addressed. On the other hand, whether and how other stimuli of CREB including cAMP and calcium might influence MEKK1-induced phosphorylation of TORC1 should also be explored. Generally, both MEKK1 and TORCs represent important hubs for signal integration and diversification. It will be of great interest to identify and characterize their upstream modulators and downstream effectors in the activation of CRE-driven transcription.
Supplementary Material
ACKNOWLEDGMENTS
We thank Melanie Cobb, Silvio Gutkind, Naofumi Mukaida, Dennis Templeton, and Charles Vinson for gifts of plasmids and Tony Chin, Abel Chun, Raven Kok, James Ng, Vincent Tang, and Chi Ming Wong for critical reading of the manuscript. This work was supported by Hong Kong Research Council (HKU 7683/05M, HKU 7486/06M, HKU 7636/07M, HKU 7661/08M, and HKU 1/06C), Fogarty International Center of National Institutes of Health (R01 TW06186-01), and Association for International Cancer Research (07-0424) grants.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E08-04-0369) on September 10, 2008.
REFERENCES
- Ahn S., Olive M., Aggarwal S., Krylov D., Ginty D. D., Vinson C. A dominant-negative inhibitor of CREB reveals that it is a general mediator of stimulus-dependent transcription of c-fos. Mol. Cell. Biol. 1998;18:967–977. doi: 10.1128/mcb.18.2.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcon-Vargas D., Tansey W. P., Ronai Z. Regulation of c-myc stability by selective stress conditions and by MEKK1 requires aa 127–189 of c-myc. Oncogene. 2002;21:4384–4391. doi: 10.1038/sj.onc.1205543. [DOI] [PubMed] [Google Scholar]
- Alessi D. R., Sakamoto K., Bayascas J. R. LKB1-dependent signaling pathways. Annu. Rev. Biochem. 2006;75:137–163. doi: 10.1146/annurev.biochem.75.103004.142702. [DOI] [PubMed] [Google Scholar]
- Amelio A. L., Miraglia L. J., Conkright J. J., Mercer B. A., Batalov S., Cavett V., Orth A. P., Busby J., Hogenesch J. B., Conkright M. D. A coactivator trap identifies NONO (p54nrb) as a component of the cAMP-signaling pathway. Proc. Natl. Acad. Sci. USA. 2007;104:20314–20319. doi: 10.1073/pnas.0707999105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett B. L., et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittinger M. A., McWhinnie E., Meltzer J., Iourgenko V., Latario B., Liu X., Chen C. H., Song C., Garza D., Labow M. Activation of cAMP response element-mediated gene expression by regulated nuclear transport of TORC proteins. Curr. Biol. 2004;14:2156–2161. doi: 10.1016/j.cub.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Bruening W., Giasson B., Mushynski W., Durham H. D. Activation of stress-activated MAP protein kinases up-regulates expression of transgenes driven by the cytomegalovirus immediate/early promoter. Nucleic Acids Res. 1998;26:486–489. doi: 10.1093/nar/26.2.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canettieri G., Koo S. H., Berdeaux R., Heredia J., Hedrick S., Zhang X., Montminy M. Dual role of the coactivator TORC2 in modulating hepatic glucose output and insulin signaling. Cell Metab. 2005;2:331–338. doi: 10.1016/j.cmet.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Chan C. P., Siu K. L., Chin K. T., Yuen K. Y., Zheng B., Jin D. Y. Modulation of the unfolded protein response by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2006;80:9279–9287. doi: 10.1128/JVI.00659-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chijiwa T., Mishima A., Hagiwara M., Sano M., Hayashi K., Inoue T., Naito K., Toshioka T., Hidaka H. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfon-amide (H-89), of PC12D pheochromocytoma cells. J. Biol. Chem. 1990;265:5267–5272. [PubMed] [Google Scholar]
- Chin K. T., Zhou H. J., Wong C. M., Lee J. M., Chan C. P., Qiang B. Q., Yuan J. G., Ng I.O.L., Jin D. Y. The liver-enriched transcription factor CREB-H is a growth suppressor protein underexpressed in hepatocellular carcinoma. Nucleic Acids Res. 2005;33:1859–1873. doi: 10.1093/nar/gki332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching Y. P., Chun A.C.S., Chin K. T., Zhang Z. Q., Jeang K. T., Jin D. Y. Specific TATAA and bZIP requirements suggest that HTLV-I Tax has transcriptional activity subsequent to the assembly of an initiation complex. Retrovirology. 2004;1:18. doi: 10.1186/1742-4690-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching Y. P., Chan S. F., Jeang K. T., Jin D. Y. The retroviral oncoprotein Tax targets the coiled-coil centrosomal protein TAX1BP2 to induce centrosome overduplication. Nat. Cell Biol. 2006;8:717–724. doi: 10.1038/ncb1432. [DOI] [PubMed] [Google Scholar]
- Choy E.Y.W., Kok K. H., Tsao S. W., Jin D. Y. Utility of Epstein-Barr virus-encoded small RNA promoters for driving the expression of fusion transcripts harboring short hairpin RNAs. Gene Ther. 2008a;15:191–202. doi: 10.1038/sj.gt.3303055. [DOI] [PubMed] [Google Scholar]
- Choy E.Y.W., Siu K. L., Kok K. H., Lung R.W.M., Tsang C. M., To K. F., Kwong D. L. W., Tsao S. W., Jin D. Y. An Epstein-Barr virus-encoded microRNA targets PUMA to promote host cell survival. J. Exp. Med. 2008b;205 doi: 10.1084/jem.20072581. 10.1084/jem.20072581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun A.C.S., Jin D. Y. Transcriptional regulation of mitotic checkpoint gene MAD1 by p53. J. Biol. Chem. 2003;278:37439–37450. doi: 10.1074/jbc.M307185200. [DOI] [PubMed] [Google Scholar]
- Clerk A., Sugden P. H. The p38-MAPK inhibitor, SB203580, inhibits cardiac stress-activated protein kinases/c-Jun N-terminal kinases (SAPKs/JNKs) FEBS Lett. 1998;426:93–96. doi: 10.1016/s0014-5793(98)00324-x. [DOI] [PubMed] [Google Scholar]
- Conkright M. D., Canettieri G., Screaton R., Guzman E., Miraglia L., Hogenesch J. B., Montminy M. TORCs: transducers of regulated CREB activity. Mol. Cell. 2003;12:413–423. doi: 10.1016/j.molcel.2003.08.013. [DOI] [PubMed] [Google Scholar]
- Coxon A., Rozenblum E., Park Y. S., Joshi N., Tsurutani J., Dennis P. A., Kirsch I. R., Kaye F. J. Mect1-Maml2 fusion oncogene linked to the aberrant activation of cyclic AMP/CREB regulated genes. Cancer Res. 2005;65:7137–7144. doi: 10.1158/0008-5472.CAN-05-1125. [DOI] [PubMed] [Google Scholar]
- Cuevas B. D., Abell A. N., Johnson G. L. Role of mitogen-activated protein kinase kinase kinases in signal integration. Oncogene. 2007;26:3159–3171. doi: 10.1038/sj.onc.1210409. [DOI] [PubMed] [Google Scholar]
- Dentin R., Hedrick S., Xie J., Yates J., III, Montminy M. Hepatic glucose sensing via the CREB coactivator CRTC2. Science. 2008;319:1402–1405. doi: 10.1126/science.1151363. [DOI] [PubMed] [Google Scholar]
- Dentin R., Liu Y., Koo S. H., Hedrick S., Vargas T., Heredia J., Yates J., III, Montminy M. Insulin modulates gluconeogenesis by inhibition of the coactivator TORC2. Nature. 2007;449:366–369. doi: 10.1038/nature06128. [DOI] [PubMed] [Google Scholar]
- Favata M. F., et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- Fuchs S. Y., Adler V., Pincus M. R., Ronai Z. MEKK1/JNK signaling stabilizes and activates p53. Proc. Natl. Acad. Sci. USA. 1998;95:10541–10546. doi: 10.1073/pnas.95.18.10541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson S., Widmann C., Johnson G. L. Differential involvement of MEK kinase 1 (MEKK1) in the induction of apoptosis in response to microtubule-targeted drugs versus DNA damaging agents. J. Biol. Chem. 1999;274:10916–10922. doi: 10.1074/jbc.274.16.10916. [DOI] [PubMed] [Google Scholar]
- Hishiki T., Ohshima T., Ego T., Shimotohno K. BCL3 acts as a negative regulator of transcription from the human T-cell leukemia virus type 1 long terminal repeat through interactions with TORC3. J. Biol. Chem. 2007;282:28335–28343. doi: 10.1074/jbc.M702656200. [DOI] [PubMed] [Google Scholar]
- Hoffmann E., Dittrich-Breiholz O., Holtmann H., Kracht M. Multiple control of interleukin-8 gene expression. J. Leukoc. Biol. 2002;72:847–855. [PubMed] [Google Scholar]
- Hong S. H., Privalsky M. L. The SMRT corepressor is regulated by a MEK-1 kinase pathway: inhibition of corepressor function is associated with SMRT phosphorylation and nuclear export. Mol. Cell. Biol. 2000;20:6612–6625. doi: 10.1128/mcb.20.17.6612-6625.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iourgenko V., et al. Identification of a family of cAMP response element-binding protein coactivators by genome-scale functional analysis in mammalian cells. Proc. Natl. Acad. Sci. USA. 2003;100:12147–12152. doi: 10.1073/pnas.1932773100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson D., Ng A. C.-H., Fu A., Depatie C., Al Azzabi M., Screaton R. A. Glucose controls CREB activity in islet cells via regulated phosphorylation of TORC2. Proc. Natl. Acad. Sci. USA. 2008;105:10161–10166. doi: 10.1073/pnas.0800796105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin D. Y., Giordano V., Kibler K. V., Nakano H., Jeang K. T. Role of adapter function in oncoprotein-mediated activation of NF-κB: human T-cell leukemia virus type I Tax interacts directly with IκB kinase γ. J. Biol. Chem. 1999;274:17402–17405. doi: 10.1074/jbc.274.25.17402. [DOI] [PubMed] [Google Scholar]
- Jin D. Y., Teramoto H., Giam C. Z., Chun R. F., Gutkind J. S., Jeang K. T. A human suppressor of c-Jun N-terminal kinase 1 activation by tumor necrosis factor α. J. Biol. Chem. 1997;272:25816–25823. doi: 10.1074/jbc.272.41.25816. [DOI] [PubMed] [Google Scholar]
- Johannessen M., Delghandi M. P., Moens U. What turns CREB on? Cell Signal. 2004;16:1211–1227. doi: 10.1016/j.cellsig.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Katoh Y., et al. Silencing the constitutive active transcription factor CREB by the LKB1-SIK signaling cascade. FEBS J. 2006;273:2730–2748. doi: 10.1111/j.1742-4658.2006.05291.x. [DOI] [PubMed] [Google Scholar]
- Kim S. J., Nian C., Widenmaier S., McIntosh C. H. Glucose-dependent insulinotropic polypeptide-mediated up-regulation of β-cell antiapoptotic Bcl-2 gene expression is coordinated by cyclic AMP (cAMP) response element binding protein (CREB) and cAMP-responsive CREB coactivator 2. Mol. Cell. Biol. 2008;28:1644–1656. doi: 10.1128/MCB.00325-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga H., Ohshima T., Shimotohno K. Enhanced activation of tax-dependent transcription of human T-cell leukemia virus type I (HTLV-I) long terminal repeat by TORC3. J. Biol. Chem. 2004;279:52978–52983. doi: 10.1074/jbc.M409021200. [DOI] [PubMed] [Google Scholar]
- Kok K. H., Ng M. H., Ching Y. P., Jin D. Y. Human TRBP and PACT directly interact with each other and associate with dicer to facilitate the production of small interfering RNA. J. Biol. Chem. 2007;282:17649–17657. doi: 10.1074/jbc.M611768200. [DOI] [PubMed] [Google Scholar]
- Koo S. H., et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature. 2005;437:1109–1111. doi: 10.1038/nature03967. [DOI] [PubMed] [Google Scholar]
- Kovács K. A., Steullet P., Steinmann M., Do K. Q., Magistretti P. J., Halfon O., Cardinaux J. R. TORC1 is a calcium- and cAMP-sensitive coincidence detector involved in hippocampal long-term synaptic plasticity. Proc. Natl. Acad. Sci. USA. 2007;104:4700–4705. doi: 10.1073/pnas.0607524104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuraishy A. I., French S. W., Sherman M., Herling M., Jones D., Wall R., Teitell M. A. TORC2 regulates germinal center repression of the TCL1 oncoprotein to promote B cell development and inhibit transformation. Proc. Natl. Acad. Sci. USA. 2007;104:10175–10180. doi: 10.1073/pnas.0704170104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen C. G., Anderson A. O., Oppenheim J. J., Matsushima K. Production of interleukin-8 by human dermal fibroblasts and keratinocytes in response to interleukin-1 or tumour necrosis factor. Immunology. 1989;68:31–36. [PMC free article] [PubMed] [Google Scholar]
- Liu J., Farmer J. D., Jr., Lane W. S., Friedman J., Weissman I., Schreiber S. L. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- Lu Z., Xu S., Joazeiro C., Cobb M. H., Hunter T. The PHD domain of MEKK1 acts as an E3 ubiquitin ligase and mediates ubiquitination and degradation of ERK1/2. Mol. Cell. 2002;9:945–956. doi: 10.1016/s1097-2765(02)00519-1. [DOI] [PubMed] [Google Scholar]
- Mayr B., Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- Mukaida N., Mahe Y., Matsushima K. Cooperative interaction of nuclear factor-κB- and cis-regulatory enhancer binding protein-like factor binding elements in activating the interleukin-8 gene by pro-inflammatory cytokines. J. Biol. Chem. 1990;265:21128–21133. [PubMed] [Google Scholar]
- Ohmae S., et al. Molecular identification and characterization of a family of kinases with homology to Ca2+/calmodulin-dependent protein kinases I/IV. J. Biol. Chem. 2006;281:20427–20439. doi: 10.1074/jbc.M513212200. [DOI] [PubMed] [Google Scholar]
- Ravnskjaer K., Kester H., Liu Y., Zhang X., Lee D., Yates J. R., 3rd, Montminy M. Cooperative interactions between CBP and TORC2 confer selectivity to CREB target gene expression. EMBO J. 2007;26:2880–2889. doi: 10.1038/sj.emboj.7601715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi D., Zlotnik A. The biology of chemokines and their receptors. Annu. Rev. Immunol. 2000;18:217–242. doi: 10.1146/annurev.immunol.18.1.217. [DOI] [PubMed] [Google Scholar]
- Roy S. K., et al. MEKK1 plays a critical role in activating the transcription factor C/EBP-β-dependent gene expression in response to IFN-γ. Proc. Natl. Acad. Sci. USA. 2002;99:7945–7950. doi: 10.1073/pnas.122075799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Screaton R. A., et al. The CREB coactivator TORC2 functions as a calcium- and cAMP-sensitive coincidence detector. Cell. 2004;119:61–74. doi: 10.1016/j.cell.2004.09.015. [DOI] [PubMed] [Google Scholar]
- See R. H., Calvo D., Shi Y., Kawa H., Luke M. P., Yuan Z., Shi Y. Stimulation of p300-mediated transcription by the kinase MEKK1. J. Biol. Chem. 2001;276:16310–16317. doi: 10.1074/jbc.M008113200. [DOI] [PubMed] [Google Scholar]
- Shaw R. J., Lamia K. A., Vasquez D., Koo S. H., Bardeesy N., Depinho R. A., Montminy M., Cantley L. C. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaywitz A. J., Greenberg M. E. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu. Rev. Biochem. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- Siu Y. T., Chin K. T., Siu K. L., Choy E.Y.W., Jeang K. T., Jin D. Y. TORC1 and TORC2 coactivators are required for tax activation of the human T-cell leukemia virus type 1 long terminal repeats. J. Virol. 2006;80:7052–7059. doi: 10.1128/JVI.00103-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu Y. T., Jin D. Y. CREB-a real culprit in oncogenesis. FEBS J. 2007;274:3224–3232. doi: 10.1111/j.1742-4658.2007.05884.x. [DOI] [PubMed] [Google Scholar]
- Sparmann A., Bar-Sagi D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell. 2004;6:447–458. doi: 10.1016/j.ccr.2004.09.028. [DOI] [PubMed] [Google Scholar]
- Takemori H., Kajimura J., Okamoto M. TORC-SIK cascade regulates CREB activity through the basic leucine zipper domain. FEBS J. 2007a;274:3202–3209. doi: 10.1111/j.1742-4658.2007.05889.x. [DOI] [PubMed] [Google Scholar]
- Takemori H., Kanematsu M., Kajimura J., Hatano O., Katoh Y., Lin X. Z., Min L., Yamazaki T., Doi J., Okamoto M. Dephosphorylation of TORC initiates expression of the StAR gene. Mol. Cell. Endocrinol. 2007b;265–266:196–204. doi: 10.1016/j.mce.2006.12.020. [DOI] [PubMed] [Google Scholar]
- Teramoto H., Crespo P., Coso O. A., Igishi T., Xu N., Gutkind J. S. The small GTP-binding protein rho activates c-Jun N-terminal kinases/stress-activated protein kinases in human kidney 293T cells: evidence for a Pak-independent signaling pathway. J. Biol. Chem. 1996;271:25731–25734. doi: 10.1074/jbc.271.42.25731. [DOI] [PubMed] [Google Scholar]
- Turjanski A. G., Vaque J. P., Gutkind J. S. MAP kinases and the control of nuclear events. Oncogene. 2007;26:3240–3253. doi: 10.1038/sj.onc.1210415. [DOI] [PubMed] [Google Scholar]
- Witowsky J. A., Johnson G. L. Ubiquitylation of MEKK1 inhibits its phosphorylation of MKK1 and MKK4 and activation of the ERK1/2 and JNK pathways. J. Biol. Chem. 2003;278:1403–1406. doi: 10.1074/jbc.C200616200. [DOI] [PubMed] [Google Scholar]
- Wu L., Liu J., Gao P., Nakamura M., Cao Y., Shen H., Griffin J. D. Transforming activity of MECT1-MAML2 fusion oncoprotein is mediated by constitutive CREB activation. EMBO J. 2005;24:2391–2402. doi: 10.1038/sj.emboj.7600719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z., Huang X., Feng Y., Handschin C., Feng Y., Gullicksen P. S., Bare O., Labow M., Spiegelman B., Stevenson S. C. Transducer of regulated CREB-binding proteins (TORCs) induce PGC-1α transcription and mitochondrial biogenesis in muscle cells. Proc. Natl. Acad. Sci. USA. 2006;103:14379–14384. doi: 10.1073/pnas.0606714103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y., Makris C., Su B., Li E., Yang J., Nemerow G. R., Karin M. MEK kinase 1 is critically required for c-Jun N-terminal kinase activation by proinflammatory stimuli and growth factor-induced cell migration. Proc. Natl. Acad. Sci. USA. 2000;97:5243–5248. doi: 10.1073/pnas.97.10.5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y., Wang J., Xu S., Johnson G. L., Hunter T., Lu Z. MEKK1 mediates the ubiquitination and degradation of c-Jun in response to osmotic stress. Mol. Cell. Biol. 2007;27:510–517. doi: 10.1128/MCB.01355-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S., Robbins D. J., Christerson L. B., English J. M., Vanderbilt C. A., Cobb M. H. Cloning of rat MEK kinase 1 cDNA reveals an endogenous membrane-associated 195-kDa protein with a large regulatory domain. Proc. Natl. Acad. Sci. USA. 1996;93:5291–5295. doi: 10.1073/pnas.93.11.5291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W., Kasper L. H., Lerach S., Jeevan T., Brindle P. K., et al. Individual CREB-target genes dictate usage of distinct cAMP-responsive coactivation mechanisms. EMBO J. 2007;26:2890–2903. doi: 10.1038/sj.emboj.7601734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan M., Dai T., Deak J. C., Kyriakis J. M., Zon L. I., Woodgett J. R., Templeton D. J. Activation of stress-activated protein kinase by MEKK1 phosphorylation of its activator SEK1. Nature. 1994;372:798–800. doi: 10.1038/372798a0. [DOI] [PubMed] [Google Scholar]
- Yasumoto K., Okamoto S., Mukaida N., Murakami S., Mai M., Matsushima K. Tumor necrosis factor α and interferon γ synergistically induce interleukin 8 production in a human gastric cancer cell line through acting concurrently on AP-1 and NF-κB-like binding sites of the interleukin 8 gene. J. Biol. Chem. 1992;267:22506–22511. [PubMed] [Google Scholar]
- Zhou Y., Wu H., Li S., Chen Q., Cheng X. W., Zheng J., Takemori H., Xiong Z. Q. Requirement of TORC1 for late-phase long-term potentiation in the hippocampus. PLoS ONE. 2006;1:e16. doi: 10.1371/journal.pone.0000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.