

Researchers, institutional review boards (IRBs), participants in human subjects research, and their families face an important but largely neglected problem — how should incidental findings (IFs) be managed in human subjects research. If researchers unexpectedly stumble upon information of potential health or reproductive significance, should they seek expert evaluation, contact the participant’s physician, tell the research participant, or respond with some combination? What should consent forms and the entire consent process say about how IFs will be handled in research? What should IRBs require?
An IF is a finding concerning an individual research participant that has potential health or reproductive importance and is discovered in the course of conducting research but is beyond the aims of the study. This means that IFs may be on variables not directly under study and may not be anticipated in the research protocol. Examples include:
an IF on a genomic microarray suggesting a genetic or chromosomal variant of potential clinical importance beyond the variants or genotypephenotype associations directly under study,
an IF of misattributed paternity or parentage in a genetic family study,
an unexpected mass or aneurysm visualized in the course of structural magnetic resonance imaging (MRI) of the brain, and
an unexpected mass at the base of the lung discovered in computed tomography (CT) colonography.
We focus here on IFs discovered in the course of research, not clinical care. A significant literature already addresses incidental or accidental findings discovered in the course of non-research clinical care or screening (e.g., an adrenal tumor serendipitously discovered, sometimes called an “incidentaloma”).1 However, attention to IFs in research is at an earlier stage. No consensus exists as yet on how to handle them.
Research IFs can arise in collecting and analyzing research images and data, but may also arise in determining whether a potential research participant qualifies for inclusion in the study population or in collecting baseline physiological information. Examples of eligibility and baseline IFs include discovery of an anomalous EKG of potential clinical concern in determining whether a potential research participant qualifies as a normal control for a cardiac study, or similar unexpected findings of abnormal blood pressure, blood chemistry, or pregnancy.
We focus here on physiological and genetic IFs, rather than social and behavioral IFs. An example of the latter would be observed signs of alcohol abuse in an adolescent serving as a research participant in a functional MRI (fMRI) study of adolescent cognition that is unrelated to alcohol use. Other examples would include signs of physical abuse or suicidality in studies unrelated to those phenomena. Social and behavioral IFs raise somewhat different issues. For instance, some may be harder to ascertain than physiological and genetic IFs, and law may compel reporting signs of abuse to authorities.
We further focus on IFs in two major research domains, genetic/genomic research and imaging
An IF is a finding concerning an individual research participant that has potential health or reproductive importance and is discovered in the course of conducting research but is beyond the aims of the study. This means that IFs may be on variables not directly under study and may not be anticipated in the research protocol.
research. Commentators are beginning to recognize the tremendous importance of IFs in genetic and genomic research, particularly as we now face a genomic revolution in medicine.2 Genetic family research actually yielded one of the earliest discussed types of IF, misattributed paternity.3 With the later growth of genomic research and now genome-wide studies, substantial debate exists on whether to return individual research results to participants.4 Less discussed is the equally important question of whether to return IFs, including IFs that emerge in reanalysis of archived datasets.
Comparison to IFs in imaging studies is instructive. Defining IFs in imaging studies tends to be easier (e.g., an extracolonic finding in CT colonography research). Further, both empirical study of IFs and normative discussion of how best to handle them are more advanced in imaging than in genetic and genomic research.5 Our imaging comparisons focus on neuroimaging research using MRI and CT colonography research, two specific domains in which progress has been made on IFs.
We consequently analyzed:
IFs in genetics/genomics, examining a range of research methodologies from (a) genetic family studies to identify genetic and/or chromosomal variants associated with phenotypic disease, susceptibility, or carrier status; to (b) large-scale genomic analysis of stretches of the human genome including whole-genome analysis (WGA) (or genome-wide association studies (GWAS)) often using some type of microarray chip in order to identify genetic, genomic, and/or chromosomal variants; to
IFs in imaging, using as our examples anatomic imaging of the brain by MRI (whether in structural studies of the brain or as a structural prelude to functional studies of brain activation in fMRI) and imaging of the colon and extracolonic torso in CT colonography.
These comparisons allowed us to explore the contrasts among these research domains shown in Table 1.
Table 1.
Research Domains Studied to Form Recommendations on Handling Research IFs
| Researcher | Subjects | Settings | IFs — key examples | |
|---|---|---|---|---|
|
Genetic family studies |
M.D. or Ph.D. |
|
Clinical or non-clinical |
|
| Genomic microarrays | M.D. or Ph.D. |
|
Clinical or non-clinical Reanalysis of archived data |
|
| MRI of brain | Ph.D. or M.D. (often non-physician Ph.D.) |
|
Often non-clinical |
|
| CT colonography | M.D. because requires invasive procedure with colon insufflation |
|
Clinical |
|
This paper thus offers recommendations for how to anticipate and manage IFs in genetic and genomic research and in imaging research, focusing on neuroimaging using MRI and CT colonography as examples. However, our analysis suggests broader application to other domains of human subjects research.
A growing literature calls for guidance on how to manage research IFs and attempts to document the prevalence of IFs in different kinds of research. Studies vary in their methodology and sample, yielding a wide range of prevalence figures, but nonetheless suggesting that researchers face the IF problem with some frequency. Thus, the literature reports:
an IF of misattributed paternity at a prevalence often cited at 10% for the general population, though this figure is hard to verify and a range of numbers have been found in different populations in both research and non-research settings,6
an IF in 13%-84% of brain fMRI or MRI scans,7 and
an IF defined as an extracolonic findings in 15%-89% of participants’ images.8
Clearly, more research is needed to clarify prevalence for different kinds of IFs in different research populations including affected participants and normal controls. However, the data to date suggest that researchers and IRBs should anticipate IFs and consider in advance how best to manage them.9
Research IFs raise difficult questions including:
Do researchers, including non-M.D. researchers, have an obligation to examine their data for IFs and recognize them?
What should researchers do once they see a suspected IF? For example, should they seek a consult with a specialist with expertise in clinically evaluating the data or scan in question (e.g., a radiologist, neuroradiologist, or a clinical geneticist)? In the case of a genetics IF, should they seek testing from a genetics laboratory approved to perform clinical tests under the Clinical Laboratory Improvements Amendments (CLIA)?10
What, if anything, should the research participant be told?
What should the guardian of the minor participant or representative of an adult participant with diminished mental capacity be told?
What should research protocols and consent forms say about how IFs will be handled and what should IRBs require?
These are some of the key questions we addressed in performing a multi-disciplinary evaluation of how IFs are being approached now and should be handled. We examined the ethical, legal, scientific, and clinical questions. This paper presents our normative conclusions, schooled by empirical analysis of the guidance on IFs currently available to researchers. Thus, we analyzed whether and how model research consent forms on the websites of the 100 universities receiving the most National Institutes of Health (NIH) research funding currently address IFs; whether and how research consent forms publicly available on the Internet currently address IFs; what federal authorities recommend, if anything (including NIH, the Food & Drug Administration (FDA), Centers for Disease Control (CDC), and Department of Veterans Affairs (VA)); and what key professional societies germane to the research domains on which we focus recommend, if anything. Those data are reported and analyzed elsewhere.11 However, they show that there is little guidance available on how to handle IFs in research and no consensus as yet on the best approach. We sought to address this problem.
I. How Do IFs Arise?
We found that IFs arise somewhat differently in each research domain on which we focused.
A. Genetic Family Studies
Individuals, couples, and families may seek genetic analysis in the course of clinical care and reproductive planning.12 Here, however, we are focusing on genetic family studies in the course of research. Researchers may identify a genetic or chromosomal variant that they suspect causes or increases susceptibility to phenotypic disease or disability; the researchers may then seek to study genetic and phenotypic patterns in an affected family or families to better understand the underlying genetics. Conversely, researchers may start with a phenotypic disease or disability and pursue genetic analysis of a family or families to seek the genetic cause or susceptibility.
Typically in such studies, researchers seek to perform genetic analysis of family members known to be affected as well as others in the family to clarify whether the latter are affected, carriers, or unaffected. The family pattern, or pedigree, may shed light on the underlying genetics. The genetics itself may be clarified through linkage analysis, molecular DNA analysis, analyzing the metabolic products of genetic variants, or chromosomal analysis (by a range of methods including karyotyping and array comparative genomic hybridization (aCGH)). Thus, although we discuss below large-scale studies using genomic microarrays, microarrays may also be used in smaller-scale family genetic studies.
In the course of performing family studies, misattributed paternity or other misattributed lineage may be discovered by the researchers.13 For example, if a child is affected by a disorder recessively transmitted and the mother but not the father is a carrier, this will suggest misattributed paternity. If neither parent is a carrier, this may suggest undisclosed adoption, embryo donation, or some other scenario in which the rearing parents are not the genetic parents. Though studies report roughly a 10% incidence of misattributed paternity, there is wide agreement that this figure is poorly supported and more data are needed.14 Perhaps the most ethically challenging finding of misattributed paternity in a genetic family study would reveal incest; we have found no incidence figures on this. Ravitsky and Wilfond also note misattributed ethnic or cultural identity in ancestry studies and a finding bearing on genetic basis of tribal affiliation.15
A second form of IF in genetic family research is unexpected discovery of a genetic or chromosomal variant of potential clinical concern that is not the variant under study. Chromosomal analysis could reveal unexpected abnormalities in chromosomal regions not under study. Similarly, linkage analysis could reveal unexpected mutations in the regions under study or nearby regions. We have found no studies reporting the incidence of this kind of research IF.
A substantial amount of genetic family research is performed in research labs that are not CLIA-certified to perform testing for clinical diagnosis. For some genetic conditions under study there simply is no CLIA-approved laboratory offering testing.16 According to Gene Tests (www.geneclinics.org), genetic testing is available for approximately 1500 diseases in its database, but for approximately 20% of those conditions, genetic testing is only available in a research laboratory.17 This means that some IFs will be found by labs that are not certified to perform clinical genetic testing and further, that for some of those genetic IFs, no confirmatory testing by a CLIA-approved laboratory will be available.
B. Large-Scale Genomic Studies Using Microarrays
While genetic studies tend to focus on one gene or a small number of genes, genomic studies focus on many genes and their interaction by studying segments of the genome. Increasingly, research involves collecting data on the entire genome (as in GWAS). Genomic research tends to involve large numbers of subjects in population-based investigation of the relationship between genomic and phenotypic variations. Typically these studies utilize genomic microarrays to allow efficient analysis of large numbers of data points in many subjects.
Genomic microarrays use chips (pieces of DNA or RNA (probes) deposited in an array on a solid surface such as a microscope slide) to analyze genetic or chromosomal variants over large stretches of the genome. There are several kinds of microarrays, including those capturing data on DNA and on RNA expression and microarrays for analysis of cytogenetic abnormalities. Microarrays perform analysis at different levels of genomic resolution depending on the probes used and how the applicable software analyzes the data points. Thus, microarray analysis may reveal the sequence of base pairs, the presence of genes, gene expression, or chromosomal variation.
A chip may target only areas under study or may not be targeted and indeed may cover the full genome. Even a targeted chip may be designed to include genomic regions bordering the region of focal concern and so may pick up IFs in those adjacent regions. The chip is coupled with computer software to perform the analysis. Even when the chip itself is not targeted to areas under study, the software may be designed to mask all except the domain to be analyzed. Here again, however, the software may analyze regions adjacent to those directly under study. Though some laboratories design their own chip and software to address their research questions, others purchase commercially available products, which may or may not be tailored to the research question at issue.
The potential for genomic microarrays to generate IFs depends on the research question under investigation. If microarray analysis is used to identify genomic patterns associated with certain phenotypic pathologies or susceptibilities, then an IF would be a genomic pattern of potential clinical concern beyond those patterns under study. This could include copy-number variants (CNVs), genetic insertions, deletions, and duplications whose meaning is not clear but whose deviation from normal is great enough to raise health concerns.18 If the chip is not targeted to the domains under study and the software does not mask other results, the opportunities for IFs will increase. However, even if the chip is targeted or the software masks other results, unexpected patterns not under study in the genetic and chromosomal regions being examined may yield IFs, as may unexpected pleiotropy (the phenomenon whereby a single gene can code for multiple phenotypic traits, such as in the case of APOE alleles, which can affect susceptibility to both cardiac and Alzheimer disease, so that research on the genetics of susceptibility to cardiac disease may thus also reveal susceptibility to Alzheimer disease). In addition, IFs may appear in analysis of boundary regions, as noted above.
Microarrays can also be used in research ranging over large stretches of the genome or the whole genome to seek associations between genetic patterns and phenotypic pathology in populations. Increasingly, tissue sample biobanks and DNA databanks are being set up to facilitate this kind of large-scale genomic epidemiology often pursued as “discovery research.”19 In such discovery research it is harder to identify what might be an IF, as any genomic pattern correlating with pathology may be captured and studied. However, if the declared aim of genomic research analysis is to study certain pathologies (e.g., cardiac illness, high blood pressure, or asthma), genomic patterns suggesting other clinical concerns for an individual may be considered IFs.
We have found almost no literature on IFs in genomic microarray analysis and no studies of incidence. One recent article argues statistically that genomic medicine using microarray analysis can be expected to produce an abundance of IFs, many of which will turn out to be false positives in normal populations.20 This study, however, did not focus on research IFs distinguished from clinical IFs.21
Much genomic analysis will be conducted in research labs that are not CLIA-approved to perform clinical genetic testing.22 Thus, IFs may be discovered in such labs and, depending on the genetic IF of concern, confirmatory testing by a CLIA-approved lab may or may not be available.
C. MRI of the Brain
Structural MRI of the brain reveals anatomical structures in the brain and elsewhere in the skull, depending on the field imaged. Although MRI is not the only neuroimaging methodology (others include CT scans, positron emission tomography (PET) scans, and single photon emission computed tomography (SPECT) scans), MRI research is sufficiently active that it has yielded the most developed discussion of IFs to date.23 This may be because, as compared to other imaging technologies, MRI is more commonly used to study normal populations due to its signal characteristics and lack of ionizing radiation. The literature on IFs in MRI research on the brain focuses on structural (as opposed to functional) anomalies of potential health concern on MRI scans. Data on what constitutes normal versus anomalous function in the brain are not yet robust enough to discriminate reliably functional anomalies of potential clinical concern. Thus, we focus on structural IFs generated by MRI, whether generated by structural or functional imaging.
Whenever the brain and other contents of the skull are imaged, anatomical malformations, masses, evidence of cranial bleed or stroke, evidence of infection, evidence of injury, and evidence of dementia may be discovered. However, research scans are typically not optimized to image these IFs; the scan sequences are different in research, yielding less detailed information about brain anatomy.24 Further, researchers investigating the brain using MRI may not be trained to interpret the scan clinically; they may not be neuroradiologists, radiologists, or even physicians. Much neuroimaging research is conducted by Ph.D.s, particularly functional MRI research. Even the broader research team may not include someone trained to read scans clinically. Thus, IFs may arise when a Ph.D. principal investigator, co-investigators, or students read a research scan but happen to notice something that looks unusual.
As noted above, studies report IF prevalence rates of 13%-84%. Prevalence may be affected by the study population, scanning protocol, and definition of IF. IFs are classified as needing immediate referral, urgent referral, routine referral, or no referral. (See Table 2.) IFs needing immediate referral were found in up to 1.2% of participants; IFs needing urgent referral were found in 0.4% to 14% of participants; IFs needing routine referral were found in 1.8% to 43% of participants; and IFs needing no referral were found in 13% to 40.4% of participants.25
Table 2.
Comparison of Classification Systems for Incidental Findings in Imaging
| Neuroimaging* | CT Colonography** |
|---|---|
| Need for immediate referral for clinical evaluation | E4 — “Potentially Important Finding”;“Communicate to referring physician” |
| Need for urgent referral | |
| Need for routine referral | E3 — “Likely Unimportant Finding, Incompletely Characterized”; “work-up may be indicated” |
| No need for referral | E2 — “Clinically Unimportant Finding”; “No work-up indicated” E1 — “Normal Exam or Anatomic Variant” |
B. Kim et al., “Incidental Findings on Pediatric MR Images of the Brain,” AJNR American Journal of Neuroradiology 23, no. 10 (2002): 1674-1677, at 1675.
M. E. Zalis et al., “CT Colonography Reporting and Data Systems: A Consensus Proposal,” Radiology 236, no. 1 (2005): 3-9, at 8. Category E0 is “Limited Exam.”
Recommendations to date on how to handle IFs in neuroimaging focus on issues including whether researchers have a duty to seek consultation from a neuroradiologist or radiologist to determine whether an IF requiring further clinical attention is present and to whom the IF should be reported.26
D. CT Colonography Research
CT colonography is an imaging technology moving rapidly into clinical use. However, significant research continues including The National CT Colonography Trial (“ACRIN 6664”).27 Unlike traditional colonoscopy, which invasively visualizes the colon and rectum after bowel cleansing and sedation by intubating the colon with an endoscope, CT colonography uses low-dose CT scans to image the colorectum following bowel preparation and air insufflation. CT colonography images the entire pelvis and abdomen as well as the lung bases. CT colonography thus has the capacity to identify IFs throughout the torso. Indeed, a recent report compiling the prevalence of extracolonic findings found in a number of studies shows that 40% of patients are recorded as having IFs.28 These IFs include anatomical malformations, masses, aneurysms, evidence of infection, and evidence of injury or trauma. However, CT colonography at a low-radiation dose and without the use of intravenous or oral contrast material may fail to detect important extracolonic low-attenuating lesions (e.g., gastric carcinoma).29
Our analysis of key ethics sources concludes that researchers have an obligation to address the possibility of discovering IFs not only in their protocol and communications with the IRB, but also in their consent forms and communications with those being recruited to the study and research participants.
Because CT colonography involves insufflation of the colon through a rectal catheter and carries a small risk of colonic perforation,30 the procedure is performed in a clinical setting. The exam itself is generally performed by a radiologic technologist and nurse (or radiologist), with the researcher-physician interpreting CT colonography datasets at an off-line computer workstation. Depending on the study design, CT results may be communicated to an endoscopist prior to subsequent colonoscopy to provide a comparison reference standard. Research participants in CT colonography studies may be at normal or at increased risk for colon cancer, symptomatic or asymptomatic, or may be referred following an incomplete endoscopy. In this research context, then, IFs generally refer to extracolonic findings (those outside of the colon) or less commonly colonic findings unrelated to colonic neoplasia (e.g., inflammatory bowel disease). Note that some CT colonography studies such as ACRIN 6664 have prospectively included evaluation of extracolonic findings as a specific sub-aim of their study, so that IFs in this research context are greatly reduced by the broad aims of the study.
Research CT colonography datasets are generally interpreted by specialized abdominal radiologists, who are familiar with the clinical implications of IFs and potential follow-up tests. Faced with a scan that images the entire torso, research colonographers routinely examine the colorectal data for neoplasia and extracolonic abdomen and pelvis for other findings of potential health significance. Extracolonic findings in research colonography are considered IFs and are usually dictated at the time of the procedure.31 IFs of high clinical significance, defined as those requiring medical or surgical attention,32 are communicated to the participant or the participant’s physician through a variety of mechanisms, including a formal clinical report, a letter or fax to the physician, or direct participant contact.33 While many colonography researchers report discussing the potential for IFs with research participants prior to CT scanning, fewer researchers have included an explanation of IFs in their written consent form.34 The cost of diagnostic imaging to follow up on IFs is estimated to be approximately $24–$34 per participant in the study.35
A scale for grading and reporting extracolonic findings (E0 to E4) based on their significance and specificity (“C-RADS”) has been developed by the Working Group on Virtual Colonography.36 (See Table 2.) An E0 indicates “limited exam” and an E1 a “normal exam or anatomic variant.” An E2 marks a “clinically unimportant finding” for which a work-up is not indicated. An E3 finding signals a “likely unimportant finding, incompletely characterized” for which a “work-up may be indicated.” Most serious is an E4 designation, for a “potentially important finding;” reporting the finding to the referring physician is required. This scale tries to tailor response to the probable gravity of the finding and avoid over-reporting of unimportant findings, as these can lead to unnecessary costs and burdens of work-up, participant anxiety, and even harm resulting from the follow-up tests.
It is tempting to define IFs in CT colonography simply on the basis of anatomical location, that is, all findings outside the colon. However, some findings within the colon may be unexpected and beyond the variables of immediate concern. This would include structural abnormalities (e.g., diverticulitis) unrelated to disease processes of concern, objects in the colon, and evidence of injury or trauma (e.g., perforation at prior endoscopy).
As noted above, studies have found IFs in 15% to 89% of study participants, depending on the population’s risk profile. Though the majority of these findings are considered clinically insignificant, approximately 10% of participants have an extracolonic finding of potential medical significance needing further clinical response.37 Thus, radiologists have begun to address the question of which IFs are insignificant and should not be reported. Some CT colonography researchers have adopted a practice of reporting only highly significant IFs, as some radiological findings are nonspecific or common.38 Further, the Fleischner Society of thoracic radiologists now recommends that for non-smokers at low risk, no follow-up surveillance
If an IF is identified and thought to merit a clinical evaluation, it may turn out to be a false-positive, once the suspected pathology is ruled out, or it may yield ambiguous results at work-up, with pathology neither verified nor ruled out. In both of these cases, identification of the IF yields burden with no clear benefit.
is needed when an incidental pulmonary nodule of 4 mm or less is discovered at CT.39 This is part of a larger emerging debate in CT colonography over the net benefit or burden of identifying specific extracolonic findings.40
E. Reanalysis of Archived Data
Each of these research domains may generate data archived for future reanalysis. In genetic family research, the blood or other cells analyzed may be preserved or the genetic data derived from samples may be archived to allow further analysis. Genomic micro-array research is particularly likely to yield datasets archived for future reanalysis. Indeed, researchers may intentionally gather more data than needed for their own research in order to permit further analysis later. A number of researchers, research institutions, and commercial laboratories are establishing DNA biobanks to permit successive analyses over time and important public databases have been established, with requirements that researchers deposit their data.41 Neuroimaging and CT colonography datasets are generally archived digitally on computer hard drives, or preserved as part of the patient record using PACS (picture archiving and communication systems) along with clinical radiological images.42
Archived data preserved for reanalysis in future research studies raise the question of what constitutes an IF in the future study — should it be defined with reference to the aims of the original study or the reanalysis or both? Further, what obligations do secondary researchers (and those following) have to identify and report IFs? Do the original researchers who collected the data retain an obligation to the research participants that would create ongoing duties with respect to subsequently identified IFs?43 Underlying these questions is the issue of whether these datasets should be anonymized and if so, to what degree.44
If the data are fully anonymized and identification of research participants is impossible, even for the original researchers, then IFs cannot be reported to individual research participants. Moreover, the research no longer meets the definition of human subjects research in the federal Common Rule setting ethics standards for human subjects research.45 However, archived data may be less fully anonymized, so that secondary researchers have access to identifying information or a code rendering subjects identifiable. Secondary researchers may have direct access to that information, access through the original researchers, or access through an independent intermediary. Controversy surrounds research on data that the secondary researcher cannot identify but that the original researchers or an independent intermediary can indeed identify because they hold the code: does this research qualify as human subjects research and impose obligations accordingly, even on the secondary researchers?46
At the bottom line, whenever archived data are not fully anonymized (that is, the original researchers, secondary researchers, or an independent intermediary can identify individual research participants), the IF problem may remain. Indeed, one can argue that data should not be anonymized simply for the purposes of avoiding the IF problem, as this may deprive research participants of potential clinical benefit.47 NIH policy for genome-wide association studies (GWAS) conducted or supported by NIH provides for later submission to a GWAS repository without identifiable information but in coded form, with the keys held by submitting institutions. Thus, “the NIH GWAS data repository and secondary data users...will not be able to return individual results directly to subjects. Secondary investigators may share their findings with primary investigators, who may determine whether... to return individual or aggregate research results to participants whose health may be affected....”48 Consequently, an IF likely to be life-saving or allow the research participant to avoid or ameliorate grave disease would raise the question of whether the institution holding the code should be notified of the IF to consider contacting the research participant. We consider this further below.
II. Framework for Recommendations
Researchers have ethical duties to research participants that derive from several primary sources. Federal regulations governing human subjects research (both the Common Rule used by a large number of federal agencies49 and variants at other agencies such as the FDA50) set substantive requirements for ethical research and a procedural system for applying them through local IRBs. However, a large literature on research ethics interprets those rules and bases further researcher obligations on additional statements of research ethics (e.g., by professional societies and international bodies)51 as well as analysis of the research participant’s vulnerability and the researcher’s special obligations toward participants.52
Our analysis of key ethics sources concludes that researchers have an obligation to address the possibility of discovering IFs not only in their protocol and communications with the IRB, but also in their consent forms and communications with those being recruited to the study and research participants. Researchers have a further obligation to establish a pathway for handling suspected IFs and to communicate that to the IRB and research participants. In many, but not all circumstances, researchers have an obligation to offer to report IFs to research participants. In the case of minor or incompetent participants, this duty obligates the researcher to offer IFs to the participant’s guardian or representative. In research on archived data not fully anonymized, IFs of high importance should prompt researchers to consider an effort to contact research participants; that effort may best be undertaken by the original researcher who had contact with the research participant rather than the secondary researcher who had none. We elaborate below.
It is important to recognize at the outset of our analysis that IFs raise the question of when the researchers should initiate evaluation and disclosure of information uncovered in research. There is a distinct debate on returning research information at the request of research participants. Federal privacy rules under HIPAA are relevant to the latter question,53 as is NIH and NHGRI policy.54 However, the rules on participant-initiated disclosure do not resolve the question of how researchers should define, evaluate, and handle IFs.
While federal regulations on human subjects research do not address IFs explicitly, a number of provisions apply. With studies increasingly documenting the prevalence of IFs, IFs are both a predictable risk and benefit of research. This dual character makes a number of protections germane.
First, federal regulations require that consent forms and the consent process address both risks and benefits of the research. Consent forms must describe “any reasonably foreseeable risks” and “any benefits.”55 For a research participant recruited as a normal control, discovery of an IF suggesting pathology may trigger anxiety, burdens, and the costs of further evaluation to verify or rule out a clinical problem. Even research participants with known pathology risk discovery of an unrelated IF, triggering the same. These risks are present whether or not discovery of the IF leads to a clinically useful diagnosis.
If an IF is identified and thought to merit a clinical evaluation, it may turn out to be a false-positive, once the suspected pathology is ruled out, or it may yield ambiguous results at work-up, with pathology neither verified nor ruled out. In both of these cases, identification of the IF yields burden with no clear benefit. Identification of such IFs is a predictable risk of research. However, some IFs will lead to diagnoses of clinical importance. Identifying an operable brain tumor in a college student serving as a normal control in an fMRI study of cognition, for example, may prove life-saving. For such a research participant, taking part in the study imposes both the risk of discovering an IF and potential benefit of discovering serious pathology in time to intervene.
These risks and potential benefits are intrinsic to research modalities that have the potential to yield information beyond the variables directly under study. Imaging research is a classic example, with fMRI neuroimaging research visualizing cranial anatomy and CT colonography research imaging extracolonic anatomy throughout the torso. But genetic family research similarly reveals genetic relatedness or nonrelatedness, even when that is not the object of the study and is unacknowledged within the family. Both genetic and genomic research may also reveal genetic or chromosomal variants of concern beyond the specific genes, chromosomes, or genomic relationships under study.
The risks and benefits of discovering IFs in research require explicit discussion in the consent process.56 Without that, research participants may not appreciate the risk of discovering an IF, being offered information they did not expect, and triggering an evaluation. They may also have unrealistic expectations on the benefit side. Here, two errors are likely. Participants may underestimate benefit by failing to appreciate that the burden of discovering an IF will in some cases be offset by ultimate clinical benefit. But more likely (given relative inattention to research IFs until recently) is that participants may overestimate benefit by expecting that any anatomy imaged or genetics/genomics being studied is thereby being screened for clinical problems. This is a new form of the well-documented therapeutic misconception, research participants’ mistaken assumption that research interventions will benefit them clinically.57 In the IFs context, the misconception is not that the research intervention (e.g., a drug under study) will yield clinical benefit, but that the research process itself (e.g., the imaging or genetic analysis) will yield such benefit. Research participants may assume that researchers will identify and report any clinical problems in anatomy imaged or genetic regions analyzed; researchers’ silence on the topic of clinical problems may be misinterpreted by research participants as a clean bill of health. Research participants may not appreciate that the MRI sequences and scans used to image their brain in a study were not optimized for clinical diagnosis, because the purpose was research. Participants in a genetic or genomic study similarly may not understand what genetic or chromosomal domains were not analyzed as part of the research and that the analytic tests used were not validated for clinical use.
Thus, it is essential to address the risks and benefits associated with IFs in the consent process. This is reinforced by regulatory provisions that IRBs may require consent forms to include a “statement that significant new findings developed during the course of research which may relate to the subject’s willingness to continue participation will be provided to the subject.”58 An IF suggesting a brain tumor or an aneurysm requiring immediate work-up may well affect a participant’s willingness to continue in neuroimaging research. In genetic or genomic research, discovery of an IF suggesting a serious genetic problem not under study may derail a participant’s willingness to continue in the research. In genetic family research, an IF revealing undisclosed adoption or other nonrelatedness may affect the participant’s willingness to continue in the study; indeed, that individual’s genetic makeup may no longer be relevant to the research.
So far, we have simply suggested that researchers should address with participants the risks and benefits associated with IFs. However, researchers need to do more, to address both with participants and with the IRB how IFs will be handled as part of the research protocol. Indeed, the pathway established for handling IFs will determine the character and magnitude of the risks and benefits involved. For example, a non-M.D. neuroimaging research team with no process for consulting a radiologist or neuroradiologist may both miss IFs of concern and misconstrue normal images as suggesting an IF. Further, with no process set up for fast consultation, they may not be able to provide the benefit of timely identification of an IF meriting immediate work-up.
Researchers have an obligation to set up a process for recognizing IFs, verifying whether there is indeed a suspicious finding of concern, and offering the finding to the research participant (or the guardian or representative of a minor or incompetent participant) for clinical evaluation and follow-up. The regulations require not only that risks to participants be communicated as part of the consent process, but that risks be minimized.59 This suggests that researchers should minimize several risks relating to IFs: the risk of failing to recognize an IF that may require clinical follow-up, the risk of identifying an IF but ignoring it or failing to offer information to the research participant, and the risk of communicating an IF to a participant causing anxiety and follow-up when more careful scrutiny shows no IF or the finding turns out to be benign. All of this suggests an obligation to set up a process for identifying, assessing, and communicating IFs.
That obligation is further supported by the regulatory requirement that risks be “reasonable in relation to anticipated benefits, if any, to subjects.”60 The risk of IFs is intrinsic to research generating any information beyond the variables directly under study. That risk must be offset by the benefits that can flow from timely identification, assessment, and communication of IFs, allowing clinical evaluation and intervention. We recommend below specific steps for managing IFs.
Recognizing researcher obligations to offer participants information on IFs of likely health or reproductive importance is consistent with an emerging view that researchers bear some clinical obligations toward research participants. Research ethics has traditionally drawn a sharp distinction between the responsibilities of health professionals rendering clinical care and the responsibilities of researchers. Clinicians’ duty of care has not been imposed on researchers. However, Richardson and Belsky have challenged this paradigm, arguing that research participants’ vulnerability and researchers’ discretion mean that researchers might owe participants a limited duty to provide ”ancillary care,” including evaluating research brain scans and then following up appropriately on life-threatening findings.61 They argue that researchers are neither personal physicians with a full-blown duty of care, nor “mere scientists” with no obligation of care, but instead occupy an intermediate category. Research participants entrust aspects of their welfare to researchers, and within that scope of entrustment, may be entitled to identification, evaluation, and communication of clinically important IFs.62
Note that this view would impose full duties of care on a personal physician acting as researcher toward the patient/participant, and lesser duties of care on the researcher who is not personal physician. Even the non-M.D. researcher would have duties by virtue
We strike a middle ground. We show respect for research participants’ objective welfare as well as their subjective interests by including IFs of likely health or reproductive importance to the participant. At the same time, we focus on participants’ health and reproductive interests, not all conceivable interests.
of the research participant’s trust, vulnerability, and dependency. The Ph.D. researcher performing fMRI research may alone have information suggesting that the participant’s brain may harbor a life-threatening aneurysm in need of work-up. Thus, that researcher may have a duty to share the scan with a neuroradiologist who can verify the presence of a suspicious finding of likely health importance, and then a duty to offer this information to the research participant for follow-up.
This view that researchers have some duties toward research participants including identifying, verifying, and communicating IFs of health or reproductive significance can be based on researcher duties to respect the autonomy and interests of research participants.63 Shalowitz and Miller, discussing research participants’ interest in being told individual research results of clinical importance, state that participants have a “presumptive entitlement to information about themselves.”64 In addition to respect for persons, Illes et al. cite reciprocity as a principle supporting an obligation to disclose IFs of potential health importance to research participants.65 Beauchamp and Childress discuss “a reciprocity-based justification for obligations of beneficence.”66 In the research context, reciprocity captures the notion that research participants are contributing to the research enterprise and are entitled to receive in return information about IFs of likely health or reproductive significance.
Recognizing a researcher duty to handle IFs responsibly and disclose them to research participants is also consistent with recent trends in the law. The Grimes case in Maryland held that researchers in that state had special obligations to research participants, citing international codes such as the Nuremberg Code and Helsinki Declaration as well as legal cases.67 Though the case has been controversial, the court concluded that a “special relationship” grows out of researchers’ knowledge of the risks that participants face and research participants’ vulnerability68 — this relationship grounds researchers’ duties toward research participants. In addition, researchers’ promises to participants in research consent forms can ground researcher duties toward participants.69 Research participants have brought a number of other lawsuits against researchers and their institutions claiming that a duty owed by the researcher was breached.70 The Office for Human Research Protections (OHRP) and other federal regulators and funders have long recognized that researchers have duties toward subjects; violation of those duties can provoke federal investigation and sanctions.71 The ethical and legal trend toward recognizing researchers’ duties toward participants is apparent.
Including among researcher duties an obligation to offer to disclose to participants IFs that have likely health or reproductive importance is consistent not only with legal recognition of researchers’ special obligations toward participants, but also with legal doctrine imposing a duty to warn of foreseeable harm.72 This doctrine is more familiar in the context of patient care, not research. However, it is based on recognizing that the physician may have unique access to information of health importance, the physician has obligations to prevent harm, and the patient is dependent upon the physician. All three of these propositions apply to researchers in their relationship to research participants (laying to one side, for now, the complex case of secondary researchers using archived data that their team did not collect). Certainly the researcher will have access to less information than the physician providing patient care, a more limited set of obligations that are grounded in averting harm in the research process, and usually a participant less dependent than is a patient relying on a physician for health care. This suggests that researcher obligations will be more limited, but that researchers do shoulder obligations that include the proper handling of unexpected information of potential health or reproductive importance, including disclosure to participants when potential harm may be averted.
Specifying how researchers should handle IFs to meet these obligations is challenging. It is instructive to compare the literature on offering individual research results to participants (as opposed to offering aggregate research results to a study population, as in a newsletter73). The literature on returning research results has evolved over time.74 (See Table 3.) In 1999, NBAC argued that disclosure of individual research results should be the exception, not the rule.75 Disclosure should occur only when findings are valid and confirmed, have significant health implications, and the health problem can be treated. In 2001 a CDC-sponsored group focusing on population-based genetic research echoed NBAC recommendations: “When the risks identified in the study are both valid and associated with a proven intervention for risk reduction, disclosure may be appropriate.”76 In 2004 an NHLBI Working Group considering return of genetic research results conditioned return on a significant risk of disease (specific relative risk >2.0), the disease having important health implications (fatal or substantial morbidity or significant reproductive implications), and the availability of therapeutic or preventive interventions.77 Debate continues on these issues. (As noted above, there is separate policy addressing research participants’ requests for research data — policy from DHHS under HIPAA78 and from NIH and NHGRI.79)
Table 3.
Comparison of Recommendations on Returning Individual Research Results
| National Bioethics Advisory Commission (NBAC)* |
Return results only if: (a) “the findings are scientifically valid and confirmed” (b) “the findings have significant implications for the subjects’ health concerns” and (c) “a course of action to ameliorate or treat these concerns is readily available.” |
| Centers for Disease Control (CDC)** |
Criteria for returning individual results in population-based genetic research: “When the risks identified in the study are both valid and associated with a proven intervention for risk reduction, disclosure may be appropriate.” |
| National Heart, Lung, and Blood Institute (NHLBI)*** |
Criteria for returning individual genetic results: (1) “The risk for the disease should be significant, i.e. relative risk>2.0. Variants with greater penetrance or associated with younger age of onset should receive priority.” Note: “Genetic test results should not be reported to study participants and their physicians as clinically valid tests unless the test(s) was performed in a CLIA certified laboratory. If the test was performed in a non-CLIA certified laboratory, a CLIA certified laboratory should be sought to confirm results by redrawing a sample and performing the test within the CLIA certified laboratory. Results reported by a research laboratory should be identified as ‘research’ results.” (2) “The disease should have important health implications, i.e. fatal or substantial morbidity or should have significant reproductive implications” and (3) “Proven therapeutic or preventive interventions should be available.” |
| National Research Council & Institute of Medicine (NRC & IOM)**** |
In human embryonic stem cell research, the duty to report individual research results “depends in large part on the reliability of the findings and the significance of the information to human health.” “CLIA regulations do not permit the return of research results to patients or subjects if the test were not conducted in a CLIA-approved laboratory.” |
| National Human Genome Re- search Institute (NHGRI)***** |
Upon their request, “[r]esearch participants should have access to experimental research data except when…[t]he research results are of unproven clinical validity, and the IRB has judged that there is no benefit to the research subjects.” |
National Bioethics Advisory Commission (NBAC), Research Involving Human Biological Materials: Ethical Issues and Policy Guidance (Rockville, MD: 1999), 1: at 72.
L. M. Beskow et al., “Informed Consent for Population-Based Research Involving Genetics,” JAMA 286, no. 18 (2001): 2315-2321, at 2320.
National Heart, Lung, and Blood Institute, NHLBI Working Group on Reporting Genetic Results in Research Studies, Meeting Summary, Bethesda, MD, July 12, 2004, available at <http://www.nhlbi.nih.gov/meetings/workshops/gene-results.htm> (last visited January 8, 2008).
National Research Council and Institute of Medicine Committee on Guidelines for Human Embryonic Stem Cell Research, Guidelines for Human Embryonic Stem Cell Research (Washington, D.C.: National Academies Press, 2005): at 89-90.
National Human Genome Research Institute, Federal Policy Recommendations Including HIPAA, available at <http://www. genome.gov/11510216> (last visited January 8, 2008).
While it is tempting to see IFs as merely a species of research results, there are key differences. As noted above, research results are on variables under study, the research aims to understand these data, and thus the researcher is likely to have whatever expertise exists to interpret those data. In contrast, because IFs are not on variables under study and may be beyond the researcher’s interpretive expertise, interpreting them may well require clinical experts beyond the research team. Second, the literature on whether to return individual research results commonly discourages returning results that lack clinical validity and clinical utility80; much of the debate focuses on results whose uncertain meaning and importance is the reason for the research. However, because IFs are not on variables under study, the key question will more often be whether the suspected anomaly (e.g., an unexpected tumor) is really there; if so, its health importance may be clear.
That said, aspects of the debate over offering research results are relevant. First, insistence on checking analytic validity and trying to establish clinical validity before offering research results suggests the importance of taking several steps when the research team identifies an IF of potential importance: (1) recheck the scan or data to confirm analytic validity (Is it this participant’s scan or analysis? Was the scan or analysis run properly? Should we run another sample?), (2) collaborate with an expert consultant (if the research team does not have adequate expertise) to confirm that there indeed is a suspicious finding and one of likely health or reproductive importance (analytic and clinical validity), and collaboratively determine whether the IF should be disclosed based on factors including its seriousness and likely importance to the research participant. (See Tables 4 and 5.) For example, a Ph.D. principal investigator on an fMRI study may well want a neuroradiologist to review the research scan and confirm the presence of a suspicious finding of likely health importance before the researchers offers this information to the research participant, triggering anxiety and follow-up. In the case of a genetic IF, step 2 may involve sending the sample to a CLIA-approved lab. Note, however, that for some conditions, testing in a CLIA-approved lab will not be available. This raises a problem: the NHLBI Working Group suggested that an IF could still be disclosed as long as it was labeled a research finding rather than a clinical finding and this was explained, but there is concern that this may not comport with CLIA’s restrictions.81 More work may need to be done to resolve this problem. As in the case of research results, a third step should involve evaluating the seriousness and likely utility of the IF to determine whether the IF should be disclosed, though we define “utility” to include informational importance to the research participant even if no treatment is available.82 We elaborate below.
Table 4.
Recommended Pathway for Handling IFs in Research
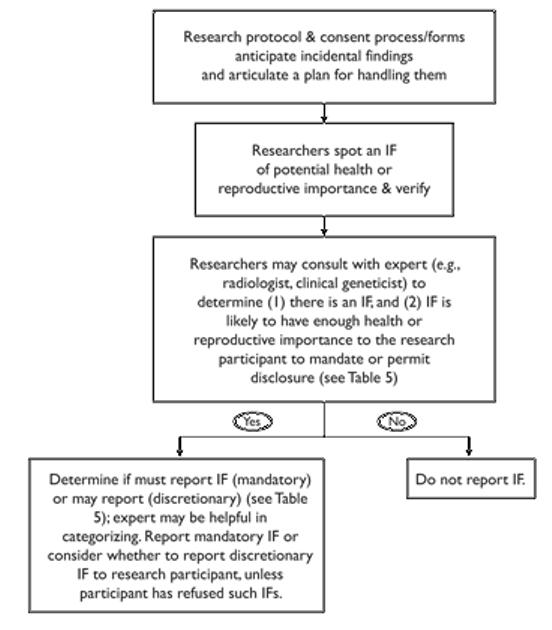 |
Table 5.
Recommended Classification of Incidental Findings
| Category | Relevant IFs | Recommended Action |
|---|---|---|
| Strong Net Benefit |
|
|
| Possible Net Benefit |
|
|
| Unlikely Net Benefit |
|
|
Second, the literature on returning research results cautions that such results should be offered to research participants, not foisted upon them.83 This is consistent with the literature on genetic testing in particular, which recognizes a right not to know results. This caution is probably appropriate for IFs as well, though researchers may understandably be hesitant to accept a research participant’s waiver of information about an IF likely to be life-threatening or grave and ameliorable, unless the participant appreciates that the information being waived may be of high health importance.
Third, the literature on offering research results states that results should have important implications for health in order to justify causing anxiety and follow-up in research participants and burdening researchers with the duty to offer these results. NHLBI’s Working Group urged that health importance includes significant reproductive implications; genetic or genomic data may lead participants to take steps to avoid serious as well as fatal genetic conditions for offspring.84 The requirement of health importance, including reproductive importance, would apply to IFs. However, determining what kind of findings would have such importance is not easy.85 Stanford’s Working Group on Reporting Results of Genetic Research distinguishes 3 categories of findings. Category I findings have “analytic validity, high clinical validity and utility and...a high probability and magnitude of harm resulting from not offering the information (i.e., life threatening, serious consequences...), and...effective preventive measures exist, or it is easy to avoid exacerbating risk factors.”86 Category II findings “do not rise to the level of Category I and do not fall into Category III.” Category III findings fail to “meet baseline analytic...or clinical validity standards.” The Working Group argues that Category I findings should be offered to participants, Category II findings may be offered, and Category III findings should not be provided even if requested by the participant. Other authors have similarly focused on analytic and clinical validity plus clinical utility or value to distinguish the kind of results that should be offered to research participants,87 though some commentators have argued for greater information sharing.88
This category framework cannot be imported wholesale to IFs. It was developed focusing on research results in genetic and genomic studies. There, the primary concern is giving research participants the results of genetic or genomic tests before the tests are validated and the phenotypic implications are understood, when most genetic and genomic results will not have immediate health implications. However, some IFs (especially from imaging studies) will indeed have immediate health implications. They will require clinical work-up and verification, at which point clinicians will often be able to use established clinical tests of clear validity. Unlike research results, whose ambiguity may be the very reason they are under study, a number of IFs may be entirely susceptible to clinical validation and management. The IF issues focus more on what duties researchers have to identify, evaluate, and communicate these findings of potential clinical importance.
Nonetheless, researchers properly note an IF of potential health or reproductive importance and then kick off the evaluation process (leading to decisions about the likely importance of the IF) when the suspected finding may affect the research participant’s health in the foreseeable future or affect the participant’s reproductive decisions. Emphasizing “health” importance means that we are addressing findings that a research participant would be likely to find important for their health care or health planning, not all findings that may change diet, lifestyle, or individual behavior. We are not suggesting that researchers become clinicians, but rather that when research unexpectedly yields information of likely importance to the participant’s health or reproductive decision-making, the researcher may have an obligation or discretionary option to communicate that information, depending on the seriousness of the finding.
In so defining those IFs that researchers may have a duty or option to disclose to research participants, we reject two extremes. At one extreme, researchers would disclose only IFs of established analytic and clinical validity, clear clinical utility, and grave health importance. This would anchor the category on what a clinician would deem highly significant to avert harm, ignoring the broader category of what a research participant might find important health or reproductive information. At the other extreme, researchers would have a duty to disclose any IF of analytic validity, so that the research participant could decide if the information was useful and important.89 This would anchor the category on what a research participant might find important for any reason. Under this definition, researchers would have a very broad duty to communicate findings, so broad that it would become difficult to distinguish some forms of research from therapeutic intervention.90
We strike a middle ground. We show respect for research participants’ objective welfare as well as their subjective interests by including IFs of likely health or reproductive importance to the participant. At the same time, we focus on participants’ health and reproductive interests, not all conceivable interests. Further, we envision that the researcher and expert consultant (e.g., a neuroradiologist reviewing a brain scan for a suspected IF) will make a determination of what a reasonable research participant would likely find relevant to their health or reproductive decisions. This determination can be individuated and guided by asking the research participant at the time of consent to participate in the research what categories of information they would like to receive.91 We thus recognize what Richardson and Belsky call researchers’ “ancillary-care responsibilities” (discussed more below), without turning researchers into clinicians.92 We also try to identify a reasonable and practical limit to researchers’ duties to identify, evaluate, and disclose IFs.
This approach is consistent with recommendations on offering research findings that recognize the importance of a finding’s validity and health or reproductive utility, but we define “utility” to include information that a research participant is likely to find important, even if clinicians cannot use that information to alter the participant’s clinical course. We thus recognize a spectrum of utility to the participant, ranging from life-saving to ameliorative to useful in heightening surveil-lance to useful in thinking and planning about health. This rejects an approach to utility grounded solely in what a clinician would find useful. We broaden “utility” to ask also what a research participant would find useful, recognizing not only treatment utility but also health or reproductive information utility. Working to harmonize recommendations of research findings and IFs is important because in some research contexts, such as discovery research using genomic microarrays, the line between research findings and IFs will be hard to discern. Our approach advances thinking in both realms by reconceiving what is properly meant by “utility,” recognizing that researchers may need to collaborate with expert colleagues to evaluate the validity and broad utility of research or incidental findings, and by recognizing some key differences between research findings and IFs, particularly the clear need to plan for prompt evaluation of IFs, possible disclosure to research participants, and clinical referral. We consider below how this chain of events should best occur to properly handle IFs, what duties devolve on researchers, and what actions are permissible even if not required.
III. Recommendations for Managing IFs
Our project group agreed on the following recommendations for managing IFs in human subjects research. Table 4 schematizes the pathway we recommend and Table 5 summarizes the categories we suggest for classifying IFs and determining what action to take.
It is unrealistic to place on researchers an affirmative duty to search for IFs. Researchers may not be qualified to screen for IFs; a Ph.D. researcher performing fMRI research cannot be expected to review scans with the expertise of a neuroradiologist. Further, the data with which the researcher is working may not allow researchers to spot the anomalies a clinician would who was using a clinically validated test.
A. Address IFs in the Consent Process
Researchers should anticipate the possibility of identifying IFs in the research process and should address this explicitly in the process of seeking research participants’ informed consent to be part of the research. Researchers should explain the potential for discovering IFs, offer examples of the kinds of IFs this type of research may yield, indicate the probability of discovering IFs when the literature or past experience yields statistics, and describe the steps researchers will follow to handle IFs (as discussed further below and indicated in Tables 4-5). By describing planned consultation to verify and evaluate IFs, researchers will be alerting research participants to the possibility of consultation with an expert beyond the research team and will be seeking the participant’s consent. Researchers should include this information concerning IFs on consent forms.
Researchers should elicit in the consent process whether each research participant wishes to be notified of IFs likely to offer strong net benefit or possible net benefit as indicated in Table 5. Thus, researchers should try to find out whether participants would want to learn of a condition (or significant genetic risk of a condition) likely to be life-threatening that can (or cannot) be treated, a condition (or significant genetic risk of a condition) likely to be grave or serious that can (or cannot) be treated, or genetic information that can be used in reproductive decision-making to avoid or to ameliorate a life-threatening, grave, or serious condition in offspring. Alternatively, researchers following our recommendations can tell research participants in the consent process what IFs the researchers intend to disclose or withhold (as indicated in Table 5) and offer research participants an opportunity to state a different preference. Research participants may assert a right not to know certain categories of information; that right is well-recognized in the genetics literature.93 However, researchers may decide to check back with a research participant in whom an IF reveals a life-threatening or grave condition that may be treated, but who has asserted at initial consent a preference not to know. Without revealing the information itself, the researchers may try to confirm that the research participant indeed wants to refuse even information of high health importance and utility.
Researchers should strive to use standard terms such as “incidental findings” in their protocol and consent documents. A confusing range of terms and definitions for IFs appear in the literature and on consent forms. Terms include “unexpected findings” and “extracolonic findings” (in colonography).94 Some discussions fail to distinguish between research results on variables under study and IFs.95 There is also a potential to confuse IFs with adverse events, though the latter more strictly refer to iatrogenic harm caused by the research intervention itself, such as morbidity or mortality resulting from taking a drug in a research protocol.96
Greater uniformity in terminology and definition would be advantageous. It would allow comparisons and meta-analysis across studies using similar research methods and across research methods. This would aid our understanding on a range of issues including the prevalence of IFs, what proportion turn out to be clinical findings of importance, and whether identifying IFs yields net benefit to research participants and at what cost.
Researchers and consent forms should define IFs as findings of potential health or reproductive importance that are beyond the aims of the study. This definition is better than “findings on variables not under study” because our definition would include IFs on variables or data points that were collected but not the focus of study (e.g., anatomy visualized on a scan but not under study, genes included in an untargeted genomic microarray but not under study, and chromosomes visualized in karyotyping but not under study). Our definition is also an improvement over “variables not planned for in the research protocol” because we urge that investigators and IRBs routinely anticipate IFs and create a plan for managing them.
What if the aims of a study evolve over time? IFs should be defined relative to the aims consented to by the research participants, as our definition suggests. It is they who will bear the health and psychological consequences if they are told of an IF, experience anxiety, potentially undergo follow-up, and benefit or suffer from identification of the IF. Similarly, it is research participants who will live with the consequences if an IF of importance is not identified or communicated to them. Thus, it is the research participant who most critically needs to understand through the consent process what this category of information is and how IFs will be handled by the investigators.
Research participants need to understand that research can uncover not only research results of potential health or reproductive importance, but also incidental findings of such importance. They need to know in the consent process how both categories of information will be handled. A given study may handle them differently. Genetic or genomic research, for example, yielding research results whose meaning is not validated and understood, may nonetheless uncover an IF of a well-understood mutation or chromosomal abnormality of clear health importance. In such a case, the investigators and IRB could reasonably decide not to return individual research results but to offer to disclose the IF to the research participant.
This suggests that research participants need to understand how both categories of information will be handled. Fairness to research participants means that the arrangement to which they consent should prevail unless and until investigators ask them to reconsent to a new arrangement.
B. Address the Potential for IFs in Future Analyses of Archived Data
What about research participants consenting to future reanalyses of their archived data? In some cases, complete anonymity of the data will make notifying participants of IFs impossible.97 Research participants should be told when they are asked to consent to future research if anonymity or anonymization will make reporting IFs impossible.98 However, the literature suggests that data should not be anonymized for the sole purpose of avoiding a possible responsibility to communicate research results or IFs,99 and some data will be archived and reanalyzed without full anonymization. An example is DNA databanks that follow research participants prospectively to correlate genotype and phenotype. Again, the terms of the research participant’s consent should prevail. If the participant consented to data collection relative to a certain set of research aims, with “research results” and “IFs” defined accordingly, then that arrangement and those definitions should prevail unless and until the research participant agrees to a modification. This means that a later study on data archived from an initial study could uncover information of potential health or reproductive importance on variables directly under study that would nonetheless be considered IFs under the terms of the first study and governing consent form.100
The logistics of identifying, evaluating, and communicating IFs will be more complex when archived data are analyzed by secondary researchers who did not collect the original data. The original researchers may have the only access to identifying information that would permit communication with individual research participants. Further, the original researchers may be the only researchers with a direct relationship with the research participants. In such cases, the original researchers may be best situated to communicate IFs to the research participants. However, there will be circumstances in which data are long-archived and it is not possible or realistic to work through the original researchers. Indeed, an intermediary — rather than the original researchers — may hold the codes allowing identification of research participants.101 In such cases, consulting with an IRB may be essential to devise a feasible way to contact research participants. It is important to plan ahead when data are first collected for archiving and future reanalysis, anticipating the possibility of later IFs.
A substantial literature discusses the ethics of recontacting research participants with research results of potential health importance.102 That literature acknowledges that recontact can be disruptive. Some research participants may wish to avoid recontact no matter how important the health or reproductive information to be conveyed. Others may wish recontact if the information may be valuable in preventing serious medical harm.103 Because individuals may differ on their willingness to be recontacted regarding IFs discovered in the future, they should be asked to consent to recontact. Most informative will be to ask
Whenever IFs are to be disclosed, they should be disclosed directly to the research participant. Some of the literature and consent forms available suggest that IFs should instead be disclosed to the research participant’s primary care physician. However, this gives the research participant no control over the information and compromises the participant’s privacy.
their recontact preferences for each category of information listed in Table 5 under “Strong Net Benefit” and “Possible Net Benefit.”
Realistically, recontacting or attempting to recontact research participants to communicate IFs discovered by secondary researchers using archived datasets may be difficult. Moreover, the passage of time from the initial data collection to discovery of an IF by secondary researchers may reduce the potential health significance of some IFs. Some limitations are appropriate. It is not unreasonable to limit attempts to recontact participants to IFs offering strong net benefit as defined in Table 5.
A standard this high may also be appropriate when considering whether to offer to disclose IFs discovered in research for which consent was never obtained. The Office for Human Research Protections (OHRP) has determined that research using previously collected specimens and data does not involve “human subjects” and so falls outside the scope of the Common Rule as long as the information was not originally collected for that research and the information is coded so that the individuals’ identifiers are not known to the investigator. 104 Such research protocols often involve patients’ data and specimens and are not required to undergo IRB scrutiny. Individuals may not even know the research is being conducted. Given the lack of consent and potential for surprise, it may be appropriate to limit attempts to contact these patients to IFs offering strong net benefit.
C. Plan for the Discovery of IFs
Researchers generally have no obligation to act as clinicians and affirmatively search for IFs. The goal of research is to seek generalizeable knowledge, not to provide health information to individuals. Thus, recommendations to date on handling IFs in neuroimaging state that researchers are not obligated to perform extra MRI scans or modify their scans to provide clinical information.105
It is unrealistic to place on researchers an affirmative duty to search for IFs. Researchers may not be qualified to screen for IFs; a Ph.D. researcher performing fMRI research cannot be expected to review scans with the expertise of a neuroradiologist. Further, the data with which the researcher is working may not allow researchers to spot the anomalies a clinician would who was using a clinically validated test.
An exception to the general proposition that researchers do not have a duty to search for IFs may occur when the researcher is also the research participant’s treating physician. A treating physician in a doctor-patient relationship has an obligation to use professional care in analyzing all information about a patient, even a patient who is also the doctor’s research participant. This does not mean that the physician-researcher is obligated to collect extra research data or do extra research scans or scans optimized for clinical diagnosis, but it does mean that in reviewing research data and scans the physician-researcher is obligated to spot IFs that a professional of his or her training would ordinarily recognize. Whether or not the researcher is also the treating physician, if the researcher or a member of the research team spots an IF of potential concern, the principal investigator bears a duty to handle this IF responsibly and promptly. An IF has the potential to reveal a condition that is serious or even life-threatening. As noted above, researchers’ duties of respect for research participants, duties to maximize benefits and minimize harms, duties to alert research participants to any developments that may affect their willingness to continue in the study, and more recently recognized duties of reciprocity toward research participants generously willing to bear the burdens of research for societal benefit all support a duty to attend to IFs rather than ignoring them.
The particular qualifications of the principal investigator (Ph.D., M.D., or other) do not change the obligation to plan for IFs and handle them responsibly. These duties fall on the principal investigator, not on a less experienced member of the team. Trainees cannot be assumed to have the expertise to handle IFs. This means that the principal investigator must instruct members of the research team to promptly communicate a suspected IF so that the principal investigator can handle the IF from that point.
In planning for the discovery of IFs, the researcher will need to consider how quickly members of the research team should bring a suspected IF to the attention of the principal investigator and how quickly the principal investigator should act to evaluate the IF. The researchers should consider what kinds of IFs the protocol may produce and how rapidly the identification and evaluation process needs to proceed to provide timely information to the research participant and avoid harm.
D. Plan to Verify and Evaluate a Suspected IF, with an Expert Consultant if Needed
Researchers should take steps to validate an IF and confirm its health or reproductive importance before communicating the finding to a research participant. Communicating an IF may provoke anxiety in the research participant and cause the participant to undertake clinical evaluation. Consequently, the researcher’s first step when faced with an IF of potential health or reproductive importance should be to examine the data or scan to confirm that an IF appears to be present, that the data or scans appear to have been created properly, and that they belong to a particular research participant. These steps begin to confirm analytic validity. The researcher may consider whether it is feasible to test another sample or compare another scan to see if the IF is still apparent.
The next step will be to seek confirmation that an IF appears to be present that is likely to have enough health or reproductive importance that its disclosure offers possible or strong net benefit, as described on Table 5. This is a reconfirmation of analytic validity and begins to address clinical validity as well as utility in the broader sense we have discussed above.
The researcher will often not have the expertise to make this assessment and will need to consult a clinical colleague who can review the research data or scan. The purpose of this review is not to generate a clinical diagnosis; the research data or scans often will not be adequate to that purpose. Instead, the limited goal of this review is to verify that there appears to be a suspicious finding that is likely to offer enough health or reproductive importance that notification of the IF may or even must be offered to the research participant. (See Table 5.) Note that the researcher will have initially identified an IF of potential importance and that the expert consultant will be helping confirm both the IF and its likely importance.
Thus, a genetics or genomics researcher may need to consult a clinical geneticist or genetic counselor, a neuroimaging researcher may need to consult a neuroradiologist, and a CT colonography researcher may need to consult an abdominal radiologist. Because some IFs will require urgent follow-up, this consultation pathway must be set up before beginning to enroll research participants and must be capable of generating a prompt consult. How quickly consultation may be needed depends on the type of IF a study may generate; neuroimaging studies that may reveal an aneurysm will require faster consultation capability than genetics studies that are unlikely to reveal an IF that requires urgent intervention. The neuroimaging IFs literature addresses planning for immediate, urgent, and routine referral. (See Table 2.)
In order to obtain consultation without compromising the research participant’s privacy and breaching confidentiality, the researcher should inform the research participant of this consultation pathway in seeking consent for participation in the study and should seek the consultation without providing information that would reveal the research participant’s identity. This means that the consultant’s conclusion will be recorded in research records, not the research participant’s clinical medical records. Handled in this way, consultation should not raise concerns under HIPAA because the research participant’s identity is protected.
The cost of compensating the consultant for IF verification and evaluation should be built into the research budget. Handling IFs responsibly is a researcher obligation. This could reasonably be regarded as either a direct cost or an infrastructure cost. Agencies funding research should support this expense as a cost of performing research ethically.106
Verifying and evaluating genetic IFs raises the question of whether to seek retesting in a CLIA-approved lab. Only such a lab is authorized to generate clinical test results.107 As previously noted, though, genetic testing in a CLIA-approved lab is not available for all genetic conditions.108 While CLIA-approved confirmation is ideal, we note the suggestion of the NHLBI Working Group that research lab results that cannot be verified in a CLIA-approved lab can nonetheless be disclosed to research participants, as long as those results are labeled “research” rather than “clinical” results, and the difference is explained. As we further note above, however, more work may be needed to resolve the question of whether this approach comports with CLIA regulations or regulatory change is needed to permit this.
As genetic, genomic, and imaging research technologies become more powerful, the IFs problem will grow. Genetic and genomic research will predictably include larger populations. Genomic research will cover larger stretches of the genome, up to the entire genome. Imaging research will increasingly incorporate functional (non-structural) information and quantification of imaging data will lead to additional information provided by even structural (i.e., anatomic) images. Data produced in all of these research domains will increasingly be archived and reanalyzed, thanks in part to federal data-sharing policies and the growing capabilities of computers and bioinformatics.
E. Plan to Determine Whether to Report IFs, Based on Likely Health or Reproductive Importance
The principal investigator rather than the consultant bears ultimate responsibility for determining whether the IF should be disclosed to the research participant, though consultation with the consultant may be helpful in making this determination. It is the researcher who undertakes duties of respect and reciprocity, minimizing risk, assuring a positive risk-benefit relationship, and alerting the research participant to any developments that may affect willingness to continue in the study. Further, discovery of an IF should not be the first occasion for researcher-participant communication about IFs; as part of agreeing to participate in the study, the research participant should receive information on the potential for IFs and should be asked to indicate on the consent form whether he or she wishes to receive such information. Information about IFs should generally be offered only to research participants who indicate at the time of consent to participate in the study that they wish to receive this information. However, if researchers identify an IF that is likely to be life-threatening or grave and can be ameliorated or treated, they should reconfirm with a research participant who indicated refusal that he or she is electing to decline information on all IFs, even those revealing conditions of high health importance that can be treated.
When should an IF be disclosed? We distinguish between 3 categories. (See Table 5.) An IF whose disclosure offers Strong Net Benefit is one revealing a condition likely to be life-threatening or a condition likely to be grave that can be avoided or ameliorated. As the label for this category suggests, these are IFs whose disclosure is likely to offer markedly more benefit than burden to the research participant. The researcher should offer to disclose an IF in this category to the participant. This gives the participant health information likely to be very important to the participant. This includes information about a condition likely to be life-threatening, even if it cannot be treated. This category would include genetic information that reveals significant risk of a condition likely to be life-threatening or that can be used to avoid or ameliorate a condition likely to be grave. It would also include genetic information that can be used in reproductive decision-making to avoid significant risk for offspring of a condition likely to be life-threatening or grave or to ameliorate in offspring such a condition.
An IF that offers Possible Net Benefit is one that may offer more benefit than burden to the research participant. An IF in this category reveals a health condition, including a grave or serious one that cannot be avoided or ameliorated, when a research participant is likely to deem that information important. This category would include genetic information revealing significant risk of a condition likely to be grave or serious, when that risk cannot be modified but a research participant is likely to deem that information important. It further includes genetic information that can be used in reproductive decision-making to avoid significant risk for offspring of a condition likely to be serious or to ameliorate a condition likely to be serious, when a research participant is likely to deem that information important. Researchers may reveal this kind of IF in order to show respect for the research participant’s informational needs and preferences, even though the information is not likely to change the participant’s own clinical course. However, researchers are not obligated to offer this information.
IFs that have Unlikely Net Benefit should not be offered to research participants, because they probably offer more burden than benefit. IFs in this category reveal a condition that is not likely to be of serious health or reproductive importance. This category also includes IFs whose likely health or reproductive importance cannot be ascertained. In this category, there is no justification for subjecting the research participant to the anxiety and burden of receiving this information.
Examples in the research domains we studied may help illuminate these categories. In genetic studies, an IF revealing alleles associated with hereditary nonpolyposis colorectal cancer (HNPCC) would be in the “strong net benefit” category, as they reveal a condi-
The problem of IFs is important and deserves broad discussion among researchers, research participants, IRBs, funders, and oversight bodies. Handling IFs responsibly requires clarity about the difference between research and clinical care, coupled with attention to the ethical duties of researchers when faced unexpectedly with information that could save a life, significantly alter clinical care, or prove important to the research participant.
tion likely to be life-threatening or to impose grave harm that may be avoided by alerting the research participant.109 However, an IF of APOE4 indicating susceptibility to Alzheimer disease at some point in the far future would be in the “possible net benefit” category, as the risk of Alzheimer disease is serious but cannot now be avoided or ameliorated. An IF of misattributed paternity would usually be in the “unlikely net benefit” category, particularly because communicating misattributed paternity may carry serious burdens for research participants. However, this IF could be in the “possible net benefit” category when learning of misattributed paternity would likely be of health or reproductive importance to the participant; an example would be an IF relieving a research participant’s anxiety about inheriting (and passing on to offspring) genes conferring significant risk of a condition likely to be serious.
In large-scale genomic microarray research, again an IF revealing alleles associated with HNPCC would be in the “strong net benefit” category. An IF revealing serious pharmacogenetic information, that a research participant is likely to suffer life-threatening or grave effects from taking certain medications or common doses of certain medications, would be another IF in this category. However, an IF for genes predisposing the research participant to schizophrenia would be in the “possible net benefit” category, as no intervention is available to reduce risk of this serious illness.
In neuroimaging research using MRI, an IF indicating a brain tumor, aneurysm, or arterio-venous malformation (AVM) would be an IF of “strong net benefit.” These are urgent medical problems requiring intervention to avert grave medical harm or death. However, an IF revealing lack of a posterior communicating cerebral artery would be an IF of “possible net benefit.” This anomaly could affect the severity of a stroke by limiting cerebral reperfusion, but nothing can be done about that. An example of an IF of “unlikely net benefit” would be unusual variation in the size of the amygdala; this cannot be avoided or ameliorated and is not likely to be of importance to the research participant at this time, because the significance of this size variation, if any, is unknown. Thus, the information is likely to impose only the burden of suggesting there is something wrong with the research participant’s brain with no corresponding benefit.
In CT colonography research, discovery of an extracolonic neoplasm or abdominal aortic aneurysm would be an IF of “strong net benefit” because the condition is likely to be life-threatening or grave and amenable to treatment. However, discovery of multilevel degenerative disk disease in the lumbar spine would be an IF of “possible net benefit” because the condition is likely to be serious but cannot be corrected and the research participant is likely to find this information of importance. Discovery of a lung nodule less than 4mm in a nonsmoker would be an IF of “unlikely net benefit” because this information is not likely to be of serious health or reproductive importance, as discussed above.
Whenever IFs are to be disclosed, they should be disclosed directly to the research participant. Some of the literature and consent forms available suggest that IFs should instead be disclosed to the research participant’s primary care physician.110 However, this gives the research participant no control over the information and compromises the participant’s privacy. The primary care physician is likely to record the information in the participant’s medical record before even consulting with the participant. This chain of events is not consistent with respect for the research participant and the participant’s decisional authority. The research participant should control the information and decide whom to consult, if anyone.
The researcher should communicate the IF to the participant in a way that is sensitive to the issues raised. Communicating a genetic IF, for example, may require the assistance of a genetic counselor or clinical geneticist.111 Similarly, disclosing an unexpected life-threatening condition or advanced cancer may require the assistance of a clinician such as an oncologist who can immediately address questions and concerns. When the possibility of finding grave IFs in a research study is expected to be high, researchers should consider asking research participants during the consent process if they will agree to disclosure of medically significant IFs to their primary care physician, with whom they have a preexisting relationship. Whether or not this is part of the initial consent process, a researcher disclosing an IF to a research participant should offer to communicate the IF directly to the research participant’s physician. If the research participant has no physician, the researcher should offer to suggest one. Depending on the specific IF revealed, the research participant may ask the researcher to suggest a specialist, such as oncologist. The researchers should respond to such requests with recommendations or referral. The goal in this process is to enable the research participant to address rapidly an IF that may be clinically important and anxiety-provoking.
Paying for clinical follow-up should not be the responsibility of the research team. Research teams are generally not set up or funded to provide significant and ongoing clinical care. The researcher should make clear to the research participant that paying for clinical follow-up will be the participant’s responsibility. What if the participant has no health insurance? The researcher should be prepared to advise the participant on available avenues for accessing follow-up or should know to whom to refer the participant for information and counseling. If the study population lacks insurance, then the researcher and IRB must address in advance how research participants with IFs will access clinical follow-up services.
F. Investigators and IRBs Should Create and Monitor a Pathway for IFs
Researchers have an obligation to anticipate IFs, set up a pathway for handling IFs responsibly, and address in the consent process with research participants the possibility of IFs and how they will be handled. IRBs have an obligation to address directly the issue of planning for IFs and handling them appropriately. IRBs should do this both in reviewing individual protocols and in crafting guidance and model consent forms for investigators. Our review of guidance and model consent forms offered on publicly available Internet sites by the IRBs of the 100 universities receiving the most NIH funding found that while some IRBs offered recommendations on how to handle IFs, many did not.112
When IRBs review protocols, they may face the question of whether a protocol that poses minimal risk and thus is eligible for expedited review under the rules governing human subjects research,113 creates a sufficient risk of uncovering IFs as to challenge the minimal risk classification and require full review.114 A protocol that threatens to yield a substantial number of IFs whose health or reproductive importance would counsel disclosure to research participants should indeed be afforded full review, particularly because the challenging ethical issues surrounding IFs have not yet been settled.
In addition to offering guidance on IFs and reviewing proposed protocols, IRBs have an important role to play in overseeing the adequacy of a study’s procedures for handling IFs. An IRB may conclude, for example, that a protocol is uncovering a large number of IFs or IFs of grave importance, without an adequate means for evaluating them promptly. IRBs may also be asked to provide consultation on difficult IF problems that arise, such as whether researchers should disclose to a research participant an IF of ambiguous clinical validity that may prove important to health decisions. IRBs may also be asked to consult on the difficult questions surrounding recontact of research participants when secondary analysis of archived data yields IFs of importance.
G. IFs in Pediatric and Adolescent Research Participants
Studies have begun to document and analyze IFs in research on children and adolescents.115 Because disclosing the potential for IFs and the plan for handling them is an important part of the consent process, it should be integrated into the consent process for pediatric and adolescent research. This information should be disclosed to the parent or guardian giving permission for the research and to the older child or adolescent giving assent under the regulations governing human subjects research.116 There are some special considerations that apply to handling IFs in pediatric and adolescent research.
First, certain categories of research on children and adolescents are approvable under the federal regulations on human subjects research only if they pose “no greater than minimal risk” to the minor research participant; involve greater than “minimal risk” but present the prospect of direct benefit; or represent “a minor increase over minimal risk” with no prospect of direct benefit, but are likely to yield generalizable knowledge about the minor participant’s disorder.117 The National Human Research Protections Advisory Committee has interpreted these terms, emphasizing that “[r]isks include all harms,...indignities, embarrassments, and potential breaches of privacy and confidentiality associated with research.”118 We recommend that both researchers and IRBs routinely address the question of whether the expected likelihood and gravity of IFs in a proposed study raise the risk of the research above “minimal risk.” As more data emerge on the prevalence of IFs in different forms of pediatric and adolescent research, it may turn out that some kinds of research pose a sufficiently high probability of uncovering IFs of significance, that this challenges the conclusion that the research poses only minimal risk or a minor increase over minimal risk and thus alters the risk-benefit calculus.
Second, we recommend above that all individuals agreeing to the research also be asked if they would like to receive information about IFs. This means that the parent or guardian would be asked, but also the older child or adolescent assenting to research participation. Ideally the parent or guardian’s answer will accord with the research participant’s. However, when they disagree, the researcher will have to consider how to handle an IF that would otherwise be disclosed. Consultation with the IRB may be necessary. When the parent or guardian wishes the information but the research participant does not, the answer may be to disclose to the parent or guardian and counsel them regarding the need for further clinical evaluation and therefore disclosure to the child or adolescent. The more difficult case will be when the minor research participant wishes the information but the parent or guardian asserts a preference not to know. This will require case-by-case evaluation. When an IF reveals a grave or life-threatening condition that requires clinical evaluation, the parent or guardian has a responsibility to learn of this information and act in a way to preserve the child or adolescent’s health. In such cases, an asserted right not to know should be overridden.
In some cases, an IF may reveal sensitive information that the child or adolescent may not want to be shared with the parent or guardian. An IF indicating substance abuse or pregnancy in a child or adolescent may fall in this category, as may an IF suggesting that the child or adolescent has suffered physical abuse. These social and behavioral IFs are beyond the scope of this paper, but researchers should consider what pathways they will follow to evaluate and disclose such findings in a way that respects the minor research participant’s interests, privacy, safety, and well-being.
Genetic and genomic IFs raise special issues for minor research participants because the literature on genetic testing in children generally urges that testing be delayed until adulthood unless the child will receive therapeutic benefit from earlier testing; accordingly carrier testing and screening as well as predictive testing for late-onset disorders are generally discouraged.119 This recommendation is based on recognition of the serious psychological and social consequences that can flow from genetic information, including stigma and discrimination as well as negative impact on the parent-child relationship.120 In keeping with this recommendation, only genetic or genomic IFs that are likely to confer therapeutic benefit and thus more benefit than risk should be offered to minor research participants, their parents, or guardians. An exception may be made for adolescents with mature decisional capacity, who indicate at consent a desire to receive IF information.
What about IFs uncovered in secondary research on archived data collected from minor research participants? By the time secondary analysis uncovers IFs, the minor may have reached the age of majority. The question of how to handle IFs in these cases is related to a larger debate under way on the proper scope of genetic and genomic research on children using identifiable samples121 and whether minor research participants, including those who have contributed samples to DNA databanks, should be asked at majority to reconsent to continued use of their samples.122 The questions do not arise when samples have been fully anonymized and research participants are no longer identifiable, but if researchers can identify participants, even through a coding system, the problems may remain. If research recommendations move toward seeking reconsent at majority, it would make sense to reconsent at that time on how IFs should be handled as well.
H. IFs in Adult Research Participants Without Decisional Capacity
We have found no studies focusing on IFs in adult research participants who lack decisional capacity. However, the prevalence figures emerging on the IFs on which we focus above would predict IFs in this research population as well. There has been persistent and wide debate on the appropriate scope of research on decisionally-incapacitated adults and the procedural protections that should attend such research.123 Recommendations limiting research in this population to that posing minimal risk or a minor increase over minimal risk would again raise the issue flagged above for children, that is, whether the expected incidence and nature of IFs in a proposed study would impose more than minimal risk or a minor increase over minimal risk.
An even more difficult problem is whether the research participant’s legally authorized representative (LAR) can and should be counted on to make appropriate decisions at consent about the return of IFs and subsequent decisions about appropriate clinical follow-up once an IF is disclosed. A “‘legally authorized representative’ is an individual or judicial or other body authorized under applicable law to consent on behalf of a prospective subject to the subject’s participation in the procedure(s) involved in the research.”124 It is not clear that current law facilitates LARs’ fulfillment of the decision-making duties envisioned in ethics recommendations.125 Further, deciding whether and how to follow up clinically on IFs discovered in research may be beyond the LAR’s role, as the LAR is supposed to make research decisions. Thus, handling IFs may require coordination between a research participant’s LAR and their surrogate for treatment decisions. This set of issues requires careful consideration by researchers and IRBs contemplating research on populations including those decisionally impaired.
I. Handling Social and Behavioral IFs
Though we did not focus on social and behavioral IFs, researchers and IRBs should consider how they will handle IFs that are social or behavioral as well as those that are genetic or physiological. Researchers in many states have mandatory reporting responsibilities under state law when faced with child abuse, elder abuse, and suicide risk. However, research interaction may unexpectedly reveal potentially important information not covered by such reporting laws, such as signs of alcohol abuse in an adolescent research participant. Potential research participants are entitled to know how researchers will respond if they discover this kind of information, as discovery poses risks to the participant as well as providing potential benefits.
IV. Process Responsibilities and Oversight
Making this approach work will involve a number of actors and oversight bodies. Investigators, IRBs, research funders, and regulatory authorities such as OHRP should take the problem of IFs seriously. Investigators should anticipate discovering IFs in their research and expect secondary researchers to discover IFs in reanalysis of archived data. Investigators should consequently plan for IFs in their research protocol. This means articulating the kinds of IFs anticipated, consulting the literature to state the expected prevalence of such IFs in their research population, considering which IFs will be referred to a consultant to determine whether they merit further clinical evaluation or may be of importance to research participants, and stating what will qualify as an IF that should be disclosed to the research participant because of its likely health or reproductive importance. Researchers should specify to the IRB and to potential research participants how IFs will be handled. They should consider including clinical consultants capable of reviewing IFs on their research team. If such consultants are not part of the team, researchers need to establish an arrangement with the consultants that will allow prompt review of IFs. Researchers will need to build into their research budget the funds needed to compensate such consultants for review of IFs and any funds needed to compensate clinical colleagues (such as genetic counselors or radiologists) who may be needed to help communicate IFs to research participants.
Clearly, the IRB has a crucial role to play in assuring that the research protocol anticipates IFs, sets up an appropriate plan and pathway for handling them, and then responds to IFs (including those not successfully anticipated in the protocol) in a responsible way. This means that the IRB must make sure investigators have in place a solid plan at the start of research, but that the IRB must also perform continuing oversight as IFs emerge, researchers respond to those IFs, and the pathway for handling IFs proves adequate or requires improvement. IRBs and programs overseeing human subjects research in and outside of universities should issue written guidance for researchers including model consent forms showing how to plan for and handle IFs in research.
Finally, federal agencies that fund and supervise human subjects research (including NIH, FDA, the Centers for Disease Control (CDC), and Department of Veterans Affairs (VA)) should address IFs in the guidance documents they issue for researchers and IRBs. These agencies perform a leadership role in setting standards for human subjects research and providing oversight. OHRP has a special role to play as a key federal body safeguarding the interests and welfare of research participants. Our research indicates that federal guidance documents do not yet adequately address IFs and attention to this problem is needed.
V. Conclusion
As genetic, genomic, and imaging research technologies become more powerful, the IFs problem will grow.126 Genetic and genomic research will predictably include larger populations. Genomic research will cover larger stretches of the genome, up to the entire genome. Imaging research will increasingly incorporate functional (non-structural) information and quantification of imaging data will lead to additional information provided by even structural (i.e., anatomic) images. Data produced in all of these research domains will increasingly be archived and reanalyzed, thanks in part to federal data-sharing policies and the growing capabilities of computers and bioinformatics.
This suggests that we are early in the development of the problem of how to handle IFs in research. Research would be helpful to clarify the types of IFs generated by different kinds of research, the statistical prevalence of these IFs, the costs of evaluating them and clinical following-up, and the positive and negative impacts on research participants. Meanwhile, we need to assure that IFs are handled responsibly and research participants understand what information they may be offered. These recommendations, generated by considering IFs in genetic, genomic, and imaging research, suggest the importance of looking at this problem comparatively across research domains and grounding ethics recommendations in the critical study of approaches currently in use.
Our recommendations are limited by considering only genetic, genomic, MRI neuroimaging, and CT colonography research. Further, we informed our recommendations by considering research consent forms, university model consent forms, statements by professional societies, and federal guidance documents publicly available on the Internet.127 We reasoned that these forms and documents are likely to exert the most influence on researchers and IRBs because these are the materials that they will most readily find when researching guidance and models. We refrained from seeking forms and university documents that were not posted on the Internet and we did not perform observational research to find out whether researchers are deviating from consent forms or supplementing those forms by discussing IFs with research participants.
The problem of IFs is important and deserves broad discussion among researchers, research participants, IRBs, funders, and oversight bodies. Handling IFs responsibly requires clarity about the difference between research and clinical care, coupled with attention to the ethical duties of researchers when faced unexpectedly with information that could save a life, significantly alter clinical care, or prove important to the research participant.
Acknowledgements
Preparation of this article was supported by the National Institutes of Health (NIH), National Human Genome Research Institute (NHGRI) grant #R01-HG003178 for a two-year project on “Managing Incidental Findings in Human Subjects Research” (Susan M. Wolf, Principal Investigator; Jeffrey P. Kahn, Frances Lawrenz, Charles A. Nelson, Co-Investigators). This project was based at the University of Minnesota’s Consortium on Law and Values in Health, Environment & the Life Sciences. Thanks to Charlisse Caga-anan, Barbara Figari, Britta Orr, Michelle Oyen, Suzanne Sobotka, and Lindsey Yock as well as Lisa Jones, Ph.D., for research assistance at various stages of the project and to Audrey Boyle for excellent project management. Special thanks to Elizabeth Thomson, D.N.Sc., R.N. at NHGRI for her support and insights. The contents of this article and symposium are solely the responsibility of the authors and do not necessarily represent the views of the NIH or NHGRI.
References
- 1.See, e.g., Green D.Incidental Findings in Computed Tomography of the Thorax Seminars in Ultrasound, CT & MR 200526, no. 114–19.Green DE, Woodward PJ.The Management of Indeterminate Incidental Findings Detected at Abdominal CT Seminars in Ultrasound, CT & MR 200526, no. 12–13.Cai Q, et al. Incidental Findings of Thickening Luminal Gastrointestinal Organs on Computed Tomography: An Absolute Indication for Endoscopy American Journal of Gastroenterology 200398, no. 81734–1737.Pickhardt PJ, et al. Computed Tomographic Virtual Colonoscopy To Screen for Colorectal Neoplasia in Asymptomatic Adults. N. Engl. J. Med. 2003;349(23):2191–2200. doi: 10.1056/NEJMoa031618., at 2196National Institutes of Health. Department of Health and Human Services State-of-the-Science Statement on Management of the Clinically Inapparent Adrenal Mass (“Incidentaloma”) 2002, available at <February 46http://consensus.nih.gov/2002/2002AdrenalIncidentalomasos021PDF.pdf> (last visited March 6, 2008)Stedman’s Medical Dictionary 2000, “incidentaloma”Lippincott Williams & Wilkins; Philadelphia: 27thRoof K, et al. Incidental Findings in a Federally-Sponsored Cancer Screening Program Journal of Community Health 199924, no. 4305–312.Westbrook JI, Braithwaite J, McIntosh JH.The Outcomes for Patients with Incidental Lesions: Serendipitous or Iatrogenic? American Journal of Roentgenology 1998171, no. 51193–1196.Chidiac RM, Aron DC.Incidentalomas: A Disease of Modern Technology Endocrinology & Metabolism Clinics of North America 199726, no. 1233–253.Parker LS, Majeske RA.Incidental Findings: Patients’ Knowledge, Rights, and Preferences Journal of Clinical Ethics 19956, no. 2176–179.
- 2.See Kohane IS, Masys DR, Altman RB.The Incidentalome: A Threat to Genomic Medicine JAMA 2006296, no. 2212–215.
- 3.National Human Research Protections Advisory Committee Working Group on Genetics IRB Guidebook Chapter on Human Genetics Research, Draft 2 2002June 27: at 12, available at <http://www.hhs.gov/ohrp/nhrpac/documents/nhrpac13.pdf> (last visited March 6, 2008) [hereinafter IRB Guidebook].
- 4.Ravitsky V, Wilfond BS.Disclosing Individual Genetic Results to Research Participants American Journal of Bioethics 20066, no. 68–17., at 8-9Ravitsky V, Wilfond BS.Response to Open Peer Commentaries on ‘Disclosing Individual Genetic Results to Research Participants’: Defining Clinical Utility and Revisiting the Role of Relationships American Journal of Bioethics 20066, no. 6W10–W12.Compare Parker LS.Rethinking Respect for Persons Enrolled in Research ASBH Exchange 20069, no. 21, 6-7, withShalowitz DI, Miller FG.Disclosing Individual Results of Clinical Research: Implications of Respect for Participants JAMA 2005294, no. 6737–740.
- 5.See, e.g., Illes J, et al. Practical Approaches to Incidental Findings in Brain Imaging Research Neurology 200870, no. 5384–390.Vernooij MW, et al. Incidental Findings in Brain MRI in the General Population N. Engl. J. Med 2007357, no. 181821–1828.Illes J, et al. Incidental Findings in Brain Imaging Research Science 2006311, no. 5762783–784.Zalis ME, et al. CT Colonography Reporting and Data System: A Consensus Proposal Radiology 2005236, no. 13–9., at 7-8.
- 6.Lucassen A, Parker M.Revealing False Paternity: Some Ethical Considerations Lancet 2001357, no. 92611033–1035., at 1035Anderlik MR, Rothstein MA.DNA-Based Identity Testing and the Future of the Family: A Research Agenda American Journal of Law & Medicine 200228, nos. 2 3215–232., at 221-222McEwen JE. Genetic Information, Ethics, and Information Relating to Biological Parenthood. In: Murray TH, Mehlman MJ, editors. Encyclopedia of Ethical, Legal, and Policy Issues in Biotechnology. Vol. 1. John Wiley & Sons; New York: 2000. pp. 356–363., at 359-360Ross L. Friedman.Disclosing Misattributed Paternity Bioethics 199610, no. 2115–130., at 116-117Macintyre S, Sooman A.Non-Paternity and Prenatal Genetic Screening Lancet 1991338, no. 8771869–871.
- 7.Vernooij, et al. supra note 5Kumra S, et al. Ethical and Practical Considerations in the Management of Incidental Findings in Pediatric MRI Studies Journal of the American Academy of Child & Adolescent Psychiatry 200645, no. 81000–1006., at 1002Alphs HH, et al. Findings on Brain MRI from Research Studies of Occupational Exposure to Known Neurotoxicants American Journal of Roentgenology 2006187, no. 41043–1047., at 1043-1044Illes J, et al. Ethical Consideration of Incidental Findings on Adult Brain MRI in Research Neurology 200462, no. 6888–890., at 888-889Kim BS, et al. Incidental Findings on Pediatric MR Images of the Brain AJNR American Journal of Neuroradiology 200223, no. 101674–1677., at 1675Yue NC, et al. Clinically Serious Abnormalities Found Incidentally at MR Imaging of the Brain: Data from the Cardiovascular Health Study Radiology 1997202, no. 141–46., at 42Katzman GL, Dagher AP, Patronas NJ.Incidental Findings on Brain Magnetic Resonance Imaging from 1000 Asymptomatic Volunteers JAMA 1999282, no. 136–39., at 37.Cf. Weber F, Knopf H.Incidental Findings in Magnetic Resonance Imaging of the Brains of Health Young Men Journal of Neurological Sciences 2006240, nos. 1 281–84., at 82-83Blomgren K.Clinical Significance of Incidental Magnetic Resonance Image Abnormalities in Mastoid Cavity and Middle Ear in Children International Journal of Pediatric Otorhinolaryngology 200367, no. 7757–760., at 758Lim W-K, et al. Incidental Magnetic Resonance Image Sinus Abnormalities in Asymptomatic Australian Children Journal of Laryngology & Otology 2003117, no. 11969–972., at 969-970.
- 8.American College of Radiology Imaging Network ACRIN 6664 National CT Colonography Trial 2006July 7: at 13 (citing earlier studies), available at <http://www.acrin.org/files/protocol_docs/A6664partial_summary.pdf> (last visited June 15, 2007)Spreng A, et al. Importance of Extracolonic Findings at IV Contrast Medium-Enhanced CT Colonography Versus Those at Non-Enhanced CT Colonography European Radiology 200515, no. 102088–2095.Yee J, et al. Extracolonic Abnormalities Discovered Incidentally at CT Colonography in a Male Population Radiology 2005236, no. 2519–526., at 520-521Hellström M, Svensson MH, Lasson A.Extracolonic and Incidental Findings on CT Colonography (Virtual Colonoscopy) American Journal of Roentgenology 2004182, no. 3631–638., at 631-634Rajapaksa RC, Macari M, Bini EJ.Prevalence and Impact of Extracolonic Findings in Patients Undergoing CT Colonography Journal of Clinical Gastroenterology 200438, no. 9767–771., at 768Pederson B. Ginnerup, Sosenkilde M, Christiansen T. Extracolonic Findings at Computed Tomography Colonography Are a Challenge. Gut. 2003;5:1744–1747. doi: 10.1136/gut.52.12.1744., at 1745Gluecker TM, et al. Extracolonic Findings at CT Colonography: Evaluation of Prevalence and Cost in a Screening Population Gastroenterology 2003124, no. 4911–916., at 912Pineau BC, et al. Prevalence of Extracolonic Findings at Virtual Colonoscopy Abstract No. 345, Abstracts Submitted for the 68th Annual Scientific Meeting of the American College of Gastroenterology, printed in American Journal of Gastroenterology, Supplement 1 200398, no. 9S117Edwards JT, Mendelson RM, Forbes GM.Extracolonic Findings at Virtual Colonoscopy: Implications for Screening Programs American Journal of Gastroenterology 200196, no. 103009–3012., at 3010-3011Hara AK, et al. Incidental Extracolonic Findings at CT Colonography Radiology 2000215, no. 2353–357., at 354.Cf. Khan KY, et al. Frequency and Impact of Extracolonic Findings Detected at Computed Tomographic Colonography in a Symptomatic Population British Journal of Surgery 200794, no. 3355–361., at 356Chin M, et al. Computed Tomographic Colonography: Prevalence, Nature, and Clinical Significance of Extracolonic Findings in a Community Screening Program. American Journal of Gastroenterology. 2005;100:2771–2776. doi: 10.1111/j.1572-0241.2005.00337.x., at 2773.
- 9.Illes, et al. Incidental Findings in Brain Imaging Research supra note 5.
- 10.42 U.S.C. § 263a 2007. (“Certification of laboratories”).
- 11.Lawrenz F, Sobotka S.Empirical Analysis of Current Approaches to Incidental Findings Journal of Law, Medicine & Ethics 200836, no. 2249–255.Wolf SM, Sobotka SP, Lawrenz FP. Managing Incidental Findings in Human Subjects Research: Analysis of Current Guidance and Consent Forms in progress.
- 12.See generally Ensenauer RE, Michels VV, Reinke SS.Genetic Testing: Practical, Ethical, and Counseling Considerations Mayo Clinic Proceedings 200580, no. 163–73.
- 13.IRB Guidebook supra note 3.
- 14.Lucassensupra note 6, at 1035McEwensupra note 6, at 359–360.Ross Friedman.supra note 6, at 116–117.Macintyre, Soomansupra note 6.
- 15.Ravitsky, WilfondDisclosing Individual Genetic Results to Research Participants supra note 4, at 12. [DOI] [PubMed]
- 16.National Heart, Lung and Blood Institute NHLBI Working Group on Reporting Genetic Results in Research Studies, Meeting Summary 2004July 12; Bethesda, MD, available at <http://www.nhlbi.nih.gov/meetings/workshops/gene-results.htm> (last visited June 15, 2007) [hereinafter NHLBI Working Group]. [Google Scholar]
- 17.Gene Tests, available at <http://genetests.org> (last visited March 28, 2008).See also Ormond KE.Disclosing Genetic Research Results: Examples from Practice American Journal of Bioethics 20066, no. 630–32., at 30.
- 18.See Daar AS, Scherer SW, Hegele RA.Implications of Copy-Number Variation in the Human Genome: A Time for Questions Nature Reviews Genetics 20067, no. 6414see generally ACMG Laboratory Practice Committee Working Group ACMG Recommendations for Standards for Interpretation of Sequence Variations Genetics in Medicine 20002, no. 5302–303.
- 19.See Maschke KJ.Navigating an Ethical Patchwork: Human Gene Banks Nature Biotechnology 200523, no. 5539–545., at 539.See also National Human Genome Research Institute (NHGRI) Reaffirmation and Extension of NHGRI Rapid Data Release Policies: Large-scale Sequencing and Other Community Resource Projects, available at <http://www.genome.gov/10506537> (last visited March 28, 2008).
- 20.See Kohane, Masys, Altmansupra note 2, at 214
- 21.Wolf SM, et al. Letter, “The Incidentalome,” JAMA 2006296, no. 232800–2801., at 2800.
- 22.See NHLBI Working Group supra note 16.
- 23.See Illes, et al. Incidental Findings in Brain Imaging Research supra note 5Kirschen MP, Jaworska A, Illes J.Subjects’ Expectations in Neuroimaging Research Journal of Magnetic Resonance Imaging 200623, no. 1205–209.National Institute of Neurological Disorders and Stroke Detection and Disclosure of Incidental Findings in Neuroimaging Research 2005January 67Bethesda, MD, available at <http://www.ninds.nih.gov/news_and_events/proceedings/ifexecsummary.htm> (last visited March 6, 2008) [hereinafter NINDS Proceedings]Illes J, et al. Discovery and Disclosure of Incidental Findings in Neuroimaging Research Journal of Magnetic Resonance Imaging 200420, no. 5743–747.Illes, et al. supra note 7Mamourian A.Incidental Findings on Research Functional MRI: Should We Look? American Journal of Neuroradiology 200425, no. 4520–522.Illes J, et al. Ethical and Practical Considerations in Managing Incidental Findings in Functional Magnetic Resonance Imaging Brain and Cognition 200250, no. 3358–365.Kim, et al. supra note 7Katzman, Dagher, Patronassupra note 7see generally, Kulynych JJ.The Regulation of MR Neuroimaging Research America Journal of Law & Medicine 200733, nos. 2 3295–317., at 313-315Kulynych J.Legal and Ethical Issues in Neuroimaging Research: Human Subjects Protection, Medical Privacy, and the Public Communication of Research Results Brain and Cognition 200250, no. 3345–357.
- 24.See Illes, et al. Incidental Findings in Brain Imaging Research supra note 5 at 784
- 25.Alphs, et al. supra note 7, at 1044Illes, et al. supra note 7, at 888–889.Katzman, Dagher, Patronassupra note 7, at 37Kim, et al. supra note 7, at 1675
- 26.Illes, et al. Incidental Findings in Brain Imaging Research supra note 5.
- 27.American College of Radiology Imaging Network ACRIN Protocol 6664 National CT Colonography Trial, available at <http://www.acrin.org/6664_protocol.html> (last visited June 15, 2007).
- 28.Xiong T, et al. Incidental Lesions Found on CT Colonography: Their Nature and Frequency British Journal of Radiology 200578, no. 92522–29., at 26.
- 29.Hara, et al. supra note 8, at 357
- 30.Limburg PJ, Fletcher JG.Making Sense of CT Colonography-Related Complication Rates Gastroenterology 2006131, no. 62023–2024. [DOI] [PubMed] [Google Scholar]
- 31.Siddiki H, et al. Incidental Findings in CT Colonography: Literature Review and Survey of Current Research Practice. Journal of Law, Medicine & Ethics. 2008;36(2):320–331. doi: 10.1111/j.1748-720X.2008.00276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.See Hara, et al. supra note 8, at 354Gluecker, et al. supra note 8, at 912
- 33.Siddiki, et al. supra note 31.
- 34.Id.
- 35.See Chin M, et al. Computed Tomographic Colonography: Prevalence, Nature, and Clinical Significance of Extracolonic Findings in a Community Screening Program American Journal of Gastroenterology 2005100, no. 122771–2776., at 2775Yee J, et al. Extracolonic Abnormalities Discovered Incidentally at CT Colonography in a Male Population Radiology 2005236, no. 2519–526., at 522Gluecker, et al. supra note 8, at 915Hara, et al. supra note 8, at 356–357.Cf. Xiong T, et al. Resources and Costs Associated with Incidental Extracolonic Findings from CT Colonography: A Study in a Symptomatic Population British Journal of Radiology 200679, no. 948948–961., at 949Wagner SC, et al. Picture Archiving and Communication System: Effect on Reporting of Incidental Findings Radiology 2002225, no. 2500–505., at 502-503.
- 36.Zalis, et al. supra note 5, at 7–8.
- 37.See Xiong, et al. supra note 28, at 23
- 38.Siddiki, et al. supra note 31.
- 39.See MacMahon H, et al. Guidelines for Management of Small Pulmonary Nodules Detected on CT Scans: A Statement from the Fleischner Society Radiology 2005237, no. 2395–400., at 398.
- 40.See Pederson Ginnerup, Sosenkilde, Christiansensupra note 8, at 1746–1747.Edwards, Mendelson, Forbessupra note 8, at 3011
- 41.See Maschkesupra note 19, at 539NHGRI supra note 19.
- 42.See generally Wagner, et al. supra note 35.
- 43.See, e.g., Verweij MF, Hamel BC.Unexpected Findings in Identifiable Stored Blood Samples After Analysis Without Consent: Moral Arguments For and Against Disclosure Genetic Counseling 200213, no. 2115–121. (discussing issues faced by original researchers when secondary analysis leads to “unexpected” finding of potential clinical significance in healthy controls).
- 44.See American Society for Human Genetics Statement on Informed Consent for Genetic Research American Journal of Human Genetics 199659, no. 2471–474., at 473Rothstein MA.Expanding the Ethical Analysis of Biobanks Journal of Law, Medicine & Ethics 200533, no. 189–101., at 94.
- 45.See 45 C.F.R. § 46.101 (b) (4) 2007 (exempting “research involving the collection or study of existing data..., [or] pathological specimens...if the information is recorded by the investigator in such a manner that subjects cannot be identified, directly or through identifiers linked to the subjects”).
- 46.Clayton EW.Informed Consent and Biobanks Journal of Law, Medicine & Ethics 200533, no. 115–21., at 15-17Office for Human Research Protections Guidance on Research Involving Coded Private Information or Biological Specimens 2004, no. August 10, available at <http://www.hhs.gov/ohrp/humansubjects/guidance/cdebiol.pdf> (last visited March 6, 2008) [hereinafter OHRP]National Bioethics Advisory Commission Research Involving Human Biological Materials: Ethical Issues and Policy Guidance 1999: at 115–16.Rockville, MD, 27-29.
- 47.See Working Group on Reporting Results of Genetic Research Offering Individual Results of Genetic Research: Report and Recommendations, April 22, 2006. draft, to be made available at <http://cirge.stanford.edu/library/reporting_results.html> [hereinafter Working Group on Reporting Results]cf. Cho MK.Understanding Incidental Findings in the Context of Genetics & Genomics Journal of Law, Medicine & Ethics 200836, no. 2280–285.
- 48.National Institutes of Health. Department of Health and Human Services Policy for Sharing of Data Obtained in NIH Supported or Conducted Genome-Wide Association Studies (GWAS) 2008January 25, available at <http://grants.nih.gov/grants/guide/notice-files/NOT-OD-07-088.html> (last visited March 27, 2008).
- 49.45 C.F.R. Part 46 2007 (“Protection of Human Subjects”).
- 50.21 C.F.R. Parts 50, 56 2007 (“Protection of Human Subjects,” “Institutional Review Boards”).
- 51.See, e.g., Emanuel EJ, Wendler D, Grady C.What Makes Clinical Research Ethical? JAMA 2000283, no. 202701–2711.
- 52.See Richardson HS, Belsky L.The Ancillary-Care Responsibilities of Medical Researchers: An Ethical Framework for Thinking About the Clinical Care that Researchers Owe Their Subjects Hastings Center Report 200434, no. 125–33., at 29-31.
- 53.See Claytonsupra note 46, at 2045 C.F.R. § 164.524 (a) 2007 (describing the right of access of individuals to protected health information).
- 54.National Institutes of Health Protecting Personal Health Information in Research: Understanding the HIPAA Privacy Rule, Access to Protected Health Information, available at <http://privacyruleandresearch.nih.gov/pr_08.asp#8j> (last visited April 7, 2008)National Human Genome Research Institute NHGRI Policy Recommendations on Research Privacy Guidelines Federal Policy Recommendations Including HIPAA, available at <http://www.genome.gov/11510216> (last visited June 17, 2007).
- 55.45 C.F.R. § 46.116 (a) 200721 C.F.R. § 50.25 (a) 2007
- 56.Rothstein MA.Tiered Disclosure Options Promote the Autonomy and Well-Being of Research Subjects American Journal of Bioethics 20066, no. 620–21., at 21 (calling for tiered disclosure during the initial informed consent process in which research subjects choose the types of individual genetic results they would want to receive).
- 57.See Appelbaum PS, et al. False Hopes and Best Data: Consent to Research and the Therapeutic Misconception Hastings Center Report 198717, no. 220–24., at 20.
- 58.45 C.F.R. § 46.116 200721 C.F.R. § 50.25 (b) (5) 2007
- 59.45 C.F.R. § 46.111 (a) (1) 200721 C.F.R. § 56.111 (a) (1) 2007
- 60.45 C.F.R. § 46.111 (a) (2) 200721 C.F.R. § 56.111 (a) (2) 2007
- 61.See Belsky L, Richardson HS.Medical Researchers’ Ancillary Clinical Care Responsibilities British Medical Journal 2004328, no. 74541494–1496.Richardson, Belskysupra note 52, at 32
- 62.See Belsky, Richardsonsupra note 61 at 1495Richardson, Belskysupra note 52, at 29–31.For ancillary care falling within the scope of entrustment, the strength of the claim to care depends on further factors: the participant’s vulnerability, their uncompensated risks or burdens (i.e., the gratitude owed to them by the researchers), their dependence on the researchers, and the depth of the researcher-participant relationship. Belsky, Richardsonsupra note 61 at 1495–1496.Richardson, Belskysupra note 52, at 30–31.
- 63.Illes, et al. Incidental Findings in Brain Imaging Research supra note 5, at 783
- 64.Shalowitz, Millersupra note 4, at 738
- 65.Illes, et al. Incidental Findings in Brain Imaging Research supra note 5, at 783
- 66.Beauchamp T, Childress J. Principles of Biomedical Ethics. 5th Oxford University Press; New York: 2001. pp. 173–176. [Google Scholar]
- 67.Grimes v. Kennedy Krieger Inst. , 782 A.2d 807, 849-50 (Md. 2001)834–35. [PubMed]
- 68.Grimes v. Kennedy Krieger Inst. , 782 A.2d 807, (Md. 2001)851.see also Blaz v. Michael Reese Hosp. Foundation , 74 F. Supp. 2d 803, (N.D. Ill. 1999)805–07.
- 69.See Grimes v. Kennedy Krieger Inst. , 782 A.2d 807, (Md. 2001)843–44.
- 70.See Sherman, Silverstein, Kohl, Rose, Podolsky Law Offices Clinical Trials Litigation, available at <http://www.sskrplaw.com/gene/index.html> (last visited June 17, 2007)Mello MM, Studdert DM, Brennan TA, “The Rise of Litigation in Human Subjects Research” Annals of Internal Medicine 2003139140–45.
- 71.See Office of Human Research Protections Overview Compliance Oversight, available at <http://www.hhs.gov/ohrp/compliance/index.html> (last visited June 17, 2007).
- 72.See Blaz v. Michael Reese Hosp. Foundation , 74 F. Supp. 2d 803, (N.D. Ill. 1999)805–07.Grimes v. Kennedy Krieger Inst. , 782 A.2d 807, (Md. 2001)852.Hoffman DE, Rothenberg KH.Whose Duty Is It Anyway? The Kennedy Krieger Opinion and Its Implications for Public Health Research Journal of Health Care Law & Policy 20026, no. 1109–147., at 130-131see also Appelbaum PS, Rosenbaum A.Tarasoff and the Researcher: Does the Duty to Protect Apply in the Research Setting? American Psychologist 198944, no. 6885–894.
- 73.See generally Fernandez CV, Kodish E, Weijer C.Informing Study Participants of Research Results: An Ethical Imperative IRB 200325, no. 112–19.Partridge AH, Winer EP.Informing Clinical Trial Participants About Study Results JAMA 2002288, no. 3363–365.
- 74.See, e.g., Caulfield T, et al. Research Ethics Recommendations for Whole-Genome Research: Consensus Statement PLoS Biology 20086, no. 30430–0435.Manolio TA.Taking Our Obligations to Research Participants Seriously: Disclosing Individual Results of Genetic Research American Journal of Bioethics 20066, no. 632–34., at 32-33.
- 75.See National Bioethics Advisory Commission supra note 46, at 72
- 76.Beskow LM, et al. Informed Consent for Population-Based Research Involving Genetics JAMA 2001286, no. 182315–2321., at 2320.
- 77.NHLBI Working Group supra note 16.
- 78.See Claytonsupra note 46, at 2045 C.F.R. § 164.524 (a) 2007Individuals generally have a right of access to their protected health information, with some exceptions. § 164.524 (a) (iii). These include protected health information that is maintained by a covered entity subject to CLIA, to the extent that providing access to this information would be prohibited by law, § 164.524 (a) (iii) (A); or when the covered entity is exempt from CLIA because it is a research lab that does “not report patient-specific results for the diagnosis, prevention or treatment of any disease or impairment of, or the assessment of the health of individual patients,” 42 C.F.R. § 493.3 (b) (2).
- 79.See National Institutes of Health supra note 54National Human Genome Research Institute supra note 54
- 80.See Bookman EB, et al. Reporting Genetic Results in Research Studies: Summary and Recommendations of an NHLBI Working Group American Journal of Human Genetics Part A 2006140A, no. 101033–1040., at 1034-1035Beskow LM.Considering the Nature of Individual Research Results American Journal of Bioethics 20066, no. 638–40., at 39-40.See also Grosse SD, Khoury MJ.What Is the Clinical Utility of Genetic Testing? Genetics in Medicine 20068, no. 7448–450. (discussing definitions of “clinical utility” from narrow (“ability...to prevent or ameliorate adverse health outcomes”) to broad (“considered important to individuals and families”)).
- 81.The CLIA regulations exempt research labs only when such labs “do not report patient-specific results for the diagnosis, prevention or treatment of any disease or impairment of, or the assessment of the health of individual patients.” Centers for Medicare & Medicaid Services (CMS), Department of Health and Human Services, Laboratory Requirements, 42 C.F.R. § 493.3 (b) (2) 2008 This may mean that under current regulations, research labs may not report “research results” when these are individual-specific and may be used to assess health or trigger such assessment.
- 82.This broad definition of “utility” comports with Grosse, Khourysupra note 80.
- 83.See Dinnett EM, et al. Letter, Offering Results to Research Participants British Medical Journal 2006332, no. 7540549–550., at 550Fernandez, Kodish, Weijersupra note 73, at 13Fernandez CV, Skedgel C, Weijer C.Considerations and Costs of Disclosing Study Findings to Research Participants Canadian Medical Association Journal 2004170, no. 91417–1419., at 1417.
- 84.NHLBI Working Group supra note 16.
- 85.See Facio FM.One Size Does Not Fit All American Journal of Bioethics 20066, no. 640–42., at 41.
- 86.Working Group on Reporting Results supra note 47.
- 87.See National Bioethics Advisory Commission supra note 46, at 72NHLBI Working Group supra note 16Ravitsky, WilfondDisclosing Individual Genetic Results to Research Participants supra note 4, at 10–11.Caulfield, et al. supra note 74.
- 88.See, e.g., Kohane IS, et al. Reestablishing the Researcher-Patient Compact Science 2007316, no. 5826836–837.Church GM. The Personal Genome Project. Molecular Systems Biology. 2005;(December 13) doi: 10.1038/msb4100040.; doi: 10.1038/msb4100040.
- 89.Cf. Lavieri RR, Garner SA.Ethical Considerations in the Communication of Unexpected Information with Clinical Implications American Journal of Bioethics 20066, no. 646–48.Rothsteinsupra note 56Fernandez CV, Weijer C.Obligations in Offering to Disclose Genetic Research Results American Journal of Bioethics 20066, no. 644–46.
- 90.See Clayton EW, Ross LF.Implications of Disclosing Individual Results of Clinical Research JAMA 2006295, no. 137.
- 91.Cf. Rothsteinsupra note 56 (discussing “disclosure options” for receipt of research results by participants before research begins).
- 92.Richardson, Belskysupra note 52.
- 93.See, e.g., IRB Guidebook supra note 3.
- 94.Lawrenz, Sobotkasupra note 11Wolf, Sobotka, Lawrenzsupra note 11
- 95.The literature on the return of individual research results does not always make this distinction. For example, Shalowitz, Millersupra note 4, at 738; imply that research participants have a right to any information about themselves. Lavieri and Garner suggest that “in all cases for which unanticipated, possibly clinically useful genetic data is obtained, investigators have a moral obligation to let the affected persons know that the results are available,” without limiting this information to research resultsLavieri RR, Garner SA.Ethical Considerations in the Communication of Unexpected Information with Clinical Implications American Journal of Bioethics 20066, no. 646–48.
- 96.See National Institutes of Health Guidance on Reporting Adverse Events to Institutional Review Boards for NIH-Supported Multicenter Clinical Trials 1999June 11, available at <http://grants2.nih.gov/grants/guide/notice-files/not99-107.html> (last visited March 6, 2008) (comparing the definitions and reporting requirements for adverse events in the DHHS and the FDA regulations on human subjects research)see generally 21 C.F.R. § 312.32 (a) 2007 (defining “unexpected adverse drug experience”)45 C.F.R. § 46.103 (b) (5) 2007 (requiring procedures for reporting “unanticipated problems involving risks to subjects”).
- 97.See Clayton EW, et al. Informed Consent for Genetic Research on Stored Tissue Samples JAMA 1995274, no. 221786–1792., at 1790.
- 98.See National Bioethics Advisory Commission supra note 46, at 63
- 99.Working Group on Reporting Results supra note 47.
- 100.An example of this type of incidental finding is described by Verweij, Hamelsupra note 43. A sample from an unaffected person in a study on inherited limb malformation was later used in a second study as a “normal” control. However, that study found that the sample contained a sequence variance in a certain gene, mutations of which may cause a primary cardiac disorder.Id. at 116.
- 101.See Rothsteinsupra note 44, at 95
- 102.See, e.g., Working Group on Reporting Results supra note 47Chosupra note 47Wade CH, Kalfoglou AL.When Do Genetic Researchers Have a Duty to Recontact Study Participants? American Journal of Bioethics 20066, no. 626–27.Ormondsupra note 17, at 31Rothsteinsupra note 44, at 95–96.Ormond KE, et al. Duty’ to Recontact Participants in a Population Based Genetic Database: The NUgene Experience Genetics in Medicine 20046, no. 4261Clayton EW, et al. Informed Consent for Genetic Research on Stored Tissue Samples JAMA 1995274, no. 221786–1792., at 1790.Cf. Hunter AGW, et al. Ethical, Legal, and Practical Concerns About Recontacting Patients to Inform Them of New Information: The Case in Medical Genetics American Journal of Medical Genetics 2001103, no. 4265–276.Knoppers BM.Duty to Recontact: A Legal Harbinger? American Journal of Medical Genetics 2001103, no. 4277.Fitzpatrick JL, et al. The Duty to Recontact: Attitudes of Genetics Service Providers American Journal of Human Genetics 199964, no. 3852–860. Hunter et al., Knoppers, and Fitzpatrick et al. consider the ethics of recontacting in a clinical genetics setting.
- 103.Cf. Wendler D, Emanuel E.The Debate Over Research on Stored Biological Samples: What Do Sources Think? Archives of Internal Medicine 2002162, no. 31457–1462. Wendler and Emanuel conducted a survey in an attempt to assess what individuals (both those who had participated in clinical research and contributed samples, and those who had not) think about research on stored samples. They conclude that 88.8% of respondents would want to be informed of results of uncertain clinical significance.Id., at 1457
- 104.See OHRP supra note 46.
- 105.Illes, et al. Incidental Findings in Brain Imaging Resarch supra note 5, at 784NINDS Proceedings, supra note 23.
- 106.Bookman, et al. supra note 80, at 1037 In their summary of the NHLBI Working Group on reporting genetic research results, Bookman et al. consider the costs involved in returning genetic research results to participants. They state that budgets for genetic research studies testing for mutations of known clinical significance should include the funds needed to offer results and to counsel on the meaning of such results.Id. at 1037
- 107.See 42 U.S.C. § 263a(a)-(b)see also Claytonsupra note 46, at 20 (stating that disclosure of research results from non-CLIA approved laboratories might be illegal if recipients “choose to act” on this information).
- 108.Ormondsupra note 17, at 30
- 109.See generally Lerman C, et al. Genetic Testing in Families with Hereditary Nonpolyposis Colon Cancer JAMA 1999281, no. 171618–1622., at 1618.
- 110.Lawrenz, Sobotkasupra note 11Wolf, Sobotka, Lawrenzsupra note 11.
- 111.Fernandez, Kodish, Weijersupra note 73, at 14
- 112.Lawrenz, Sobotkasupra note 11Wolf, Sobotka, Lawrenzsupra note 11
- 113.See 45 C.F.R. § 46.110 200721 C.F.R. § 56.110 2007Office for Protection from Research Risks Protection of Human Subjects: Categories of Research that May Be Reviewed by the Institutional Review Board (IRB) Through an Expedited Review ProcedureFederal Register, 63, no. 216 1998November 960364–60367., at 60366-60367, available at <http://www.hhs.gov/ohrp/humansubjects/guidance/expedited98.htm> (last visited March 6, 2008).
- 114.See Sharp HM, Orr RD.When ‘Minimal Risk’ Research Yields Clinically-Significant Data, Maybe the Risks Aren’t So Minimal American Journal of Bioethics 20044, no. 2W32–W36. (arguing that research that can yield data with implications for the participant’s health and welfare may present higher than “minimal risk”).
- 115.Downie J, Marshall J.Pediatric Neuroimaging Ethics Cambridge Quarterly of Healthcare Ethics 200716, no. 2147–160., at 152-153Kumra S, et al. Ethical and Practical Considerations in the Management of Incidental Findings in Pediatric MRI Studies Journal of the American Academy of Child & Adolescent Psychiatry 200645, no. 81000–1006.Kim, et al. supra note 7.
- 116.See 45 C.F.R. § 46.408 2007 (“Requirements for permission by parents or guardians and for assent by children.”).
- 117.45 C.F.R. §§ 46.404-06 2007
- 118.National Human Research Protections Advisory Committee Children’s Workgroup Clarifying Specific Portion of 45 CFR 46 Subpart D that Governs Children’s Research 2002: at 1, available at <http://www.hhs.gov/ohrp/nhrpac/documents/nhrpac16.pdf> (last visited March 6, 2008).
- 119.See American Academy of Pediatrics Committee on Bioethics Ethical Issues with Genetic Testing in Pediatrics Pediatrics 2001107, no. 61451–1455., at 1453-1454.
- 120.See id., at 1453
- 121.See Annas GJ.Rules for Research on Human Genetic Variation: Lessons from Iceland N. Engl. J. Med 2000342, no. 241830–1833., at 1832-1833.
- 122.See Knoppers BM, et al. Children and Incompetent Adults in Genetic Research: Consent and Safeguards Nature Reviews Genetics 20023, no. 3221–224., at 223.
- 123.See National Human Research Protections Advisory Committee Workgroup on Decisional Incapacity Report from NHRPAC on Informed Consent and the Decisionally Impaired 2002, available at <http://www.hhs.gov/ohrp/nhrpac/documents/nhrpac10.pdf> (last visited March 6, 2008) [hereinafter NHRPAC Report]National Bioethics Advisory Commission . Research Involving Persons with Mental Disorders that May Affect Decisionmaking Capacity. Rockville, MD: 1998.
- 124.45 C.F.R. § 46.102 (c) 2007
- 125.See NHRPAC Report, supra note 123.
- 126.Kohane, Masys, Altmansupra note 2.
- 127.Lawrenz, Sobotkasupra note 11Wolf, Sobotka, Lawrenzsupra note 11.