

Abstract
TNF is a pleiotropic cytokine that activates both anti- and proapoptotic signaling pathways, with cell fate determined by the balance between these two pathways. Activation of ErbB family members, including EGF receptor (EGFR/ErbB1), promotes cell survival and regulates several signals that overlap with those stimulated by TNF. This study was undertaken to determine the effects of TNF on EGFR and ErbB2 activation and intestinal epithelial cell survival. Mice, young adult mouse colon epithelial cells, and EGFR knockout mouse colon epithelial cells were treated with TNF. Activation of EGFR, ErbB2, Akt, Src, and apoptosis were determined in vivo and in vitro. TNF stimulated EGFR phosphorylation in young adult mouse colon epithelial cells, and loss of EGFR expression or inhibition of kinase activity increased TNF-induced apoptosis, which was prevented in WT but not by kinase-inactive EGFR expression. Similarly, TNF injection stimulated apoptosis in EGFR-kinase-defective mice (EGFRwa2) compared with WT mice. TNF also activated ErbB2, and loss of ErbB2 expression increased TNF-induced apoptosis. Furthermore, Src-kinase activity and the expression of both EGFR and ErbB2 were required for TNF-induced cell survival. Akt was shown to be a downstream target of TNF-activated EGFR and ErbB2. These findings demonstrate that EGFR and ErbB2 are critical mediators of TNF-regulated antiapoptotic signals in intestinal epithelial cells. Given evidence for TNF signaling in the development of colitis-associated carcinoma, this observation has significant implications for understanding the role of EGFR in maintaining intestinal epithelial cell homeostasis during cytokine-mediated inflammatory responses.
TNF first was identified by its ability to induce hemorrhagic tumor necrosis (1). However, extensive research has shown conflicting roles of TNF as both an apoptotic and a growth-promoting factor (2). In fact, we and others have reported that this pleiotropic cytokine regulates both anti- and proapoptotic signal transduction with cell fate determined by the balance between these two pathways (3–5). TNF-regulated antiapoptotic pathways include Akt/protein kinase B, ERK/MAPK, and NF-κB (4, 6), whereas proapoptotic TNF-initiated signals include p38 and stress-activated protein kinase/JNK (6–8). The molecular switch that determines TNF regulation of these two different functions is not well characterized, however.
EGF receptor (EGFR) is a member of the ErbB family, which consists of four tyrosine-kinase receptors: EGFR (ErbB1) and ErbB2–4. These receptors modulate cell survival, proliferation, and differentiation in many tissue types (9). EGFR and other ErbB family members can be activated by direct interaction with EGF-like ligands initiating formation of homo- and/or heterodimers and increased kinase activity (10, 11). In addition, EGFR can be transactivated by various extracellular stimuli, such as agonists for G protein-coupled receptors (GPCR) and cytokine receptors (12). TNF, IL-8, IL-1α, IL-1β, and IFN-γ have been reported to transactivate EGFR in hepatocytes (13), mammary epithelial cells (14), and cancer-derived cell lines (15, 16). Members of the Src family of nonreceptor intracellular tyrosine kinases have been implicated as targets of GPCRs in EGFR transactivation via direct phosphorylation of cytoplasmic domains of EGFR or via stimulation of matrix metalloproteinase (MMP) activity to promote the release of the membrane-bound EGFR ligand (17, 18). Src and MMPs (14–16) also have been reported for cytokine-stimulated EGFR transactivation in several cell types.
TNF activates a number of signal transduction pathways in intestinal epithelial cells, such as Akt and MAPK, which overlap with those of EGFR activation. Therefore, we hypothesize that transactivation of EGFR and ErbB2 is a mechanism by which TNF regulates signaling pathways to promote survival of colon epithelial cells. Here we report that EGFR and ErbB2 transactivation promote the survival responses of intestinal epithelial cells to TNF in vitro and in vivo. Furthermore, TNF transactivation of both EGFR and ErbB2 requires Src-kinase activity with Akt as a downstream target in this process. These findings identify an important relationship between TNF and EGFR/ErbB signal transduction pathways that permits maintenance of intestinal-epithelial-cell survival in a cytokine-enriched environment during acute injury response.
Results
TNF Stimulates EGFR and ErbB2 Transactivation in Intestinal Epithelial Cells.
Because previous studies have shown overlap between TNF- and EGFR/ErbB-regulated cellular responses and antiapoptotic signal transduction pathways (9, 19), we tested whether TNF transactivates EGFR/ErbB family members in young adult mouse colon (YAMC) epithelial cells. TNF transactivation of EGFR was concentration dependent with maximal activation at concentrations of 10–100 ng/ml (Fig. 1A). In addition, TNF-stimulated EGFR phosphorylation occurred within 5 min and was sustained for at least 4 h (Fig. 1B). Therefore, we used 120-min treatment of TNF at 100 ng/ml for subsequent experiments.
Fig. 1.

TNF stimulates EGFR and ErbB2 activation in intestinal epithelial cells. YAMC cells were treated with TNF at the indicated concentrations for various times. EGFR and ErbB2 phosphorylation was detected by Western blot analysis of cellular lysates with anti-EGFR-Y1068 and anti-ErbB2-Y1248 antibodies, respectively. Anti-actin antibody was used as a loading control. Data in this and subsequent figures are representative of at least three separate experiments.
Because EGFR can form heterodimers with ErbB2 to amplify EGFR biological responses (20), we determined the effect of TNF on ErbB2 activation and found that TNF increased ErbB2 phosphorylation in YAMC cells in a pattern similar to EGFR activation (Fig. 1).
To determine the role of EGFR tyrosine-kinase activity on TNF transactivation of EGFR, we reconstituted EGFR signaling in EGFR knockout mouse colon epithelial (EGFR−/−MCE) cells by stably expressing WT and kinase-inactive (ki) EGFR (Fig. 2A). TNF transactivated WT EGFR but not kiEGFR (Fig. 2A), suggesting that transactivation requires EGFR-kinase activity. Furthermore, the EGFR-kinase inhibitor, AG1478, blocked TNF-induced EGFR phosphorylation (Fig. 2B).
Fig. 2.

TNF transactivation of EGFR and ErbB2 requires both intact EGFR and ErbB2 in intestinal epithelial cells. Cells were treated with TNF (100 ng/ml) for 2 h or EGF (10 ng/ml) for 5 min in the presence or absence of 1-h pretreatment with EGFR receptor tyrosine kinase inhibitor AG1478 (150 nM) or vehicle DMSO. AG1478 and DMSO were maintained during the entire course of cytokine treatment. EGFR and ErbB2 phosphorylation was detected as described in Fig. 1. Anti-EGFR and anti-ErbB2 antibodies were used to detect EGFR and ErbB2 expression levels, respectively.
Interestingly, loss of EGFR suppressed TNF activation of ErbB2, which was rescued by WT EGFR but not by kiEGFR expression (Fig. 2A). Similarly, the EGFR tyrosine-kinase inhibitor, AG1478, blocked ErbB2 phosphorylation in response to TNF (Fig. 2B). Suppressing ErbB2 protein expression by siRNA in YAMC cells prevented activation of EGFR by TNF but not by EGF (Fig. 2C). These findings indicate that TNF-stimulated transactivation of EGFR and ErbB2 depends on the expression and activity of both receptors.
TNF-Induced EGFR and ErbB2 Transactivation Is Antiapoptotic in Intestinal Epithelial Cells.
To investigate the role of EGFR transactivation, YAMC and HT29 human colon carcinoma cell lines were treated with TNF (100 ng/ml), IL-1α (10 ng/ml), or IFN-γ (100 ng/ml) for 8 h in the presence or absence of an EGFR tyrosine-kinase inhibitor, AG1478. TNF-induced apoptosis was significantly enhanced in the presence of AG1478 in both YAMC cells (Fig. 3 A and B) and HT29 cells (Fig. 3C). Interestingly, AG1478 treatment did not increase apoptosis in either IL-1α- or IFN-γ-treated HT29 cells (Fig. 3C), suggesting that the antiapoptotic role of EGFR is specific for TNF-induced signaling. We further studied apoptosis by detecting caspase activity, the major regulator of apoptosis. TNF-induced caspase activity was increased 10-fold over the control in EGFR−/−MCE cells expressing kiEGFR or vector-only cells, a response completely reversed by WT EGFR expression (Fig. 3 D and E).
Fig. 3.

EGFR-kinase activity is required for survival of intestinal epithelial cells exposed to TNF. Cells were treated with TNF (100 ng/ml), IL-1α (10 ng/ml), or IFN-γ (100 ng/ml) as indicated for 8 h in the presence or absence of 1-h pretreatment with EGFR receptor kinase inhibitor AG1478 (150 nM). (A) Cells were fixed for TUNEL assay, with apoptotic nuclei labeled with FITC (green) and nuclei labeled with DAPI (blue). FITC- and DAPI-labeled images were taken from the same field. Arrowheads indicate apoptotic nuclei. (B and C) Apoptosis was determined by counting at least 500 cells. The percentage of cells undergoing apoptosis is shown. *, P < 0.05 compared with control, the treatment with AG1478, or TNF. (D) Caspase activity in living cells was detected by using the sulforhodamine multicaspase activity kit with caspase-active cells stained as red. (E) The percentage of cells with active caspase.
We used siRNA directed against EGFR to reduce EGFR expression in YAMC cells (Fig. 4A), which enhanced TNF-induced apoptosis, compared with nontargeting control siRNA transfection (Fig. 4B). Taken together, these results demonstrate that EGFR expression and EGFR tyrosine-kinase activity are required for survival of intestinal epithelial cells exposed to TNF.
Fig. 4.
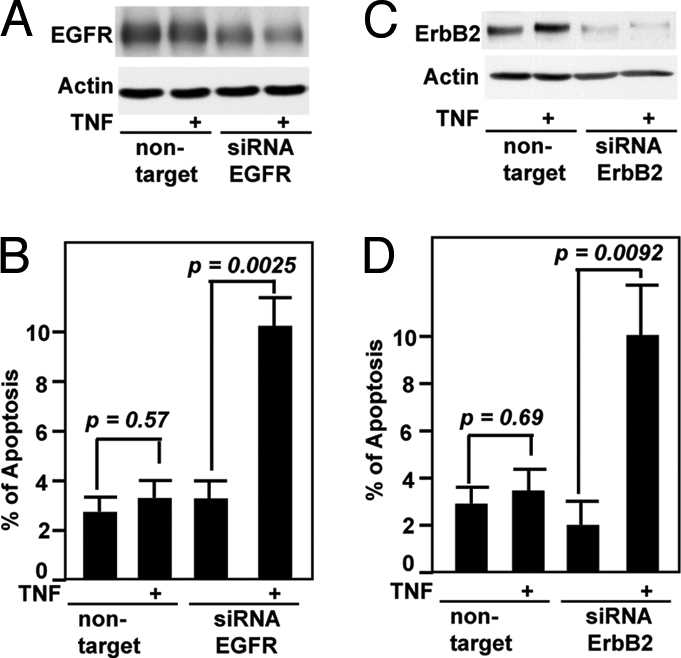
Suppression of EGFR and ErbB2 expression promotes apoptosis in intestinal epithelial cells treated with TNF. YAMC cells transfected with EGFR (A and B) or ErbB2 siRNA or nontargeting siRNA (C and D) for 24 h were treated with TNF (100 ng/ml) for 8 h to detect apoptosis by using TUNEL assay (B and D), as described in Fig. 3. EGFR (A) or ErbB2 (C) protein levels were detected as described in Fig. 2.
We transfected YAMC cells with siRNA directed against ErbB2 to study the role of ErbB2 activation in regulating cell fate in response to TNF treatment. TNF significantly enhanced apoptosis in cells with reduced ErbB2 expression (Fig. 4 C and D).
Loss of EGFR Kinase Activity Enhances TNF-Induced Colon Epithelial Apoptosis in Vivo.
Injection of 105 units of TNF into mice induces enteropathy, significant apoptosis, and intestinal epithelial cell sloughing (21–23). To determine whether TNF-induced antiapoptotic responses in vivo require EGFR kinase activity, we studied the effects of a lower TNF concentration (104 units per mouse) in EGFR-kinase-defective (EGFRwa2) mice by using i.p. injection of TNF as we described previously (5). TNF stimulated EGFR and ErbB2 phosphorylation in WT but not in EGFRwa2 mice (Fig. 5A). Immunostaining showed increased EGFR phosphorylation in cells that stained positive for E-cadherin (an epithelial cell adherens junctional marker), indicating that TNF stimulated EGFR activation in colon epithelial cells (Fig. 5B). Apoptosis, detected by ApopTag in situ oligo ligation kit (Millipore) staining, was increased up to 3-fold in TNF-treated EGFRwa2 compared with WT mice (Fig. 5 C and D). Similarly, activated-caspase-3 staining was increased in TNF-treated EGFRwa2 mice within 24 h and was limited to the epithelial cell layer (Fig. 5E), a pattern identical to the findings with ISOL staining. Thus, these data indicate that survival of intestinal epithelial cells exposed to TNF in vivo requires functional EGFR.
Fig. 5.

Loss of EGFR kinase activity promotes colon epithelial cell apoptosis in mice injected with TNF. WT and EGFRwa2 (EGFR-kinase-defective) mice were injected with TNF (104 units) or PBS for 24 h. (A) Total colon mucosal lysates were collected for Western blot analysis with indicated antibodies. Paraffin-embedded colon tissues were double-stained with rabbit anti-EGFR-Tyr-1068 (green in B) and mouse anti-E-cadherin (red in B) or with anti-active-caspase-3 antibodies (red in E) or DAPI (blue in B and E). Images were taken from the same field by using fluorescence microcopy and overlaid in B and C (40× magnification). Arrowheads (B) indicate costaining of EGFR-Tyr-1068 and E-cadherin. Arrows (E) indicate activated caspase-3-stained cells. (C) Paraffin-embedded colon tissues were studied for apoptosis by using ISOL staining, and apoptotic nuclei labeled with peroxidase were visualized by using differential interference contrast microscopy. Arrowhead indicates ISOL-labeled apoptotic nuclei. (D) The number of apoptotic nuclei per 100 colonic glands is shown. Shown is one representative of seven mice in each treatment group.
Src Activity Is Required for TNF-Stimulated EGFR and ErbB2 Transactivation and the Antiapoptotic Response in Intestinal Epithelial Cells.
One mechanism of EGFR transactivation includes activation of Src-family protein kinases, leading to MMP-dependent ligand release (17). Alternatively, activated Src is able to phosphorylate and activate EGFR directly (18).
In YAMC cells, TNF stimulated Src activation (Fig. 6A). Because the Src family members Src, Fyn, and Yes are expressed in YAMC cells (24), we transfected cells with a siRNA mixture that reduced expression of all three of these Src family members and attenuated EGFR and ErbB2 transactivation by TNF but did not attenuate EGF-stimulated EGFR activation (Fig. 6B). Likewise, treatment of cells with pharmacological inhibitors of Src family kinases, CGP77675, PP1, or PP2, similarly inhibited TNF transactivation of both EGFR and ErbB2 [supporting information (SI) Fig. S1A]. These findings suggest that Src regulates TNF-dependent activation of EGFR and ErbB2 in intestinal epithelial cells.
Fig. 6.

Src mediates TNF transactivation of EGFR and ErbB2 and cell survival in intestinal epithelial cells. YAMC cells (A) and cells transfected with a siRNA mixture for Src family members (Src, Fyn, and Yes) siRNA or nontargeting siRNA for 24 h (B) were treated with TNF (100 ng/ml) for 2 h or as indicated in A or EGF (10 ng/ml) for 5 min. Cellular lysates were prepared to detect Src activation by Western blot analysis using anti-phospho-Tyr-416 (P) Src antibody, and EGFR and ErbB2 phosphorylation was detected as described in Fig. 1. Anti-Src, anti-Fyn, and anti-Yes antibodies were used to detect Src, Fyn, and Yes protein levels, respectively. YAMC cells were treated with TNF (100 ng/ml) for 8 h in the presence or absence of 1-h pretreatment with Src inhibitors, CGP (0.2 μM), PP1 (1 μM), or PP2 (1 μM). Apoptosis was detected by using TUNEL assay, as described in Fig. 3. (C) The percentage of cells undergoing apoptosis is shown.
To determine the role of MMPs in transactivation of EGFR and ErbB2, we used the pharmacological inhibitors TAPI-1 and GM6001. Neither the ADAM-17-specific inhibitor TAPI-1 (Fig. S1B) nor the broad-spectrum MMP inhibitor GM6001 (data not shown) reduced TNF-stimulated EGFR and ErbB2 transactivation.
To determine whether Src activation by TNF regulates the fate of intestinal epithelial cells exposed to TNF, YAMC cells were pretreated with Src inhibitors and TNF-induced apoptosis was detected by TUNEL assay. All of the Src inhibitors tested enhanced TNF-induced apoptosis (Fig. 6C), suggesting that Src activation is required not only for EGFR and ErbB2 transactivation but also for the survival of intestinal epithelial cells in response to TNF treatment.
Akt Is a Potential Target of TNF-activated EGFR in Intestinal Epithelial Cells.
One of the major downstream signaling targets of EGFR is Akt, a pleiotropic antiapoptotic kinase. TNF activates Akt in a PI3K-dependent manner in intestinal epithelial cells (4), and PI3K inhibitors enhance apoptosis in TNF-treated cells (6). Therefore, we tested whether EGFR and ErbB2 are required for activation of Akt by TNF. Loss of EGFR expression suppressed TNF activation of Akt, which was rescued by WT EGFR (Fig. 7A). Likewise, TNF-stimulated Akt activation was inhibited by reducing ErbB2 expression (Fig. 7B). Furthermore, Src inhibitors blocked TNF-activation of Akt (Fig. S2). Interestingly, although both EGF and TNF stimulate ERK1/2 activation in intestinal epithelial cells, loss of EGFR or ErbB2 expression did not block TNF-induced activation of ERK1/2 (Fig. 7) or IκB degradation (Fig. S3).
Fig. 7.

EGFR and ErbB2 are required for Akt activation in intestinal epithelial cells treated with TNF. EGFR−/−MCE cells stably expressing WT EGFR or vector only (A) and YAMC cells transfected with ErbB2 siRNA or nontargeting siRNA (B) were treated with TNF (100 ng/ml) for 2 h or with EGF (10 ng/ml) for 5 min. Akt and ERK1/2 activation were detected by Western blot analysis of cellular lysates with anti-phospho (P)-Ser 473 Akt and anti-P-ERK1/2 antibodies, with total expression levels detected by using anti-Akt and anti-ERK1/2 antibodies, respectively.
Discussion
This study demonstrates an important mechanism of TNF promotion of intestinal epithelial cell survival through transactivation of EGFR and ErbB2, with the expression of both EGFR and ErbB2 required for TNF transactivation of either tyrosine kinase. Although specific mechanisms of EGFR and ErbB2 transactivation by TNF are not yet clear, this study provides evidence that this pathway requires Src-kinase activity and seems to be independent of MMP activity and that Akt activation is a common target downstream of EGFR/ErbB2 transactivation. These findings support the transactivation of EGFR and ErbB2 as a “molecular switch” determining the survival response of intestinal epithelial cells exposed to TNF.
Elucidating the downstream targets of EGFR activation by TNF is important to develop a clear understanding of the cellular responses induced by TNF. We showed that Akt/PI3K is a potential key target of TNF-initiated EGFR transactivation. We also found that TNF stimulates ERK1/2 MAPK activation in the absence of EGFR expression (Fig. 7). Thus, although EGFR is not necessary for TNF-stimulated MAPK activation in intestinal epithelial cells, neither is MAPK activation sufficient to promote cell survival after TNF stimulation (6). Our previous studies showed that TNF regulates proliferation through ERK1/2 activation (25, 26). Together these findings suggest that EGFR/ErbB2 may not be required for TNF-stimulated MAPK activation to regulate intestinal epithelial cell proliferation. Our finding is consistent with results from two recently published reports. The first report showed that gefitinib, an EGFR kinase inhibitor, increases apoptosis stimulated by recombinant TNF-related apoptosis-inducing ligand in a bladder cancer cell line in an Akt-dependent and ERK/MAPK-independent manner (27). The second paper showed that TNF activates MAPKs when EGFR is blocked in HeLa cells (28). Other studies, however, indicate ERK/MAPK as a target of TNF-transactivated EGFR (13, 29).
We have reported previously that TNF initiates antiapoptotic signaling pathways through kinase suppressor of Ras (KSR) in intestinal epithelial cells (4, 5) with KSR required for TNF activation of ERK1/2 MAPK and IκB degradation in YAMC cells (4, 5). Our findings here, however, show that neither ERK1/2 MAPK (Fig. 7) nor IκB (Fig. S3) is a downstream target of TNF-activated EGFR transactivation. Thus, TNF may regulate cell survival through multiple signaling pathways including both EGFR-transactivation-dependent and independent mechanisms.
Interestingly, Akt seems to be a target for both KSR- and EGFR-regulated antiapoptotic signaling pathways because TNF-induced Akt activation is inhibited, but not completely blocked, in KSR−/−MCE (5) (Fig. S3) and EGFR−/−MCE cells (Fig. 7 and Fig. S3). Our studies suggest that TNF-initiated EGFR and KSR signaling pathways are parallel in colon epithelial cells because (i) TNF stimulates EGFR transactivation and Akt activation in KSR1−/−MCE cells; (ii) the stronger inhibitory effects on Akt activation (as compared with YAMC cells) are observed in EGFR−/−MCE cells at 1, 5, 10, and 120 min of TNF treatment and at 1, 5, 60, and 120 min in KSR1−/−MCE cells; (iii) the peak of Akt activation by TNF is at 60 min treatment in EGFR−/−MCE cells and at 10 min in KSR1−/−MCE cells.
TNF production is increased in a number of gastrointestinal disorders, such as inflammatory bowel disease (30) and Helicobacter pylori-induced gastritis (31), which are coincident with EGFR/ErbB activation and an increased risk of tumorigenesis (32). Furthermore, dysregulated production of TNF is a mediator of tumor growth (19). Therefore, the mechanisms of TNF-regulated antiapoptotic responses are important in understanding the role of TNF in the pathogenesis of inflammation and inflammation-associated tumorigenesis. Activation of EGFR and other ErbB family members by cytokines has been reported in several cell types (13–16). TNF stimulates rapid EGFR phosphorylation on Tyr-1068 in HeLa cells, although at a lower level of phosphorylation than with EGF treatment (28), consistent with our finding that EGF is more effective than TNF in stimulating increased EGFR phosphorylation in YAMC cells (Figs. 2 and 6). Thus, this evidence of the complex relationship of cross-talk between TNF and EGFR/ErbB2 in modulating cellular functions has significant implications for the clinical manipulation of TNF signaling in inflammatory diseases and tumor development.
Another finding is that TNF-transactivation of either EGFR or ErbB2 requires expression of both EGFR and ErbB2 (Fig. 2). TNF may increase EGFR/ErbB2 heterodimer formation, because ligand-induced formation of ErbB2-containing heterodimers is preferred over other ErbB combinations (33). Furthermore, heterodimers, especially those containing ErbB2, induce more potent signaling than EGFR homodimers (34). Indeed, ligand-induced EGFR and ErbB2 phosphorylation depends on the dimerization partner, and the presence of ErbB2 modifies EGFR-regulated signaling pathways (35). Overexpression of ErbB2 enhances ligand-independent EGFR activation and EGFR recycling to the cell surface and inhibits down-regulation of activated EGFR (34, 36). In addition, ErbB2 activation by TNF may be amplified by heterodimer formation with EGFR because ErbB2 can be phosphorylated directly by EGFR (37); alternatively, formation of the heterodimer induces a conformational change that activates the intrinsic tyrosine kinase domain of ErbB2, leading to autophosphorylation of ErbB2 (38).
Src family members seem to regulate TNF-stimulated EGFR and ErbB2 transactivation in intestinal epithelial cells. Thus, the tyrosine kinase activity of Src family members seems to be necessary for maximal TNF-stimulated EGFR/ErbB2 transactivation and the antiapoptotic response. We cannot exclude the involvement of MMPs that are not sensitive to either GM6001 or TAPI-1, however. We have generated ADAM-17/TACE-deficient MCE cells, and TNF stimulates EGFR transactivation in the absence of this well characterized MMP, which is known to initiate processing of several EGFR ligands (data not shown). Further studies, such as screening for TNF-stimulated EGFR/ErbB2 ligand production, will be needed to define a role for MMP activity in TNF transactivation of EGFR.
In summary, our study demonstrates that TNF stimulates EGFR and ErbB2 transactivation with Akt as a potential target to promote an antiapoptotic response in intestinal epithelial cells in vitro and in vivo. Interestingly, recent reports show that TNF signaling is required for the development of colitis-associated carcinoma (39), and PI3K/Akt activation in tumor suppressor phosphatase and tensin homolog (PTEN)-deficient mice plays a significant role in intestinal stem cell activation and proliferation to initiate intestinal polyposis (40). Therefore, these observations advance our understanding of the possible role of EGFR and other ErbB family members in maintaining intestinal epithelial cell homeostasis in TNF-induced inflammation with important implications for inflammation-associated carcinogenesis.
Materials and Methods
Information regarding antibodies, cytokines, growth factors, inhibitors, reagents, and the detailed methods for cell line preparation, cell culture, cellular lysates preparation, immunohistochemistry, and apoptosis are provided in the SI Materials and Methods.
Cell Culture.
YAMC cells were isolated from the colonic epithelium of H-2Kb-tsA58 mice (Immortomouse; Charles River Laboratories) (41). EGFR−/−MCE cells were isolated from the colonic epithelium of EGFR-null heterozygous mice crossed with the Immortomouse (42).
Generation of Stable Cell Lines.
EGFR−/−MCE cells were transfected with pcDNA3.1/Zeo vector control, pcDNA3.1/Zeo/WT EGFR, or pcDNA3.1/Zeo/kiEGFR (K721R), which blocks ATP binding, by using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Zeocin-selected pools of cells were stained with anti-EGFR528-PE antibody (50 ml/5 × 106 cells) and sorted by using Becton-Dickinson FACSAria flow cytometry. Stable pools of cells expressing EGFR were maintained in medium containing 200 μg of Zeocin per milliliter.
Transient Transfection of siRNA.
YAMC cells were transiently transfected with 50 nM nontargeting siRNA, 50 nM mouse EGFR, or ErbB2 SMARTpool siRNA at 70% confluence by using Lipofectamine 2000 according to the manufacturer's instructions. After 24 h of transfection, cells were cultured in serum-starved medium for 16–18 h before treatment.
Mice, TNF Injection, and Tissue Preparations.
All animal experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee at Vanderbilt University. EGFRwa2 mice with a naturally occurring EGFR-kinase-defective mutation (Val to Gly mutation at residue 743 in the kinase domain) (43) were obtained from David Threadgill (University of North Carolina, Chapel Hill, NC) on a C57BL/6 background. PCR primers specific for the EGFR sequence containing the point mutation were used for genotyping (sequences available upon request). Mice (8–10 weeks old, 25–30 g) were anesthetized and then were injected i.p. either with TNF (104 units in PBS containing 2% FBS) or with PBS with 2% FBS only in a total volume of 200 μl (5).
Colon tissue was fixed in 10% neutral-buffered formalin and was paraffin-embedded before sectioning. The colonic mucosa was scraped into homogenization buffer and tissue lysed (44) for Western blot analysis.
Statistical Analysis.
Statistical significance in each study was determined by using the Student's t test with a confidence level of 0.05. All data presented are representative of at least three repeat experiments and are presented as mean ± SEM.
Supplementary Material
Acknowledgments.
We thank Dr. Robert Whitehead (Vanderbilt University) for helping generate the colon epithelial cell line, Dr. Philip Dube (Vanderbilt University) for suggestions for manuscript preparation, and the Vanderbilt University Digestive Disease Research Center Cell and Animal Modeling, Cell Imaging, and Flow Cytometry Cores. This work was supported by National Institutes of Health Grants DK56008 and DK02212 (to D.B.P.) and DK58404 (to Vanderbilt University Digestive Disease Research Center).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0801463105/DCSupplemental.
References
- 1.Carswell EA, et al. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci USA. 1975;72:3666–3670. doi: 10.1073/pnas.72.9.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 3.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 4.Yan F, John SK, Polk DB. Kinase suppressor of Ras determines survival of intestinal cells exposed to tumor necrosis factor. Cancer Res. 2001;61:8668–8675. [PubMed] [Google Scholar]
- 5.Yan F, et al. Kinase suppressor of Ras protects intestinal epithelium from cytokine-mediated apoptosis during inflammation. J Clin Invest. 2004;114:1272–1280. doi: 10.1172/JCI21022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yan F, Polk DB. Probiotic bacterium prevents cytokine-induced apoptosis in intestinal epithelial cells. J Biol Chem. 2002;277:50959–50965. doi: 10.1074/jbc.M207050200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 8.Xia Z, Dickens M, Rainegeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 Map kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 9.Citri A, Yarden Y. EGF-ERBB signalling: Towards the systems level. Nat Rev Mol Cell Biol. 2006;7:505–516. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- 10.Citri A, Skaria KB, Yarden Y. The deaf and the dumb: The biology of ErbB-2 and ErbB-3. Exp Cell Res. 2003;284:54–65. doi: 10.1016/s0014-4827(02)00101-5. [DOI] [PubMed] [Google Scholar]
- 11.Carpenter G. ErbB-4: Mechanism of action and biology. Exp Cell Res. 2003;284:66–77. doi: 10.1016/s0014-4827(02)00100-3. [DOI] [PubMed] [Google Scholar]
- 12.Zwick E, Hackel PO, Prenzel N, Ullrich A. The EGF receptor as central transducer of heterologous signalling systems. Trends Pharmacol Sci. 1999;20:408–412. doi: 10.1016/s0165-6147(99)01373-5. [DOI] [PubMed] [Google Scholar]
- 13.Argast GM, Campbell JS, Brooling JT, Fausto N. Epidermal growth factor receptor transactivation mediates tumor necrosis factor-induced hepatocyte replication. J Biol Chem. 2004;279:34530–34536. doi: 10.1074/jbc.M405703200. [DOI] [PubMed] [Google Scholar]
- 14.Chen WN, et al. Induced autocrine signaling through the epidermal growth factor receptor contributes to the response of mammary epithelial cells to tumor necrosis factor alpha. J Biol Chem. 2004;279:18488–18496. doi: 10.1074/jbc.M310874200. [DOI] [PubMed] [Google Scholar]
- 15.Itoh Y, et al. IL-8 promotes cell proliferation and migration through metalloproteinase-cleavage proHB-EGF in human colon carcinoma cells. Cytokine. 2005;29:275–282. doi: 10.1016/j.cyto.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 16.Tanida S, et al. The mechanism of cleavage of EGFR ligands induced by inflammatory cytokines in gastric cancer cells. Gastroenterology. 2004;127:559–569. doi: 10.1053/j.gastro.2004.05.017. [DOI] [PubMed] [Google Scholar]
- 17.Fischer OM, Hart S, Gschwind A, Ullrich A. EGFR signal transactivation in cancer cells. Biochem Soc Trans. 2003;31:1203–1208. doi: 10.1042/bst0311203. [DOI] [PubMed] [Google Scholar]
- 18.Tice DA, Biscardi JS, Nickles AL, Parsons SJ. Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Proc Natl Acad Sci USA. 1999;96:1415–1420. doi: 10.1073/pnas.96.4.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szlosarek P, Charles KA, Balkwill FR. Tumour necrosis factor-alpha as a tumour promoter. Eur J Cancer. 2006;42:745–750. doi: 10.1016/j.ejca.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 20.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 21.Garside P, Bunce C, Tomlinson RC, Nichols BL, Mowat AM. Analysis of enteropathy induced by tumour necrosis factor α. Cytokine. 1993;5:24–30. doi: 10.1016/1043-4666(93)90020-6. [DOI] [PubMed] [Google Scholar]
- 22.Garside P, Mowat AM. Natural killer cells and tumour necrosis factor-α-mediated enteropathy in mice. Immunology. 1993;78:335–337. [PMC free article] [PubMed] [Google Scholar]
- 23.Kiesslich R, et al. Identification of epithelial gaps in human small and large intestine by confocal endomicroscopy. Gastroenterology. 2007;133:1769–1778. doi: 10.1053/j.gastro.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 24.Frey MR, Golovin A, Polk DB. Epidermal growth factor-stimulated intestinal epithelial cell migration requires Src family kinase-dependent p38 MAPK signaling. J Biol Chem. 2004;279:44513–44521. doi: 10.1074/jbc.M406253200. [DOI] [PubMed] [Google Scholar]
- 25.Kaiser GC, Polk DB. Tumor necrosis factor α regulates proliferation in a mouse intestinal cell line. Gastroenterology. 1997;112:1231–1240. doi: 10.1016/s0016-5085(97)70135-5. [DOI] [PubMed] [Google Scholar]
- 26.Kaiser GC, Yan F, Polk DB. Conversion of TNFα from antiproliferative to proliferative ligand in mouse intestinal epithelial cells by regulating mitogen-activated protein kinase. Exp Cell Res. 1999;249:349–358. doi: 10.1006/excr.1999.4488. [DOI] [PubMed] [Google Scholar]
- 27.Shrader M, et al. Gefitinib reverses TRAIL resistance in human bladder cancer cell lines via inhibition of AKT-mediated X-linked inhibitor of apoptosis protein expression. Cancer Res. 2007;67:1430–1435. doi: 10.1158/0008-5472.CAN-06-1224. [DOI] [PubMed] [Google Scholar]
- 28.Singhirunnusorn P, et al. Transient suppression of ligand-mediated activation of epidermal growth factor receptor by tumor necrosis factor-alpha through the TAK1–p38 signaling pathway. J Biol Chem. 2007;282:12698–12706. doi: 10.1074/jbc.M608723200. [DOI] [PubMed] [Google Scholar]
- 29.Ueno Y, et al. Selective inhibition of TNF-alpha-induced activation of mitogen-activated protein kinases and metastatic activities by gefitinib. Br J Cancer. 2005;92:1690–1695. doi: 10.1038/sj.bjc.6602548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Breese EJ, et al. Tumor necrosis factor α-producing cells in the intestinal mucosa of children with inflammatory bowel disease. Gastroenterology. 1994;106:1455–1466. doi: 10.1016/0016-5085(94)90398-0. [DOI] [PubMed] [Google Scholar]
- 31.Lindholm C, Quiding-Jarbrink M, Lonroth H, Hamlet A, Svennerholm AM. Local cytokine response in Helicobacter pylori-infected subjects. Infect Immun. 1998;66:5964–5971. doi: 10.1128/iai.66.12.5964-5971.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malecka-Panas E, et al. Differential activation of total and EGF receptor (EGF-R) tyrosine kinase (tyr-k) in the rectal mucosa in patients with adenomatous polyps, ulcerative colitis and colon cancer. Hepatogastroenterology. 1997;44:435–440. [PubMed] [Google Scholar]
- 33.Graus-Porta D, Beerli RR, Daly JM, Hynes NE. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 1997;16:1647–1655. doi: 10.1093/emboj/16.7.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lenferink AE, et al. Differential endocytic routing of homo- and hetero-dimeric ErbB tyrosine kinases confers signaling superiority to receptor heterodimers. EMBO J. 1998;17:3385–3397. doi: 10.1093/emboj/17.12.3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Olayioye MA, et al. ErbB-1 and ErbB-2 acquire distinct signaling properties dependent upon their dimerization partner. Mol Cell Biol. 1998;18:5042–5051. doi: 10.1128/mcb.18.9.5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Worthylake R, Opresko LK, Wiley HS. ErbB-2 amplification inhibits down-regulation and induces constitutive activation of both ErbB-2 and epidermal growth factor receptors. J Biol Chem. 1999;274:8865–8874. doi: 10.1074/jbc.274.13.8865. [DOI] [PubMed] [Google Scholar]
- 37.Qian X, LeVea CM, Freeman JK, Dougall WC, Greene MI. Heterodimerization of epidermal growth factor receptor and wild-type or kinase-deficient Neu: A mechanism of interreceptor kinase activation and transphosphorylation. Proc Natl Acad Sci USA. 1994;91:1500–1504. doi: 10.1073/pnas.91.4.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Worthylake R, Wiley HS. Structural aspects of the epidermal growth factor receptor required for transmodulation of erbB-2/neu. J Biol Chem. 1997;272:8594–8601. doi: 10.1074/jbc.272.13.8594. [DOI] [PubMed] [Google Scholar]
- 39.Popivanova BK, et al. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest. 2008;118:560–570. doi: 10.1172/JCI32453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He XC, et al. PTEN-deficient intestinal stem cells initiate intestinal polyposis. Nat Genet. 2007;39:189–198. doi: 10.1038/ng1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Whitehead RH, VanEeden PE, Noble MD, Ataliotis P, Jat PS. Establishment of conditionally immortalized epithelial cell lines from both colon and small intestine of adult H-2Kb-tsA58 transgenic mice. Proc Natl Acad Sci USA. 1993;90:587–591. doi: 10.1073/pnas.90.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dise RS, Frey MR, Whitehead RH, Polk DB. Epidermal growth factor stimulates Rac activation through Src and phosphatidylinositol 3-kinase to promote colonic epithelial cell migration. Am J Physiol Gastrointest Liver Physiol. 2008;294:G276–G285. doi: 10.1152/ajpgi.00340.2007. [DOI] [PubMed] [Google Scholar]
- 43.Luetteke NC, et al. The mouse waved-2 phenotype results from a point mutation in the EGF receptor tyrosine kinase. Genes Dev. 1994;8:399–413. doi: 10.1101/gad.8.4.399. [DOI] [PubMed] [Google Scholar]
- 44.Polk DB. Ontogenic regulation of PLCγ1 activity and expression in the rat small intestine. Gastroenterology. 1994;107:109–116. doi: 10.1016/0016-5085(94)90067-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.