

Abstract
Background
A gene regulatory module (GRM) is a set of genes that is regulated by the same set of transcription factors (TFs). By organizing the genome into GRMs, a living cell can coordinate the activities of many genes in response to various internal and external stimuli. Therefore, identifying GRMs is helpful for understanding gene regulation.
Results
Integrating transcription factor binding site (TFBS), mutant, ChIP-chip, and heat shock time series gene expression data, we develop a method, called Heat-Inducible Module Identification Algorithm (HIMIA), for reconstructing GRMs of yeast heat shock response. Unlike previous module inference tools which are static statistics-based methods, HIMIA is a dynamic system model-based method that utilizes the dynamic nature of time series gene expression data. HIMIA identifies 29 GRMs, which in total contain 182 heat-inducible genes regulated by 12 heat-responsive TFs. Using various types of published data, we validate the biological relevance of the identified GRMs. Our analysis suggests that different combinations of a fairly small number of heat-responsive TFs regulate a large number of genes involved in heat shock response and that there may exist crosstalk between heat shock response and other cellular processes. Using HIMIA, we identify 68 uncharacterized genes that may be involved in heat shock response and we also identify their plausible heat-responsive regulators. Furthermore, HIMIA is capable of assigning the regulatory roles of the TFs that regulate GRMs and Cst6, Hsf1, Msn2, Msn4, and Yap1 are found to be activators of several GRMs. In addition, HIMIA refines two clusters of genes involved in heat shock response and provides a better understanding of how the complex expression program of heat shock response is regulated. Finally, we show that HIMIA outperforms four current module inference tools (GRAM, MOFA, ReMoDisvovery, and SAMBA), and we conduct two randomization tests to show that the output of HIMIA is statistically meaningful.
Conclusion
HIMIA is effective for reconstructing GRMs of yeast heat shock response. Indeed, many of the reconstructed GRMs are in agreement with previous studies. Further, HIMIA predicts several interesting new modules and novel TF combinations. Our study shows that integrating multiple types of data is a powerful approach to studying complex biological systems.
Background
Single-cell organisms such as yeasts constantly face changing or even harsh environments such as high temperature that threaten their survival or, at least, prevent them from performing optimally [1]. By organizing the genome into gene regulatory modules (GRMs), a yeast cell can coordinate the activities of many genes and carry out complex functions in response to high temperature. Therefore, identifying GRMs of heat response is instrumental for understanding cellular responses to heat shock.
The advances of high-throughput tools such as DNA microarray [2,3] and chromatin immunoprecipitation-DNA chip (ChIP-chip) [4,5] have made the computational reconstruction of GRMs of a yeast cell possible. Several module inference methods have been proposed. Lee et al. [6] performed ChIP-chip experiments on 106 TFs in yeast and discovered six types of network motifs in yeast gene regulation. Using microarray data, Segal et al. [7] developed a probabilistic model to identify yeast GRMs. Later, three studies were conducted to combine ChIP-chip and gene expression data to identify yeast GRMs. First, Xu et al. [8] extended Segal et al.'s probabilistic model to incorporate ChIP-chip data. Second, Bar-Joseph et al. [9] developed GRAM to identify rich medium gene regulatory modules. Third, Wu et al. [10] developed MOFA to identify GRMs of yeast cell cycle. More data sources were used in more recent studies. Kato et al. [11] identified GRMs of yeast cell cycle by combining sequence, ChIP-chip and gene expression data. Lemmens et al. [12] developed ReMoDiscovery to identify GRMs of the yeast cell cycle and yeast stress response by combining motif information, ChIP-chip and gene expression data. Tanay et al. [13] applied a graph theoretic approach and developed SAMBA to reveal the modular organization of the yeast regulatory system by combining protein interactions, growth phenotype data, ChIP-chip, and gene expression data. However, all these module inference algorithms are statistics-based methods, treating dynamic time series gene expression data the same way as static steady state gene expression data. That is, they do not consider the dependency between different time points of a time series and thus do not utilize the dynamic nature of time series data. Because many time series gene expression data sets are now available in the public domain [14-18], it is desirable to develop a module inference method that can utilize the dynamic nature of time series gene expression data. The aim of this study is to develop a module inference method that suits this need.
By combining current transcription factor binding site (TFBS) [19,20], mutant [20], ChIP-chip [21], and heat shock time series gene expression data [18], we developed a module inference method, called Heat-Inducible Module Identification Algorithm (HIMIA), to reconstruct heat-inducible GRMs in yeast (see Figure 1). HIMIA is divided into five steps. First, three independent data sources (ChIP-chip, mutant, and TFBS data) are used to construct a high-confidence TF-promoter binding matrix (see Methods for details). From the TF-promoter binding matrix, all the TFs that bind to any specific target gene can be inferred. Second, using heat shock time series gene expression data, a dynamic system model of gene regulation is applied to describe how a target gene's expression under heat shock is controlled by the TFs that bind to its promoter (see Appendix for details). A dynamic system model is capable of utilizing the dynamic nature of time series gene expression data, making it different from the static statistics-based models in previous studies [7-13]. After the dynamic system modeling, the TFs that have significant regulatory effects on the target gene's expression can be extracted from all TFs that bind to the target gene. From this procedure, a high-confidence TF-gene regulatory matrix is constructed. Each TF-gene regulatory relationship in this matrix is supported by at least three independent data sources (gene expression and TFBS data plus ChIP-chip or/and mutant data). Third, candidate heat-inducible genes are identified. A gene is said to be heat-inducible if at least two time points of its gene expression profile measured under heat shock are induced by at least three folds compared to that under the unstressed condition. Causton et al. [18] defined a candidate heat-inducible gene by the criterion that at least one time point of its gene expression profile shows a change fold of at least three. In order to reduce the false positives, we use a criterion that requires at least two time points with a change fold of at least three. Fourth, using the list of candidate heat-inducible genes and the high-confidence TF-gene regulatory matrix, heat-responsive TF sets can be inferred by statistical methods. A TF set is said to be heat-responsive only if a significant portion of the targets that are co-regulated by all the TFs in the TF set is heat-inducible. The hypergeometric distribution is used to test the statistical significance. Fifth, for each heat-responsive TF set, we collect all their regulatory targets that are heat-inducible to form a candidate heat-inducible GRM. That is, a heat-inducible GRM consists of a set of heat-inducible genes that are regulated by the same set of heat-responsive TFs. Because genes in the same GRM are regulated by the same set of TFs, their gene expression profiles should be more similar to each other than those of a set of genes that are not in a single GRM. Therefore, for each candidate GRM we further extract a subset of genes whose gene expression profiles are more coherent than that of the set of all heat-inducible genes which are regulated by different sets of TFs. Finally, this subset of highly coherent genes forms a heat-inducible GRM that is regulated by the same set of heat-responsive TFs.
Figure 1.
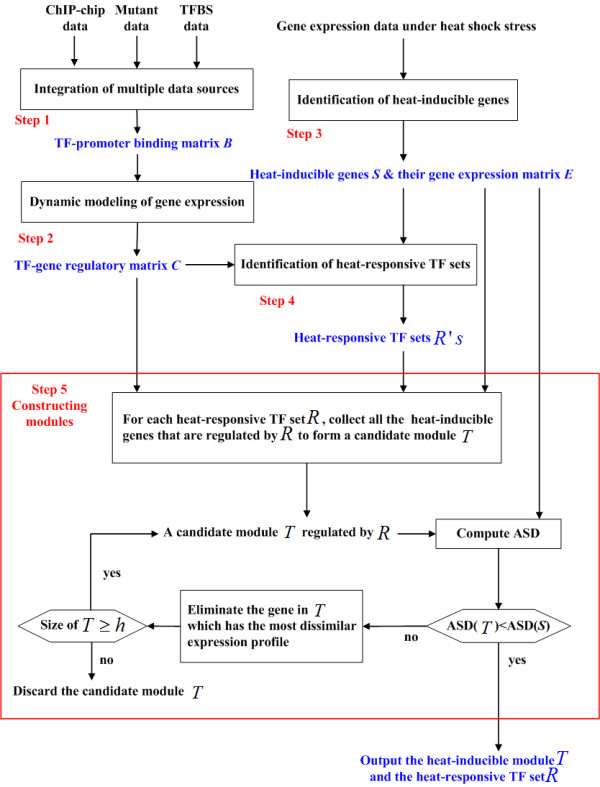
The flowchart of HIMIA.
Results
By integrating TFBS, mutant, ChIP-chip, and gene expression data, HIMIA identified 29 GRMs, which in total contain 182 heat-inducible genes regulated by 12 heat-responsive TFs (see Figure 2 and Additional file 1). According to the literature [19-23,25-33,35], 108 of the 182 genes and 7 of the 12 TFs are known to be involved in heat shock response.
Figure 2.
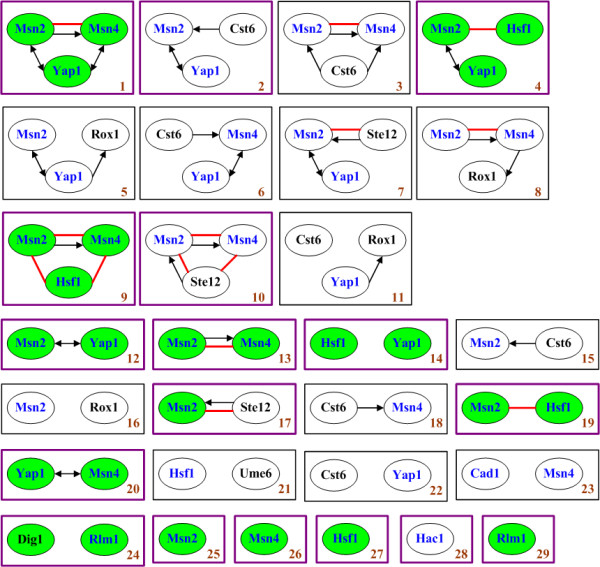
The 29 GRMs identified in this study. Each rectangle stands for a module and the ovals in a rectangle indicate the TFs that regulate the module. A TF name is colored blue if it is known to be involved in heat shock response but black otherwise. The periphery of a rectangle is colored purple if the module has at least one enriched MIPS functional category but black otherwise. An oval is colored green if the TF's function is consistent with at least one of the module's enriched MIPS functional categories. Two ovals are connected by an undirected red line if these two TFs have physical interactions indicated by the current protein-protein interaction data [22]. Two ovals are connected by a directed black line if the two TFs have genetic interactions indicated by mutant or ChIP-chip data [20,21]. For example, Msn2→Yap1 means that either TF Msn2 binds to the promoter of gene YAP1 or the disruption of TF Msn2 results in a significant change of the expression of gene YAP1.
Validation of the identified modules
Several lines of evidence support that HIMIA identifies biologically relevant heat-inducible GRMs. First, by the virtue of our method, each TF-gene regulatory relationship in a module is of high confidence because it is supported by at least three independent data sources (gene expression and TFBS data plus ChIP-chip or/and mutant data). Second, each module is assigned to at least one TF that is known to be involved in heat shock response (see Figure 2). Third, in the 24 modules that are controlled by more than one TF, 70% (32/46 counting multiplicity) of the TFs in the same module have physical or genetic interactions [20-22] (see Figure 2). Fourth, on average, 67% of genes in a heat-inducible GRM are known to be involved in heat shock response (see Additional file 1). Fifth, 59% (17/29) of the identified modules include groups of genes that function in the same cellular process: each of these modules contains at least one over-represented MIPS functional category [23] with the adjusted p-value < 0.05 (after the Bonferroni correction for multiple tests [24]) using the cumulative hypergeometric distribution (see Figure 2 and Additional file 1). Finally, the modules are generally accurate in assigning TFs to sets of genes whose functions are consistent with the TFs' known roles. For the 17 modules that contain groups of genes enriched in the same cellular process, we found that the regulatory functions of the 76% (26/34 counting multiplicity) of the TFs are consistent with one of their modules' over-represented functional categories (see Figure 2). Taken together, these results provide evidence that HIMIA identifies not only sets of biologically related heat-inducible genes, but also heat-responsive TFs that individually or collectively regulate these genes.
Identification of important heat-responsive TFs
We identified 12 heat-responsive TFs, 7 (Msn2, Msn4, Hsf1, Yap1, Hac1, Rlm1 and Cad1) of which are known to be involved in heat shock response (see Table 1). Our findings are supported by the literature. First, Msn2 and Msn4 are known to regulate the general stress response in yeast. They regulate the expression of many genes in response to several stresses, including heat shock, osmotic shock, oxidative stress, low pH, glucose starvation, sorbic acid and high ethanol concentrations, by binding to the stress response element (STRE) located in the promoters of these genes [25-27]. Second, Hsf1 is the well-known heat shock factor that binds to the heat shock element (HSE) to regulate the transcription of many heat-inducible genes, including genes involved in protein folding, detoxification, energy generation, carbohydrate metabolism, and cell wall organization [25,27,28]. All these gene products are important for cell to counteract the deleterious effects of heat. Third, Yap1, a well-known oxidative shock factor, is also known to be involved in heat shock response [29]. For example, Yap1 induces the expression of GSH1 and GSH2 to synthesize glutathione in heat shock response [30]. Fourth, heat stress can cause unfolded proteins to accumulate in the endoplasmic reticulum (ER), triggering the unfolded protein response (UPR). Hac1 binds to the UPR element (UPRE) to regulate genes that are involved in UPR [31]. Fifth, heat stress causes a weakening of cell wall and membrane stretching which stimulates the protein kinase C (PKC) pathway [25]. Rlm1, a component of the PKC pathway, is then activated to perform the function of maintaining the cell wall integrity [32]. Finally, Cad1, an AP-1 like bZIP transcriptional activator involved in stress responses (e.g. heat shock response, pleiotropic drug resistance), controls a set of genes involved in stabilizing proteins [33]. The involvement of Cad1 in stress responses was also identified by Segal et al. [7].
Table 1.
The number of inferred heat-inducible target genes regulated by each of the 12 heat-responsive TFs
| TF name | # of inferred heat-inducible target genes |
| Msn2 | 73 |
| Msn4 | 67 |
| Hsf1 | 64 |
| Yap1 | 48 |
| Hac1 | 29 |
| Rlm1 | 23 |
| Cst6 | 14 |
| Ste12 | 14 |
| Rox1 | 13 |
| Ume6 | 8 |
| Dig1 | 7 |
| Cad1 | 6 |
Seven well-known heat-responsive TFs are bold-faced and colored blue. The TFs are ordered by the number of their inferred heat-inducible target genes.
In addition to the above seven known heat-responsive TFs, five novel heat-responsive TFs (Rox1, Cst6, Ume6, Ste12 and Dig1) have also been identified by HIMIA. Rox1 contains a high-mobility group (HMG) domain that is responsible for DNA bending activity [29]. Cst6 and Ume6 regulate genes involved in the cell cycle and DNA processing [23]. Ste12 and Dig1 are involved in the regulation of mating-specific genes and the invasive growth pathway [29]. Identification of these five TFs as heat-responsive TFs suggests that heat shock response may have crosstalk with other cellular processes. It is known that the cell cycle transiently arrests during a heat shock stress [25], validating our proposal that heat shock response may have crosstalk with the cell cycle process. Moreover, these five novel heat-responsive TFs and the other seven known heat-responsive TFs form a highly connected network of interactions, supporting our prediction that these five novel heat-responsive TFs may play a role in heat shock response (see Figure 3). This dense network of interactions also suggests that different combinations of a fairly small number of heat-responsive TFs may be sufficient to regulate a large number of genes involved in heat shock response.
Figure 3.
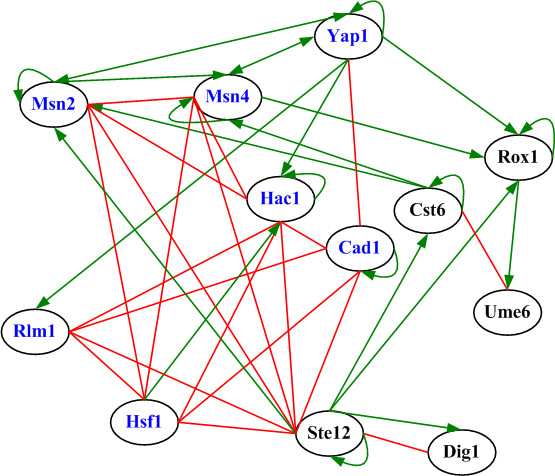
The interaction network formed by the 12 identified heat-responsive TFs. An oval indicates an identified heat-responsive TF. A TF name is colored blue if it is known to be involved in heat shock response but black otherwise. Two ovals are connected by an undirected red line if the two TFs have physical interactions indicated by the current protein-protein interaction data [22]. Two ovals are connected by a directed green line if these two TFs have genetic interactions indicated by the mutant or ChIP-chip data [20,21]. Note that the 12 identified heat-responsive TFs form a highly connected network of interactions.
Taken together, these results suggest that HIMIA is effective for identifying TFs that play a role in heat shock response or are involved in other cellular processes that may crosstalk with heat shock response.
Identification of important genes involved in heat shock response
Disruption of a large number of cellular assemblies and processes, an increased protein unfolding and aggregation, and membrane structure alterations are paramount in cells exposed to high temperature. Heat shock response serves to counteract these deleterious effects. Through it cells increase their thermotolerance or ability to withstand heat stress [25].
Many events occur in yeast cells during heat shock response. First, heat shock proteins (HSPs) are rapidly synthesized. Many HSPs function as protein chaperones, so named because of their ability to bind to partially unfolded proteins to protect them from degradation or aggregation [1]. HIMIA successfully identified heat shock protein genes HSP10 and HSP60 (both in module 27) to be regulated by heat-shock transcription factor Hsf1 and HSP12 and HSP78 (both in module 25) to be regulated by the general stress factor Msn2. In addition, HSP31, HSP104, SSA1, SSA4 and SSE2 (all in module 19) are identified to be regulated by both Hsf1 and Msn2. Other genes involved in the protein folding or refolding are also found. For example, EUG1, LHS1, SCJ1 and ERO1 (all in module 28) are regulated by Hac1, a TF known to be involved in protein unfolding response [31]. Second, heat shock causes the extremely rapid accumulation of a large cytoplasmic pool of trehalose. Trehalose is one of the most effective substances known for preservation of membranous structures and enzyme activities during heating. One of the major roles for Msn2/4 in heat shock response is to regulate the expression of genes required for the synthesis of trehalose [25]. HIMIA successfully identified three genes for trehalose synthease subunits: TSL1 (in module 25), TSP1, and NTH1 (both in module 26) are indeed regulated by Msn2 or Msn4. Third, heat shock increases the synthesis of certain components of the ubiquitination system for intracellular protein turnover, indicating a much greater requirement for turnover of abnormal proteins in cells recovering from heat stress [25]. HIMIA successfully identified several genes that are known to be involved in protein degradation. DER1 and PBI2 (both in module 28) are identified to be regulated by Hac1. APG12, UBC8, JEM1 and UBI4 (all in module 27) are regulated by Hsf1. LAP4 and ASI1 (both in module 25) are regulated by Msn2. ATG8 and YSP3 (both in module 29) are regulated by Rlm1. Fourth, high temperature causes the weakness of cell walls and induces the expression of genes that are involved in cell wall biogenesis and maintenance [25]. HIMIA successfully identified seven genes (CWP1, GFA1, KTR2, FLC2, CHS3, HKR1 and SLT2) known to be involved in the cell wall biogenesis and maintenance [29]. All these seven genes are in module 29 and regulated by Rlm1, an important cell integrity maintenance factor [32]. Finally, it is known that during heat shock cells induce a variety of genes related to carbohydrate metabolism, fatty acid metabolism, respiration and others [25]. HIMIA successfully identified genes that are involved in these cellular processes. For example, HXK1, GLK1 and GPH1 (all in module 25) involved in glucose metabolism are identified to be regulated by Msn2. FAA1 and FAA4 (both in module 27) involved in fatty acid metabolism are identified to be regulated by Hsf1. CYC7 and COX20 (both in module 26) involved in respiration are identified to be regulated by Msn4. The known functions of these genes hint the cellular processes that may be affected in response to heat shock, and suggest mechanisms the cell uses to protect itself in the face of a heat stress [25].
Annotating uncharacterized genes
Among the 182 identified heat-inducible genes, 68 genes have unknown function according to the Saccharomyces Genome Database [29]. We suggest most if not all of these genes are involved in heat shock response. Our predictions are supported by the fact that all these 68 genes are induced by more than three folds at least at two time points of their expression profiles under heat shock. Moreover, all these 68 genes are regulated by known heat-responsive TFs and the TF-gene regulatory relationships are supported by at least three independent data sources (gene expression and TFBS plus ChIP-chip or/and mutant data). As shown in Additional file 1, 14 of these genes are regulated by Hac1, 25 by Hsf1, 24 by Msn2, 20 by Msn4, 17 by Yap1, and so on.
As an example, it is known that the weakening of cell wall during high temperature stimulates the cell integrity pathway [25]. PRM5 is known to be induced in the cell integrity pathway but its molecular function is unknown [29]. HIMIA identified PRM5 (in module 29) to be regulated by Rlm1, an important cell wall integrity maintenance factor, suggesting that PRM5 is indeed involved in the cell wall maintenance during heat shock. Moreover, we successfully identified the putative heat shock protein gene YRO2 (in module 26) to be regulated by Msn4. In addition, putative genes that may be involved in protein degradation are also found. For example, HBT1 and RIM20 (both in module 26) are identified to be regulated by Msn4. MGR1 and ADD37 (both in module 28) are identified to be regulated by Hac1. All these examples show that these uncharacterized genes may play a role in heat shock response. However, further experimental validations are needed to confidently annotate these uncharacterized genes as heat-inducible genes.
Assigning regulatory roles of TFs that regulate a GRM
We can assign the regulatory roles of TFs that regulate a module. A TF is said to be an activator of a module if the p-value for observing so many TF-gene pairs in the module each with a significant positive correlation (within the 5% right tail of the distribution of correlation) is less than 0.001. The p-value is the probability that an observation would be made by chance, and is calculated using the cumulative binomial distribution [10]:
where N is the total number of genes in a module, n0 is the number of genes in the module that show a significant positive correlation in expression with the TF, and p0 is the probability of observing an arbitrary gene in the genome that has a significant positive correlation in expression with the TF. As shown in Table 2, we assigned Cst6 as an activator of four modules, Hsf1 an activator of three modules, Msn2 an activator of five modules, Msn4 an activator of two modules, Rlm1 an activator of two modules and Yap1 an activator of four modules.
Table 2.
Identified activators of GRMs
| Activator identified | Module # (p-value) |
| Cst6 | #2 (3.7 × 10-4), #3 (3.7 × 10-4), #15 (6.4 × 10-5), #18 (1 × 10-3) |
| Hsf1 | #14 (6.8 × 10-5), #21 (3.7 × 10-4), #27 (2.4 × 10-13) |
| Msn2 | #12 (8.5 × 10-5), #13 (4.1 × 10-5), #17 (3.3 × 10-5), #19 (8.6 × 10-4), #25 (5.8 × 10-13) |
| Msn4 | #13 (1.2 × 10-8), #26 (<1 × 10-13) |
| Rlm1 | #24 (1.9 × 10-4), #29 (2 × 10-10) |
| Yap1 | #1 (9.7 × 10-6), #12 (9 × 10-9), #14 (5.3 × 10-7), #20 (3.3 × 10-7) |
A TF is said to be an activator of a module if the p-value for observing so many TF-gene pairs in the module each with a significant positive correlation is less than 0.001.
Refining the clusters of the genes involved in the same cellular process
Heat shock causes an increased protein unfolding and aggregation in a cell. Thus, many genes that are involved in protein (re)folding are induced to bind to partially unfolded proteins to protect them from degradation or aggregation [25]. Among the 182 identified heat-inducible genes, 20 genes are known to be involved in protein (re)folding. Although these genes are functionally similar, they may be under different transcriptional controls. Indeed, HIMIA assigns these 20 genes into five modules (see Figure 4a). For example, HSP10 and HSP60 are identified to be regulated by Hsf1, HSP78 by Msn2/4, HSP31 and HSP104 by both Hsf1 and Msn2/4, and so on.
Figure 4.
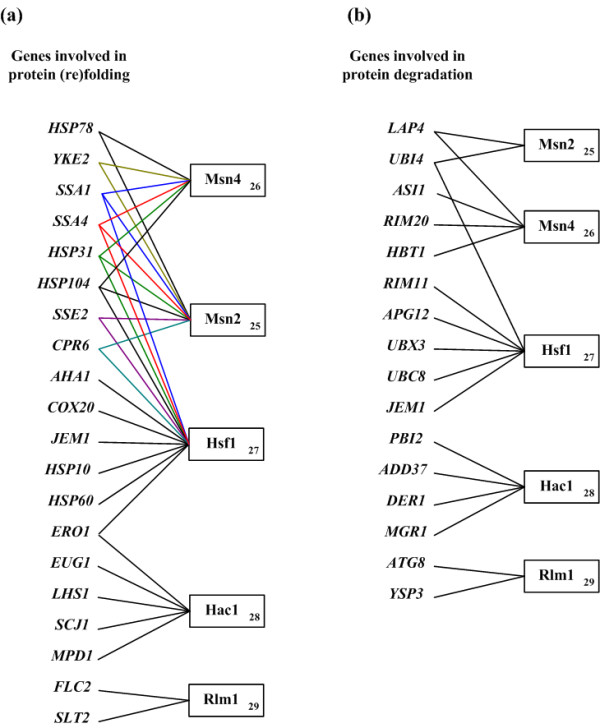
Refining clusters of genes involved in the same cellular process. (A) Refining the cluster of genes involved in protein (re)folding. HIMIA assigns the 20 identified genes that are involved in protein (re)folding into five modules. (B) Refining the cluster of genes involved in protein degradation. HIMIA assigns the 16 identified genes that are involved in protein degradation into five modules.
Denatured proteins that cannot be properly refolded are targeted for degradation by ubiquitination, so it is not surprising that genes involved in protein degradation are induced under heat shock [25]. Among the 182 identified heat-inducible genes, 16 genes are known to be involved in protein degradation. Although these genes are functionally similar, they may be under different transcriptional controls. HIMIA assigns these 16 genes into five modules (see Figure 4b). For example, PBI2, ADD37,DER1 and MGR1 are identified to be regulated by Hac1, ATG8 and YSP3 by Rlm1, and so on.
Therefore, HIMIA can refine the cluster of genes involved in protein (re)folding and degradation and can provide a better understanding of how the cell regulates the complex expression program of these genes.
Discussion
Performance comparison with existing methods
Several module inference methods have been proposed. GRAM [9] and MOFA [10] are intuitive algorithms to identify yeast GRMs by combining ChIP-chip and gene expression data. ReMoDiscovery [12] is an intuitive algorithm to identify yeast GRMs by combining motif information, ChIP-chip, and gene expression data. SAMBA [13] uses a graph theoretic approach to identify GRMs by combining four data sources (ChIP-chip, gene expression, protein interactions, and growth phenotype data). All these four module inference algorithms are statistics-based methods. On the contrary, HIMIA is a dynamic system model-based method that integrates four data sources (ChIP-chip, time series gene expression, mutant, and TFBS data). Since HIMIA is different from the four pervious methods, a performance comparison was conducted. We tested the ability of each of these five algorithms to retrieve the known stress-responsive TFs annotated in the MIPS database [23]. Performance comparison was based on the Jaccard similarity score [12,34], which scores the overlap between an algorithm's output and the list of known stress-responsive TFs (i.e., the true answers). Therefore, the higher the Jaccard similarity score, the better the ability of an algorithm to retrieve the known stress-responsive TFs. Table 3 shows that HIMIA has the highest Jaccard similarity score among the five tested algorithms.
Table 3.
Performance comparison of five module inference tools
| Algorithm name | TP | FP | FN | Jaccard similarity score |
| HIMIA | 7 | 5 | 25 | 0.19 |
| ReMoDiscoery | 11 | 38 | 21 | 0.16 |
| GRAM | 13 | 56 | 19 | 0.15 |
| MOFA | 8 | 32 | 24 | 0.13 |
| SAMBA | 10 | 54 | 22 | 0.12 |
Performance comparison was based on the Jaccard similarity score [12,34], which scores the overlap between an algorithm's output and the list of known stress-responsive TFs. Specifically, the score is defined as TP/(TP+FP+FN) [12,34], where TP stands for true positives, FP for false positives, and FN for false negatives. Note that the higher the Jaccard similarity score, the better the ability of an algorithm to retrieve the known stress-responsive TFs.
Randomization test and comparison with a null model
Two randomization tests were performed to show that the output of HIMIA is statistically meaningful. First, we randomly permuted the gene expression data 1000 times. Second, we randomly permuted the ChIP-chip data 1000 times. When either of the two randomized data sets was used as the input, HIMIA could no longer effectively identify the known stress-responsive TFs (see Table 4). This means that the output of HIMIA is statistically meaningful and is far beyond random expectation.
Table 4.
Randomization tests of HIMIA
| Jaccard similarity score | |
| HIMIA (using original input data) | 0.19 |
| HIMIA (using randomized gene expression data) | 0.025 (mean), 0.023 (standard deviation) |
| HIMIA (using randomized ChIP-chip data) | 0.009 (mean), 0.016 (standard deviation) |
Two randomization tests were performed. First, we randomly permuted the gene expression data 1000 times. Second, we randomly permuted the ChIP-chip data 1000 times. When either of the two randomized data sets was used as the input, HIMIA could no longer effectively identify the known stress-responsive TFs, indicating that the output of HIMIA is statistically meaningful and is far beyond random expectation.
In addition, we compared the performance of HIMIA and the null model to retrieve the known stress-responsive TFs annotated in the MIPS database [23]. The null model was defined as using the same input data (mutant, ChIP-chip, TFBS, and gene expression data) but only the overlap was calculated instead of going through the iterative procedure described on Figure 1. The simulation result shows that HIMIA has a better ability in retrieving the known stress-responsive TFs than does the null model (Jaccard similarity scores 0.19 v.s. 0.17).
Parameter settings of HIMIA
The choice of the relaxing the p-value to 0.01 (in Step 1 of HIMIA) has a biological meaning. Two previous papers [6,21] used a statistical error model to assign a p-value of the binding relationship of a TF-promoter pair. They found that if p ≤ 0.001, the binding relationship of a TF-promoter pair is of high confidence and can usually be confirmed by promoter-specific PCR. If p > 0.01, the binding relationship of a TF-promoter pair is of low confidence and cannot be confirmed by promoter-specific PCR most of the time. However, if 0.001 <p ≤ 0.01, the binding relationship of a TF-promoter pair is ambiguous and can be confirmed by promoter-specific PCR in some cases but not in the other cases. One of our aims in this study was to solve this ambiguity, so we chose 0.01 to be the relaxed p-value. However, we added the requirement that the promoter must contain one or more binding sites of the TF.
In the original paper, Causton et al. [18] defined a candidate heat-inducible gene by the criterion that at least one time point of its gene expression profile showed a change fold of at least three. In order to reduce the chance of including false positives, we used a criterion that at least two time points showed a change fold of at least three. Using this criterion, 528 genes were identified as candidate heat-inducible genes. Of course, some false positives might still exist among these 528 genes. In the subsequent steps, however, HIMIA further refined this set of candidate heat-inducible genes using four independent data sources (mutant, ChIP-chip, TFBS, and time series gene expression data). Finally, only 182 genes were identified heat-inducible genes, which were classified into 29 heat-inducible GRMs regulated by heat-responsive TFs. Among the 182 identified genes, 108 are known to be involved in heat shock response. We provided several lines of evidence to support the biological relevance of the 29 identified heat-inducible GRMs (see Results section for details). Therefore, the possible existence of false positives should not be a serious problem in HIMIA.
Genes in the heat-inducible GRMs
HIMIA identified 29 heat-inducible GRMs. On average, 67% of the genes in a heat-inducible GRM are known to be involved in heat shock response, 17% of the genes in a GRM are uncharacterized genes (i.e., proteins with unknown functions), and 16% of the genes in a GRM are annotated in other cellular processes. Although we cannot claim that all genes in the last two categories are heat-inducible genes, our predictions are supported by three observations. First, all these genes are induced by more than three folds at least at two time points of their expression profiles under heat shock. Second, all these genes are regulated by at least one known heat-responsive TFs in the corresponding GRMs, and the TF-gene regulatory relationships are supported by at least three independent data sources (gene expression and TFBS plus ChIP-chip or/and mutant data). Third, it is known that during heat shock yeast cells express a variety of genes related to carbohydrate metabolism, fatty acid metabolism, energy generation, respiration, signaling pathways (e.g. PKC and cell integrity pathways) and others [1,25]. For the known genes in a GRM that have not been annotated in heat shock response, most of them are related to one of the cellular processes mentioned above. This provides indirect evidence for these genes to be possibly triggered by heat shock response. However, further experimental validations are needed to reliably claim that these predicted heat-inducible genes do play a role in heat shock response.
We also found that genes with diverse functions may be in the same GRM. As an example, Hsf1, Msn2, and Yap1 are known heat-responsive TFs [1,23,25-30,35] and eight genes (HSP31, HSP104, SSA4, SSE2, CPR6, SPI1, TKL2, and YHR087W) are in the module {Hsf1, Msn2, Yap1} (module 4). HSP31, HSP104, SSA4 and SSE2 are genes encoding heat shock proteins that function as protein chaperones [29]. CPR6 encodes a protein that binds to Hsp82 and contributes to chaperone activity [29]. SPI1 encodes a protein that is involved in cell wall biogenesis [35]. TKL2 is involved in carbon metabolism and is induced in response to heat shock [27]. YHR087W is an uncharacterized gene with unknown function [29]. It is not surprising that we find genes encoding HSPs, cell wall biogenesis protein, and carbon metabolic proteins in a GRM because they are all needed when yeast cells are subjected to heat shock [1,25]. HSPs function as protein chaperones, so named because of their ability to bind to partially unfolded proteins to protect them from degradation or aggregation. High temperature causes the weakness of cell wall and induces the expression of genes that are involved in cell wall biogenesis and maintenance. It is known that heat stress imposes large demands for energy (ATP) generation by the cell [25]. In short, heat shock response is multifaceted, involving the expression of genes with diverse functions.
Conclusion
We developed a method, called Heat-Inducible Module Identification Algorithm (HIMIA), for reconstructing GRMs of heat shock response in yeast by integrating current ChIP-chip, mutant, TFBS, and time series gene expression data. Unlike previous module inference tools which are static statistics-based methods, HIMIA is a dynamic system model-based method that utilizes the dynamic nature of time series gene expression data. HIMIA identified 29 GRMs, which in total contain 182 heat-inducible genes regulated by 12 heat-responsive TFs. The literature indicates that 108 of the 182 genes and 7 of the 12 TFs are known to be involved in heat shock response. The biological relevance of each inferred GRM was validated by using the literature, enrichment for genes in the same MIPS functional category, protein-protein interaction data, and so on. Our analysis suggests that different combinations of a fairly small number of heat-responsive TFs may be responsible for regulating a large number of genes involved in heat shock response and that there may exist crosstalk between heat shock response and other cellular processes. In addition, HIMIA suggested that 68 uncharacterized genes may be involved in heat shock response and it also identified their plausible heat-responsive regulators. Furthermore, HIMIA is capable of assigning the regulatory roles of the TFs that regulate GRMs and Cst6, Hsf1, Msn2, Msn4, and Yap1 are found to be activators of several GRMs. In addition, HIMIA refined two clusters of genes that are involved in heat shock response and provided a better understanding of how the complex expression program of heat shock is regulated. Finally, we showed that HIMIA outperformed four current module inference tools (GRAM, MOFA, ReMoDisvovery, and SAMBA), and we conducted two randomization tests to show that the output of HIMIA is statistically meaningful.
Methods
Data sets and data preprocessing
We use four kinds of data in this study. First, the ChIP-chip data are from Harbison et al. [21]. They used genome-wide location analysis to determine the genomic occupancy of 203 TFs in rich media conditions and, for 84 of these TFs, in at least one of 12 other environmental conditions. Second, the TFBS data are from MacIsaac et al.'s study [19] and the YEASTRACT database [20]. MacIsaac et al. used evolutionarily conservative criteria to computationally identify the binding sites of many TFs. The YEASTRACT database includes a set of computational tools that can be used to identify complex motifs over-represented in the promoters of co-regulated genes. Third, the mutant data are from the YEASTRACT database [20]. The mutant data can tell us which gene's expression was changed significantly owing to the deletion (or mutation) of the gene that encodes a TF. The evidence may come from detailed gene by gene analysis or genome-wide expression analysis. Finally, the time series gene expression data under heat shock stress are from Causton et al.'s study [18]. Samples for all genes in the yeast genome are collected at six time points (0, 15, 30, 45, 60, 120 minute). That is, each gene has a 6-timepoint gene expression profile. The cubic spline method [36] is then used to interpolate extra data points into the original time profile. Note that genes that have missing values in their original time profiles are discarded in our study. We did not use Gasch et al.'s gene expression data under heat shock stress [17] because 41% (2509/6152) of the genes in the yeast genome had missing values in their time profiles.
Average standard deviation (ASD)
To check the expression coherence of a set of genes, ASD is used [11]: ASD ≜ m(σ(eg,i)), where eg,i is the normalized expression level of gene g at time i, σ is the standard deviation over genes, and m is the average over time. The lower the ASD is, the closer the expression levels are to the average, i.e., the more coherent in expression profiles of the genes in a set are.
Heat-Inducible Module Identification Algorithm (HIMIA)
HIMIA are divided into five steps.
Step 1
Construction of a high-confidence TF-promoter binding matrix B = [bi,j]
In this matrix, bi,j = 1 if (1) the p-value for TFj to bind the promoter of gene i is ≤ 0.01 in the ChIP-chip data and the promoter of gene i contains one or more binding sites of TFj or (2) the disruption of TFj results in a significant change of the expression of gene i and the promoter of gene i contains one or more binding sites of TFj. Otherwise, bi,j = 0
Step 2
Construction of a high-confidence TF-gene regulatory matrix C = [ci,j]
In this matrix, ci,j = 1 if bi,j = 1 and if TFj is shown by the dynamic model to have a large regulatory effect on the expression of gene i (see Appendix for details). Otherwise, ci,j = 0
Step 3
Identification of heat-inducible genes and construction of their gene expression matrix E = [ep,q]
A gene is called a heat-inducible gene if at least two time points of its gene expression profile measured under heat shock are induced by at least three folds compared to that under the unstressed condition. We then collect all the time profiles of the identified heat-inducible genes to form a matrix E = [ep,q], where ep,q is the expression value of the p-th heat-inducible gene at time point q.
Step 4
Identification of heat-responsive TF sets
The number of TFs in a TF set could be one, two or more. A TF set is said to be heat-responsive if a statistically significant portion of the target genes that are co-regulated by all the TFs in the TF set is heat-inducible. The hypergeometric distribution is used to test the statistical significance. For example, let R ={TFu, TFv, TFw} be a TF set, G = {gene k | ck,u = ck,v = ck,w = 1} be the set of genes that are regulated by all the TFs in R, S be the set of heat-inducible genes in the yeast genome identified in Step 3, T = G ∩ S be the set of heat-inducible genes that are regulated by all the TFs in R, and Y be the set of all genes in the yeast genome. Then the p-value for rejecting the null hypothesis (H0: R is not a heat-responsive TF set) is calculated as [37]:
where |G| means the number of genes in set G. This p-value is then adjusted by the Bonferroni correction to represent the true alpha level in the multiple hypothesis testing [24]. Finally, R is said to be a heat-responsive TF set if the adjusted p-value padjusted ≤ 0.01
Step 5
Construction of heat-inducible GRMs
For each heat-responsive TF set R identified in Step 4, T forms a heat-inducible candidate GRM. That is, a heat-inducible candidate GRM T consists of a set of heat-inducible genes that are regulated by a heat-responsive TF set R. Because genes in the same GRM are regulated by the same set of TFs, their gene expression profiles should be more similar to each other than those of a set of genes that are not in a single GRM. Therefore, we require that the gene expression profiles of the genes in the same GRM be more coherent than those of the set of all heat-inducible genes, which are regulated by different sets of TFs. For measuring the expression coherence of a set of genes, ASD is used. The lower the ASD is, the more coherent the expression profiles of the genes in a set are. Therefore, if ASD(T) ≥ ASD(S) HIMIA iteratively eliminates genes in T starting from the one with the most dissimilar expression profile until ASD() <ASD(S), where is the set of the remaining genes and S is the set of all heat-inducible genes identified in Step 3. That is, HIMIA tries to identify a subset of co-regulated heat-inducible genes whose gene expression profiles are more coherent than those of the set of all heat-inducible genes which are regulated by different sets of TFs. Finally, HIMIA outputs a GRM M(R) ≜ if contains more than a certain number of genes, say five. The above procedure goes over all heat-responsive TF sets R's identified in Step 4.
Jaccard similarity score
The Jaccard similarity score [12,34] was used to score the overlap between an algorithm's output and the list of known stress-responsive TFs. Specifically, it is defined as TP/(TP+FP+FN), where TP stands for true positives, FP for false positives, and FN for false negatives. Clearly, the higher the Jaccard similarity score, the better the ability of an algorithm to retrieve the known stress-responsive TFs.
Appendix
Dynamic system model of gene regulation
We consider the transcriptional regulatory mechanism of a target gene as a system with the regulatory profiles of several TFs as the inputs and the gene expression profile of the target gene as the output. The transcriptional regulation of a target gene is described by the following stochastic dynamic equation [38-40]:
| (1) |
where y[t] represents the expression profile of the target gene at time point t, N denotes the number of TFs that bind to the promoter of the target gene inferred from the TF-promoter binding matrix B, di indicates the regulatory ability of TF i, xi[t] represents the regulatory profile of TFi at time point t, k represents the basal level induced by RNA polymerase II, λ indicates the degrading effect of the target gene's expression at present time point y[t] on the target gene's expression at next time point y[t + 1] and ε[t] denotes the stochastic noise due to the modeling error and the measuring error of the target gene's expression profile. ε[t] is assumed to be a Gaussian noise with mean zero and unknown standard deviation σ. The biological meaning of Equation (1) is that y[t + 1] (the target gene's expression value at next time point) is determined by (the production effect of the N TFs at present time point and RNA polymerase II) and -λ·y[t] (the degradation effect of the target gene at present time point).
It has been shown that TF binding usually affects gene expression in a nonlinear fashion: below some level it has no effect, while above a certain level the effect may become saturated. This type of binding behavior can be modeled using a sigmoid function [10,39-42]. Therefore, we define xi[t] (the regulatory profile of TFi at time point t) as a sigmoid function of zi[t] (the gene expression profile of TFi at time point t):
| (2) |
where r denotes the transition rate of the sigmoid function and Ai denotes the mean of the gene expression profile of TFi.
Estimating the parameters of the dynamic system model
We rewrite Equation (1) into the following regression form:
| (3) |
where φ[t] = [x1[t] ⋯ xN[t] 1 - y[t]] denotes the regression vector and θ = [d1 ⋯ dN k λ]T is the parameter vector.
From the gene expression data under heat shock in Caustion et al.'s study [18], it is easy to get the values of {xi[tv], y[tv]} for i ∈ {1,2, ⋯, N}, v ∈ {1, 2, ⋯, M}, where M is the number of the time points of a target gene's expression profile. Equation (3) at different time points can be put together as follows:
| (4) |
For simplicity, we can further define the notations Y Φ and e to represent Equation (4) as follows:
| Y = Φ·θ + e | (5) |
The parameter vector θ can be estimated by the maximum likelihood (ML) method as follows [38]:
| (6) |
Since di stands for the regulatory ability of TFi, a small absolute value of di means that TFi only has a small effect on the target gene's expression, while a large absolute value means that TFi has a large regulatory effect on the target gene's expression. We regard TFi to be a true regulator of the target gene if its regulatory ability di is statistically significantly different from zero (i.e. |di| ≫ 0). The test statistic a t-distribution with degree of freedom (M - 1) - (N + 2) is used to assign a p-value for rejecting the null hypothesis H0: di = 0, where uii is the ith diagonal element of the matrix (ΦTΦ)-1 and is an unbiased estimator of σ (the standard deviation of the stochastic noise ε[t]) [24]. The p-value computed by the i-distribution is then adjusted by the Bonferroni correction to represent the true α level in the multiple hypothesis testing [24]. Then, TFi is said to be a true regulator of the target gene if the adjusted p-value padjusted ≤ 0.01.
Authors' contributions
WSW developed the algorithm, performed the simulation and wrote the manuscript. WHL conceived the research topic, provided essential guidance and revised the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Additional file 1 contains the detailed information of the 29 identified heat-inducible GRMs, the 182 identified heat-inducible genes, the 68 identified uncharacterized genes and the performance comparison of five module inference tools.
Acknowledgments
Acknowledgements
This study was supported by the Taiwan National Science Council NSC-096-2917-I-564-122 and NIH grants GM30998 and GM081724.
Contributor Information
Wei-Sheng Wu, Email: wessonwu@gmail.com.
Wen-Hsiung Li, Email: wli@uchicago.edu.
References
- Hohmann S, Mager WH. Yeast Stress Responses. Berlin: Springer-Verlag; 2003. [Google Scholar]
- Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- DeRisi JL, Iyer VR, Brown PO. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- Ren B, Robert F, Wyrick JJ, Aparicio O, Jennings EG, Simon I, Zeitlinger J, Schreiber J, Hannett N, Kanin E, Volkert TL, Wilson CJ, Bell SP, Young RA. Genome-wide location and function of DNA binding proteins. Science. 2000;290:2306–2309. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]
- Iyer VR, Horak CE, Scafe CS, Botstein D, Snyder M, Brown PO. Genomic binding sites of the yeast cell-cycle transcription factors SBF and MBF. Nature. 2001;409:533–538. doi: 10.1038/35054095. [DOI] [PubMed] [Google Scholar]
- Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z, Gerber GK, Hannett NM, Harbison CT, Thompson CM, Simon I, Zeitlinger J, Jennings EG, Murray HL, Gordon DB, Ren B, Wyrick JJ, Tagne JB, Volkert TL, Fraenkel E, Gifford DK, Young RA. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
- Segal E, Shapira M, Regev A, Pe'er D, Botstein D, Koller D, Friedman N. Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data. Nat Genet. 2003;34:166–176. doi: 10.1038/ng1165. [DOI] [PubMed] [Google Scholar]
- Xu X, Wang L, Ding D. Learning module networks from genome-wide location and expression data. FEBS Lett. 2004;578:297–304. doi: 10.1016/j.febslet.2004.11.019. [DOI] [PubMed] [Google Scholar]
- Bar-Joseph Z, Gerber GK, Lee TI, Rinaldi NJ, Yoo JY, Robert F, Gordon DB, Fraenkel E, Jaakkola TS, Young RA, Gifford DK. Computational discovery of gene modules and regulatory networks. Nat Biotechnol. 2003;21:1337–1342. doi: 10.1038/nbt890. [DOI] [PubMed] [Google Scholar]
- Wu WS, Li WH, Chen BS. Computational reconstruction of transcriptional regulatory modules of the yeast cell cycle. BMC Bioinformatics. 2006;7:421. doi: 10.1186/1471-2105-7-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Hata N, Banerjee N, Futcher B, Zhang MQ. Identifying combinatorial regulation of transcription factors and binding motifs. Genome Biology. 2004;5:R56. doi: 10.1186/gb-2004-5-8-r56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmens K, Dhollander T, De Bie T, Monsieurs P, Engelen K, Smets B, Winderickx J, De Moor B, Marchal K. Inferring transcriptional modules from ChIP-chip, motif and microarray data. Genome Biol. 2006;7:R37. doi: 10.1186/gb-2006-7-5-r37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanay A, Sharan R, Kupiec M, Shamir R. Revealing modularity and organization in the yeast molecular network by integrated analysis of highly heterogeneous genomewide data. Proc Natl Acad Sci USA. 2004;101:2981–2986. doi: 10.1073/pnas.0308661100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu S, DeRisi J, Eisen M, Mulholland J, Botstein D, Brown PO, Herskowitz I. The transcriptional program of sporulation in budding yeast. Science. 1998;282:699–705. doi: 10.1126/science.282.5389.699. [DOI] [PubMed] [Google Scholar]
- Cho RJ, Campbell MJ, Winzeler EA, Steinmetz L, Conway A, Wodicka L, Wolfsberg TG, Gabrielian AE, Landsman D, Lockhart DJ, Davis RW. A genome-wide transcriptional analysis of the mitotic cell cycle. Mol Cell. 1998;2:65–73. doi: 10.1016/S1097-2765(00)80114-8. [DOI] [PubMed] [Google Scholar]
- Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Anders K, Eisen MB, Brown PO, Botstein D, Futcher B. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell. 1998;9:3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Causton HC, Ren B, Koh SS, Harbison CT, Kanin E, Jennings EG, Lee T, True HL, Lander ES, Young RA. Remodeling of Yeast Genome Expression in Response to Environmental Changes. Mol Biol Cell. 2001;12:323–337. doi: 10.1091/mbc.12.2.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIsaac KD, Wang T, Gordon DB, Gifford DK, Stormo GD, Fraenkel E. An improved map of conserved regulatory sites for Saccharomyces cerevisiae. BMC Bioinformatics. 2006;7:113. doi: 10.1186/1471-2105-7-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira MC, Monteiro P, Jain P, Tenreiro S, Fernandes AR, Mira NP, Alenquer M, Freitas AT, Oliveira AL, Sá-Correia I. The YEASTRACT database: a tool for the analysis of transcription regulatory associations in Saccharomyces cerevisiae. Nucl Acids Res. 2006;34:D446–D451. doi: 10.1093/nar/gkj013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbison CT, Gordon DB, Lee TI, Rinaldi NJ, Macisaac KD, Danford TW, Hannett NM, Tagne JB, Reynolds DB, Yoo J, Jennings EG, Zeitlinger J, Pokholok DK, Kellis M, Rolfe PA, Takusagawa KT, Lander ES, Gifford DK, Fraenkel E, Young RA. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431:99–104. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Zhu L, Guo J, Fu C, Zhou H, Dong D, Li Z, Zhang DY, Lin K. SPIDer: Saccharomyces protein-protein interaction database. BMC Bioinformatics. 2006;7:S16. doi: 10.1186/1471-2105-7-S5-S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mewes HW, Frishman D, Guldener U, Mannhaupt G, Mayer K, Mokrejs M, Morgenstern B, Munsterkotter M, Rudd S, Weil B. MIPS: a database for genomes and protein sequences. Nucleic Acids Res. 2002;30:31–34. doi: 10.1093/nar/30.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendenhall W, Sincich T. Statistics for Engineering and the Sciences. 4. Englewood Cliffs: Prentice-Hall; 1995. [Google Scholar]
- Hohmann S, Mager WH. Yeast Stress Responses. New York: Chapman & Hall; 1997. [Google Scholar]
- Martinez-Pastor MT, Marchler G, Schüller C, Marchler-Bauer A, Ruis H, Estruch F. The Saccharomyces cerevisiae zinc finger proteins Msn2p and Msn4p are required for transcriptional induction through the stress response element (STRE) EMBO J. 1996;15:2227–2235. [PMC free article] [PubMed] [Google Scholar]
- Boy-Marcotte E, Lagniel G, Perrot M, Bussereau F, Boudsocq A, Jacquet M, Labarre J. The heat shock response in yeast: differential regulations and contributions of the Msn2p/Msn4p and Hsf1p regulons. Molecular Microbiology. 1999;33:274–283. doi: 10.1046/j.1365-2958.1999.01467.x. [DOI] [PubMed] [Google Scholar]
- Eastmond DL, Nelson HC. Genome-wide analysis reveals new roles for the activation domains of the Saccharomyces cerevisiae heat shock transcription factor (Hsf1) during the transient heat shock response. J Biol Chem. 2006;281:32909–32921. doi: 10.1074/jbc.M602454200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JM, Adler C, Ball C, Chervitz SA, Dwight SS, Hester ET, Jia Y, Juvik G, Roe T, Schroeder M, Weng S, Botstein D. SGD: Saccharomyces Genome Database. Nucleic Acids Res. 1998;26:73–80. doi: 10.1093/nar/26.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama K, Izawa S, Inoue Y. The Yap1p-dependent induction of glutathione synthesis in heat shock response of Saccharomyces cerevisiae. J Biol Chem. 2000;275:15535–15540. doi: 10.1074/jbc.275.20.15535. [DOI] [PubMed] [Google Scholar]
- Mori K, Kawahara T, Yoshida H, Yanagi H, Yura T. Signalling from endoplasmic reticulum to nucleus: transcription factor with a basic-leucine zipper motif required for the unfolded protein-repsonse pathway. Genes Cells. 1996;1:803–817. doi: 10.1046/j.1365-2443.1996.d01-274.x. [DOI] [PubMed] [Google Scholar]
- Jung US, Sobering AK, Romeo MJ, Levin DE. Regulation of the yeast Rlm1 transcription factor by the Mpk1 cell wall integrity MAP kinase. Mol Microbiol. 2002;46:781–789. doi: 10.1046/j.1365-2958.2002.03198.x. [DOI] [PubMed] [Google Scholar]
- Miyahara K, Hirata D, Miyakawa T. yAP-1- and yAP-2-mediated, heat shock-induced transcriptional activation of the multidrug resistance ABC transporter genes in Saccharomyces cerevisiae. Curr Genet. 1996;29:103–105. doi: 10.1007/BF02221572. [DOI] [PubMed] [Google Scholar]
- Shakhnovich BE, Reddy TE, Galinsky K, Mellor J, Delisi C. Comparisons of predicted genetic modules: identification of co-expressed genes through module gene flow. Genome Inform Ser Workshop Genome Inform. 2004;15:221–228. [PubMed] [Google Scholar]
- Güldener U, Münsterkötter M, Kastenmüller G, Strack N, van Helden J, Lemer C, Richelles J, Wodak SJ, García-Martínez J, Pérez-Ortín JE, Michael H, Kaps A, Talla E, Dujon B, André B, Souciet JL, De Montigny J, Bon E, Gaillardin C, Mewes HW. CYGD: the Comprehensive Yeast Genome Database. Nucleic Acids Res. 2005;33:D353–368. doi: 10.1093/nar/gki053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faires JD, Burden R. Numerical Methods. 2. Pacific Grove: Brooks/Cole Publishing Company; 1998. [Google Scholar]
- Wu WS, Chen BS. Identifying stress transcription factors using gene expression and TF-gene association data. Bioinformatics and Biology Insights. 2007;1:9–17. doi: 10.4137/bbi.s292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson R. System modeling and identification. Englewood Cliffs: Prentice-Hall; 1993. [Google Scholar]
- Chang YH, Wang YC, Chen BS. Identification of transcription factor cooperativity via stochastic system model. Bioinformatics. 2006;22:2276–2282. doi: 10.1093/bioinformatics/btl380. [DOI] [PubMed] [Google Scholar]
- Wu WS, Li WH, Chen BS. Reconstructing a network of stress-response regulators via dynamic system modeling of gene regulation. Gene Regulation and Systems Biology. 2008;2:53–62. doi: 10.4137/grsb.s558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu WS, Li WH, Chen BS. Identifying regulatory targets of cell cycle transcription factors using gene expression and ChIP-chip data. BMC Bioinformatics. 2007;8:188. doi: 10.1186/1471-2105-8-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HC, Lee HC, Lin TY, Li WH, Chen BS. Quantitative characterization of the transcriptional regulatory network in the yeast cell cycle. Bioinformatics. 2004;20:1914–1927. doi: 10.1093/bioinformatics/bth178. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1 contains the detailed information of the 29 identified heat-inducible GRMs, the 182 identified heat-inducible genes, the 68 identified uncharacterized genes and the performance comparison of five module inference tools.