

Abstract
Several Sec proteins including a guanosine diphosphate/guanosine triphosphate exchange factor for Sar1p have been implicated in autophagy. In this study, we investigated the role of Sar1p in pexophagy by expressing dominant-negative mutant forms of Sar1p in Pichia pastoris. When expressing sar1pT34N or sar1pH79G, starvation-induced autophagy, glucose-induced micropexophagy, and ethanol-induced macropexophagy are dramatically suppressed. These Sar1p mutants did not affect the initiation or expansion of the sequestering membranes nor the trafficking of Atg11p and Atg9p to these membranes during micropexophagy. However, the lipidation of Atg8p and assembly of the micropexophagic membrane apparatus, which are essential to complete the incorporation of the peroxisomes into the degradative vacuole, were inhibited when either Sar1p mutant protein was expressed. During macropexophagy, the expression of sar1pT34N inhibited the formation of the pexophagosome, whereas sar1pH79G suppressed the delivery of the peroxisome from the pexophagosome to the vacuole. The pexophagosome contained Atg8p in wild-type cells, but in cells expressing sar1pH79G these organelles contain both Atg8p and endoplasmic reticulum components as visualized by DsRFP-HDEL. Our results demonstrate key roles for Sar1p in both micro- and macropexophagy.
INTRODUCTION
Autophagy is a lysosomal degradation pathway meaning “self-eating.” Autophagy can be subclassified based on substrate selectivity and mode of sequestration (for reviews, see Levine and Klionsky, 2004; Dunn et al., 2005; Yorimitsu and Klionsky, 2005). Nonselective micro- and macroautophagy can be stimulated by nitrogen and amino acid starvation. During microautophagy, sequestration occurs by invaginations of the lysosomal membrane. Sequestration of macromolecules and organelles into autophagosomes proceeds by macroautophagy. These autophagosomes eventually fuse with lysosomes, thereby delivering their cargo for degradation. The selective degradation of peroxisomes has been referred to as pexophagy. Micropexophagy is the sequestration of peroxisomes by the vacuole for degradation. When Pichia pastoris are subjected to glucose adaptation, clusters of peroxisomes are surrounded by finger-like protrusions or septations originating from the vacuole. The peroxisome cluster is then incorporated into the vacuolar lumen where it is subsequently degraded. During adaptation from methanol to ethanol, individual peroxisomes are sequestered into autophagosomes and delivered to the vacuole for degradation. This process is called macropexophagy.
During glucose-induced micropexophagy, initiation of the sequestering membranes proceeds from the vacuole. Membrane initiation requires Atg18p, Vac8p, and Vps15p (Stasyk et al., 1999; Guan et al., 2001; Fry et al., 2006). Unless stated by designations for other species, we refer to the P. pastoris homologues of the Sar1, Atg, and Sec proteins in this article. Afterward, the sequestering membranes expand to surround the peroxisomes. Membrane expansion requires Atg2p, Atg9p, and Atg11p (Kim et al., 2001; Strømhaug et al., 2001; Chang et al., 2005). We demonstrated that Atg9p, an integral membrane protein, traffics from a unique peripheral compartment to perivacuolar structures (PVSs) abutting the vacuole, and finally to the sequestering membranes (SMs). The movement of Atg9p to the sequestering membranes requires Atg2p and Atg11p. Mukaiyama et al. (2004) characterized the micropexophagic membrane apparatus (MIPA) required for expansion and completion events of micropexophagy. The de novo assembly of the MIPA is positioned between expanding sequestering membranes. The assembly of the MIPA from a preautophagosome structure (PAS) requires Atg7p, Atg8p, and Atg9p (Mukaiyama et al., 2004; Chang et al., 2005). Atg8p, Atg9p, and Atg17p localize to the PAS, with Atg8p also being found at the MIPA (Mukaiyama et al., 2004; Chang et al., 2005). Finally, completion of micropexophagy proceeds upon the fusion of MIPA with the adjacent sequestering membranes and incorporation of the peroxisomes into the vacuole for degradation. These fusion events require Vac8p and Atg24p (Ano et al., 2005; Fry et al., 2006; Oku et al., 2006).
During ethanol-induced macropexophagy, peroxisomes are enwrapped by two or more membranes of unknown origin, forming pexophagosomes (Dunn et al., 2005). The molecular events responsible for the formation of the pexophagosome remain rather vague. These sequestering membranes contain Atg8p and Atg25p and seem to arise from the PAS that contains several Atg proteins, including Atg8p, Atg9p, and Atg17p (Chang et al., 2005; Oku et al., 2006). The completion of the pexophagosome requires Atg8p and Atg21p. The outer membrane of the pexophagosome then fuses with the vacuole, delivering the peroxisomes for degradation. In addition to Atg25p at the pexophagosome, fusion requires Vam7p present at the vacuole and Atg24p present at the vertices of the fusion complex (Ano et al., 2005; Monastyrska et al., 2005; Stevens et al., 2005). Within the vacuole, the inner membrane of the pexophagosome is ruptured by the lipase Atg15p, and the released peroxisomes are rapidly degraded by proteinases (Teter et al., 2001).
Recent data suggest that many molecular events of the secretory pathway may be shared by autophagy. For example, in Saccharomyces cerevisiae, ScAtg8p interacts with the vesicle-soluble N-ethylmaleimide-sensitive factor attachment protein receptor (v-SNARE) proteins ScBet1p, ScSec22p, and ScNyv1p, which are required for endoplasmic reticulum (ER)-to-Golgi trafficking and vacuolar protein trafficking (Legesse-Miller et al., 2000). In addition, studies have shown that several Sec proteins are required for autophagy, namely, ScSec16p, ScSec17p, ScSec23p, ScSec24p, ScSec12p, ScSec18p, and ScSec7p (Ishihara et al., 2001; Hamasaki et al., 2003; Reggiori et al., 2004). The roles for ScSec16p, ScSec17p, and ScSec23p have yet to be clearly defined (Ishihara et al., 2001). ScSec12p facilitates the formation of the autophagosome from the PAS (Reggiori et al., 2004). When sec24ts mutants are exposed to nonpermissive temperatures in the presence of rapamycin, ScAtg8p is found at multiple cytosolic structures presumed to be PAS (Hamasaki et al., 2003). Finally, ScSec18p and ScSec7p seem to be required for the fusion of the autophagosome with the vacuole (Ishihara et al., 2001; Reggiori et al., 2004). Hence, many Sec proteins are indispensable for both protein secretion and autophagy.
Some of the secretory proteins required for autophagosome formation have a role in coat protein complex II (COPII)-mediated trafficking (Ishihara et al., 2001; Hamasaki et al., 2003; Reggiori et al., 2004). ScSec12p is a GDP/GTP transmembrane exchange factor which influences ScSar1p activity (for review, see Sato and Nakano, 2007). ScSar1p, stimulated by the exchange of GTP for GDP, binds to the ER membrane. ScSec23p interacts with ScSec24p, which bind to ScSar1p. ScSec13p binds to the ScSar1p-ScSec23p/ScSec24p coat protein complex, promoting vesicular budding. The data suggest that COPII-dependent protein trafficking is essential for ongoing autophagy and may implicate the ER in the assembly of the autophagosomes (Ishihara et al., 2001; Hamasaki et al., 2003; Reggiori et al., 2004).
Studies in S. cerevisiae have shown that the COPII complex of proteins is required for starvation-induced autophagy. However, the role of this protein complex in pexophagy has not been evaluated. In this study, we characterized the essential role of Sar1p in micro- and macropexophagy in P. pastoris. We demonstrated that when either dominant-negative form of Sar1p, sar1pT34N, or sar1pH79G is expressed, both micro- and macropexophagy are repressed. The initiation and amplification of the sequestering membranes seemed to proceed normally during micropexophagy. However, the assembly of the MIPA was impaired when these Sar1p mutants were expressed. The failure of MIPA to form was not due to incorrect sorting of Atg2p, Atg9p, or Atg11p or to an inactivation of Atg7p. However, the lipidation of Atg8p seemed to be altered. During macropexophagy, the pexophagosome failed to assemble when sar1pT34N was expressed. To the contrary, the expression of sar1pH79G did not seem to alter the formation of the pexophagosome, but it disrupted its delivery of the peroxisomes to the vacuole. Our data demonstrate that Sar1p is essential for the membrane sequestration events of micro- and macropexophagy.
MATERIALS AND METHODS
Yeast Strains and Media
The yeast strains used in this study are listed in Table 1. They were routinely cultured at 30°C in YPD (1% Bacto yeast extract, 2% Bacto peptone, and 2% dextrose). P. pastoris was grown in YNM (0.67% yeast nitrogen base, 0.4 mg/l biotin, and 0.5% methanol) to induce peroxisome biogenesis. The degradation of peroxisomes was induced when cells were transferred from YNM to YND (0.67% yeast nitrogen base, 0.4 mg/l biotin, and 2% glucose) or YNE (0.67% yeast nitrogen base, 0.4 mg/l biotin, and 0.5% ethanol). Nitrogen starvation medium, SD(-N), contained 0.17% yeast nitrogen base (without amino acids and NH4SO4) and 2% glucose. All media contained 2% agar when made as plates. Histidine and arginine were added at 40 μg/ml when needed. Vector amplification was done in Escherichia coli (DH5α) cultured at 37°C in LB (0.5% Bacto yeast extract, 1% Bacto tryptone, and 1% NaCl) with ampicillin (100 μg/ml). Zeocin was added at 25 μg/ml when culturing DH5α and 100 μg/ml when culturing P. pastoris.
Table 1.
P. pastoris strains
| Name | Genotype | Reference |
|---|---|---|
| GS115 | his4 | Cregg et al. (1985) |
| PPF1 | arg4 his4 | Yuan et al. (1997) |
| DMM1 | GS115::pDM1 (PAOX1BFP-SKL, ZeocinR) | Kim et al. (2001) |
| SJCF257 | arg4 his4 atg8Δ | Farre et al. (2007) |
| WDKO7 | PPF1 gsa7Δ::ARG4 | Yuan et al. (1999) |
| S7-GFPx3 | PPY12 arg4 his4::pPOP-S7-GFPX3 (PSec7SEC7-GFPx3) | Bevis et al. (2002) |
| AJM18 | S7-GFPx3 arg4 his4::pAJM6 (PGAPDH mRFP-ATG9, ZeocinR) | This study |
| AJM38 | GS115 his4::pWD18 (PATG7atg7-HA (C518S), HIS4) | This study |
| WDY64 | PPF1 his4 arg4::pWD16-Sar1(T34N) (PCUPsar1(T34N), ARG4) | This study |
| WDY65 | PPF1 his4 arg4::pWD16-Sar1(H79G) (PCUPsar1(H79G), ARG4) | This study |
| WDY66 | WDY64 his4::pWD21 (PGAPDH GFP-ATG8, HIS4) | This study |
| WDY67 | WDY65 his4::pWD21 (PGAPDH GFP-ATG8, HIS4) | This study |
| WDY70 | DMM1 his4::pWD21 (PGAPDH GFP-ATG8, HIS4) | This study |
| WDY111 | SJCF257 arg4 his4 atg8Δ::pWD18(PATG7atg7-HA (C518S), HIS4) | This study |
| WDY120 | LAM44::pAJM6 (PGAPDH mRFP-ATG9, ZeocinR) | This study |
| WDY121 | LAM45::pAJM6 (PGAPDH mRFP-ATG9, ZeocinR) | This study |
| WDY122 | WDY66::pAJM6 (PGAPDH mRFP-ATG9, ZeocinR) | This study |
| WDY123 | WDY67::pAJM6 (PGAPDH mRFP-ATG9, ZeocinR) | This study |
| WDY126 | WDY64 his4::pWD18(PATG7atg7-HA (C518S), HIS4) | This study |
| WDY127 | WDY65 his4::pWD18(PATG7atg7-HA (C518S), HIS4) | This study |
| WDY132 | AJM18 arg4 his4::pWD16-Sar1(T34N) (PCUPsar1(T34N), ARG4) | This study |
| WDY133 | AJM18 arg4 his4::pWD16-Sar1(H79G) (PCUPsar1(H79G), ARG4) | This study |
| WDY138 | WDY66::pDM1 (PAOX1BFP-SKL, ZeocinR) | This study |
| WDY139 | WDY67::pDM1 (PAOX1BFP-SKL, ZeocinR) | This study |
| WDY150 | WDKO7 his4::pJCF403(PATG8 3 × HA-ATG8, HIS4) | This study |
| WDY152 | WDY64 his4::pJCF403(PATG8 3 × HA-ATG8, HIS4) | This study |
| WDY153 | WDY64 his4::pJCF404(PATG8 3 × HA-atg8 (G116A), HIS4) | This study |
| WDY154 | WDY65 his4::pJCF403(PATG8 3 × HA-ATG8, HIS4) | This study |
| WDY155 | WDY65 his4::pJCF404(PATG8 3 × HA-atg8 (G116A), HIS4) | This study |
| LAM13 | DMM1 his4::pSar1(T34N) (PCUPsar1(T34N), HIS4) | This study |
| LAM14 | DMM1 his4::pSar1(H79G) (PCUPsar1(H79G), HIS4) | This study |
| LAM40 | WDY64 his4::pTC1 (PGAPDH GFP-ATG9, HIS4) | This study |
| LAM41 | WDY65 his4::pTC1 (PGAPDH GFP-ATG9, HIS4) | This study |
| LAM42 | WDY64 his4::pPS64 (PGAPDH GFP/HA-ATG11, HIS4) | This study |
| LAM43 | WDY65 his4::pPS64 (PGAPDH GFP/HA-ATG11, HIS4) | This study |
| LAM44 | WDY64 his4::pPS69 (PGAPDH GFP-ATG2, HIS4) | This study |
| LAM45 | WDY65 his4::pPS64 (PGAPDH GFP-ATG2, HIS4) | This study |
| LAM46 | WDY64 his4::pPS55-G12 (PGAPDH GFP/HA-ATG18, HIS4) | This study |
| LAM47 | WDY65 his4::pPS55-G12 (PGAPDH GFP/HA-ATG18, HIS4) | This study |
| LAM54 | WDY64 his4::pWD24 (PGAPDH GFP-ATG17, HIS4) | This study |
| LAM55 | WDY65 his4::pWD24 (PGAPDH GFP-ATG17, HIS4) | This study |
| LAM63 | WDY64 his4::pIB2-dsRED-HDEL (PGAPDH dsRFP-HDEL, HIS4) | This study |
| LAM64 | WDY65 his4::pIB2-dsRED-HDEL (PGAPDH dsRFP-HDEL, HIS4) | This study |
Quantitative Assessment of Alcohol Oxidase (AOX) Degradation
Cells were grown in 20 ml of YNM with methanol as sole carbon and energy source. At 40 h, 0.4 g of glucose was added with or without 0.1 mM copper sulfate. Aliquots (2 ml) of cells (2–4 OD600) at 0 and 6 h of glucose adaptation were pelleted and resuspended in 1 ml of 20 mM Tris, pH 7.5, containing 50 mM NaCl, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 μg/ml pepstatin A, and 0.5 μg/ml leupeptin. The cells were then lysed by vortexing in the presence of 0.5-ml glass beads (425–600 μm). The glass beads and cellular debris were removed by centrifugation, and AOX was measured by adding 50 μl of this extract to 3 ml of reaction mix containing 3.4 U/ml horseradish peroxidase and 0.53 mg/ml 2,2′-azino-bis(3-ethylbenz-thazoline-6-sulfonic acid) in 33 mM potassium phosphate buffer, pH 7.5. The reaction was initiated by adding 10 μl of methanol and terminated after 20 min by adding 200 μl of 4 N HCl. The absorbance was read at 410 nm.
Western Blot Analysis
Cells were prepared for SDS-polyacrylamide gel electrophoresis (PAGE) and Western analysis as described previously (Chang et al., 2005). The cells (2–4 OD600) were pelleted, washed with water, and lysed in 150 μl of SDS sample buffer (67 mM Tris, pH 6.8, 2% SDS, 10% glycerol, 0.1% bromphenol blue, 1.5% dithiothreitol [DTT] solution containing 4 mM PMSF, 4 mM pepstatin A, and 2 mM leupeptin) by vortexing with glass beads (425–600 μm in diameter). Just before SDS-PAGE, the samples were incubated at 100°C for 5 min, and cell debris and glass beads were removed by centrifugation. Five to 20 μl of sample was loaded onto 7.5, 10, or 15% polyacrylamide gels. After electrophoresis, the proteins were transferred to nitrocellulose using the Trans-Blot SemiDry Transfer Cell (Bio-Rad Laboratories, Hercules, CA). The blots were blocked in 5% nonfat dried milk in phosphate-buffered saline (PBS) containing 0.1% Tween 20 (PBST) and incubated with the primary antibodies rabbit anti-Sar1p (gift from Dr. Ben Glick, University of Chicago), mouse anti-carboxypeptidase Y (Invitrogen, Carlsbad, CA), rabbit anti-AOX (Tuttle and Dunn, 1995), rabbit anti-hemagglutinin (HA) (Covance, Emeryville, CA), or rabbit anti-green fluorescent protein (GFP) (Sigma-Aldrich, St. Louis, MO). The blots were then washed in PBST, followed by incubation with secondary antibodies conjugated to horseradish peroxidase. After washing with PBST, the antibodies were detected using enhanced chemiluminescence (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom). The films were then scanned and protein bands quantified using ImageJ software (http://rsbweb.nih.gov/ij/).
Construction of Conditional Expression Vectors
Expression vectors containing sar1(T34N) and sar1(H79G) behind the copper-inducible CUP1 promoter were constructed as follows. The PpARG4 gene was amplified by polymerase chain reaction (PCR) from pYM30 (kind gift from Dr. Jim Cregg, Keck Graduate Institute) and inserted into the SspI and NdeI sites of pUC19. The resulting plasmid, pUC19-PpArg4, was amplified, cut with NdeI, blunt-ended with T4 DNA polymerase, and digested with EcoRI. The S. cerevisiae CUP1 promoter was excised from pCAJ3 vector with BamHI (blunt-ended with T4 DNA polymerase) and EcoRI and then ligated into the pUC19-PpArg4. Next, the poly-A sequence from pIB2 was excised and inserted into the HindIII and AflII site, yielding the pWD16 conditional expression vector. Finally, sar1(T34N) and sar1(H79G) kindly provided by Dr. Ben Glick were amplified by PCR and inserted into the EcoRI and HindIII sites of pWD16. GFP-ATG17 was constructed by inserting ATG17 amplified by PCR from genomic DNA into the KpnI and XhoI sites of pPS55 following similar procedures used to construct expression vectors containing GFP-ATG2, GFP-ATG8, GFP-ATG9, GFP-ATG11, and GFP-ATG18 (Guan et al., 2001; Kim et al., 2001; Strømhaug et al., 2001; Chang et al., 2005). DsRFP-HDEL was provided by Dr. Ben Glick, and pWD18 (PATG7 atg7-HA (C518S), HIS4) was constructed as described previously (Yuan et al., 1999; Chang et al., 2005).
Yeast Transformation
Cells grown overnight in YPD to a density of OD600 = 1.0 were harvested and treated with 10 mM DTT in YPD containing 25 mM HEPES, pH 8, for 15 min at 30°C. The cells were washed twice in ice-cold water and once in 1 M sorbitol and then resuspended in 1 M sorbitol. Cells (40 μl) were mixed with 0.2–1 μg of linearized vector and transferred to a 0.2-cm gap cuvette (Bio-Rad, Hercules, CA). The DNA was introduced by electroporation at 1.5 kV, 25 μF, and 400 Ω (Gene Pulser; Bio-Rad). The cells transformed with vectors containing the HIS4 gene were transferred to plates containing 0.67% yeast nitrogen base without amino acids, 2% glucose, 1 M sorbitol, 0.4 mg/l biotin, and 2% agar and incubated at 30°C for 3–5 d before colonies formed. For antibiotic selection, Zeocin (Invitrogen) was added to a final concentration of 100 μg/ml.
Fluorescence Microscopy
Cells expressing GFP-tagged proteins or DsRFP-HDEL were grown in YNM for 20 h. N-(triethlyammoniumpropyl)-4-(p-diethylaminophenylhexatrienyl) pyridinium dibromide (FM4-64; Invitrogen) was added to a final concentration of 20 μg/ml, and cells incubated for 12–16 h. Cells were then transferred to YND or YNE with or without 0.1 mM copper sulfate. After 2–4 h, the cells were examined live using an Axiophot fluorescence microscope (Carl Zeiss, Jena, Germany). Image capture was done using a SPOT camera (Diagnostics Instruments, Sterling Heights, MI) interfaced with IPLab software (BD Biosciences, San Jose, CA).
Electron Microscopy
Upon glucose or ethanol adaptation, cells were harvested by centrifugation, washed in water, and fixed with 1.5% KMnO4 in veronal-acetate buffer (30 mM sodium acetate, 30 mM sodium barbital, pH 7.6) for 20 min at room temperature. The samples were dehydrated by washing with increasing concentrations of ethanol followed by two washes with 100% propylene oxide. The cells were then infiltrated with a 50:50 mix of propylene oxide and POLY/BED 812 (Polysciences, Warrington, PA) for 16 h at 4°C and another 24 h at 22°C under vacuum. Afterward, the cells were infiltrated with 100% POLY/BED with accelerator 2,4,6-tri(dimethylaminomethyl)phenol (DMP-30; Polysciences) for another 2 d at 22°C under vacuum. The cells were pelleted and incubated at 60°C for 2 d to complete the polymerization of the resin. The cell pellets were mounted on blocks, sectioned, and prepared for examination on a 100CX transmission electron microscope (JEOL, Tokyo, Japan). Quantification of the endoplasmic reticulum was done using ImageJ software by measuring the linear length of the ER relative to the cell surface.
RESULTS
Expression of Dominant-Negative Mutants of Sar1p in P. pastoris
ScSec12p, the guanine nucleotide exchange factor for the G protein ScSar1p, has been shown to be required for autophagy in S. cerevisiae (Ishihara et al., 2001; Hamasaki et al., 2003; Reggiori et al., 2004). Because Sar1p is essential for cell growth (Nakano and Muramatsu, 1989), the role of Sar1p in pexophagy has been difficult to evaluate. To circumvent this technical problem, we have constructed dominant-negative forms of Sar1p whose expression is regulated by a copper-inducible CUP1 promoter of S. cerevisiae. One mutant was sar1pT34N, a protein analogous to mammalian Sar1[T39N], which has a high-affinity for GDP and blocks COPII vesicle formation (Altan-Bonnet et al., 2004). A second mutant was sar1pH79G, a protein analogous to mammalian Sar1[H79G], which is defective in hydrolyzing GTP (Altan-Bonnet et al., 2004). At 2 h (data not shown) and 4 h (Figure 1A) of copper induction, the expression of the mutant Sar1p proteins in WDY64 and WDY65 cells was approximately twofold over levels observed in the absence of copper and fourfold greater than basal wild-type (WT) levels. Furthermore, we observed that cell growth was dramatically reduced when these proteins were expressed (Supplemental Figure 1). The data suggest that copper promotes the expression of sar1pT34N and sar1pH79G proteins, thereby suppressing cell growth.
Figure 1.
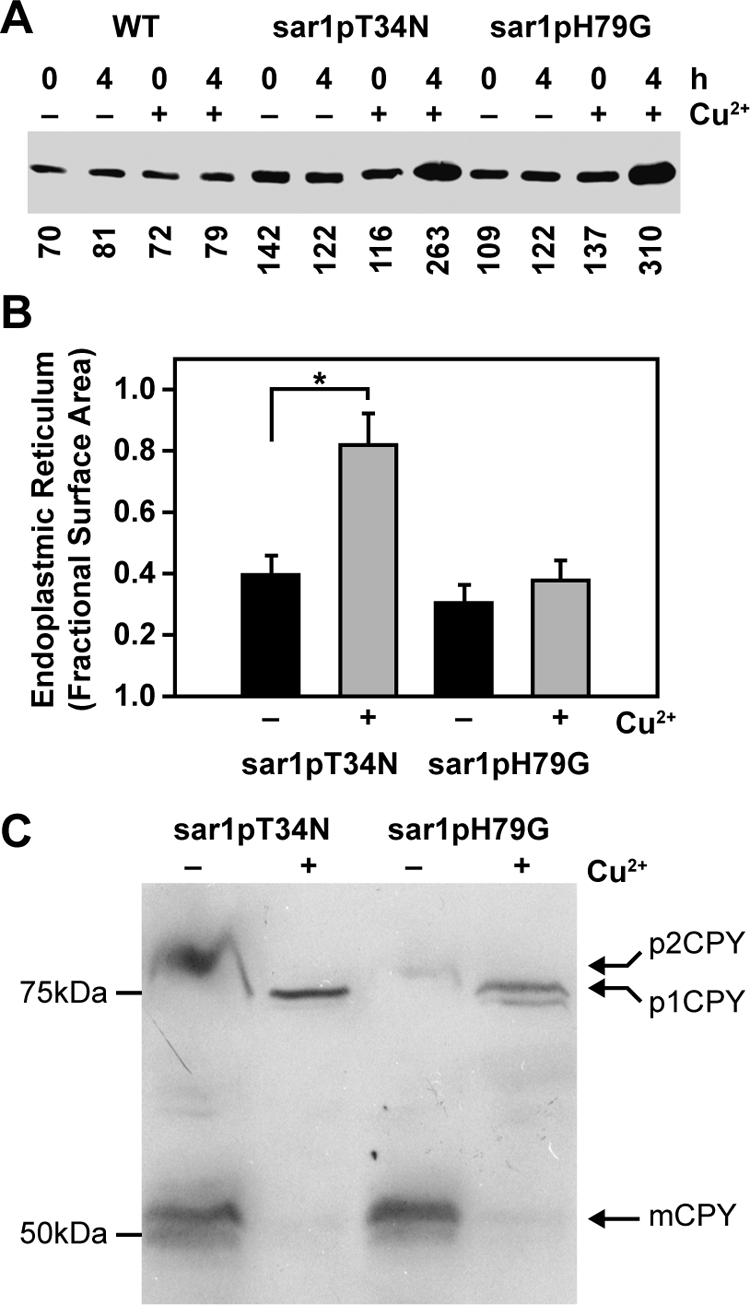
Effects of sar1pT34N and sar1pH79G on the morphology of the endoplasmic reticulum and Golgi apparatus. (A) WT, WDY64, and WDY65 cells were grown in YNM and then switched to YND (± CuSO4) for 0 and 4 h. Equal amounts of SDS solubilized cells (OD600 = 0.3) were then evaluated by Western blotting using rabbit anti-Sar1p antibodies (gift from Dr. Ben Glick). The relative amounts of Sar1p were quantified using ImageJ software. The data represent one of three trials. (B) WDY64 and WDY65 cells were adapted from YNM to YND (± CuSO4) for 4 h. The cells were processed for electron microscopy, and images were taken on a JEOL 100CX microscope. The surface area of the endoplasmic reticulum was quantified using morphometric protocols and ImageJ software. The data represent mean ± SE (n = 12–14 cells). The statistical differences relative to untreated control were determined by Student's t test (*p = 0.001). (C) WDY64 and WDY65 cells were grown in minimal YNDH medium and then switched to nitrogen-starvation SD(-N) medium (± CuSO4) for 8 h. The cells were solubilized and precursor (p1CPY and p2CPY) and mature (mCPY) forms of carboxypeptidase Y, identified by their molecular sizes on Western blots.
Given the role of Sar1p in protein trafficking out of and into the endoplasmic reticulum, we examined the effects of the mutant Sar1p proteins on the morphology of the endoplasmic reticulum (Figure 1B) and on the processing of carboxypeptidase Y (Figure 1C). Without copper, the endoplasmic reticulum appeared to be discontinuous sheets at the cell periphery (Figure 3A). When sar1pT34N expression is enhanced with copper, the ER surface area increased twofold (Figure 1B), looking like a continuous sheet at the cell periphery and throughout the cell (Figure 3B). These changes were not observed when sar1pH79G was expressed. Carboxypeptidase Y (CPY) is synthesized as a precursor (p1CPY) at the ER and transported to the Golgi apparatus in which it is further glycosylated (p2CPY) (Nakano and Muramatsu, 1989; Deitz et al., 2000). From the Golgi apparatus, p2CPY is transported to the vacuole and proteolytically activated into the mature form (Nakano and Muramatsu, 1989; Deitz et al., 2000). Nakano and Muramatsu have shown that p1CPY accumulates in cells lacking ScSar1p (Nakano and Muramatsu, 1989). To evaluate the effects of the Sar1p mutants on the processing of CPY, we examined on Western blots the presence of precursor and mature forms of CPY in cells starved for nitrogen, a condition known to enhance CPY synthesis and activation in S. cerevisiae (Gasch et al., 2000). The Golgi precursor (p2CPY) and proteolytically activated mature CPY (mCPY) was observed in nitrogen-starved P. pastoris not expressing the Sar1p mutants (Figure 1C). Mature CPY was not found in cells expressing either sar1pT34N or sar1pH79G mutant. Instead, the ER-associated p1CPY was present in these cells. In mammalian cells, Sar1[H79G] has been shown to promote the release of Golgi peripheral membrane proteins (golgin97 and COPI) (Miles et al., 2001; Yoshimura et al., 2004). Therefore, we next examined the cellular distribution of Sec7p, a peripheral membrane protein present exclusively at the Golgi apparatus, in cells not expressing sar1pT34N or sar1pH79G (Supplemental Figure 2). In the presence of sar1pT34N or sar1pH79G, Sec7p was visualized not only at the Golgi apparatus but also in the cytosol. In some cells, Sec7p could be observed only in the cytosol. The data suggest that these mutant proteins, when expressed, alter the morphology of the ER and the trafficking of newly synthesized CPY consistent with the role of Sar1p in protein movements between the ER and Golgi apparatus.
Figure 3.

The expression of sar1pT34N or sar1pH79G suppresses a late sequestration event of micropexophagy. WDY64 (A and B), WDY65 (E and F), LAM13 (C and D), and LAM14 (G and H) cells were adapted from YNM to YND in the absence (A and E) and presence (B, C, D, F, G, and H) of CuSO4 for 2–3 h. Cells were then fixed and prepared for viewing by electron microscopy (A, B, E, and F) or viewed in situ by fluorescence microscopy (C, D, G, and H). In cells not treated with CuSO4, the peroxisomes (P) could be found within the vacuole (A) or surrounded by membranes derived from the vacuole (E). When cells expressed either sar1pT34N (B) or sar1pH79G, the peroxisomes were partially surrounded by the vacuole and sequestering membranes but not observed within the vacuole. An extensive, ribbon-like ER (ER) was observed in cells expressing sar1pT34N, whereas a multilamellar Golgi (Go) apparatus was evident with sar1pH79G expression. By fluorescence microscopy, the vacuole and those sequestering membranes derived from the vacuole were observed by staining with FM4-64, and the peroxisomes were visualized by expressing BFP-SKL. In cells expressing either sar1pT34N or sar1pH79G, the peroxisomes were observed to be almost completely surrounded by sequestering membranes. These membranes stained with FM4-64 and occurred as vesiculated extensions of the vacuole.
Using the aforementioned cells, we examined the effects of these Sar1p mutants on starvation-induced autophagy (Supplemental Figure 3). The degradation of endogenous proteins in cells prelabeled with [14C]valine was measured in nitrogen-starved cells in the absence and presence of copper. Nitrogen starvation induces the nonselective delivery of cellular components to the vacuole by autophagy in yeast. We compared endogenous proteolysis in WT cells with cells expressing the Sar1p mutant proteins (Supplemental Figure 3). We observed a 25% reduction of proteolysis in starved WT cells exposed to copper. This was compared with the >60% reduction in starvation-induced proteolysis in cells whose expression of sar1pT34N or sar1pH79G had been induced with copper. The data reveal that like ScSec12p, Sar1p is required for starvation-induced macroautophagy.
Effects of Dominant-Negative Mutants of Sar1p on Pexophagy
We examined the effects of the Sar1p mutant proteins on glucose-induced micropexophagy (Figure 2A) and ethanol-induced macropexophagy (Figure 2B). Cells were grown in methanol, adapted to glucose in the absence and presence of copper for 6 h, and the remaining AOX activity was quantified. Less than 20% of the AOX remained in the WT cells regardless of the presence of copper. In the absence of copper, 80% of the AOX was degraded by the WDY64 and WDY65 cells. However, when these cells were incubated with copper, only 10% of the AOX was lost during glucose adaptation. Next, cells were grown in methanol, adapted to ethanol in the absence and presence of copper for 0–24 h, and the remaining AOX was detected by Western blotting. We observed that in the absence of copper, AOX protein was almost completely degraded within 9 h. However, when sar1pT34N or sar1pH79G was expressed, a substantial amount of AOX protein remained for up to 24 h. The data reveal that Sar1p is essential for both micro- and macropexophagy.
Figure 2.
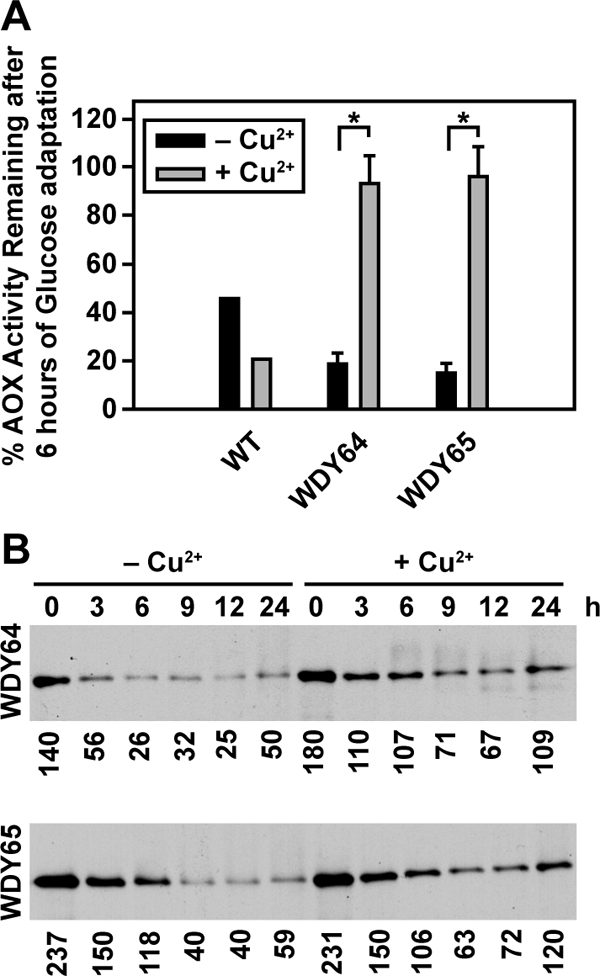
Sar1p is essential for pexophagy. Glucose-induced micropexophagy (A) and ethanol-induced macropexophagy (B) were evaluated in wild-type cells and cells expressing sar1pT34N and sar1pH79G. (A) GS115, WDY64, and WDY65 cells were grown in YNM and then switched to YND (± CuSO4), at which time the loss of AOX activity was measured over 6 h. The data represent the mean ± SE of four to six trials. The statistical differences relative to untreated controls were determined by Student's t test (*p < 0.001). (B) WT, WDY64, and WDY65 cells were grown in YNM and then switched to YNE (± CuSO4). At 0 to 24 h, samples were prepared for SDS-PAGE, and the remaining AOX was visualized by Western blotting. The relative amounts of AOX were quantified using ImageJ software. The data are a representative blot of three independent trials.
Suppression of Glucose-induced Micropexophagy by Dominant-Negative Mutants of Sar1p Occurs at a Late Sequestration Event
We have shown that the expression of sar1pT34N or sar1pH79G dramatically inhibits the degradation of peroxisomes induced by glucose adaptation. We will now evaluate whether these proteins inhibit the events of peroxisome sequestration, degradation, or both. We first examined the morphology of those cells expressing sar1pT34N and sar1pH79G by electron microscopy. WDY64 and WDY65 were adapted from YNM to YND for 4 h in the absence or presence of copper (Figure 3). During glucose adaptation in cells not expressing the Sar1p mutants, peroxisomes were surrounded by the vacuole or within the vacuole (Figure 3, A and E). In cells expressing sar1pT34N (Figure 3, B–D) or sar1pH79G (Figure 3, F–H), the peroxisome clusters were situated outside the vacuole, suggesting that Sar1p was required for delivery of the peroxisomes to the vacuole and not vacuolar degradation.
The events of micropexophagy include signaling, sequestration, and degradation. The above-mentioned data suggest that the Sar1p mutants were suppressing an event before degradation, most likely sequestration. The engulfment of peroxisomes by SMs that arise from the vacuole proceeds via SM initiation, expansion, and completion. These events can be visualized by observing the SM with FM4-64 staining incorporating peroxisomes expressing BFP-SKL (Supplemental Figure 4). For these studies, we constructed cell lines expressing BFP-SKL behind the AOX promoter in cells expressing the Sar1p mutants. During initiation, the sequestering membranes extend less than halfway around the peroxisomes. Expansion of the sequestering membranes can be observed when the sequestering membranes extend 50–90% around the peroxisomes. The completion events are characterized by peroxisomes being completely surrounded by the sequestering membranes, but the membranes seem segmented and not completely fused. Finally, the degradative stage is characterized by a continuous ring of FM4-64–labeled membranes surrounding peroxisomes that were no longer clearly delineated due to degradation. At 2 h of glucose adaptation, wild-type cells were observed at all phases of micropexophagy, with ∼80% being in the expansion and completion stages, suggesting that these events are likely rate limiting. Of those cells expressing sar1pT34N, two-thirds were at the expansion (Figure 3C) and completion (Figure 3D) stages. A similar finding was observed in cells expressing sar1pH79G (Figure 3, G and H). These results, combined with the inability of these dominant-negative mutants to support AOX degradation (Figure 2), suggest that Sar1p was not required for initiation but likely for expansion events, completion events, or a combination.
Sar1p Is Essential for the Formation of the MIPA
Our data suggest that Sar1p is critical for the expansion phases, completion phases, or both of peroxisome sequestration during glucose-induced micropexophagy. Expansion of the sequestering membranes requires Atg2p, Atg9p, and Atg11p and the assembly of the MIPA, which requires Atg8p. Completion requires MIPA and other proteins, including Vac8p and Atg24p. During expansion, Atg9p traffics from a unique peripheral compartment to become associated with the sequestering membranes. This trafficking requires Atg2p and Atg11p. Also at this time, Atg8p becomes associated with the newly assembled MIPA, which is positioned between the expanding sequestering membranes. Therefore, we decided to examine the effects of these Sar1p mutants on the trafficking of Atg2p, Atg9p, Atg11p, and Atg8p during glucose adaptation. Cell lines expressing sar1pT34N or sar1pH79G and GFP-Atg2p, GFP-Atg9p, GFP-Atg11p, or GFP-Atg8p were constructed (Table 1). The cells were adapted from YNM to YND in the absence and presence of copper sulfate and examined by fluorescence microscopy (Figures 4 and 5).
Figure 4.

The trafficking of Atg11p, Atg2p, and Atg9p during glucose-induced micropexophagy is not dependent upon Sar1p. WDY64 and WDY65 cells expressing GFP-Atg11p, GFP-Atg2p, GFP-Atg9p were adapted from YNM to YND in the absence and presence of CuSO4. At 2 h of glucose adaptation, the cells were visualized by fluorescence microscopy. The cellular localizations of Atg11p, Atg2p, and Atg9p were unaltered in cells expressing sar1pT34N or sar1pH79G. GFP-Atg11p was present at the vacuole and sequestering membranes. GFP-Atg2p was visualized at peripheral foci. However, in the presence of the Sar1p mutants, these foci seemed to be greater in number and sometimes coalesced forming irregular structures (yellow arrows). GFP-Atg9p was observed at the vacuole and sequestering membranes labeled with FM4-64. Atg9p could also be visualized at peripheral foci (white arrowheads) and perivacuolar structures (white arrows).
Figure 5.

Sar1p is essential for the formation of the MIPA. WDY64 (sar1pT34N) and WDY65 (sar1p H79G) cell lines expressing GFP-Atg8 were adapted from YNM to YND in the absence (A and C) and presence (B and D) of CuSO4. At 2 h of glucose adaptation, the cells were visualized in situ by fluorescence microscopy. The vacuole and those sequestering membranes derived from the vacuole were observed by staining with FM4-64. GFP-Atg8p was observed at the PAS, foci situated at the tip of the sequestering membranes (white arrowhead) and at the MIPA, crescent-shaped structure adjoining the sequestering membranes (white arrows). In the absence of copper, the MIPA was observed bridging the FM4-64–labeled membranes. However, when the expression of either sar1pT34N or sar1pH79G was induced with CuSO4, GFP-Atg8p localized to one or more dots or a flattened MIPA-like structure, but not situated near sequestering membranes (yellow arrowheads). (E) Percentage of WDY64 and WDY65 cells with MIPA was quantified in the absence and presence of CuSO4. The data represent mean ± SE (n = 5 trials of ≥100 cells counted per trial). The statistical differences were determined by Student's t test (*p = 0.026; **p = 0.014).
We first examined the trafficking of Atg11p. During micropexophagy, Atg11p colocalizes with Atg9p at the PVS and SM, and it is required for the trafficking of Atg9p to the PVS (Chang et al., 2005). In the absence of copper, Atg11p was found at the PVS, vacuole and SM at 2 h post glucose adaptation. The distribution of Atg11p was unaltered when either sar1pT34N or sar1pH79G was expressed (Figure 4). Next, we examined the distribution of GFP-Atg2p in cells. On glucose adaptation, Atg2p becomes associated with foci known as the Atg2p peripheral compartment. Its role in the expansion of the SM is not yet defined, but Atg2p is required for the transit of Atg9p to the SM. When LAM44 and LAM45 cells were adapted from YNM to YND in the absence of copper, Atg2p was found in the cytosol and at foci situated at the cell periphery and near the vacuole. On the copper-induced expression of sar1pT34N or sar1pH79G, Atg2p was virtually absent from the cytosol and was instead observed in foci and larger irregularly shaped structures (Figure 4, yellow arrows). These coalescing structures may represent an expansion of this peripheral compartment due to the expression of the Sar1p mutants.
We have previously shown that during the membrane expansion events, Atg9p transits from peripheral vesicles, to the PVS, and finally to the SM (Chang et al., 2005). We next examined the effects of sar1pT34N or sar1pH79G on the cellular distribution of GFP-Atg9p during glucose adaptation. In the absence of copper, GFP-Atg9p was found at the peripheral vesicles, PVS, and SM (Figure 4). Atg9p was also present at the vacuole membrane. When copper was added, the distribution of GFP-Atg9p was not dramatically altered. In addition to the peripheral foci (Figure 4, white arrowhead), Atg9p was present at the PVS (Figure 4, white arrow) and the SM (colocalization with FM4-64).
We have reported that Atg2p and Atg9p localize to distinct peripheral compartments (Chang et al., 2005). However, because Atg2p requires Atg9p for its localization, we suggest that these compartments may transiently interact. Based on our data, it is possible that Sar1p may influence the assembly of both Atg2p and Atg9p peripheral compartments. Indeed, the structure of the Atg2p compartment was altered, and there seemed to be more Atg9p-containing foci present in cells expressing the Sar1p mutants. However, Atg9p did not colocalize with Atg2p when either sar1pT34N or sar1pH79G was expressed (unpublished observations).
Finally, we examined the cellular distribution of GFP-Atg8p in glucose-adapted cells expressing the mutant forms of Sar1p (Figure 5). Under normal conditions, Atg8p localizes to the PAS from which it transits to the newly assembled MIPA. At 2 h of glucose adaptation without copper, GFP-Atg8p was present throughout the cytosol and localized specifically to the PAS (Figure 5, white arrowhead) and the MIPA situated between the adjoining sequestering membranes labeled with FM4-64 (Figure 5, white arrows). In the absence of copper, ∼10% of the cells contained MIPA structures (Figure 5E). When the expression of sar1pT34N or sar1pH79G is enhanced by copper, only 2% or less of the cells contained a MIPA (Figure 5E). In fact, GFP-Atg8p was observed predominantly in multiple punctate and flattened structures (Figure 5, yellow arrowheads). These structures were not associated with the FM4-64–labeled sequestering membranes, but they were distributed throughout the cell. Our data suggest that in the presence of sar1pT34N or sar1pH79G, MIPA fails to assemble, thereby inhibiting the expansion and completion events.
We also noticed that the PAS labeled with GFP-Atg8p, which normally occurs as one or two foci, was structurally different when sar1pT34N or sar1pH79G was expressed. Therefore, we examined the cellular distribution of GFP-Atg17p, a structural component of the PAS not present at the MIPA (Ano et al., 2005). During glucose adaptation in the absence of copper, GFP-Atg17p was found at one or two distinct foci juxtaposed to the vacuole or sequestering membranes (Supplemental Figure 5). However, in the presence of copper, GFP-Atg17p was present in foci and large structures of irregular shapes that were distant from the vacuole and SM. The data suggest that Sar1p may play a role in the maintenance of the PAS.
Lipidation of Atg8p during Glucose-induced Micropexophagy Is Altered upon Expression of Dominant-Negative Sar1p Mutants
We have determined that the MIPA fails to assemble when either sar1pT34N or sar1pH79G is expressed. Atg8p not only localizes to the MIPA but also is essential for its formation (Mukaiyama et al., 2004). On proteolytic activation by Atg4p, the C-terminal glycine of Atg8p becomes conjugated by a thio-ester linkage to cysteine 518 of Atg7p (Yuan et al., 1999). Atg8p is then conjugated to phosphatidylethanolamine (PE) by the actions of Atg3p. We have shown previously that Atg7p-HA forms a DTT-sensitive thio-ester conjugate with a protein whose size we estimated at 25 kDa (Yuan et al., 1999). When the active cysteine of Atg7p is mutated to a serine (atg7pC518S-HA), this conjugation becomes DTT resistant (Yuan et al., 1999). Therefore, when atg7pC518S-HA was expressed in WT cells, we observed two protein bands at 70 and 95 kDa (Figure 6A). The upper band was not observed in cells lacking Atg8p, suggesting that the 95-kDa band represents atg7pC518S-HA conjugated by an ester linkage to Atg8p. To assess the effects of sar1pT34N or sar1pH79G on Atg7p activity, we expressed atg7pC518S-HA in these mutants. These cells were grown in YNM and switched to YND for 3 and 6 h with and without copper sulfate. Afterward, the protein conjugate of Atg7p and Atg8p was visualized by Western blotting (Figure 6A). In the absence and presence of copper, both unconjugated (70 kDa) and conjugated (95 kDa) forms of atg7pC518S-HA were observed, suggesting that the formation of the high-energy conjugate of Atg8p with Atg7p proceeds normally when these Sar1p mutants are expressed.
Figure 6.

Effects of Sar1p on the lipidation of Atg8p. (A) atg7pC518S-HA was expressed in GS115, SJCF257 (atg8Δ), WDY64, and WDY65 cells. The cells were adapted from YNM to YND in the absence and presence of CuSO4 for 3 and 6 h. At that time, the cells were solubilized in SDS sample buffer, and the 70-kDa Atg7p and 95-kDa Atg7p protein–protein ester conjugate visualized by Western blotting. The protein–protein conjugates of atg7pC518S were observed in wild-type cells and in cells expressing sar1pT34N or sar1pH79G, but not in cells lacking Atg8p. (B) atg7Δ, WDY64 (sar1pT34N), and WDY65 (sar1pH79G) cells expressing HA-Atg8p were adapted from YNM to YND in the absence and presence of CuSO4 for 2 or 4 h. The cells were then solubilized, proteins separated on 12% SDS-PAGE 6 M urea gels, and the molecular forms of Atg8p were detected by Western blotting using mouse anti-HA antibodies. During 2–4 h of glucose adaptation, two forms of HA-Atg8p were apparent in the sar1pT34N and sar1pH79G cells. The faster migrating form of HA-Atg8p was absent in atg7Δ cells and in cells expressing HA-Atg8p(G116A). HA-Atg8p(G116A) cannot be lipidated because of its inability to be proteolytically processed and thus migrates slower than HA-Atg8p (Mukaiyama et al., 2004). Based on these findings, we have identified this faster migrating band as the lipidated form of Atg8p (HA-Atg8p-PE).
We next examined the effects of sar1pT34N or sar1pH79G on the lipidation of Atg8p. This was done by examining the processing of HA-Atg8p on Western blots given that the lipidated form of Atg8p migrates faster on SDS-PAGE (Strømhaug et al., 2004). WDY152 and WDY154 were grown in YNM and switched to YND in the absence or presence of copper sulfate for 2 and 4 h. In the absence of copper, we observed two forms of HA-Atg8p. The faster migrating form was consistent with the lipidation of Atg8p. That is, this form was absent in cells lacking Atg7p and in cells expressing a lipidation defective HA-Atg8p(G116A) (Figure 6B). In the presence of copper, both forms of Atg8p were observed, suggesting that the lipidation of Atg8p is unaltered by the expression of sar1pT34N or sar1pH79G.
Sar1p Is Required for Early and Late Events of Macropexophagy
We have shown that sar1pT34N and sar1pH79G suppress the degradation of AOX during ethanol-induced macropexophagy. During macropexophagy, individual peroxisomes are engulfed by sequestering membranes that can be visualized by GFP-Atg8p. Like the MIPA, these membranes are presumed to be organized from the PAS, which occur as single distinct foci per cell (Dunn et al., 2005). In cells not expressing sar1pT34N or sar1pH79G, GFP-Atg8p was present at the PAS (Figure 7, white arrowheads), pexophagosomes (Figure 7, white arrows), and within the vacuole (Figure 7, A and C). At 2 h of ethanol adaptation, >10% of the cells contained pexophagosomes, and 40–60% of the vacuoles contained the GFP signal. We next examined the effects of sar1pT34N and sar1pH79G on the assembly of pexophagosomes, as visualized by GFP-Atg8p labeling (Figure 7) and ultrastructural morphology (Supplemental Figure 6). When sar1pT34N was expressed, only 2% of the cells contained pexophagosomes, and 5% of the vacuoles were labeled with GFP. GFP-Atg8p localized to the “abnormal” PAS, which occurred as two or more clusters of foci within each cell (Figure 7B, yellow arrowhead). Occasionally, we visualized membranes near the peroxisome, which may represent the initiation of the sequestering membranes (Supplemental Figure 6, arrow). In cells expressing sar1pH79G, we observed pexophagosomes within 12% of the cells counted (Figure 7D, white arrow), but only 5% of the vacuoles contained GFP. At 3 h of ethanol adaptation, the number of cells with pexophagosomes increased to 15% compared with 8% in cells not expressing the Sar1p mutant (data not shown). In addition, GFP-Atg8p localized to unknown structural clusters instead of a single PAS foci (Figure 7D, yellow arrowheads). These abnormal PAS were also observed in sar1pT34N cells and had similar morphology to structures containing Atg17p (Supplemental Figure 5). Ultrastructural analyses revealed that these pexophagosomes contained a single peroxisome and were bound by multiple membranes (Supplemental Figure 6, arrowheads). These observations, combined with our biochemical data (Figure 2A), suggest that the pexophagosomes present in cells expressing sar1pH79G may fuse poorly with the vacuole. We have shown that sar1pT34N suppresses the formation of the pexophagosome, whereas the delivery of the peroxisome from the pexophagosome to the vacuole for degradation seems to be defective in cells expressing sar1pH79G.
Figure 7.

Effects of sar1pT34N and sar1pH79G on ethanol-induced macropexophagy. WDY64 (sar1pT34N) and WDY65 (sar1pH79G) cell lines expressing GFP-Atg8p and BFP-SKL were adapted from YNM to YNE in the absence (A and C) and presence (B and D) of CuSO4. At 2 h of ethanol adaptation, the cells were visualized in situ by fluorescence microscopy. In the absence of CuSO4, GFP-Atg8p was found at the PAS (white arrowheads) and pexophagosomes (arrows). When sar1pT34N is expressed, GFP-Atg8p was observed almost exclusively at multiple foci that were larger than the PAS (yellow arrowheads). To the contrary, when sar1pH79G is expressed, GFP-Atg8p was observed at the pexophagosome (arrows) and multiple large foci (yellow arrowheads). (E) Percentage of WDY64 and WDY65 cells with pexophagosomes was quantified in the absence and presence of CuSO4. The data represent mean ± SE (n = 5 trials of ≥100 cells counted per trial). The statistical differences were determined by Student's t test (*p = 0.003).
DsRFP-HDEL Localizes to Pexophagosomes When sar1pH79G Is Expressed
These dominant-negative Sar1p mutants have been shown to effectively alter the trafficking of ER proteins in mammalian cells (Ward et al., 2001). Because the ER has been implicated in the formation of autophagosomes in mammalian cells (Dunn, 1990), we decided to examine the localization of DsRFP-HDEL during macropexophagy (Figure 8). DsRFP-HDEL with its amino-terminal signal sequence and carboxy-terminal HDEL retrieval signal is targeted to the ER lumen by the receptor Erd2p, which binds the HDEL sequence (Bevis et al., 2002). On ethanol adaptation, DsRFP-HDEL was found exclusively at the ER located adjacent to the cell periphery and around the nucleus (Figure 8, A and D). The distribution of DsRFP-HDEL was unaltered in ethanol-adapted cells expressing sar1pT34N (Figure 8, B and C). When sar1pH79G is expressed during 2 h of ethanol adaptation, we observed DsRFP-HDEL at the ER and labeling the SM around individual peroxisomes (Figure 8E, arrows). At times >3 h, the red fluorescent protein (RFP) was observed within the vacuole (Figure 8F, arrowheads). These pexophagosomes did not contain Sec7p, a protein of the Golgi apparatus (unpublished observations). The results suggest that components of the ER are observed with the pexophagosomes when ER recycling is inhibited by sar1pH79G.
Figure 8.

Components of the endoplasmic reticulum are observed at the pexophagosome when cells express sar1pH79G. WDY64 (A–C) and WDY65 (D–F) cell lines expressing DsRFP-HDEL were adapted from YNM to YNE in the absence (A and D) and presence (B, C, E, and F) of CuSO4. At 2 h (A, B, D, and E) and 3 h (C and F) of ethanol adaptation, the cells were visualized by fluorescence microscopy (top) and by phase contrast microscopy (bottom). DsRFP-HDEL was localized solely to the ER situated near the cell periphery and around the nucleus. This distribution was unaltered during macropexophagy (A and D). When sar1pT34N was expressed, the cellular distribution of DsRFP-HDEL was restricted to the ER (B and C). At 2 h of ethanol adaptation in the presence of sar1pH79G, DsRFP-HDEL was visualized not only at the ER but also at vacuoles containing phase-dense peroxisomes (E, arrows). At 3 h, DsRFP-HDEL could be detected within the yeast vacuole (F, arrowheads).
DISCUSSION
Earlier reports have demonstrated that some Sec proteins are required for autophagy in S. cerevisiae. Three of these proteins, ScSec12p, ScSec23p, and ScSec24p, are essential for ScSar1p activity and trafficking of proteins out of and into the ER. We examined the role of Sar1p in the selective degradation of peroxisomes by pexophagy by regulating the expression of two dominant-negative mutants of Sar1p. The sar1pT34N mutant has a high-affinity for GDP and suppresses COPII-dependent protein exit from the ER (Ward et al., 2001; Altan-Bonnet et al., 2004). The sar1pH79G mutant has a defective GTPase, thereby inhibiting the recycling of proteins back to the ER (Ward et al., 2001; Altan-Bonnet et al., 2004). In agreement with studies of cells lacking Sar1p (Nakano and Muramatsu, 1989), we have shown that cell growth is retarded when either mutant Sar1p protein is expressed. Furthermore, the expression of sar1pT34N or sar1pH79G results in an accumulation of p1CPY, suggesting that it fails to transit from the ER to the Golgi apparatus. In addition, sar1pT34N expression results in a dramatic increase in the surface area of the ER consistent with the inability of proteins to exit this compartment. The data suggest that the overexpression of dominant-negative sar1pT34N and sar1pH79G alters the protein movements between the ER and Golgi apparatus. Using these dominant-negative mutants of Sar1p, we have shown that Sar1p is essential for starvation-induced autophagy, glucose-induced micropexophagy, and ethanol-induced macropexophagy.
Role of Sar1p in Micropexophagy
Our data suggest that the degradation of peroxisomal AOX is dramatically reduced when either sar1pT34N or sar1pH79G is expressed. Given the role of Sar1p in the trafficking of vacuolar proteinases, it is possible that peroxisome degradation would be suppressed. However, we observed that the peroxisomes were almost completely engulfed by sequestering membranes, but they remained outside the vacuole. This suggests that the blockage occurs at a late sequestration event (e.g., expansion, completion, or both) and not vacuole degradation. Moreover, MIPA formation did not occur when either sar1pT34N or sar1pH79G was expressed. The MIPA is a crescent-shaped structure that joins the apposing sequestering membranes to initiate their fusion, thereby completing peroxisome sequestration. This structure contains Atg8p and Atg26p, and its formation requires several Atg proteins, including Atg9p and Atg11p. Therefore, we examined the trafficking of Atg9p and Atg11p, and we showed that their trafficking to the sequestering membranes was unaffected by the Sar1p mutants.
The MIPA assembles from the PAS or initiation complex, a structure that contains Atg8p and Atg17p (Oku et al., 2006; Yamashita et al., 2006). The formation of the MIPA is influenced by many Atg proteins, including Atg8p and Atg26p and the availability of lipids such as PE and phosphatidylinositol 4′-monophosphate (PI4P) (Yamashita et al., 2007). The availability of PE has been shown to influence Atg8p localization to the PAS and starvation-induced autophagy (Nebauer et al., 2007). Atg8p is conjugated to PE through the actions of Atg4p, Atg7p, and Atg3p and then assembled into the forming MIPA. Our data suggest that the inability of cells expressing the Sar1p mutants to assemble the MIPA is unrelated to the lipidation of Atg8p (Figure 6). This is consistent with the association of GFP-Atg8p with multiple PAS-like structures (Figure 5, B and C). Therefore, the inability to form the MIPA may be related directly or indirectly to COPII-dependent vesicular trafficking of proteins and/or lipids required for the assembly of the MIPA from the initiation complex. We have shown that the movements of Atg9p, Atg11p, and Atg18p, which are required for MIPA formation (unpublished observations), to the sequestering membranes do not require Sar1p (Figure 4 and Supplemental Figure 5). The trafficking of lipids such as PE is thought to be mediated by both vesicular and nonvesicular means (Levine, 2004). The post-Golgi trafficking of PE and autophagy-like transport of cytosolic prApeIp (proaminopeptidase I) to the vacuole is suppressed in cells lacking ScVps4p or ScVps36p (Nebauer et al., 2007). Meanwhile, it has been demonstrated that Sar1p-GTP binds to liposomes composed of phosphatidylcholine and phosphatidylethanolamine, thereby stimulating COPII assembly at the ER (Supek et al., 2002). However, the effects of Sar1p and COPII on the trafficking of PE remain to be studied.
Atg26p is also required for MIPA formation. PI4P transits from the ER or Golgi apparatus to the PAS or initiation complex, thereby recruiting Atg26p by interacting with its GRAM domain (Yamashita et al., 2006). The sterol conversion of Atg26p is essential for the formation of the MIPA. It is possible that the trafficking of PI4P from the ER or to the Golgi apparatus may be altered in cells expressing the Sar1p mutants. Indeed, Sar1p has been implicated in the exit of glycosylphosphatidylinositol-anchored GFP from the ER (Stephens and Pepperkok, 2004). However, we have no data to support that PI4P transport is influenced by Sar1p.
The expression of sar1pT34N or sar1pH79G inhibits glucose-induced micropexophagy by suppressing the assembly of the MIPA. A probable scenario is that Sar1p mediates directly membrane trafficking, which is essential for MIPA formation. Nevertheless, the suppression of vesicular movements between the ER and Golgi apparatus will affect organelle structure and influence the trafficking of proteins through the Golgi apparatus, thereby indirectly affecting MIPA formation. Indeed, we have observed a dissociation of Sec7p from the Golgi apparatus in the presence of the Sar1p mutants. The vesicular trafficking mediated by Sar1p may also govern the movements of PE and PI4P. Further studies are needed to better define the role of Sar1p in MIPA formation.
Role of Sar1p in Macropexophagy
The data from sec mutants in S. cerevisiae provides circumstantial evidence that the autophagosome membranes may originate from the ER (Ishihara et al., 2001; Hamasaki et al., 2003; Reggiori et al., 2004). In this study, we have characterized the essential role of Sar1p, a protein essential for ER function, in macropexophagy in P. pastoris. Ethanol-induced peroxisome degradation by macropexophagy is suppressed when cells express either sar1pT34N or sar1pH79G (Figure 2). However, these proteins inhibit different events of macropexophagy. We have shown that pexophagosomes do not form when sar1pT34N is expressed. ScSec12p, a GDP/GTP exchange factor that influences ScSar1p activity, is also required for the formation of autophagosomes in nitrogen-starved S. cerevisiae (Ishihara et al., 2001; Reggiori et al., 2004).
Brucella abortus is an intracellular pathogen that infiltrates the autophagosome to replicate in vacuoles that contain the ER protein calnexin (Celli et al., 2005). When mammalian Sar1[T39N] is expressed, this bacterium fails to replicate or localize to calnexin-positive autophagosomes, but instead it is found in lysosome-associated membrane protein 1-positive phagolysosomes (Celli et al., 2005). These results substantiate our findings that mammalian Sar1[T39N] and its P. pastoris analogue sar1pT34N suppress autophagosome formation. In B. abortus, because the replication vacuole is not formed, this bacterium transits to the phagolysosome where it is killed and degraded. In contrast, bacterial replication within calnexin-positive autophagosomes was not altered by sar1pH79G. This is consistent with our findings that sar1pH79G does not affect formation of the pexophagosome.
Pexophagosomes with GFP-Atg8p at their delimiting membranes are observed in cells expressing sar1pH79G. Their morphology is consistent with that of fully formed pexophagosomes (Supplemental Figure 6). The data suggest that sar1pH79G may interrupt fusion of the pexophagosome with the vacuole. It is possible that the trafficking to the vacuole of the fusion machinery (Ypt7, Vti1, and Vam3) or the degradative lipases (Atg15) or proteinases (Pep4 and Prb1) may be altered by sar1pH79G (Levine and Klionsky, 2004). In contrast, alterations in the membranes of the pexophagosome may interrupt fusion and lysis. Indeed, we have observed that the delimiting membranes of the pexophagosomes also contained DsRFP-HDEL, a marker for Erd2 (HDEL receptor), an ER membrane protein. At later times of macropexophagy, the RFP was found within the vacuole, suggesting that there was some delivery of the pexophagosome to the vacuole. At no time in wild-type cells did we observe DsRFP-HDEL delimiting the membranes of the pexophagosome or within the vacuole. These results are consistent with sar1pH79G inhibiting retrograde trafficking of the DsRFP-HDEL and possibly other ER components from the pexophagosome to the ER. The presence of these ER components may ultimately suppress pexophagosome fusion with the vacuole and provide resistance to vacuolar lipases and proteinases. Another possible explanation for the presence of Erd2 at the pexophagosome membrane is that Sar1p acts by altering the ER and Golgi trafficking of newly synthesized membrane proteins to a compartment that assembles the pexophagosome (e.g., PAS). Although we do not observe DsRFP-HDEL at the PAS, our data suggest that the morphology of this compartment is dramatically altered when expressing the Sar1p mutants. Our data, based on the use of dominant-negative mutants and a single ER marker, do not prove unambiguously that the pexophagosome is derived from the ER, but only that Sar1p seems to modulate the formation and maturation of the pexophagosome. In contrast, these mutant Sar1p proteins may act indirectly by altering the structure and function of the ER and Golgi apparatus. Nevertheless, the ER has been implicated in the formation of the autophagosome in mammalian (Dunn, 1990, 1993; Ueno et al., 1991) and yeast (Ishihara et al., 2001; Reggiori et al., 2004) cells. Therefore, we propose that Sar1p-dependent recycling of ER components from the pexophagosome is critical for pexophagosome maturation and delivery of the peroxisomes to the vacuole for degradation. Further studies using additional ER markers are required to better ascertain a role for the ER in pexophagosome formation and maturation.
In summary, we have used Sar1p mutants defective in anterograde (sar1pT34N) and retrograde (sar1pH79G) transport of ER proteins to demonstrate that Sar1p is essential for pexophagy. These mutants suppress micropexophagy by inhibiting the formation of the MIPA and the delivery of the peroxisomes to the vacuole. In macropexophagy, sar1pT34N inhibits the formation of the pexophagosome. Meanwhile, sar1pH79G has no effect on pexophagosome formation but instead suppresses pexophagosome fusion with the vacuole. Our results demonstrate that protein trafficking, lipid trafficking, or both as directed by Sar1p are essential for micro- and macropexophagy.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. B. S. Glick for generously providing the pSar1T34N, pSar1H79G, pDsRFP-HDEL, and pIB2 vectors and Sar1p antibodies. We also thank Dr. S. Subramani for providing the SJCF257 cells and the expression vectors containing 3xHA-ATG8 and 3xHA-ATG8(G116A). Finally, we thank Todd Barnash for help in assembling the figures and Debra Akin for helpful discussions and editing. This work was supported by National Cancer Institute (National Institutes of Health) grant CA-95552 (to W.A.D.).
Abbreviations used:
- AOX
alcohol oxidase
- COPII
coat protein complex II
- CPY
carboxypeptidase Y
- p1CPY
endoplasmic reticulum-associated precursor of CPY
- p2CPY
Golgi-associated precursor of CPY
- mCPY
vacuole-associated mature carboxypeptidase Y
- ER
endoplasmic reticulum
- FM4-64
N-(triethlyammoniumpropyl)-4-(p-diethylaminophenylhexatrienyl) pyridinium dibromide
- MIPA
micropexophagy-specific membrane apparatus
- PAS
preautophagosome structure
- PI4P
phosphatidylinositol 4′-monophosphate
- PE
phosphatidylethanolamine
- PVS
perivacuolar structure
- SM
sequestering membrane
- TCA
trichloroacetic acid
- and v-SNARE
vesicle-soluble N-ethylmaleimide-sensitive factor attachment protein receptor.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E07-09-0868) on September 3, 2008.
REFERENCES
- Altan-Bonnet N., Sougrat R., Lippincott-Schwartz J. Molecular basis for Golgi maintenance and biogenesis. Curr. Opin. Cell Biol. 2004;16:364–372. doi: 10.1016/j.ceb.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Ano Y., Hattori T., Oku M., Mukaiyama H., Baba M., Ohsumi Y., Kato N., Sakai Y. A sorting nexin PpAtg24 regulates vacuolar membrane dynamics during pexophagy via binding to phosphatidylinositol-3-phosphate. Mol. Biol. Cell. 2005;16:446–457. doi: 10.1091/mbc.E04-09-0842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevis B. J., Hammond A. T., Reinke C. A., Glick B. S. De novo formation of transitional ER sites and Golgi structures in Pichia pastoris. Nat. Cell Biol. 2002;4:750–756. doi: 10.1038/ncb852. [DOI] [PubMed] [Google Scholar]
- Celli J., Salcedo S. P., Gorvel J. P. Brucella coopts the small GTPase Sar1 for intracellular replication. Proc. Natl. Acad. Sci. USA. 2005;102:1673–1678. doi: 10.1073/pnas.0406873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang T., Schroder L. A., Thomson J. M., Klocman A. S., Tomasini A. J., Strømhaug P. E., Dunn W. A., Jr PpATG9 encodes a novel membrane protein that traffics to vacuolar membranes, which sequester peroxisomes during pexophagy in Pichia pastoris. Mol. Biol. Cell. 2005;16:4941–4953. doi: 10.1091/mbc.E05-02-0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregg J. M., Barringer K. J., Hessler A. Y., Madden K. R. Pichia pastoris as a host system for transformations. Mol. Cell. Biol. 1985;5:3376–3385. doi: 10.1128/mcb.5.12.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deitz S. B., Rambourg A., Kepes F., Franzusoff A. Sec7p directs the transitions required for yeast Golgi biogenesis. Traffic. 2000;1:172–183. doi: 10.1034/j.1600-0854.2000.010209.x. [DOI] [PubMed] [Google Scholar]
- Dunn W. A. Mechanism and regulation of autophagic degradation of cellular proteins. Adv. Cell. Mol. Biol. Memb. 1993;1:117–138. [Google Scholar]
- Dunn W. A., Jr Studies on the mechanisms of autophagy: formation of the autophagic vacuole. J. Cell Biol. 1990;110:1923–1933. doi: 10.1083/jcb.110.6.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn W. A., Jr, Cregg J. M., Kiel J. A., van der Klei I. J., Oku M., Sakai Y., Sibirny A. A., Stasyk O. V., Veenhuis M. Pexophagy: the selective autophagy of peroxisomes. Autophagy. 2005;1:75–83. doi: 10.4161/auto.1.2.1737. [DOI] [PubMed] [Google Scholar]
- Farre J. C., Vidal J., Subramani S. A cytoplasm to vacuole targeting pathway in P. pastoris. Autophagy. 2007;3:230–234. doi: 10.4161/auto.3905. [DOI] [PubMed] [Google Scholar]
- Fry M. R., Thomson J. M., Tomasini A. J., Dunn W. A., Jr Early and late molecular events of glucose-induced pexophagy in Pichia pastoris require Vac8. Autophagy. 2006;2:280–288. doi: 10.4161/auto.3164. [DOI] [PubMed] [Google Scholar]
- Gasch A. P., Spellman P. T., Kao C. M., Carmel-Harel O., Eisen M. B., Storz G., Botstein D., Brown P. O. Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan J., Strømhaug P. E., George M. D., Habibzadegah-Tari P., Bevan A., Dunn W. A., Jr, Klionsky D. J. Cvt18/Gsa12 is required for cytoplasm-to-vacuole transport, pexophagy, and autophagy in Saccharomyces cerevisiae and Pichia pastoris. Mol. Biol. Cell. 2001;12:3821–3838. doi: 10.1091/mbc.12.12.3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamasaki M., Noda T., Ohsumi Y. The early secretory pathway contributes to autophagy in yeast. Cell Struct. Funct. 2003;28:49–54. doi: 10.1247/csf.28.49. [DOI] [PubMed] [Google Scholar]
- Ishihara N., Hamasaki M., Yokota S., Suzuki K., Kamada Y., Kihara A., Yoshimori T., Noda T., Ohsumi Y. Autophagosome requires specific early Sec proteins for its formation and NSF/SNARE for vacuolar fusion. Mol. Biol. Cell. 2001;12:3690–3702. doi: 10.1091/mbc.12.11.3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Kamada Y., Strømhaug P. E., Guan J., Hefner-Gravink A., Baba M., Scott S. V., Ohsumi Y., Dunn W. A., Klionsky D. J. Cvt9/gsa9 functions in sequestering selective cytosolic cargo destined for the vacuole. J. Cell Biol. 2001;153:381–396. doi: 10.1083/jcb.153.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legesse-Miller A., Sagiv Y., Glozman R., Elazar Z. Aut7p, a soluble autophagic factor, participates in multiple membrane trafficking processes. J. Biol. Chem. 2000;275:32966–32973. doi: 10.1074/jbc.M000917200. [DOI] [PubMed] [Google Scholar]
- Levine B., Klionsky D. J. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev. Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- Levine T. Short-range intracellular trafficking of small molecules across endoplasmic reticulum junctions. Trends Cell Biol. 2004;14:483–490. doi: 10.1016/j.tcb.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Miles S., McManus H., Forsten K. E., Storrie B. Evidence that the entire Golgi apparatus cycles in interphase HeLa cells: sensitivity of Golgi matrix proteins to an ER exit block. J. Cell Biol. 2001;155:543–555. doi: 10.1083/jcb.200103104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monastyrska I., Kiel J. A., Krikken A. M., Komduur J. A., Veenhuis M., van der Klei I. J. The Hansenula polymorpha ATG25 gene encodes a novel coiled-coil protein that is required for macropexophagy. Autophagy. 2005;1:92–100. doi: 10.4161/auto.1.2.1832. [DOI] [PubMed] [Google Scholar]
- Mukaiyama H., Baba M., Osumi M., Aoyagi S., Kato N., Ohsumi Y., Sakai Y. Modification of a ubiquitin-like protein Paz2 conducted micropexophagy through formation of a novel membrane structure. Mol. Biol. Cell. 2004;15:58–70. doi: 10.1091/mbc.E03-05-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano A., Muramatsu M. A novel GTP-binding protein, Sar1p, is involved in transport from the endoplasmic reticulum to the Golgi apparatus. J. Cell Biol. 1989;109:2677–2691. doi: 10.1083/jcb.109.6.2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebauer R., Rosenberger S., Daum G. Phosphatidylethanolamine, a limiting factor of autophagy in yeast strains bearing a defect in the carboxypeptidase Y pathway of vacuolar targeting. J. Biol. Chem. 2007;282:16736–16743. doi: 10.1074/jbc.M611345200. [DOI] [PubMed] [Google Scholar]
- Oku M., Nishimura T., Hattori T., Ano Y., Yamashita S., Sakai Y. Role of Vac8 in formation of the vacuolar sequestering membrane during micropexophagy. Autophagy. 2006;2:272–279. doi: 10.4161/auto.3135. [DOI] [PubMed] [Google Scholar]
- Reggiori F., Wang C. W., Nair U., Shintani T., Abeliovich H., Klionsky D. J. Early stages of the secretory pathway, but not endosomes, are required for Cvt vesicle and autophagosome assembly in Saccharomyces cerevisiae. Mol. Biol. Cell. 2004;15:2189–2204. doi: 10.1091/mbc.E03-07-0479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K., Nakano A. Mechanisms of COPII vesicle formation and protein sorting. FEBS Lett. 2007;581:2076–2082. doi: 10.1016/j.febslet.2007.01.091. [DOI] [PubMed] [Google Scholar]
- Stasyk O. V., van der Klei I. J., Bellu A. R., Shen S., Kiel J. A., Cregg J. M., Veenhuis M. A Pichia pastoris VPS15 homologue is required in selective peroxisome autophagy. Curr. Genet. 1999;36:262–269. doi: 10.1007/s002940050499. [DOI] [PubMed] [Google Scholar]
- Stephens D. J., Pepperkok R. Differential effects of a GTP-restricted mutant of Sar1p on segregation of cargo during export from the endoplasmic reticulum. J. Cell Sci. 2004;117:3635–3644. doi: 10.1242/jcs.01269. [DOI] [PubMed] [Google Scholar]
- Stevens P., Monastyrska I., Leao-Helder A. N., van der Klei I. J., Veenhuis M., Kiel J. A. Hansenula polymorpha Vam7p is required for macropexophagy. FEMS Yeast Res. 2005;5:985–997. doi: 10.1016/j.femsyr.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Strømhaug P. E., Bevan A., Dunn W. A., Jr GSA11 encodes a unique 208-kDa protein required for pexophagy and autophagy in Pichia pastoris. J. Biol. Chem. 2001;276:42422–42435. doi: 10.1074/jbc.M104087200. [DOI] [PubMed] [Google Scholar]
- Strømhaug P. E., Reggiori F., Guan J., Wang C. W., Klionsky D. J. Atg21 is a phosphoinositide binding protein required for efficient lipidation and localization of Atg8 during uptake of aminopeptidase I by selective autophagy. Mol. Biol. Cell. 2004;15:3553–3566. doi: 10.1091/mbc.E04-02-0147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F., Madden D. T., Hamamoto S., Orci L., Schekman R. Sec16p potentiates the action of COPII proteins to bud transport vesicles. J. Cell Biol. 2002;158:1029–1038. doi: 10.1083/jcb.200207053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teter S. A., Eggerton K. P., Scott S. V., Kim J., Fischer A. M., Klionsky D. J. Degradation of lipid vesicles in the yeast vacuole requires function of Cvt17, a putative lipase. J. Biol. Chem. 2001;276:2083–2087. doi: 10.1074/jbc.C000739200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuttle D. L., Dunn W. A., Jr Divergent modes of autophagy in the methylotrophic yeast Pichia pastoris. J. Cell Sci. 1995;108:25–35. doi: 10.1242/jcs.108.1.25. [DOI] [PubMed] [Google Scholar]
- Ueno T., Muno D., Kominami E. Membrane markers of endoplasmic reticulum preserved in autophagic vacuolar membranes isolated from leupeptin-administered rat liver. J. Biol. Chem. 1991;266:18995–18999. [PubMed] [Google Scholar]
- Ward T. H., Polishchuk R. S., Caplan S., Hirschberg K., Lippincott-Schwartz J. Maintenance of Golgi structure and function depends on the integrity of ER export. J. Cell Biol. 2001;155:557–570. doi: 10.1083/jcb.200107045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita S., Oku M., Sakai Y. Functions of PI4P and sterol glucoside are necessary for the synthesis of a nascent membrane structure during pexophagy. Autophagy. 2007;3:35–37. doi: 10.4161/auto.3311. [DOI] [PubMed] [Google Scholar]
- Yamashita S., Oku M., Wasada Y., Ano Y., Sakai Y. PI4P-signaling pathway for the synthesis of a nascent membrane structure in selective autophagy. J. Cell Biol. 2006;173:709–717. doi: 10.1083/jcb.200512142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorimitsu T., Klionsky D. J. Autophagy: molecular machinery for self-eating. Cell Death Differ. 2005;12(suppl 2):1542–1552. doi: 10.1038/sj.cdd.4401765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura S., Yamamoto A., Misumi Y., Sohda M., Barr F. A., Fujii G., Shakoori A., Ohno H., Mihara K., Nakamura N. Dynamics of Golgi matrix proteins after the blockage of ER to Golgi transport. J. Biochem. 2004;135:201–216. doi: 10.1093/jb/mvh024. [DOI] [PubMed] [Google Scholar]
- Yuan W., Strømhaug P. E., Dunn W. A., Jr Glucose-induced autophagy of peroxisomes in Pichia pastoris requires a unique E1-like protein. Mol. Biol. Cell. 1999;10:1353–1366. doi: 10.1091/mbc.10.5.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W., Tuttle D. L., Shi Y. J., Ralph G. S., Dunn W. A., Jr Glucose-induced microautophagy in Pichia pastoris requires the alpha-subunit of phosphofructokinase. J. Cell Sci. 1997;110:1935–1945. doi: 10.1242/jcs.110.16.1935. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.