

Abstract
Saccharomyces cerevisiae cells growing in the outdoor environment must adapt to sudden changes in temperature and other variables. Many such changes trigger stress responses that delay bud emergence until the cells can adapt. In such circumstances, the morphogenesis checkpoint delays mitosis until a bud has been formed. Mitotic delay is due to the Wee1 family mitotic inhibitor Swe1p, whose degradation is linked to bud emergence by the checkpoint kinase Hsl1p. Hsl1p is concentrated at the mother-bud neck through association with septin filaments, and it was reported that Hsl1p activation involved relief of autoinhibition in response to septin interaction. Here we challenge the previous identification of an autoinhibitory domain and show instead that Hsl1p activation involves the phosphorylation of threonine 273, promoted by the septin-associated kinase Elm1p. We identified elm1 mutants in a screen for defects in Swe1p degradation and show that a phosphomimic T273E mutation in HSL1 bypasses the need for Elm1p in this pathway.
INTRODUCTION
The G2/M transition in the eukaryotic cell cycle is regulated by several checkpoint pathways that assess the cell's readiness to proceed through mitosis. Under adverse conditions, mitosis is delayed by inhibitory tyrosine phosphorylation of Cdk1 catalyzed by Wee1 family kinases. Near the G2/M transition, Wee1 is degraded by a pathway that includes sequential Wee1 phosphorylation by Cdk1 and a Polo family kinase (Asano et al., 2005; Watanabe et al., 2005). This degradation pathway is blocked by the DNA replication checkpoint in vertebrate cells (Michael and Newport, 1998; Yamada et al., 2004) and by the morphogenesis checkpoint in yeast (Keaton and Lew, 2006).
Yeast cells growing in the wild are subject to frequent changes in their environment due to the weather and other organisms. Many environmental changes trigger stress responses that include actin cytoskeleton depolarization, temporarily halting bud growth. In these circumstances, the morphogenesis checkpoint delays mitosis until cells have successfully constructed a bud (Lew, 2003). The mitotic delay requires at least two pathways: one that inhibits the Cdc25-family phosphatase Mih1p (Harrison et al., 2001) and one that blocks degradation of the Wee1 family kinase Swe1p (Sia et al., 1998).
Degradation of Swe1p has been studied in some detail, and requires the assembly of a cytoskeletal structure composed of septin filaments at the mother-bud neck (Keaton and Lew, 2006; Weirich et al., 2008). The checkpoint kinase Hsl1p is localized to this structure (Barral et al., 1999; Longtine et al., 2000) and recruits the conserved Swe1p-interacting protein Hsl7p to that site (Shulewitz et al., 1999; Longtine et al., 2000). Hsl7p in turn tethers Swe1p to the mother-bud neck (Longtine et al., 2000), where it is phosphorylated by the Polo family kinase Cdc5p (Sakchaisri et al., 2004). The mechanism whereby Cdc5p-phosphorylated Swe1p is targeted for degradation remains unknown.
Regulation of Swe1p degradation by stresses could occur at many points in the pathway described above, but recent work has focused attention on Hsl1p. Hsl1p kinase activity is required for effective Hsl7p recruitment and Swe1p degradation, and several checkpoint-inducing conditions (such as delaying bud emergence or disrupting the septin structure) appear to inactivate Hsl1p (Barral et al., 1999; Theesfeld et al., 2003). Moreover, osmotic shock (which also triggers Swe1p stabilization; Sia et al., 1998) is thought to act through phosphorylation of Hsl1p by the osmostress mitogen-activated kinase Hog1p (Clotet et al., 2006).
Recent work suggested a mechanistic basis for Hsl1p regulation by septins. A domain (residues 987-1100) located in the large C-terminal noncatalytic region was found to bind both to the Hsl1p kinase domain (residues 1–450) and to septins (Hanrahan and Snyder, 2003). It was proposed that septin binding relieved autoinhibition by this domain, unleashing Hsl1p kinase activity (Hanrahan and Snyder, 2003). Although attractive, this model does not explain why only an assembled septin structure, and not free septin complexes, can activate Hsl1p. Moreover, even after a ring of septins forms at the cell cortex and Hsl1p is localized to that ring, Hsl1p remains inactive if bud formation is delayed (Theesfeld et al., 2003). Thus, important aspects of Hsl1p regulation remain to be elucidated.
In this article, we report the results of a genetic screen to identify factors important for Swe1p degradation. We identified the kinase Elm1p, which (like Hsl1p) is localized to the mother-bud neck. Our results suggest that Elm1p phosphorylates Hsl1p at Thr273, activating Hsl1p to promote Swe1p degradation.
MATERIALS AND METHODS
Yeast Strains and Plasmids
All yeast strains used in this study (Table 1) are in the BF264–15DU background (ade1, his2, leu2–3112, trp1–1a, and ura3Δns; Richardson et al., 1989). The following disruptions and alleles are described in the cited publications: cla4::TRP1 (Benton et al., 1997); gin4-Δ9 (Longtine et al., 1998a); hsl1::URA3 (Ma et al., 1996); mih1::LEU2 (Russell et al., 1989); bed1::URA3 (Mondesert and Reed, 1996); nap1::kanr, HSL7-HA:kanr, and cdc12-6::LEU2 (Longtine et al., 2000); HSL1–13myc::URA3 (McMillan et al., 1999); hsl1K110R-13myc (Theesfeld et al., 2003); and CDC3-GFP (Caviston et al., 2003). elm1::LEU2 and mih1::TRP1 disruptions were generated by the one-step PCR-based method (Baudin et al., 1993) using pRS305 and pRS304 (Sikorski and Hieter, 1989) as template.
Table 1.
Yeast strains used in this study
| Strain | Relevant genotype | Source |
|---|---|---|
| DLY1 | abar1 | Richardson et al. (1989) |
| DLY4410 | α gin4::kanR | Gladfelter et al. (2003) |
| DLY5000 | abar1 HSL1–13myc:URA3 HSL7-HA:kanR | Theesfeld et al. (2003) |
| DLY5336 | abar1 HSL1–13myc:URA3 cdc12–6:LEU2 HSL7-HA:kanR | This study |
| DLY5390 | abar1 hsl1K110R-13myc:URA3 HSL7-HA:kanR | Theesfeld et al. (2003) |
| DLY5844 | ahsl1::URA3 HSL7-HA:kanR | This study |
| DLY6940 | α ade1 ade3 mih1::LEU2+ pDLB2064 (MIH1 ADE3) | This study |
| DLY7395a | a3xHSL1T273E-13myc:TRP1 cdc12–6:LEU2 hsl1::URA3 HSL7-HA:kanR | This study |
| DLY7400 | aHSL1–13myc::TRP hsl1::URA3 HSL7-HA:kanRcdc12–6:LEU2 | This study |
| DLY7451 | aHSL1T273E-13myc::TRP1 hsl1::URA3 HSL7-HA:kanR | This study |
| DLY7601 | a/α elm1::URA3/ELM1, gin4::kanR/GIN4, cla4::TRP1/CLA4 mih1::LEU2/MIH1 | This study |
| DLY7697 | a/α mih1::TRP1/MIH1 elm1::LEU2/ELM1 | This study |
| DLY7704 | α elm1::LEU2 | This study |
| DLY7841 | α hsl1T273A-13myc:TRP1 hsl1::URA3 HSL7-HA:kanR | This study |
| DLY7980 | ahsl1T273A-13myc:TRP1 hsl1::URA3 mih1::LEU2 HSL7-HA:kanR | This study |
| DLY8113 | aHSL1–13myc:TRP1 hsl1::URA3 HSL7-HA:kanR | This study |
| DLY8116 | ahsl1D239A-13myc:TRP1 hsl1::URA3 HSL7-HA:kanR | This study |
| DLY8117 | ahsl1K110R-13myc:TRP1 hsl1::URA3 HSL7-HA:kanR | This study |
| DLY8119 | ahsl1N244A-13myc:TRP1 hsl1::URA3 HSL7-HA:kanR | This study |
| DLY8684 | aHSL1Δ987–1100-13myc::TRP1 hsl1::URA3 HSL7-HA:kanR | This study |
| DLY8709 | aHSL1Δ987–1100-13myc::TRP1 cdc12–6:LEU2 hsl1::URA3 HSL7-HA:kanR | This study |
| DLY8745 | aHSL1–13myc:TRP1 cdc12–6:LEU2 hsl1::URA3 HSL7-HA:kanR | This study |
| DLY9391a | a3xHSL1T273E-13myc::TRP1 hsl1::URA3 HSL7-HA:kanR | This study |
| DLY9406 | ahsl1T273A-13myc::TRP1 elm1::LEU2 mih1::LEU2+ pDLB2729 (ELM1) | This study |
| DLY9407 | aHSL1T273E-13myc::TRP1 elm1::LEU2 mih1::LEU2+ pDLB2729 (ELM1) | This study |
| DLY9409 | aHSL1–13myc:TRP1 elm1::LEU2 mih1::LEU2+ pDLB2729 (ELM1) | This study |
| DLY9410 | aelm1::LEU2 mih1::LEU2+ pDLB2729 (ELM1)+ pDLB2510 (HSL1M177V-13myc) | This study |
| DLY9543 | ahsl1::LEU2:GAL1-GST-HSL1987–1518-13myc:TRP1 hsl1::URA3 | This study |
| DLY9583 | ahsl1::LEU2:GAL1-GST-HSL1987–1518:URA3 HSL1–13myc:TRP1 | This study |
| DLY9587 | aGAL1-GST:TRP1 | This study |
| DLY9691 | aHSL1Δ987–1100-13myc:TRP1 elm1::LEU2 mih1::LEU2+ pDLB2729 (ELM1) | This study |
| DLY9803 | ahsl1K110R-13myc:TRP1 hsl1::URA3 elm1::LEU2 | This study |
| DLY9804 | aHSL1T273E-13myc:TRP1 hsl1::URA3 elm1::LEU2 | This study |
| DLY9806 | aHSL1–13myc:TRP1 hsl1::URA3 elm1::LEU2 | This study |
| DLY9820 | ahsl1T273A-13myc:TRP1 hsl1::URA3 elm1::LEU2 | This study |
| DLY9863 | aGAL1-GST-HSL1987–1100:TRP1 | This study |
| DLY9882 | abar1 hsl1::URA3 mih1::LEU2 HSL1–13myc:TRP1 | This study |
| DLY9883 | abar1 hsl1::URA3 mih1::LEU2 swe1::LEU2 HSL1–13myc:TRP1 | This study |
| DLY10072 | aHSL1T273E-13myc:TRP1 hsl1::URA3 HSL7-HA:kanR | This study |
| DLY10076 | ahsl1T273A-13myc:TRP1 gin4::kanRhsl1::URA3 | |
| DLY10077 | aHSL1T273E-13myc:TRP1 hsl1::URA3 elm1::LEU2 | This study |
| DLY10097 | aCDC3-GFP:URA3 GAL1-GST:TRP1 | This study |
| DLY10098 | aCDC3-GFP:URA3 GAL1-GST-HSL1987–1100:TRP1 | This study |
| DLY11002 | aCDC3-GFP:URA3 hsl1::LEU2:GAL1-GST-HSL1987–1518-13myc:TRP1 | This study |
| DLY11005 | α HSL1T273E-13myc:TRP hsl1::URA3 HSL7-HA:kanRmih1::LEU2 | This study |
| DLY11006 | α HSL1T273E-13myc:TRP hsl1::URA3 HSL7-HA:kanRmih1::LEU2 swe1::LEU2 | This study |
| DLY11007 | ahsl1T273A-13myc:TRP hsl1:URA3 HSL7-HA:kanRmih1::LEU2 swe1::LEU2 | This study |
| DLY11008 | aHSL1T273E-13myc:TRP1 hsl1::URA3 gin4::kanR | This study |
| JMY3–58 | α hsl1::URA3 | This study |
a These strains derive from a transformant that expressed more Hsl1p (threefold by quantitative Western blotting) than wild-type or other mutant transformants, suggesting that three copies of the plasmid had integrated at TRP1.
To generate the MIH1 ADE3 plasmid for the sectoring screen, we first assembled full-length MIH1 (from 550 base pairs upstream to 140 base pairs downstream of the ORF) using a PCR-amplified promoter and N-terminal sequences joined at the internal SalI site with C-terminal coding and downstream sequences from a MIH1 library plasmid (Liu et al., 1992), cloned between the EcoRI (5′) and NotI (3′) sites of pRS316 (Sikorski and Hieter, 1989). This assembly introduced a XhoI site (encoding Leu Glu) immediately downstream of the initiator methionine. A 5.2-kb BamHI-SalI fragment containing the ADE3 gene was then excised from pSW198 (Murphy et al., 1996), blunted, and cloned into the blunted NotI site, generating pDLB2064 containing tandem MIH1 and ADE3 genes in the same direction.
An integrating TRP1 plasmid containing full-length Hsl1p-13myc expressed from the HSL1 promoter (587 base pairs upstream of the ORF) and followed by ADH1 terminator sequences from the pFA6 plasmids (Longtine et al., 1998b), with the sequence CGGATCCCCGGGTTAATTAAC (BamHI and PacI sites underlined) encoding Arg Ile ProGly Leu Ile Asn between the Hsl1p and 13myc coding sequences, was assembled by cloning and gap repair from fragments of pNE30 (Edgington et al., 1999) and HSL1–13myc::kanr (Longtine et al., 2000). The resulting plasmid, pDLB2548, contains those sequences inserted between the PstI and SacI sites in pRS304 (Sikorski and Hieter, 1989), with all intervening polylinker sites absent. Digestion at the unique BstXI site was used to target integration at TRP1.
The Hsl1p T273A and T273E mutants change the T273 ACT codon to TCT (T273A) or GAG (T273E: this mutant also has a silent substitution for diagnostic purposes changing codon L271 from TTA to TTG to destroy a DraI site). The Hsl1p K110R mutant was previously described (Theesfeld et al., 2003). The Hsl1p N244A mutant changes the N244 AAT codon to GCT. The Hsl1p D239A mutant changes the D239 GAT codon to GCA. The Hsl1p Δ987–1100 mutant replaces the coding sequences for residues 987–1100 with the sequence CGGATCTGG, encoding Arg Ile Trp. All mutations were introduced into pDLB2548, confirmed by sequencing, and integrated as described above.
To express glutathione S-transferase (GST)-Hsl7p in E. coli, the HSL7 ORF was cloned into the EcoRI site of pGEX-KG (GE Healthcare Life Sciences, Piscataway, NJ), generating pDLB2211.
To express GST-fusions with pieces of HSL1, we first introduced the GAL1 promoter (EcoRI-BamHI fragment) into YIplac204 and YIplac128 (Gietz and Sugino, 1988) and used linkers to change the remaining polylinker to AAAATGGTCGACACCATGGCGCATATGGAGCTC (start codon bold, SacI and SacI sites underlined), yielding pDLB1473 (TRP1) and pDLB1474 (LEU2). Then GST was cloned in from pGEX as a SalI-SacI fragment, yielding pDLB2492 (TRP1) and pDLB2493 (LEU2). Then a PCR product encoding HSL1987–1100 flanked by BamHI and SacI sites was cloned in, fusing GST to HSL1987–1100 via the sequence CTGGTTCCGCGTGGATCC (BamHI underlined) encoding Leu ProLeu Arg Gly Ser (from pGEX). This yielded integrating LEU2 (pDLB2502) and TRP1 (pDLB2506) GAL1-GST-HSL1987–1100 plasmids. Integration was targeted to the marker gene by digestion at the unique BstXI (TRP1) or BstEII (LEU2) sites to express GST-Hsl1p987–1100. To express GST-Hsl1p987–1518, the same plasmids were targeted to integrate at HSL1 by digestion at the unique BsaBI site. Integration in this manner results in a truncated HSL1 (residues 1–1100 followed by a stop codon) followed by GAL1-GST-HSL1987–1518.
Media, Growth, and Cell Cycle Synchrony Procedures
Yeast strains were grown in YEPD (1% yeast extract 2% Bacto Peptone, 2% dextrose, 0.012% adenine, and 0.01% uracil) or in synthetic dextrose complete media lacking relevant amino acids at 30°C unless otherwise noted. Bacterial strains were grown in Difco LB broth (BD Biosciences, San Jose, CA) supplemented with 50 μg/ml ampicillin at 37°C, with the exception of those bacteria used to produce recombinant proteins, which were grown at 18°C.
For galactose inductions, yeast were grown at 24°C in YEPS (as YEPD but with 2% sucrose and 0.05% dextrose), galactose was added to a final concentration of 2%, and cells were collected 20 h later.
For cell cycle synchrony analyses, cells were grown to exponential phase (∼5 × 106 cells/ml) and arrested in G1 by incubation with 40 ng/ml α-factor for 3 h at 30°C. Cells were released from the arrest by harvesting and resuspension in fresh YEPD.
Screen for mih1Δ Synthetic Lethal Mutants
mih1Δ ade1 ade3 cells (DLY6940) harboring the CEN-MIH1-ADE3-URA3 plasmid pDLB2064 were grown in media lacking uracil to a density of 2 × 107 cells/ml, diluted in water, and plated on YEPD at 5000 cells/plate. Plates were subjected to UV irradiation for 31 s (90% kill) and then allowed to grow in the dark for 4 d at 30°C. Nonsectoring (n = 350; all red) colonies were picked and checked by restreaking, and 20 confirmed mutants were frozen.
To identify the mutant lesion, mutants were transformed with a TRP1-marked genomic library (see below), grown for 4 d at 30°C, and replica plated onto −Trp plates containing 5-fluoro-orotic Acid (5-FOA; Toronto Research Chemicals, Toronto, ON, Canada), which kills cells containing the original URA3 plasmid. Viable colonies should therefore contain library plasmids with either MIH1 or the genes that gave rise to synthetic lethality with mih1Δ. Plasmids were retransformed into the mutant strains to test complementation of the nonsectoring and 5-FOA–sensitive phenotypes, and the inserts in non-MIH1 complementing plasmids were sequenced. ELM1 and NDD1 plasmids complemented not only the lethality but also the morphology defects of their respective mutants.
To generate the genomic library, 100 μg of genomic DNA from DLY1 was partially digested with 30 U of Sau3A (New England Biolabs, Beverly, MA) for 4 min and separated on a 1% agarose gel, and 6–10-kb fragments were purified and ligated into the BamHI site of YCplac22 (Gietz and Sugino, 1988). The ligation reaction was transformed into ElectroMAX DH10B electrocompetent cells (Invitrogen, Carlsbad, CA) and 20 transformations were plated on separate 140-mm Petri plates. After overnight growth, each plate's transformants were inoculated into 200 ml of media and grown for 2 h at 37°C. Plasmid DNA was extracted using Rapid Plasmid Maxiprep System (Marligen, Ijamsville, MD). Ten independent transformants had inserts >4 kb.
Microscopy and Immunofluorescence
For analysis of cell morphology and DNA staining, cells were fixed in 70% ethanol for >1 h, resuspended in phosphate-buffered saline (PBS) containing 0.2 μg/ml 4–6-diamidino-2-phenylindole (Sigma, St. Louis, MO), and sonicated. The cells were then resuspended in a drop of mounting medium (90% glycerol in PBS 9.2 mM p-phenylenediamine; Sigma), and stored at −80°C.
For immunofluorescence analysis, cells were fixed in 4.1% formaldehyde for 1 h (to visualize Hsl7p-HA) or 1.5 h (to visualize Hsl1p-13myc) at room temperature, then washed in solution B (0.1 M KPO4, pH 7.5, 1.2 M sorbitol), and treated with 10 μl lyticase (Sigma) and 143 mM β-mercaptoethanol (Sigma) for 10–15 min to digest the cell wall. Spheroplasts were washed twice in PBS and applied to precleaned 10-well microscope slides (Polysciences, Warrington, PA) coated with 0.1% polyethylenimine (Sigma). After they settled, the wells were washed once with TBS containing 5% goat serum (Invitrogen) and 0.1% Tween-20 (Bio-Rad, Richmond, CA), and incubated with 1:200 mouse anti-myc 9E10 (Santa Cruz Biotechnology, Santa Cruz, CA), 1:200 mouse anti-HA 12CA5 (Roche), or rabbit anti-Cdc11 sc7170 (Santa Cruz Biotechnology) for 1 h. The wells were washed 20 times and incubated with 1:200 goat anti-mouse Alexafluor 488 or goat anti-rabbit Alexafluor 568 (Molecular Probes, Eugene, OR) for another hour. After another 20 washes, 2 μl of mounting media was added to each well, and a coverslip was gently deposited.
For DIC imaging, cells were fixed in 70% ethanol, washed with PBS, and resuspended in mounting medium. For imaging Cdc3p-GFP, cells were fixed in 2% paraformaldehyde (Sigma) for 10 min at room temperature, washed once with solution B and once with PBS, and resuspended in mounting medium.
Cells were examined on a Zeiss Axioimager (Thornwood, NY) and photographed with a Hamamatsu Orca ER monochrome cooled-CCD camera (Bridgewater, NJ). Images were viewed using MetaMorph software (Universal Imaging, West Chester, PA).
Biochemical Procedures
Yeast cell lysis was performed by the TCA method (Keaton 2008) for Western blotting or the NP40 method (Bose et al., 2001) for immunoprecipitation. Samples were subjected to PAGE using 6% low-bis polyacrylamide (Barral et al., 1999) to better resolve Hsl1p-13myc for Western blotting or 8% polyacrylamide gels in all other cases.
For Western blotting, proteins were transferred to nitrocellulose membranes (Pall Life Sciences, Ann Arbor, MI), blocked with PBS containing 0.1% Tween 20 (PBST) and 3% nonfat dry milk for 1 h, and then probed with 1:1000 mouse anti-myc 9E10 (Santa Cruz Biotechnology), 1:1000 mouse anti-HA 12CA5 (Roche), 1:1000 mouse anti-GST (Santa Cruz Biotechnology), or 1:25,000 rabbit anti-Cdc11 (Santa Cruz Biotechnology) for 1 h, and washed five times in PBST. Blots were then probed with 1:7500 goat anti-mouse IRdye800 (Rockland Biosciences, Gilbertsville, PA) or anti-rabbit AlexaFluor680 (Molecular Probes) for 1 h in TBST with 3% milk and 0.01% SDS, washed as above, and visualized using an Odyssey scanner (Li-Cor Biosciences, Lincoln, NE).
For Cdc28p and phospho-Cdc28p detection, membranes were blocked in a 1:1 solution containing Odyssey Blocking Buffer (Li-Cor Biosciences) and TBS. The blots were probed with 1:10,000 mouse anti-PSTAIRE (for Cdc28p) and 1:2000 rabbit anti-phospho-tyr15-Cdc2 (for phospho-Cdc28p, Cell Signaling Technology, Beverly, MA), which were diluted in blocking buffer with 0.1% Tween-20 and incubated overnight. The blots were then washed five times in TBST. The same secondary antibodies described above were diluted to the same concentration in blocking buffer with 0.1% Tween-20 and 0.01% SD, incubated for 1 h, and then washed and visualized as above.
For immunoprecipitation, 2 mg of total protein was diluted to 300 μl in NP40 lysis buffer and mixed with 100 μl of a 1:5 suspension of anti-c-Myc-agarose affinity gel beads (Sigma) prewashed three times with NP40 lysis buffer. After 2 h, beads were precipitated, washed as above, and either prepared for PAGE (2/3 of beads) or used for kinase assays (1/3 of beads).
For kinase assays, beads were washed two more times with kinase buffer (10 mM Tris-HCl, pH 7.5, 25 mM MgCl2) and resuspended in 10 μl of kinase buffer containing GST-Hsl7p. Kinase reactions were started by mixing in 10 μl of kinase buffer with 10 μM ATP and 10 μCi of γ-32P-ATP (3000 Ci/mmol; Perkin Elmer-Cetus Life Sciences, NEN EasyTides, Boston, MA), incubated at 30°C for 30 min, and stopped with 20 μl of 95°C 2× sample buffer. After PAGE, the radioactive gel was boiled in 5% wt/vol TCA for 15 min, dried for 2 h, developed for 24 h with a Phosphor Screen (Molecular Dynamics, Sunnyvale, CA), scanned using a Storm840 phosphoimager (Amersham Biosciences, Piscataway, NJ), and analyzed using Storm scanning and quantitation software.
To express recombinant GST-Hsl7p, 1L DH10B cells harboring pDLB2211 were grown at 37°C to OD 0.8 and induced with 1 mM IPTG overnight at 18°C. Cells were collected using an Avanti J-25I centrifuge (Beckman Instruments, Fullerton, CA) at 6000 rpm for 30 min at 4°C, washed with 20 ml 0.9% NaCl, and frozen at −80°C. Buffer A (2.5 ml; 2.3 M sucrose, 50 mM Tris-HCl, pH 7.5, and 1 mM EDTA) and 25 μl of 100 mM PMSF were added to the frozen cell pellet. As the cells began to thaw, 10 ml of buffer B (50 mM Tris-HCl, pH 7.5, 10 mM KCl, 1 mM EDTA, and 1 mM DTT), and another 100 μl more of 100 mM PMSF was added together with 4 mg lysozyme. This mixture was kept on ice for 1 h with frequent mixing using a rubber policeman to break up the frozen bacterial pellet. One hundred seventy-five microliters of 10% sodium deoxycholate, 263 μl of 1 M MgCl2, and 25 μl of 5 mg/ml DNaseI (Worthington Biochemical, Lakewood, NJ) were mixed in and allowed to sit on ice for 15 min. The lysate was clarified by centrifugation 12000 rpm for 30 min at 4°C. Glutathione Sepharose (Amersham Biosciences) beads were prewashed two times in 10 ml of buffer C (10 mM HEPES, pH 8.0, 1 mM DTT), added to the lysate, and mixed gently overnight at 4°C. Beads were washed three times in buffer C with 300 mM NaCl and placed in a 20-ml column for elution with 2.5 ml of 5 mM glutathione, pH 8 (Sigma), and mixed gently at 4°C for 1 h. Five 0.5-ml fractions were collected and GST-Hsl7p levels were determined by PAGE and Coomassie blue staining. Fractions with pure GST-Hsl7p were pooled and dialyzed against kinase reaction buffer (10 mM Tris-HCl, pH 7.5, and 25 mM MgCl2).
RESULTS
Screen for Regulators of Swe1p Degradation Identifies Elm1p
Previous work indicated that mutations (e.g., hsl1Δ or hsl7Δ) that inactivate the Swe1p degradation pathway are well tolerated by unstressed cells, because the Mih1p phosphatase remains able to counteract the stabilized Swe1p. However, in the absence of Mih1p, Swe1p stabilization leads to a lethal G2 arrest (McMillan et al., 1999). To identify other genes involved in Swe1p degradation, we conducted a screen for mutants that are viable in the presence of Mih1p, but inviable in its absence (Figure 1). In addition to multiple isolates with mutations in HSL1 and HSL7, the screen produced five isolates with mutations in the ELM1 gene and two with mutations in the NDD1 gene. Ndd1p is a transcription factor known to stimulate both mitotic cyclin and Cdc5p expression (Zhu et al., 2000; Darieva et al., 2003; Reynolds et al., 2003). Elm1p, a protein kinase localized at the mother-bud neck (Bouquin et al., 2000), is the subject of this report. Sequencing of one of the elm1 mutants revealed a frameshift mutation within the kinase domain (altering the sequence after F313 to FCTRAMSFGEFEYRFCDGStop), suggesting that it was a null allele, and we confirmed that elm1Δ mih1Δ mutants were synthetically lethal (Table 2).
Figure 1.

Screen for regulators of Swe1p degradation identifies Elm1p. (A) In the starting strain for the screen (DLY 6940), the only copy of MIH1 is carried on a plasmid that also confers a red colony color by virtue of the ADE3 gene (pDLB2064). Loss of the plasmid during colony growth yields sectored red and white colonies (left: white sectors are mih1Δ). However, if the strain acquires a mutation disabling Swe1p degradation (mutX), the mih1Δ cells will die, so white sectors will not develop and the colony will be uniformly red (right: illustrated for an hsl1 isolate). (B) Results of the screen.
Table 2.
elm1 mutants are unique among septin-perturbing mutations in their synthetic lethality with mih1
| Genotype | Viable | Inviable |
|---|---|---|
| elm1Δ | 30 | 1 |
| mih1 | 48 | 0 |
| gin4Δ | 14 | 0 |
| cla4Δ | 13 | 0 |
| nap1Δ | 13 | 0 |
| mih1Δ elm1Δ | 0 | 31 |
| mih1Δ gin4Δ | 18 | 0 |
| mih1Δ cla4Δ | 13 | 0 |
| mih1Δ nap1Δ | 7 | 0 |
| mih1Δ gin4 cla4Δ | 7 | 0 |
| elm1Δ gin4Δ | 9 | 0 |
| elm1Δ cla4Δ | 13 | 0 |
| elm1Δ gin4Δ cla4Δ | 21 | 0 |
Crosses (diploids DLY7601 and DLY7697) were sporulated and tetrads dissected. Spore colonies were scored for the markers associated with each mutation, and the number of viable or inviable spore colonies recovered for each genotype is shown.
Elm1p was first discovered by Alan Myers and colleagues in a genetic screen for mutations conferring an elongated cell morphology (Blacketer et al., 1993). In a subsequent screen, they identified a dominant mutation in HSL1 as a suppressor of the elm1 phenotype, suggesting that Elm1p acts upstream of Hsl1p (Edgington et al., 1999; Thomas et al., 2003). Further work indicated that Elm1p is important for assembling a properly organized septin structure at the mother-bud neck (septins are mislocalized to the bud tip in elm1Δ mutants), and a recent study indicated that it does so (at least in part) by phosphorylating and activating the septin-organizing kinase Gin4p (Bouquin et al., 2000; Asano et al., 2006). Thus, a possible explanation for both the Myers lab genetics and the identification of elm1 mutants in our screen is that by perturbing septin organization, elm1 mutants indirectly reduce Hsl1p activity and stabilize Swe1p.
Although plausible, the above hypothesis does not explain why we did not isolate any mutations in other septin-organizing genes in our screen. To ask whether such mutations would (like elm1 mutants) be synthetically lethal in combination with deletion of MIH1, we performed directed crosses between mih1Δ strains and strains bearing mutations in the septin-organizing genes GIN4, CLA4, and NAP1 (Longtine et al., 1998a, 2000). As shown in Table 2, only elm1Δ mih1Δ mutants were synthetic lethal: even gin4Δ cla4Δ mih1Δ triple mutants were recovered at the expected frequencies in our crosses. Because gin4Δ cla4Δ mutants display much greater septin disorganization than do elm1Δ mutants (Gladfelter et al., 2004), these findings suggest that the mih1Δ synthetic lethality is not simply a consequence of septin defects, but rather indicates a more specific role for Elm1p in down-regulating Swe1p.
Elm1p-dependent Hsl1p Phosphorylation
In addition to its role in septin organization, Elm1p is one of three upstream kinases for the yeast AMPK homolog Snf1p (Sutherland et al., 2003). Specifically, Elm1p phosphorylates Snf1p at T210 in the kinase activation loop. We noticed that the Snf1p and Hsl1p activation loops are very similar in sequence (see Figure 3A: Hsl1p T273 occupies a position analogous to Snf1 T210), suggesting that Elm1p might similarly phosphorylate Hsl1p. However, we were unsuccessful in demonstrating phosphorylation of kinase-dead Hsl1p by Elm1p in vitro, similar to findings in a recent report from another lab (Asano et al., 2006). Although this negative result may indicate that Elm1p does not phosphorylate Hsl1p, it was also possible that our in vitro conditions lacked an important factor (e.g., the septin collar) and that Elm1p does phosphorylate Hsl1p in vivo.
Figure 3.

Phosphorylation of Hsl1p T273 is important for Hsl1p function. (A) Alignment of Snf1p and Hsl1p activation loop sequences. (B) Hsl1p, Hsl1pT273A, and Hsl1pT273E are expressed at similar levels in vivo. Untagged (DLY1), HSL1-myc HSL7-HA (DLY8113), hsl1T273Amyc HSL7-HA (DLY7841), and HSL1T273Emyc HSL7-HA (DLY10072) cells were grown to exponential phase at 30°C, lysed by TCA precipitation, and Hsl1p-myc, and Hsl7p-HA were detected by Western blotting. (C) Hsl1pT273A is less biologically active than Hsl1p or Hsl1pT273E. HSL1-myc mih1Δ (DLY9882), HSL1T273Emyc mih1Δ (DLY 11005), hsl1T273Amyc mih1Δ (DLY7980), HSL1-myc mih1Δ swe1Δ (DLY9883), HSL1T273Emyc mih1Δ swe1Δ (DLY 11006), and hsl1T273Amyc mih1Δ swe1Δ (DLY11007) cells were grown to exponential phase at 30°C and examined by DIC microscopy. (D) Cell cycle profiles of the cells from C were determined by scoring the budding and nuclear division status of DAPI-stained cells (n = 250). (E) Hsl1pT273A is localized to the bud neck, but does not effectively recruit Hsl7p. HSL1-myc HSL7-HA (DLY8113), hsl1T273Amyc HSL7-HA (DLY7841), HSL1T273Emyc HSL7-HA (DLY7451), and hsl1K110Rmyc HSL7-HA (DLY8117) cells were grown to exponential phase at 30°C and processed for immunofluorescence to visualize the septin Cdc11p together with either Hsl1p-myc (top pairs) or Hsl7p-HA (bottom pairs). Representative cells are shown (left) together with the results of scoring 250 cells with a septin collar for the presence of detectable Hsl1p or Hsl7p (right).
Previous studies showed that Hsl1p undergoes a heterogeneous mobility shift upon analysis by SDS-PAGE that can be collapsed to a tight band by phosphatase treatment (Barral et al., 1999; McMillan et al., 1999). The shift was thought to occur exclusively through autophosphorylation, because kinase-dead Hsl1p did not undergo a shift in the previously used lysis and gel conditions. However, using a TCA precipitation method to better preserve phosphorylation, we found that even kinase-dead Hsl1pK110R undergoes a partial mobility shift, suggesting that Hsl1p can be phosphorylated by upstream kinases (Figure 2A). Because preservation of the full shift-inducing modification required denaturing extraction, we were unable to confirm that the additional shift is phosphatase-collapsible, though it seems very likely to be due to phosphorylation. Similar partial shifts were observed with three distinct kinase-dead alleles that showed only background in vitro autophosphorylating activity (Figure 2, B and C), indicating that the shift is not due to residual Hsl1p activity. The shift was greatly reduced when Hsl1p was expressed in cells lacking organized septins entirely (cdc12-6 cells shifted to 37°C for 1 h; Figure 2A), indicating that phosphorylation by upstream kinases is septin-dependent. Neither Hsl1p nor Hsl1pK110R exhibited the shift when expressed in elm1Δ mutants (Figure 2D). In cells synchronized by pheromone arrest and release, both wild-type and kinase-dead Hsl1p exhibited mobility shifts at the time of bud emergence (Figure 2E). These results suggest that Hsl1p undergoes an initial Elm1p- and septin-dependent phosphorylation at the time of bud emergence, followed rapidly by more extensive autophosphorylation, consistent with the hypothesis that the initial trans-phosphorylation stimulates Hsl1p activity.
Figure 2.

Hsl1p is regulated by upstream kinases. (A) Catalytically inactive Hsl1p undergoes a partial mobility shift. HSL1-myc HSL7-HA (DLY5000), hsl1K110Rmyc HSL7-HA (DLY5390), and HSL1-myc HSL7-HA cdc12-6 (DLY5336) cells were grown at 24°C to exponential phase and shifted to 37°C for 1 h. Cells were lysed by TCA precipitation and Hsl1p-myc and Hsl7p-HA were detected by Western blotting. (B) Three different mutations in the kinase domain reduce Hsl1p autophosphorylation in vitro. HSL1-myc (DLY8113), hsl1K110Rmyc (DLY8117), hsl1D293Amyc (DLY8116), hsl1N244Amyc (DLY8119), and hsl1Δ (DLY5844) cells were grown to exponential phase at 30°C, harvested, and lysed. Hsl1p was immunoprecipitated and incubated with γ-32P-ATP for 30 min at 30°C before SDS-PAGE and autoradiography. Bottom, a Western blot of the proteins used for the kinase assay. (C) The catalytic mutants all display a partial mobility shift. The same strains as in B were lysed by TCA precipitation and Hsl1p-myc was detected by Western blotting. (D) The mobility shift associated with both wild-type Hslp1 and Hsl1pK110R is lost in the absence of Elm1p. Untagged (DLY1), HSL1-myc ELM1 (DLY8113), HSL1-myc elm1Δ (DLY9806), hsl1K110Rmyc ELM1 (DLY8117), and hsl1K110Rmyc elm1Δ (DLY9803) cells were grown to exponential phase at 30°C and lysed by TCA precipitation, and Hsl1p-myc was detected by Western blotting. Cdc11p (septin) was used as a loading control. (E) The mobility shift of catalytically inactive Hsl1p occurs at the time of bud emergence. HSL1-myc HSL7-HA (DLY5000) and hsl1K110Rmyc HSL7-HA (DLY5390) cells were synchronized in G1 with α-factor and released into fresh media at 30°C. The % of cells that had budded or undergone nuclear division were scored at the indicated times after release (n = 100 cells scored from DAPI-stained samples). Cells were lysed by TCA precipitation and Hsl1p-myc and Hsl7p-HA were detected by Western blotting.
Hsl1p T273 Phosphorylation and Hsl1p Function
Many kinases are activated by phosphorylation of the activation loop threonine (Rubenstein and Schmidt, 2007). To assess whether this was the case for Hsl1p, we generated T273A (nonphosphorylatable) and T273E (phosphomimic) mutants and expressed them (with C-terminal myc tags) from the HSL1 promoter in yeast. The mutants were expressed at levels similar to wild-type Hsl1p (Figure 3B).
To assess how Hsl1p T273 phosphorylation affects Hsl1p function, we crossed strains expressing T273A or T273E mutants as the only source of Hsl1p with mih1Δ strains. Although HSL1T273E mih1Δ cells exhibited a growth rate, cell morphology (Figure 3C), and cell cycle profile (Figure 3D) similar to the HSL1 mih1Δ controls, the hsl1T273A mih1Δ cells were markedly larger and more elongated, indicative of a prolonged G2 delay. This phenotype was Swe1p-dependent (Figure 3, C and D). Immunofluorescence microscopy revealed that both Hsl1pT273A and Hsl1pT273E were properly localized to the septin cortex, but only Hsl1pT273E was able to effectively localize Hsl7p to that site (Figure 3E). Thus, the nonphosphorylatable Hsl1pT273A displays reduced ability to recruit Hsl7p and down-regulate Swe1p in vivo. Because the phosphomimic Hsl1pT273E appears as active as wild-type Hsl1p, it seems likely that the reduced Hsl1pT273A function reflects its inability to be phosphorylated, rather than a structural requirement for threonine at that position.
Hsl1p T273 Phosphorylation Stimulates Hsl1p Kinase Activity
Immunoprecipitated Hsl1pT273A displayed reduced autophosphorylation and dramatically reduced Hsl7p phosphorylation, compared with wild-type Hsl1p in vitro (Figure 4A). Surprisingly, Hsl1pT273E was also less active than wild-type Hsl1p in this assay, though considerably more active than Hsl1pT273A (Figure 4A).
Figure 4.
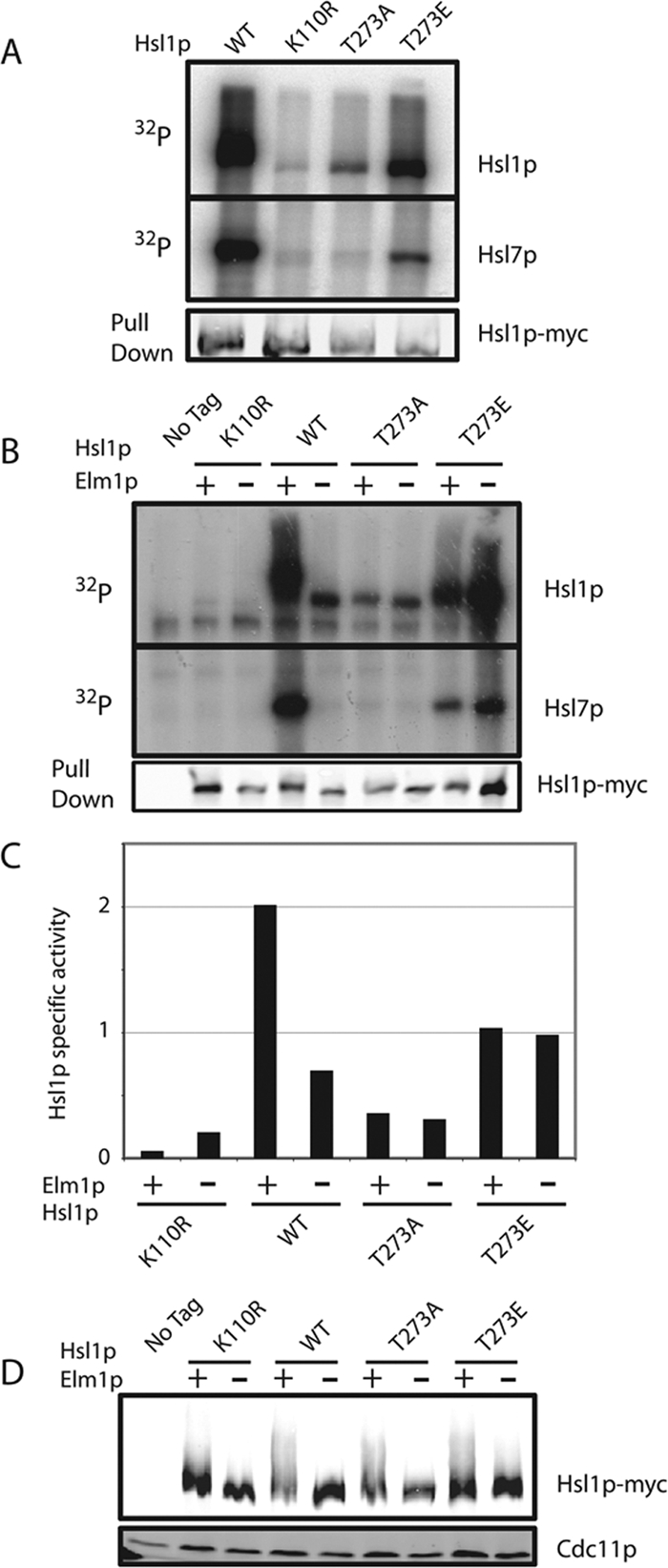
Hsl1p is less active when Elm1p is absent or Hsl1p T273 is not phosphorylatable. (A) Autophosphorylation and Hsl7p phosphorylation is reduced upon mutation of Hsl1p T273. Untagged (DLY1), HSL1-myc (DLY8113), hsl1T273Amyc (DLY7841), HSL1T273Emyc (DLY7451), and hsl1K110Rmyc (DLY8117) cells were grown to exponential phase at 30°C, harvested, and lysed. Hsl1p was immunoprecipitated and incubated with γ-32P-ATP and GST-Hsl7p for 30 min at 30°C before SDS-PAGE and autoradiography. Although the Hsl1p and Hsl7p bands are from the same gel, different exposures are shown. Bottom, a Western blot of the Hsl1p proteins used for the kinase assay. (B) Hsl1p kinase activity depends on Elm1p, but the activity of T273 nonphosphorylatable mutants does not. The same strains as in A together with HSL1-myc elm1Δ (DLY9806), hsl1T273Amyc elm1Δ (DLY9820), HSL1T273Emyc elm1Δ (DLY9804), and hsl1K110Rmyc elm1Δ (DLY9803) were grown and processed as above. Note that the last lane is overloaded. (C) Quantitation of the kinase assay in B. 32P incorporation into Hsl1p in each lane was quantitated by phosphorimager and divided by the amount of Hsl1p in the relevant immunoprecipitate, quantitated using Li-Cor Odyssey software. The ratio is plotted in arbitrary units. (D) The phosphomimic mutation T273E partially restores the Hsl1p mobility shift in elm1Δ cells. The same strains as in B were lysed by TCA precipitation, and Hsl1p-myc and Cdc11p were detected by Western blotting.
Wild-type Hsl1p expressed in elm1Δ mutants displayed dramatically reduced kinase activity in vitro, comparable to that of Hsl1pT273A (Figure 4B). Strikingly, the reduced activity of Hsl1pT273A and the higher activity of Hsl1pT273E were both unaffected by deletion of ELM1 (Figure 4B). These findings suggest that phosphorylation of Hsl1p T273 stimulates Hsl1p kinase activity and that T273 phosphorylation is Elm1p-dependent.
Data presented above showed that a mobility shift indicative of phosphorylation of Hsl1p by upstream kinases in vivo depends on Elm1p. We found that the mobility shift was partly restored in elm1Δ mutants expressing Hsl1pT273E (Figure 4C). The simplest hypothesis that explains all of these observations is that Elm1p both phosphorylates Hsl1p at T273 and improves septin organization. Single defects in either Hsl1p phosphorylation (as in the ELM1 hsl1T273A mutant; Figure 3A) or septin organization (as in the elm1Δ HSL1T273E mutant; Figure 4C) do not abrogate Hsl1p autophosphorylation, but the combined deficit (as in elm1Δ HSL1 cells) precludes autophosphorylation.
Phosphomimic Hsl1pT273E Is Able to Rescue the elm1Δ mih1Δ Synthetic Lethality
If phosphorylation of Hsl1p at T273 accounts for Elm1p's special role (beyond helping to organize septins) in the Swe1p degradation pathway, then a phosphomimic Hsl1pT273E should bypass this defect and allow elm1Δ mih1Δ mutants to proliferate. To test this prediction, we generated an elm1Δ mih1Δ strain that was kept alive by a URA3-marked plasmid expressing ELM1. The presence of the plasmid makes cells sensitive to 5-FOA, which is converted to a toxic product by Ura3p (Boeke et al., 1987). Loss of the plasmid makes cells resistant to 5-FOA, but leads to G2 arrest of this strain, so that cells cannot proliferate on 5-FOA–containing plates. However, elm1Δ mih1Δ HSL1T273E cells were able to grow without the plasmid (Figure 5A), indicating that Swe1p down-regulation proceeds without Elm1p when Hsl1p is pseudophosphorylated at T273. To a lesser degree, the elm1Δ mih1Δ growth defect was also suppressed by the HSL1M177V allele described by Myers and colleagues (Edgington et al., 1999; Figure 5A).
Figure 5.
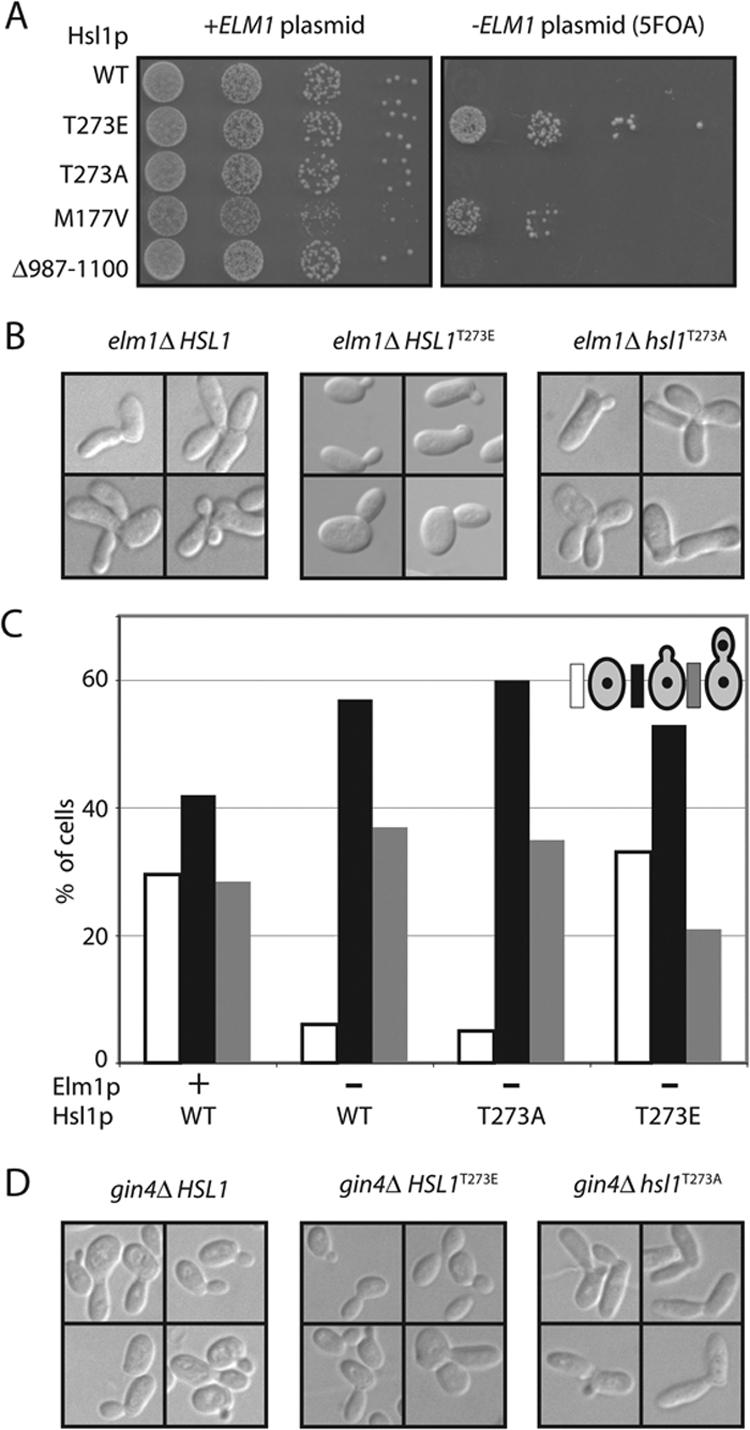
Pseudophosphorylation of Hsl1p T273 suppresses mih1Δ elm1Δ synthetic lethality. (A) A mih1Δ elm1Δ strain kept alive with a URA3-marked plasmid carrying ELM1 was transformed with plasmids expressing the indicated allele of Hsl1p. On 5-FOA media only cells that have lost the ELM1 plasmid can live. Strains: HSL1 (DLY9409), hsl1T273A (DLY9406), HSL1T273E (DLY9407), HSL1M177V (DLY9410), and HSL1Δ987–1100 (DLY9691). Cells were spotted onto YEPD with and without 5-FOA. (B) elm1Δ elongated cell morphology is suppressed by Hsl1pT273E. DIC images of representative cells from proliferating asynchronous cultures of elm1Δ (DLY7704), hsl1T273A elm1Δ (DLY9820), and HSL1T273E elm1Δ (DLY10077) strains. (C) The G2/M cell cycle delay in elm1Δ cells is suppressed by Hsl1pT273E. Cell cycle profiles were determined as described in Figure 3D for the following strains: WT (DLY8113), elm1Δ (DLY9806), hsl1T273A elm1Δ (DLY9820), and HSL1T273E elm1Δ (DLY10077). (D) gin4Δ elongated bud morphology is not suppressed by Hsl1pT273E, but is exacerbated by Hsl1pT273A. DIC images of representative cells from proliferating asynchronous cultures of gin4Δ (DLY4410), hsl1T273A gin4Δ (DLY10076), and HSL1T273E gin4Δ (DLY11008), strains.
elm1Δ single mutants exhibit a notably elongated cell morphology associated with a Swe1p-dependent G2 delay (Edgington et al., 1999; Figure 5B). Introduction of Hsl1pT273E suppressed this defect (Figure 5B). Interestingly, Hsl1pT273E did not suppress the elongated morphology of gin4Δ mutants (Figure 5C), suggesting that the suppression is Elm1p-specific. Moreover, Hsl1pT273A exacerbated the gin4Δ elongated morphology (Figure 5C), suggesting that Elm1p-dependent Hsl1p T273 phosphorylation occurs to some degree in gin4Δ mutants.
Comparison of “Activated” Versions of Hsl1p
Our results suggest a pathway for Hsl1p activation via phosphorylation of T273 by Elm1p, and previous work (Hanrahan and Snyder, 2003) suggested a pathway for Hsl1p activation via septin binding to relieve autoinhibition by the 987–1100 domain. An “activated” Hsl1pT273E could bypass the requirement for Elm1p (Figure 5), whereas an “activated” Hsl1pΔ987–1100 was reported to bypass the requirement for assembled septins and to prevent Swe1p-mediated bud elongation (Hanrahan and Snyder, 2003). These findings raise several questions regarding the relationship between these pathways (e.g., might 987-1100 autoinhibition block T273 phosphorylation?) and their contribution to Hsl1p activity in different circumstances (e.g., do they always work together or respond to distinct inputs?). To begin to address these issues, we first compared the capabilities of the activated versions of Hsl1p in assays to determine the degree to which they could bypass Elm1p (Figure 5) and septin (Figure 6) requirements. We found that Hsl1pΔ987–1100 could not rescue the viability of elm1Δ mih1Δ cells (Figure 5A), indicating that deletion of the 987-1100 domain could not compensate for the loss of Elm1p.
Figure 6.

Neither Hsl1pT273E nor Hsl1pΔ987–1100 suppress G2 delay caused by septin disassembly. (A) HSL1 CDC12 (DLY8113), HSL1-myc cdc12-6 (DLY8745), HSL1Δ987–1100 CDC12 (DLY8684), HSL1Δ987–1100 cdc12-6 (DLY8709), HSL1T273E CDC12 (DLY9391), and HSL1T273E cdc12-6 (DLY7395) cells were grown to exponential phase at 18°C and shifted to 37°C for 3 h before fixation and DIC microscopy. (B) Cells from the same strains as in A were shifted from 18°C to 37°C for a total of 3 h. After the first hour, cells were treated with nocodozole to increase the amount of Cdc28p/Clb2. Total cell lysates were separated by SDS-PAGE and Cdc28p Tyr19 phosphorylation was assessed by Western blotting. (C) Cells from the same strains as in A were shifted from 18°C to 37°C for 3 h and lysed by TCA precipitation, and Hsl1p-myc and Cdc11p were detected by Western blotting.
To assess whether the activated versions could compensate for loss of organized septins, we introduced them into a cdc12-6 strain in which the Cdc12p septin is temperature-sensitive. After a 30-min shift to 37°C the septin structure completely disassembles from the neck and after 3 h at 37°C the cells display elongated buds (Figure 6A) and elevated Cdk1 tyrosine phosphorylation (Figure 6B), indicative of Swe1p-mediated cell cycle delay. We found that septin mutants containing Hsl1pT273E or Hsl1pΔ987–1100 were indistinguishable from those containing wild-type Hsl1p: neither form was able to reduce Swe1p-mediated arrest (Figure 6). Thus, it appears that neither “activated” allele can bypass the need for septins in Swe1p down-regulation.
The role of Hsl1p in Swe1p degradation is thought to involve bringing Swe1p and Cdc5p together at the septin cortex (Sakchaisri et al., 2004). Thus, it seemed possible that even if Hsl1pΔ987–1100 were to remain active as a kinase after septin disruption, it might be unable to promote Swe1p degradation. Because Hsl1p mobility during SDS-PAGE is (at least in part) dependent on Hsl1p kinase activity, it has been used as a surrogate measure for Hsl1p activity in vivo. After shift of cdc12-6 mutants to 37°C, the phosphorylation-shifted mobility of wild-type Hsl1p collapsed rapidly (Figure 6C). Both Hsl1pT273E and Hsl1pΔ987–1100 displayed a similarly rapid collapse to the hypophosphorylated form (Figure 6C). Hsl7p also undergoes a mobility shift due to Hsl1p-mediated phosphorylation, and we found that the Hsl7p shift also disappeared after septin temperature shift, regardless of which Hsl1p allele was present (Figure 6C). Thus, Hsl1pΔ987–1100 does not maintain autophosphorylation, or Hsl7p phosphorylation, or the ability to down-regulate Swe1p after septin disassembly.
Our failure to reproduce a critical result reported for Hsl1pΔ987–1100 prompted us to reexamine other experiments underlying the hypothesis that 987–1100 is an autoinhibitory domain. One such experiment showed that overexpression of Hsl1p 987–1100 or 987–1518 fragments caused cells to grow elongated buds, which was presumed to reflect trans-inhibition of endogenous Hsl1p (Hanrahan and Snyder, 2003). We confirmed that overexpression of GST-Hsl1p987–1518 caused cells to develop elongated buds, though we were less successful with GST-Hsl1p987–1100 (Figure 7A). However, because this region also binds to septins (Hanrahan and Snyder, 2003), it seemed possible that the bud elongation was due to septin perturbation rather than inhibition of endogenous Hsl1p. Indeed, we noted aberrant septin structures (including bud tip staining and diffuse or bar-like neck staining) upon overexpression of GST-Hsl1p987–1518 (Figure 7B). Furthermore, overexpression of GST-Hsl1p987–1518 induced additional bud lengthening even in hsl1Δ strains (Figure 7C), indicating that it cannot act solely by inhibiting endogenous Hsl1p. Previous work suggested that one role for septins is to cause bulging on the bud side of the mother-bud neck: without septins, elongated buds emerge as cylinders rather than initially broadening at the base of the bud (Gladfelter et al., 2005). We found that whereas the buds of hsl1Δ mutants bulged normally at the base, many of the buds in cells overexpressing GST-Hsl1p987–1518 did not (Figure 7), suggesting that the bud morphology is due to septin perturbation, rather than direct Hsl1p inactivation.
Figure 7.

Overexpression of the Hsl1p C-terminal region perturbs septin organization. (A) Overexpression of GST-Hsl1p987–1518 causes development of elongated buds. Cells expressing GAL1-GST (DLY9587), GAL1-GST-HSL1987–1100 (DLY9863), and GAL1-GST-HSL1987–1518 (DLY9583) were grown on galactose media to induce overexpression, fixed, and imaged by DIC microscopy. (B) Overexpression of GST-Hsl1p987–1518 causes septin disorganization. Cells containing GFP-CDC3 and GAL1-GST (DLY10097) or GAL1-GST-HSL1987–1518 (DLY11002) were imaged by DIC and fluorescence microscopy. (C) Overexpression of GST-Hsl1p987–1518 causes more severe bud elongation and neck defects in hsl1Δ cells. DIC images of hsl1Δ (JMY3–58) and hsl1Δ GAL1-GST-HSL1987–1518 (DLY9543) cells. Arrows indicate aberrant necks. (D) Western blot showing expression of GST-Hsl1p fragments in strains DLY10097, DLY10098, and DLY11002. Blots were probed with anti-GST and anti-Cdc11 (loading control) antibodies.
DISCUSSION
Screen for Regulators of Swe1p Degradation
Swe1p degradation is thought to involve Swe1p recruitment to the mother-bud neck, where septin filaments bring together the checkpoint kinase Hsl1p, the Swe1p-binding protein Hsl7p, and the Polo kinase Cdc5p (Lew, 2003; Keaton and Lew, 2006). This allows phosphorylation of Swe1p by Cdc5p at multiple sites, but subsequent steps in Swe1p degradation remain unclear. Our initial goal in devising a screen for Swe1p degradation mutants was to identify new downstream factors, potentially including an ubiquitin ligase, but instead we identified a transcription factor (Ndd1p) and yet another kinase (Elm1p). It may be that downstream ligases are redundant, or that (like Cdc5p) they play other essential roles that make them difficult to identify in a loss-of-function screen.
Our screen was for mutations that would be synthetically lethal with mih1Δ. The Ndd1p transcription factor forms part of the Swi5 factor complex responsible for the induction of a suite of genes whose transcripts peak in abundance during G2/M (Darieva et al., 2003; Reynolds et al., 2003). Among the transcripts are CDC5 and the mitotic cyclins CLB1 and CLB2 (Zhu et al., 2000). Because Swe1p degradation involves sequential phosphorylation by mitotic cyclin/Cdk1 complexes and Cdc5p (Asano et al., 2005), reduced expression of these transcripts likely explains the identification of ndd1 mutants in our screen. Moreover, even if some Swe1p degradation continues to occur in ndd1 mutants, there would be fewer cyclin/Cdk1 complexes for the remaining Swe1p to inhibit, leading to G2 arrest.
A Novel Role for the Elm1p Kinase
The other novel hit emerging from our screen was Elm1p, a neck-localized kinase that is known to promote assembly of a normal septin collar (Bouquin et al., 2000). The fact that other septin organization mutants did not emerge from the screen, combined with directed tests showing that even quite severe septin perturbation does not result in synthetic lethality with mih1Δ, suggested that Elm1p plays an additional role responsible for the phenotype. Based on the previous finding that Elm1p phosphorylates the activation loop threonine in the kinase Snf1p (Sutherland et al., 2003) and the similarity in activation loop sequence between Snf1p and Hsl1p, we suspected that Elm1p might phosphorylate Hsl1p at that position (T273) to promote Swe1p degradation. We present several lines of evidence suggesting that this hypothesis is correct. Hsl1p isolated from elm1Δ mutants displayed dramatically reduced phosphorylation and in vitro kinase activity compared with Hsl1p from wild-type cells. Mutation of T273 to A severely reduced Hsl1p kinase activity and function and rendered the activity insensitive to the presence or absence of Elm1p, and most strikingly, mutation of Hsl1p T273 to E (to mimic the phosphorylated state) was able to bypass the requirement for Elm1p to promote Hsl1p phosphorylation, kinase activity, and function, rescuing the elm1Δ mih1Δ synthetic lethality.
On the other hand, we were unable to demonstrate direct phosphorylation of Hsl1p by Elm1p in vitro. A similar result was reported by others (Asano et al., 2006). It is therefore possible that Elm1p promotes Hsl1p T273 phosphorylation indirectly via an intermediary kinase. An obvious candidate is Gin4p, which is activated by Elm1p and localized to the neck (Longtine et al., 1998a; Asano et al., 2006). However, whereas the phosphomimic Hsl1pT273E effectively suppressed the elm1Δ elongated bud phenotype, it was not able to suppress the gin4Δ elongated bud phenotype, arguing against this hypothesis. We currently favor the hypothesis that Elm1p directly phosphorylates Hsl1p at T273 in the context of the yeast bud neck, but that our in vitro assay is inadequate to recapitulate this event. Consistent with the view that neck localization enables Hsl1p phosphorylation by Elm1p, a previous study found that a delocalized truncated form of Elm1p was still competent to phosphorylate Snf1p but was unable to suppress bud elongation in vivo (Rubenstein et al., 2006). In summary, our findings indicate that full Hsl1p activation involves phosphorylation of T273 by an upstream kinase, most likely Elm1p.
Hsl1p and Septin Roles in Swe1p Degradation
In a previous study it was suggested that residues 987–1100 of Hsl1p constitute an autoinhibitory domain whose inhibition of the Hsl1p kinase domain is relieved upon binding of septins (Hanrahan and Snyder, 2003). Key support for that model came from the finding that a “constitutively active” Hsl1pΔ987–1100 was able to suppress the Swe1p-dependent bud elongation after shift of septin mutants to restrictive temperature (Hanrahan and Snyder, 2003). This finding implied that the only role of septins in promoting Swe1p degradation is to activate Hsl1p. However, as Hsl1p is thought to function by targeting Swe1p and Cdc5p to the septins, it is not obvious how even a constitutively active Hsl1p could bypass the requirement for assembled septins in Swe1p degradation. We reexamined the issue and were unable to reproduce the key finding from the previous study. In a variety of assays, Hsl1pΔ987–1100 behaved indistinguishably from wild-type Hsl1p. Moreover, Hsl1pT273E, which behaves as an “activated” version in the elm1Δ context, was also unable to suppress bud elongation in a septin mutant. Thus, we suggest that septins are important to provide a scaffold upon which Swe1p can be targeted for degradation and that simply activating Hsl1p cannot bypass that function.
Other support for the Hsl1p autoinhibition hypothesis came from the observation that overexpression of an Hsl1p C-terminal domain led to the development of an elongated bud morphology, which was interpreted as evidence for transinhibition of endogenous Hsl1p kinase (Hanrahan and Snyder, 2003). However, we showed that unlike deletion of HSL1, overexpression of the Hsl1p C-terminal domain led to septin disorganization and an accompanying alteration in bud morphology. In addition, overexpression of the domain promoted more severe bud elongation even in an hsl1Δ strain. These findings are most consistent with the hypothesis that the domain perturbs septins, rather than directly inhibiting Hsl1p (and indeed two-hybrid data support a septin interaction; Hanrahan and Snyder, 2003). Together with the other findings mentioned above, this means that the 987–1100 domain autoinhibition hypothesis has no strong in vivo support, though autoinhibition is a common regulatory strategy and the possibility remains that such a mechanism may contribute to Hsl1p regulation.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E08-06-0663) on September 3, 2008.
REFERENCES
- Asano S., Park J. E., Sakchaisri K., Yu L. R., Song S., Supavilai P., Veenstra T. D., Lee K. S. Concerted mechanism of Swe1/Wee1 regulation by multiple kinases in budding yeast. EMBO J. 2005;24:2194–2204. doi: 10.1038/sj.emboj.7600683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano S., Park J. E., Yu L. R., Zhou M., Sakchaisri K., Park C. J., Kang Y. H., Thorner J., Veenstra T. D., Lee K. S. Direct phosphorylation and activation of a Nim1-related kinase Gin4 by Elm1 in budding yeast. J. Biol. Chem. 2006;281:27090–27098. doi: 10.1074/jbc.M601483200. [DOI] [PubMed] [Google Scholar]
- Barral Y., Parra M., Bidlingmaier S., Snyder M. Nim1-related kinases coordinate cell cycle progression with the organization of the peripheral cytoskeleton in yeast. Genes Dev. 1999;13:176–187. doi: 10.1101/gad.13.2.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudin A., Ozier-Kalogeropoulos O., Denouel A., Lacroute F., Cullin C. A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. Nucleic Acids Res. 1993;21:3329–3330. doi: 10.1093/nar/21.14.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton B. K., Tinkelenberg A., Gonzalez I., Cross F. R. Cla4p, a Saccharomyces cerevisiae Cdc42p-activated kinase involved in cytokinesis, is activated at mitosis. Mol. Cell. Biol. 1997;17:5067–5076. doi: 10.1128/mcb.17.9.5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blacketer M. J., Koehler C. M., Coats S. G., Myers A. M., Madaule P. Regulation of dimorphism in Saccharomyces cerevisiae: involvement of the novel protein kinase homolog Elm1p and protein phosphatase 2A. Mol. Cell. Biol. 1993;13:5567–5581. doi: 10.1128/mcb.13.9.5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeke J. D., Trueheart J., Natsoulis G., Fink G. R. 5-Fluoroorotic acid as a selective agent in yeast molecular genetics. Methods Enzymol. 1987;154:164–173. doi: 10.1016/0076-6879(87)54076-9. [DOI] [PubMed] [Google Scholar]
- Bose I., Irazoqui J. E., Moskow J. J., Bardes E. S., Zyla T. R., Lew D. J. Assembly of scaffold-mediated complexes containing Cdc42p, the exchange factor Cdc24p, and the effector Cla4p required for cell cycle-regulated phosphorylation of Cdc24p. J. Biol. Chem. 2001;276:7176–7186. doi: 10.1074/jbc.M010546200. [DOI] [PubMed] [Google Scholar]
- Bouquin N., Barral Y., Courbeyrette R., Blondel M., Snyder M., Mann C. Regulation of cytokinesis by the Elm1 protein kinase in Saccharomyces cerevisiae. J. Cell Sci. 2000;113:1435–1445. doi: 10.1242/jcs.113.8.1435. [DOI] [PubMed] [Google Scholar]
- Caviston J. P., Longtine M., Pringle J. R., Bi E. The role of Cdc42p GTPase-activating proteins in assembly of the septin ring in yeast. Mol. Biol. Cell. 2003;14:4051–4066. doi: 10.1091/mbc.E03-04-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clotet J., Escote X., Adrover M. A., Yaakov G., Gari E., Aldea M., de Nadal E., Posas F. Phosphorylation of Hsl1 by Hog1 leads to a G(2) arrest essential for cell survival at high osmolarity. EMBO J. 2006;25:2338–2346. doi: 10.1038/sj.emboj.7601095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darieva Z., Pic-Taylor A., Boros J., Spanos A., Geymonat M., Reece R. J., Sedgwick S. G., Sharrocks A. D., Morgan B. A. Cell cycle-regulated transcription through the FHA domain of Fkh2p and the coactivator Ndd1p. Curr. Biol. 2003;13:1740–1745. doi: 10.1016/j.cub.2003.08.053. [DOI] [PubMed] [Google Scholar]
- Edgington N. P., Blacketer M. J., Bierwagen T. A., Myers A. M. Control of Saccharomyces cerevisiae filamentous growth by cyclin-dependent kinase Cdc28. Mol. Cell. Biol. 1999;19:1369–1380. doi: 10.1128/mcb.19.2.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz R. D., Sugino A. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six base pair restriction sites. Gene. 1988;74:527–534. doi: 10.1016/0378-1119(88)90185-0. [DOI] [PubMed] [Google Scholar]
- Gladfelter A. S., Kozubowski L., Zyla T. R., Lew D. J. Interplay between septin organization, cell cycle and cell shape in yeast. J. Cell Sci. 2005;118:1617–1628. doi: 10.1242/jcs.02286. [DOI] [PubMed] [Google Scholar]
- Gladfelter A. S., Zyla T. R., Lew D. J. Genetic interactions among regulators of septin organization. Eukaryot. Cell. 2004;3:847–854. doi: 10.1128/EC.3.4.847-854.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanrahan J., Snyder M. Cytoskeletal activation of a checkpoint kinase. Mol. Cell. 2003;12:663–673. doi: 10.1016/j.molcel.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Harrison J. C., Bardes E. S., Ohya Y., Lew D. J. A role for the Pkc1p/Mpk1p kinase cascade in the morphogenesis checkpoint. Nat. Cell Biol. 2001;3:417–420. doi: 10.1038/35070104. [DOI] [PubMed] [Google Scholar]
- Keaton M. A., Lew D. J. Eavesdropping on the cytoskeleton: progress and controversy in the yeast morphogenesis checkpoint. Curr. Opin. Microbiol. 2006;9:540–546. doi: 10.1016/j.mib.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Keaton M. A., Szkotnicki L., Marquitz A. R., Harrison J., Zyla T. R., Lew D. J. Nucleocytoplasmic trafficking of G2/M regulators in yeast. Mol. Biol. Cell. 2008;19:4006–4018. doi: 10.1091/mbc.E08-03-0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew D. J. The morphogenesis checkpoint: how yeast cells watch their figures. Curr. Opin. Cell Biol. 2003;15:648–653. doi: 10.1016/j.ceb.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Liu H., Krizek J., Bretscher A. Construction of a GAL1-regulated yeast cDNA expression library and its application to the identification of genes whose overexpression causes lethality in yeast. Genetics. 1992;132:665–673. doi: 10.1093/genetics/132.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine M. S., Fares H., Pringle J. R. Role of the yeast Gin4p protein kinase in septin assembly and the relationship between septin assembly and septin function. J. Cell Biol. 1998a;143:719–736. doi: 10.1083/jcb.143.3.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine M. S., McKenzie A., III, DeMarini D. J., Shah N. G., Wach A., Brachat A., Philippsen P., Pringle J. R. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998b;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Longtine M. S., Theesfeld C. L., McMillan J. N., Weaver E., Pringle J. R., Lew D. J. Septin-dependent assembly of a cell-cycle-regulatory module in Saccharomyces cerevisiae. Mol. Cell. Biol. 2000;20:4049–4061. doi: 10.1128/mcb.20.11.4049-4061.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X.-J., Lu Q., Grunstein M. A search for proteins that interact genetically with histone H3 and H4 amino termini uncovers novel regulators of the Swe1 kinase in Saccharomyces cerevisiae. Genes Dev. 1996;10:1327–1340. doi: 10.1101/gad.10.11.1327. [DOI] [PubMed] [Google Scholar]
- McMillan J. N., Longtine M. S., Sia R.A.L., Theesfeld C. L., Bardes E.S.G., Pringle J. R., Lew D. J. The morphogenesis checkpoint in Saccharomyces cerevisiae: cell cycle control of Swe1p degradation by Hsl1p and Hsl7p. Mol. Cell. Biol. 1999;19:6929–6939. doi: 10.1128/mcb.19.10.6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael W. M., Newport J. Coupling of mitosis to the completion of S phase through Cdc34-mediated degradation of Wee1. Science. 1998;282:1886–1889. doi: 10.1126/science.282.5395.1886. [published erratum appears in Science 1999; 1283, 1835] [DOI] [PubMed] [Google Scholar]
- Mondesert G., Reed S. I. BED1, a gene encoding a galactosyltransferase homologue, is required for polarized growth and efficient bud emergence in Saccharomyces cerevisiae. J. Cell Biol. 1996;132:137–151. doi: 10.1083/jcb.132.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy R., Watkins J. L., Wente S. R. GLE2, a Saccharomyces cerevisiae homologue of the Schizosaccharomyces pombe export factor RAE1, is required for nuclear pore complex structure and function. Mol. Biol. Cell. 1996;7:1921–1937. doi: 10.1091/mbc.7.12.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds D., Shi B. J., McLean C., Katsis F., Kemp B., Dalton S. Recruitment of Thr 319-phosphorylated Ndd1p to the FHA domain of Fkh2p requires Clb kinase activity: a mechanism for CLB cluster gene activation. Genes Dev. 2003;17:1789–1802. doi: 10.1101/gad.1074103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson H. E., Wittenberg C., Cross F., Reed S. I. An essential G1 function for cyclin-like proteins in yeast. Cell. 1989;59:1127–1133. doi: 10.1016/0092-8674(89)90768-x. [DOI] [PubMed] [Google Scholar]
- Rubenstein E. M., McCartney R. R., Schmidt M. C. Regulatory domains of Snf1-activating kinases determine pathway specificity. Eukaryot. Cell. 2006;5:620–627. doi: 10.1128/EC.5.4.620-627.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein E. M., Schmidt M. C. Mechanisms regulating the protein kinases of Saccharomyces cerevisiae. Eukaryot. Cell. 2007;6:571–583. doi: 10.1128/EC.00026-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell P., Moreno S., Reed S. I. Conservation of mitotic controls in fission and budding yeast. Cell. 1989;57:295–303. doi: 10.1016/0092-8674(89)90967-7. [DOI] [PubMed] [Google Scholar]
- Sakchaisri K., Asano S., Yu L. R., Shulewitz M. J., Park C. J., Park J. E., Cho Y. W., Veenstra T. D., Thorner J., Lee K. S. Coupling morphogenesis to mitotic entry. Proc. Natl. Acad. Sci. USA. 2004;101:4124–4129. doi: 10.1073/pnas.0400641101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulewitz M. J., Inouye C. J., Thorner J. Hsl7 localizes to a septin ring and serves as an adapter in a regulatory pathway that relieves tyrosine phosphorylation of Cdc28 protein kinase in Saccharomyces cerevisiae. Mol. Cell. Biol. 1999;19:7123–7137. doi: 10.1128/mcb.19.10.7123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sia R.A.L., Bardes E.S.G., Lew D. J. Control of Swe1p degradation by the morphogenesis checkpoint. EMBO J. 1998;17:6678–6688. doi: 10.1093/emboj/17.22.6678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski R. S., Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland C. M., Hawley S. A., McCartney R. R., Leech A., Stark M. J., Schmidt M. C., Hardie D. G. Elm1p is one of three upstream kinases for the Saccharomyces cerevisiae SNF1 complex. Curr. Biol. 2003;13:1299–1305. doi: 10.1016/s0960-9822(03)00459-7. [DOI] [PubMed] [Google Scholar]
- Theesfeld C. L., Zyla T. R., Bardes E. G., Lew D. J. A monitor for bud emergence in the yeast morphogenesis checkpoint. Mol. Biol. Cell. 2003;14:3280–3291. doi: 10.1091/mbc.E03-03-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C. L., Blacketer M. J., Edgington N. P., Myers A. M. Assembly interdependence among the S. cerevisiae bud neck ring proteins Elm1p, Hsl1p and Cdc12p. Yeast. 2003;20:813–826. doi: 10.1002/yea.1003. [DOI] [PubMed] [Google Scholar]
- Watanabe N., Arai H., Iwasaki J., Shiina M., Ogata K., Hunter T., Osada H. Cyclin-dependent kinase (CDK) phosphorylation destabilizes somatic Wee1 via multiple pathways. Proc. Natl. Acad. Sci. USA. 2005;102:11663–11668. doi: 10.1073/pnas.0500410102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirich C. S., Erzberger J. P., Barral Y. The septin family of GTPases: architecture and dynamics. Nat. Rev. Mol. Cell Biol. 2008;9:478–489. doi: 10.1038/nrm2407. [DOI] [PubMed] [Google Scholar]
- Yamada A., Duffy B., Perry J. A., Kornbluth S. DNA replication checkpoint control of Wee1 stability by vertebrate Hsl7. J. Cell Biol. 2004;167:841–849. doi: 10.1083/jcb.200406048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G., Spellman P. T., Volpe T., Brown P. O., Botstein D., Davis T. N., Futcher B. Two yeast forkhead genes regulate the cell cycle and pseudohyphal growth. Nature. 2000;406:90–94. doi: 10.1038/35017581. [DOI] [PubMed] [Google Scholar]