

Abstract
A tricyclic substructure of the tetracyclic nitrogen core of the daphniglaucins was formed by an oxidative activation of the allyl side chain of a bicyclo[1.1.0]butylmethylamine, a spontaneous intramolecular formal Alder-ene reaction, and a selective cyclization of a triol intermediate.
Among complex alkaloids, quaternary ammonium salts occupy a unique position because of their frequently intricate architecture. Some of these compounds are used medicinally as anticancer and antimicrobial agents,2 and they have also found utility as chiral phase transfer catalysts.3 These polycyclic alkaloids constitute challenging targets for natural product total synthesis (Figure 1).4 For example, the daphniglaucins are cytotoxic quaternary Daphniphyllum alkaloids isolated in 2003 by Kobayashi et al. from the leaves of Daphniphyllum glaucescens and have an unusual framework consisting of a novel fused-polycyclic skeleton assembled around a 1-azoniatetracyclo[5.2.2.0.1,60.4,9]undecane ring system.4c–e Despite these attractive structural features, a total synthesis or a partial synthetic approach have not yet been reported.
Figure 1.

Representative polycyclic quaternary alkaloids.
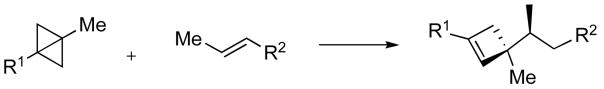
In this communication, we describe our studies on the use of a bicyclo[1.1.0]butane building block in the synthesis of the cyclic quaternary ammonium scaffold of daphniglaucins. To the best of our knowledge, this sequence represents the first use of this highly strained hydrocarbon in alkaloid synthesis.5 We have previously reported on the preparations and ring transformations of bicyclobutylmethylamines.6,7 An intramolecular ene reaction of N-allylated derivatives of 1 provides a stereoselective access to spirocyclic pyrrolidines 2 under remarkably mild phase transfer conditions (Scheme 1).
Scheme 1.

Cascade N-allylation/formal Alder-ene sequence.
In a continuation of our explorations of the novel intramolecular cycloaddition chemistry of bicyclo[1.1.0]-butanes,6,7 we intended to demonstrate their utility for the synthesis of biologically interesting cyclic quaternary ammonium salts. We envisioned that a tricyclic substructure of the core heterocycle of daphniglaucin A, i.e. the 1-azoniatricyclo[5.2.2.01,6]undecane 3, could be derived from a triol 4, which could be obtained from spirocycle 5 via oxidative cleavage and reduction (Scheme 2). An intramolecular formal Alder-ene reaction of bicyclobutylmethylamine 6 would produce 5. The aldehyde group in the nitrogen side chain in 6 was found to be essential for the thermal ene reaction of bicyclo[1.1.0]-butanes lacking conjugated aromatic substituents. The need for an electron-poor enophile in this process was supported by preliminary experimental as well as theoretical studies (Scheme 3 and Table 1).
Scheme 2.

Retrosynthetic strategy toward cyclic quaternary ammonium salts from bicyclobutanes. The corresponding core structure of daphniglaucins is highlighted.
Scheme 3.

Formal Alder-ene reactions of terminally unsubstituted bicyclobutylmethylamines require electron-deficient enophiles.
Table 1.
Frontier molecular orbital calculations of the HOMO-LUMO gapa) as a function of R1 and R2 substituents on ene and enophile
 | |||||
|---|---|---|---|---|---|
| entry | R1 | HOMO of bicylobutane [eV] | R2 | LUMO of alkene [eV] | ΔE (LUMO- HOMO) [eV] |
| 1 | Ph | −7.95 | H | 5.25 | 13.20 |
| 2 | H | −9.37 | H | 5.25 | 14.62 |
| 3 | CHO | −9.73 | H | 5.25 | 14.98 |
| 4 | H | −9.37 | CHO | 2.95 | 12.32 |
calculated with a HF/6-31G* basis set using MacSpartan 06.
Even upon heating unactivated allylic and propargylic bicyclobutylmethylamines to 150 °C, only starting material was isolated (Scheme 3, equations (1) and (2)). More vigorous conditions led to decomposition, but ene-products could not be identified. In contrast, an electron-deficient ester function provided the cycloaddition product in 51% yield at 50 °C (Scheme 3, equation (3)). In situ oxidation of a propargyl alcohol to the ynal with Dess-Martin periodinane (DMP) led to the analogous cycloaddition product at room temperature and in good yield (Scheme 3, equation (4)).
Table 1 provides an overview of HOMO-LUMO energies calculated at the HF/6-31G* level for substituted enophiles and bicyclobutanes. In order to minimize the HOMO-LUMO gap, the ene reaction is expected to proceed via the interaction of the HOMO of the bicyclobutane with the LUMO of the alkene. A large frontier orbital energy difference was observed between electron-rich alkenes and terminal bicyclobutanes or electron deficient bicyclobutanes bearing a formyl group (entries 2 and 3). In contrast, a phenyl substituent on the bicyclobutane or a carbonyl substituent at the alkene considerably enhances the reactivity by diminishing the HOMO-LUMO gap by ca. 1.4 and 2.3 eV, respectively (entries 1 and 4). In practice, all intramolecular ene reactions that we attempted with analogs of 6 that did not contain an electron-withdrawing aldehyde or ester function on the allylic amide side chain failed under the classical thermal conditions.
The synthesis of an appropriately substituted 1-azoniatricyclo[5.2.2.01,6]undecane segment of daphniglaucin is outlined in Schemes 4 and 5. 1,5-Pentanediol (7) was selectively O-tritylated, oxidized to the aldehyde and then condensed with N,N-diphenylphosphinamide in the presence of p-toluenesulfinic acid to afford the imine precursor 8.8,9 Treatment of 1,1-dibromo-2-chloromethylcyclopropane (9) with MeLi followed by t-BuLi generated bicyclo[1.1.0]butan-1-yllithium in situ,10 which, upon addition of 8, eliminated the sulfinylate and added to the resulting imine to give amide 10. This labile 1,2-adduct was immediately subjected to N-alkylation with allylbromide 1111 under phase-transfer conditions to give bicyclobutane 12 in 58% overall yield from 8. Desilylation of 12 with TBAF afforded the allylic alcohol 13. Several oxidants, solvents and reaction temperature conditions were examined in our attempts to optimize the conversion of 13 to 14. The best result was obtained when TPAP in DCM was used as the oxidant at room temperature for 1 h; aldehyde 14 was not isolated from the reaction mixture but was immediately heated after addition of benzene as a cosolvent, thus affording the formal ene product 15 in 67% yield as a 6:1 mixture of diasteromers.12,13,14 The major isomer was isolated by chromatography on SiO2 and its relative configuration was secured by an X-ray structure analysis of the hydrazone derivative 16 (Figure 2).6b
Scheme 4.

Reagents and conditions: (i) TrCl, Et3N, DMAP, CH2Cl2, rt, 71%; (ii) (COCl)2, DMSO, Et3N, CH2Cl2, −78 °C, 92%; (iii) H2NP(O)Ph2, p-TolSO2H, Et2O, rt, 94%; (iv) MeLi, t-BuLi, Et2O, THF, −78 °C; 8; (v) Bu4NHSO4, 50% aq. NaOH, toluene, rt, 58% (2 steps); (vi) TBAF, THF, rt, 85%; (vii) TPAP, NMO, CH2Cl2, benzene, rt to reflux, 67% (6 : 1 dr); (viii) 2,4-dinitrophenylhydrazine, MeOH, rt, 86%.
Scheme 5.

Reagents and conditions: (i) NaBH4, MeOH, rt, 79%; (ii) BnBr, KOH, 18-crown-6, THF, H2O, rt, 86%; (iii) conc. HCl, THF, rt; (iv) (Boc)2O, NaHCO3, MeCN, H2O, rt, 69% (2 steps); (v) MsCl, Et3N, CH2Cl2, rt; (vi) TFA, CH2Cl2, rt; (vii) Et3N, MeCN, rt, 75% (3 steps); (viii) OsO4, NaIO4, 2,6-lutidine, t-BuOH, H2O, rt; (ix) NaBH4, MeOH, rt, 45% (2 steps); (x) MsCl, NaHCO3, CH2Cl2, H2O, rt, 61%.
Figure 2.

Stereoview of the X-ray crystal structure of hydrazone 16.
The major diastereomer 15 was used in the subsequent transformation to the quaternary ammonium salt 21 (Scheme 5). Reduction of the aldehyde with NaBH4 and protection of the resulting alcohol with BnBr provided benzyl ether 17. We determined empirically that the best strategy for formation of the tricyclic ammonium ion was to close the fused six-membered ring before installing the second, bridged six-membered ring. Accordingly, 17 was converted to the primary alcohol 18 by concurrent solvolysis of N,N-diphenylphosphinoyl and trityl groups, followed by N-Boc protection. Mesylation of the primary hydroxyl group of 18, cleavage of the Boc group with TFA and cyclization in the presence of triethylamine furnished indolizidine 19. Oxidative ring opening of the cyclobutene was readily accomplished by a Johnson-Lemieux oxidation in the presence of 2,6-lutidine, and the resulting dialdehyde was reduced to diol 20 with NaBH4.15 Finally, double mesylation of diol 20 with excess MsCl in NaHCO3/H2O/DCM resulted in spontaneous cyclization followed by in situ hydrolysis of the unreacted neopentyl mesylate to give the tricyclic quaternary ammonium salt 21.16,17 Target compound 21 was isolated as an approximately 4:1 mixture of chloride and mesylate salts based on 1H NMR integration of the mesylate methyl group.
In conclusion, we have successfully extended the utility of the bicyclobutane strained ring system to alkaloid synthesis. The key reaction for the construction of the tricyclic quaternary ammonium core of daphniglaucin was based on the thermal intramolecular formal Alder-ene reaction of the N-allylated bicyclo[1.1.0]butylmethylamine 14. Further studies toward the total synthesis of daphniglaucins and other applications of bicyclobutanes in target-directed synthesis are currently in progress in our laboratories.
Acknowledgments
This work was supported in part by NIH grant P50-GM067082 and Merck Research Laboratories. We thank Dr. Steve Geib for an X-ray crystallographic analysis. M.U. is grateful for support from Hyogo Science and Technology Association.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Present address: Kobe Pharmaceutical University, Motoyamakita, Higashinada, Kobe 658-8558, Japan.
- 2.For recent examples, see. Hung TM, Ngoc TM, Youn UJ, Min BS, Na MK, Thuong PT, Bae KH. Biol Pharm Bull. 2008;31:159. doi: 10.1248/bpb.31.159.Wu YR, Ma YB, Zhao YX, Yao SY, Zhou J, Zhou Y, Chen JJ. Planta Med. 2007;73:787. doi: 10.1055/s-2007-981549.Bai LP, Zhao ZZ, Cai Z, Jiang ZH. Bioorg Med Chem. 2006;14:5439. doi: 10.1016/j.bmc.2006.05.012.Jayasuriya H, Herath KB, Ondeyka JG, Polishook JD, Bills GF, Dombrowski AW, Springer MS, Siciliano S, Malkowitz L, Sanchez M, Guan Z, Tiwari S, Stevenson DW, Borris RP, Singh SB. J Nat Prod. 2004;67:1036. doi: 10.1021/np049974l.
- 3.For reviews, see. Hashimoto T, Maruoka K. Chem Rev. 2007;107:5656. doi: 10.1021/cr068368n.Maruoka K, Ooi T, Kano T. Chem Commun. 2007:1487. doi: 10.1039/b613049f.O’Donnell M. Acc Chem Res. 2004;37:506. doi: 10.1021/ar0300625.
- 4.(a) Kubota T, Sunaura T, Morita H, Mikami Y, Hoshino T, Obara Y, Nakahata N, Kobayashi J. Heterocycles. 2006;69:469. [Google Scholar]; (b) Ishiyama H, Matsumoto M, Sekiguchi M, Shigemori H, Ohsaki A, Kobayashi J. Heterocycles. 2005;66:651. [Google Scholar]; (c) Kobayashi J, Takatsu H, Shen YC, Morita H. Org Lett. 2003;5:1733. doi: 10.1021/ol034388p. [DOI] [PubMed] [Google Scholar]; (d) Takatsu H, Morita H, Shen YC, Kobayashi J. Tetrahedron. 2004;60:6279. [Google Scholar]; (e) Morita H, Takatsu H, Shen YC, Kobayashi J. Tetrahedron Lett. 2004;45:901. [Google Scholar]; (f) Kobayashi J, Sekiguchi M, Shimamoto S, Shigemori H, Ishiyama H, Ohsaki A. J Org Chem. 2002;67:6449. doi: 10.1021/jo025854b. [DOI] [PubMed] [Google Scholar]; (g) Penelle J, Tits M, Christen P, Molgo J, Brandt V, Frédérich M, Angenot L. Phytochemistry. 2000;53:1057. doi: 10.1016/s0031-9422(00)00033-9. [DOI] [PubMed] [Google Scholar]; (h) Penelle J, Tits M, Christen P, Brandt V, Frédérich M, Angenot L. J Nat Prod. 1999;62:898. doi: 10.1021/np9804738. [DOI] [PubMed] [Google Scholar]
- 5.For the use of an 1-(arylsulfonyl)bicyclobutane in the synthesis of two isoprenoids, see: Gaoni Y, Tomazic A. J Org Chem. 1985;50:2948.
- 6.(a) Wipf P, Stephenson CRJ, Okumura K. J Am Chem Soc. 2003;125:14694. doi: 10.1021/ja038623a. [DOI] [PubMed] [Google Scholar]; (b) Wipf P, Walczak MAA. Angew Chem Int Ed. 2006;45:4172. doi: 10.1002/anie.200600723. [DOI] [PubMed] [Google Scholar]
- 7.Walczak MAA, Wipf P. J Am Chem Soc. 2008;130:6924. doi: 10.1021/ja802906k. and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Angehrn P, Buchmann S, Funk C, Goetschi E, Gmuender H, Hebeisen P, Kostrewa D, Link H, Luebbers T, Masciadri R, Nielsen J, Reindl P, Ricklin F, Schmitt-Hoffmann A, Theil F. J Med Chem. 2004;47:1487. doi: 10.1021/jm0310232. [DOI] [PubMed] [Google Scholar]
- 9.Côté A, Boezio AA, Charette AB. Proc Natl Acad Sci USA. 2004;101:5405. doi: 10.1073/pnas.0307096101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weber J, Haslinger U, Brinker UH. J Org Chem. 1999;64:6085. [Google Scholar]
- 11.DeBoef B, Counts WR, Gilbertson SR. J Org Chem. 2007;72:799. doi: 10.1021/jo0620462. [DOI] [PubMed] [Google Scholar]
- 12.Experimental protocol for the formation of 15 from 13: TPAP (96 mg, 0.27 mmol) was added to a stirred solution of alcohol 13 (1.8 g, 2.7 mmol), powdered 4 Å sieves (2 g) and 4-methylmorpholine N-oxide (651 mg, 5.39 mmol) in CH2Cl2 (30 mL) at rt. The reaction mixture was stirred for 1 h at rt, diluted with benzene (200 mL) heated at reflux for 4 h and filtered through a thin pad of Celite. The filtrate was concentrated and purified by chromatography on SiO2 (AcOEt) to afford a diastreomeric mixture (6:1) of spirocycles (1.21 g, 67%) as a colorless foam, which was carefully re-purified by chromatography on SiO2 (AcOEt) to give the major isomer 15 (942 mg, 52%).
- 13.Spectral data of 15: IR (neat) 3413, 3057, 2937, 2869, 1723, 1489, 1439, 1190, 1121, 1073, 1047, 909, 729 cm−1; 1H NMR (300 MHz, CDCl3) δ 9.74 (s, 1 H), 7.95-7.88 (m, 2 H), 7.78-7.71 (m, 2 H), 7.54-7.20 (m, 21 H), 6.47 (d, J = 3.0 Hz, 1 H), 6.11 (d, J = 3.0 Hz, 1 H), 3.52-3.37 (m, 2 H), 3.30-2.78 (m, 4 H), 2.58 (dd, J = 18.0, 3.0 Hz, 1 H), 2.45 (d, J = 13.5 Hz, 1 H), 2.44-2.38 (m, 1 H), 2.22 (d, J = 13.5 Hz, 1 H), 1.93-1.76 (m, 1 H), 1.55-1.18 (m, 4 H), 1.02-0.88 (m, 1 H); 13C NMR (75 Hz, CDCl3) δ 201.4, 144.3, 141.2, 134.5, 133.0, 132.8 (d, J = 9.0 Hz), 132.2 (d, J = 9.8 Hz), 131.7 (d, J = 2.3 Hz), 131.4 (d, J = 3.0 Hz), 131.3, 131.1, 128.6, 128.4, 128.2, 128.1, 127.7, 126.7, 86.2, 64.2, 63.1, 59.7 (d, J = 1.5 Hz), 50.3, 44.5, 33.7 (d, J = 4.5 Hz), 34.7, 33.8 (d, J = 5.3 Hz), 29.9, 23.8; MS (ESI) m/z (rel. intensity) 688 ([M+Na]+, 90), 527 (25), 443 (37), 243 (100); HRMS (ESI) calc for C44H44NO3PNa (M+Na) 688.2957, found 688.2964.
- 14.For reactions of bicyclo[1.1.0]butanes with benzyne that afforded a mixture of ene and other cycloaddition products, see: Pomerantz M, Gruber GW, Wilke RN. J Am Chem Soc. 1968;90:5040.Pomerantz M, Wilke RN, Gruber GW, Roy U. J Am Chem Soc. 1972;94:2752.Gassman PG, Richmond GD. J Am Chem Soc. 1968;90:5637.Gassman PG, Richmond GD. J Am Chem Soc. 1970;92:2090.
- 15.Yu W, Mei Y, Kang Y, Hua Z, Jin Z. Org Lett. 2004;6:3217. doi: 10.1021/ol0400342. [DOI] [PubMed] [Google Scholar]
- 16.Experimental protocol for the formation of 21 from 20: To a solution of diol 20 (18 mg, 0.054 mmol) in CH2Cl2 (3 mL) were added NaHCO3 (10 mg, 0.12 mmol), H2O (0.12 mL) and MsCl (0.009 mL, 0.12 mmol) at 0 °C. The reaction mixture was stirred for 5 h at rt and concentrated in vacuo. The residue was dissolved in CH2Cl2 and an insoluble solid was removed by filtration. After the solvent was evaporated under reduced pressure, the residue was dissolved in water. The solution was filtered through small pad of Celite and the filtrate was concentrated in vacuo to form white solid, which was recrystallized in CH2Cl2 and hexane to afford the ammonium salt 21 (12 mg, 61%) as 4:1 mixture of Cl− and MsO− counterions.
- 17.Spectral data of 21: white solid; IR (neat) 3224, 2932, 2844, 1452, 1360, 1195, 1171, 1098, 1072, 740 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 7.37-7.29 (m, 5 H), 5.60-5.10 (br, 1 H), 4.47, 4.44 (AB, J = 12.0 Hz, 2 H), 3.84 (t, J = 11.5 Hz, 1 H), 3.75 (t, J = 8.5 Hz, 1 H), 3.62 (d, J = 11.5 Hz, 1 H), 3.53 (d, J = 11.5 Hz, 1 H), 3.52 (t, J = 11.5 Hz, 1 H), 3.45-3.23 (m, 5 H), 3.19-3.12 (m, 1 H), 2.34 (s, 0.6 H, CH3SO3−), 2.18-2.11 (m, 1 H), 2.08 (td, J = 12.0, 5.5 Hz, 1 H), 1.98-1.91 (m, 1 H), 1.84-1.66 (m, 5 H), 1.63-1.55 (m, 1 H), 1.51 (q, J = 11.5 Hz, 1 H), 1.41-1.29 (m, 1 H); 13C NMR (75 Hz, CD3OD) δ 139.8, 129.6, 129.1, 128.9, 74.4, 74.0, 71.2, 69.5, 60.8, 55.4 (2C), 54.2, 41.6, 32.1, 31.7, 22.3, 21.8, 20.7; MS (ESI) m/z (rel. intensity) 316 ([M-Cl]+ or [M-OMs]+, 100), 224 (30); HRMS (ESI) calc for C20H30NO2 (M-Cl or M-OMs) 316.2277, found 316.2265.