

Abstract
Peptides are emerging as a new class of biomaterials due to their unique chemical, physical, and biological properties. The development of peptide-based biomaterials is driven by the convergence of protein engineering and macromolecular self-assembly. This review covers the basic principles, applications, and prospects of peptide-based biomaterials. We focus on both chemically synthesized and genetically encoded peptides, including poly-amino acids, elastin-like polypeptides, silk-like polymers and other biopolymers based on repetitive peptide motifs. Applications of these engineered biomolecules in protein purification, controlled drug delivery, tissue engineering, and biosurface engineering are discussed.
Keywords: biomaterials, elastin-like polypeptides, drug delivery, protein purification, tissue engineering, surface engineering
1 Introduction
Synthetic and natural biopolymers are finding their way into a variety of applications in materials science and biointerface engineering, such as tissue engineering scaffolds, drug delivery matrices, and as detectors and transducers in biosensors. Commonly used natural biopolymers include cellulose, collagen, hyaluronic acid, and fibrin gels. In contrast to these naturally occurring biopolymers, “engineered” peptide-based biopolymers have recently attracted much attention as a new class of materials. Prototypical examples of engineered peptide-based biomaterials include poly-amino acids, elastin-like polypeptides, silk-like proteins, coiled-coil domains, tropoelastin-based peptides, leucine zipper based peptides, peptide amphiphiles, beta-sheet forming ionic oligopeptides, and beta-hairpin peptides.
This explosion of new peptide-based materials is driven by two scientific developments. The first is our increasingly sophisticated understanding of protein structure-function, which provides peptide motifs that are useful for the design of repetitive, polypeptide based materials. The second is the maturation of recombinant DNA technologies, which allows these materials to be synthesized in large yields with precise control over the chain length, stereochemistry, and monodispersity.
This article aims to provide the materials science community a primer on the design principles, synthesis techniques, and characterization methodologies of biopolymers, as well as an overview of current trends in the field of peptide-based biomaterials. The current limitations, challenges, and prospects of peptide-based biopolymers are compared to their synthetic counterparts. This review also summarizes the current applications of peptide-based biopolymers as protein purification tags, targeted drug delivery carriers and controlled release depots, self-assembled and chemically crosslinked tissue engineering scaffolds, and as components for biosensing and bioanalytical devices.
2 Peptide-based biopolymers
2.1 Overview
2.1.1 Structural description
Our basic understanding of amino acids, peptides, and proteins forms the foundation for the current development of peptide-based biomaterials. A discussion of the properties and applications of peptide-based biomaterials would be incomplete without an understanding of how their structure influences their physical, chemical, and biological properties. The primary amino acid sequence, the secondary structure (e.g., alpha-helix or beta-sheet motifs), and the tertiary structure (i.e., the actual three dimensional structure of the peptide) determine the functionality of these materials. Furthermore, for peptides that undergo self-assembly, the interactions between individual molecules also affects the structure and thus the function of peptide-based materials. For more details, readers are referred to other reviews on the structure-function relationship of peptide-based materials [1-5].
2.1.2 Physical, chemical, and biological properties
Peptide-based biomaterials are “soft” and “wet”, while most other materials used in biomedical sciences and biointerface engineering, such as metals, ceramics, and polymers (with the exception of hydrogel polymers), are regarded as “hard” and “dry”. Hence, hydration is particularly important in maintaining the structural and functional integrity of peptide-based biomaterials. The constraint of hydration is both a blessing and curse: it makes these materials unsuitable for applications that cannot tolerate water, but in applications that necessitate bringing these materials into contact with an aqueous environment, the requirement of hydration can be profitably exploited in materials design.
Polypeptide-based materials have other important advantages and disadvantages as compared to synthetic polymers that determine their applications. The primary disadvantage of peptide-based polymers is the limited number of building blocks; compared to synthetic polymers, which can be synthesized via an ever-expanding set of monomers, peptide based polymers are largely restricted to the 20 natural amino acids (though we note that this limit is expanding through new synthetic and biological methods to incorporate unnatural amino acids into peptides and proteins [6, 7]). Despite this important limitation, peptide based materials offer many potential advantages over synthetic materials. First, short peptide motifs such as RGD, KNEED, and IKVAV that are ubiquitous ligands for cell receptors and mediate various cell behaviors such as attachment and spreading [8-12] can be appended to or embedded within repetitive polypeptide materials with greater ease than in synthetic materials. Second, self-assembly and directed-assembly of peptides have recently gained research interest as viable ways to generate functional biomaterials. Leucine zipper-based materials, peptide amphiphiles, beta-sheet forming ionic oligopeptides, and beta-hairpin peptides are a few examples of self-assembling biomaterials that can be generated from peptides. Third, many peptide-based biomaterials are easily degraded by the body, thus making them desirable as drug delivery vehicles and tissue engineering scaffolds. Fourth, biology is replete with peptide sequences that exhibit structural transitions in response to the binding of metal ions and other biological ligands [13-26]; these sequences offer enormous possibilities in the design of biologically responsive materials.
In this discussion, it is important to note that peptide-based materials are often touted as biocompatible. This statement, in the absence of hard experimental evidence, must be greeted with some skepticism. There is no intrinsic reason for peptide based materials to be biocompatible other than the somewhat naïve notion that they must be, simply because they are composed of building blocks that are native to all organisms. As an important counter-example, it should be noted that many peptides and protein drugs are immunogenic, so the potential immunogenicity of all peptide based materials must be considered, especially peptides that are “non-self”.
2.2 Materials design and synthesis
There are two general approaches to designing polypeptides. In the synthetic approach, standard polymerization techniques are employed using amino acids or their derivatives as monomers, such as the solid-phase polypeptide synthesis [27, 28] or synthesis by NCA polymerization [29, 30]. This approach is especially useful if the goal is to design hybrid materials that combine peptide motifs within a non-peptidic macromolecular architecture (e.g., peptide side chains on a synthetic polymer backbone). On the other hand, the chain length and stereochemistry of these polymers are often difficult to control. In contrast, recombinant DNA techniques offer an alternative, genetically encoded approach for the synthesis of polypeptides. This approach offers the advantage of high specificity in sequence, stereochemistry, and molecular weight. However, not all polypeptides can be expressed well in a heterologous host, nor can the polypeptide, during expression, be combined with non-peptidic moieties, other than by post-expression modification of the polypeptide.
In this review, we refer to polypeptides made via chemical methods as synthetic polypeptides, and we refer to polypeptides made by recombinant DNA methodologies as genetically encoded polypeptides. With this distinction clarified, we next summarize two examples of peptide-based materials that have been synthesized either by chemical methods or genetically encoded synthesis: elastin-like polypeptides and silk-like polymers.
2.2.1 Elastin-like polypeptides
Elastin is one of the most abundant extracellular matrix proteins, along with glycosaminoglycans (GAGs) and collagens, and it has the unique mechanical property of allowing repeated extensibility followed by elastic recoil. It is commonly found in large arteries, lung parenchyma, and elastic ligaments [31-35]. Elastin is an insoluble protein, and it is synthesized as the soluble precursor tropoelastin (MW 70 kDa), which is composed of alternating hydrophobic and hydrophilic domains containing lysine crosslinking sites. Tropoelastin is secreted from cells during elastogenesis, and then forms fibrils that are enzymatically crosslinked [31].
Elastin-like polypeptides are derived from a repeating motif within a hydrophobic domain of mammalian tropoelastin: the most common motif has the sequence (VPGXG)m, where X can be any amino acid other than proline, and m is the number of repeats [36]. There are many other variants of ELPs that range from other pentapeptides with the repeat sequences KGGVG [37] or LGGVG [38] to heptapeptides with the sequence LGAGGAG and nonapeptides with the sequence LGAGGAGVL [39]. All of these elastin analogues appear to exhibit elastin-like properties. Here, we largely confine our discussion to the most widely studied pentapeptide motifs.
ELPs composed of the (VPGXG)m repeat are thermally responsive polypeptides that undergo a reversible, inverse temperature phase transition [40]. ELPs are highly soluble in an aqueous solution below their transition temperature (Tt), but aggregate rapidly above their Tt. In applications where control of the Tt of ELPs is important, there are several design constraints, as follows: first, thermally responsive ELPs must retain the Val-Pro-Gly-Xaa-Gly repeat unit, where Xaa is a guest residue; a variant that contravenes this rule is IPGVG [41]. Second, proline at the guest residue position must be avoided, as its presence at the fourth position prevents helix formation. Third, the guest residue composition strongly affects the transition temperature, making its selection the primary focus in ELP design. Fourth, the transition temperature shows a 1/L dependence on the chain length (L) of an ELP. In addition to ELPs solely consisting of pentapeptide repeats, side groups capable of adding functionality to the ELP can be added without disrupting the transition. For example, Cys has been added to the 3’-terminus of some ELPs to allow for conjugation, while lysine residues have been added to allow for crosslinking.
The strict amino acid sequence requirements make it attractive to design ELPs at the genetic level. A good starting point for the design of ELPs comes from the studies of Urry and co-workers, who quantified the effects of each of the guest residues on transition temperature by determining the free energy change involved in the transition of ELPs containing a single guest residue [36, 42] (Table 1). These studies clearly illustrated the effects of hydrophobicity on the Tt of an ELP. A similar approach has been used to successfully predict the Tt of ELP fusion proteins y quantifying the effect that the solvent accessible residues on a protein have on the Tt of the fused ELP. Trabbic-Carlson et al. calculated the solvent accessible hydrophobic surface area of proteins with known crystal structures using PROBE [43], and showed that the change in Tt of an ELP fusion protein inversely scales with this parameter [44] (Figure 2). This modeling approach allows the Tt of an ELP fusion protein to be predicted from that of the ELP that is appended to.
Table 1.
The effects of guest residue composition on the Tt of ELP homopolymers. The Tt’s reported in this table are a useful starting point for choosing which guest residues to include in the ELP gene in order to achieve a desired LCST, but are only approximate as they do not take into account the effect of MW on the Tt of the ELP [36].
| Residue | R Group | Abbreviation | Letter | Tta | ΔHt kcal/mold ± 0.05 | ΔSt kcal/mold ± 0.05 |
|---|---|---|---|---|---|---|
| Tryptophan |

|
Trp | W | −90°C | 2.10 | 7.37 |
| Tyrosine |

|
Tyr | Y | -55°C | 1.87 | 6.32 |
| Phenylalanine |

|
Phe | F | −30°C | 1.93 | 6.61 |
| Histidine |
|
His | H | −10°C | ||
| Proline(calc.)b | −CH2CH2CH2− | Pro | P | (−8°C) | ||
| Leucine | −CH2CH(CH3)2 | Leu | L | 5°C | 1.51 | 5.03 |
| Isoleusine | −CH(CH3)CH2CH3 | Ile | I | 10°C | 1.43 | 4.60 |
| Methionine | −CH2CH2SCH3 | Met | M | 20°C | 1.00 | 3.29 |
| Valine | −CH(CH3)2 | Val | V | 24°C | 1.20 | 3.90 |
| Histidine |

|
His+ | H+ | 30°C | ||
| Glutamic Acid | −CH2CH2COOH | Glu | E | 30°C | 0.96 | 3.14 |
| Cysteine | −CH2SH | Cys | C | 30°C | ||
| Lysine | −CH2CH2CH2CH2NH2 | Lys+ | K+ | 35°C | 0.71 | 2.26 |
| Proline(exptl)c | −CH2CH2CH2− | Pro | P | 40°C | 0.92 | 2.98 |
| Alanine | −CH3 | Ala | A | 45°C | 0.85 | 2.64 |
| Aspartic Acid | − CH2COOH | Asp | D | 45°C | 0.78 | 2.57 |
| Threonine | −CH(OH)CH3 | Thr | T | 50°C | 0.82 | 2.60 |
| Asparagine | −CH2CONH2 | Asn | N | 50°C | 0.71 | 2.29 |
| Serine | −CH2OH | Ser | S | 50°C | 0.59 | 1.86 |
| Glycine | −H | Gly | G | 55°C | 0.70 | 2.25 |
| Arginine | −CH2CH2CH2NHC(NH)NH2 | Arg | R | 60°C | ||
| Glutamine | −CH2CH2CONH2 | Gln | Q | 60°C | 0.55 | 1.76 |
| Lysine | −CH2CH2CH2CH2NH3+ | Lys | K | 120°C | ||
| Tyrosinate |

|
Tyr- | Y- | 120°C | 0.31 | 0.94 |
| Asporate | −CH2COO- | Asp- | D+ | 120°C | ||
| Glutamate | −CH2CH2COO- | Glu- | E- | 250°C |
Figure 2.
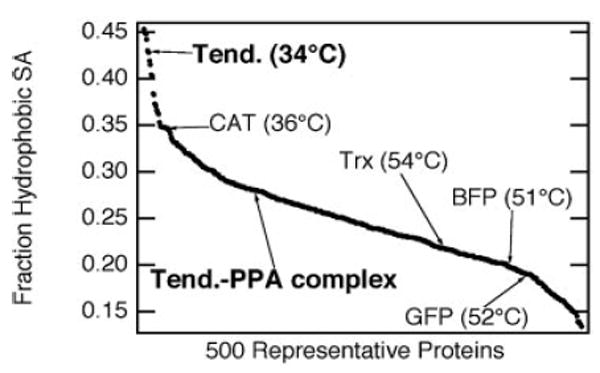
Effect of the hydrophobic fraction of solvent-accessible surface area (SAShydrophobic/SAStotal) on the Tt of ELP fusion proteins. Hydrophobic fractions of solvent-accessible surface for 500 proteins (having > 40 amino acids) selected from the protein database (PDB) are shown. The positions of five specific proteins are plotted along with their corresponding ELP fusion protein transition temperatures (at 25 μM in PBS) to show the inverse relationship between SAShydrophobic/SAStotal and Tt. Green fluorescent protein (GFP), blue fluorescent protein (BFP), thioredoxin (Trx), chloramphenicol acetyltransferase (CAT), and tendamistat (Tend) are shown. This plot also predicts that when a Tend-ELP fusion protein binds its protein partner, porcine pancreatic α-amylase (PPA), the transition temperature will increase due to the increase in hydrophobic surface area of the bound protein complex [44].
2.2.2 Silk-like polypeptides
Silk is a natural protein fiber that is produced by spiders of the class Arachnida as well as several worms of the order Lepidoptera. Spider silk is a remarkable biomaterial that is lightweight, extremely strong and elastic, and has excellent impact resistance. Silks from the silkworm Bombyx mori and the orb-weaving spider Nephila clavipes have been investigated to understand their structure and processing mechanisms and to exploit the properties of these proteins for use as biomaterials [45, 46]. These native silk proteins contain highly repetitive crystalline domains periodically interrupted by less crystalline or amorphous regions. The crystalline region from B. mori fibroin is a 59 amino acid repeat [GAGAGSGAAG[SGAGAG]8Y], with a 3:2:1 ratio of glycine, alanine, and serine [47]. Repeated motifs from N. clavipes dragline silk are less conserved, but a 13 amino acid repeat [YGGLGSQGAGRGG] has been identified [48, 49]. Many synthetic genes encoding B. mori silk-like sequences have been cloned and expressed in E. coli. A six amino acid repeat [GAGAGS] controls crystallinity in films and fibers [50], and repeats such as [(AG)3PEG] [51] and [(AG)3EG] [52] have been used in efforts to control crystalline order in protein-based materials.
Capello et al. demonstrated that silk-like proteins (SLPs) based on the repeating motif [GAGAGS] produce crystalline structures similar to the β-sheet structures of native silk proteins. Their degree of crystallinity can be controlled by periodically including blocks of amino acids such as GAAGY. They also showed that the addition of elastin-like blocks within the SLPs to make silk-elastin-like proteins (SELPs) further disrupts the crystalline structure. While SLPs are generally insoluble in aqueous solution, the ELP blocks influence the molecular chain properties of the protein so that the SELPs are water soluble [50]. This is an important property, as water solubility is essential for the manufacturing, purifying, and processing of silk-like biopolymers. A second method for increasing the water solubility of SLPs is the addition of sterical crystallization triggers. Winkler et al. added methionine residues flanking the β-sheet forming polyalanine sequences of a recombinant silk protein. The oxidized methionine residues sterically hinder β-sheet formation and increase the protein’s hydrophilicity, while in the reduced state, the added residues have no effect on crystallization [53]. They also modified a recombinant silk protein to include a phosphorylation site for cyclic AMP-dependent protein kinase (cAPK). By introducing charged phosphate groups, the hydrophobic interactions required for β-sheet formation were interrupted, also preventing insolubility [53].
One of the greatest hurdles in producing silk in large quantities is the level of expression. The highly repeating gene sequence that codes for silk is not efficiently translated in E. coli, the most extensively used expression host. Some alternative expression hosts have proven more successful, although they still present challenges of their own. Insect cells show lower-than-desired levels of expression. Transgenic species such as tobacco and potato have shown high levels of expression (2% of all soluble protein) [54], and yeast cells have shown high levels of expression, but the resultant protein has lower purity. Partial gene sequences cloned into mammalian epithelial cells have produced proteins with mechanical properties close to that of native silk fibers [55].
2.3 Synthesis
2.3.1 Synthetic polymers
Given the time and effort required for the synthesis of genetially encoded polypeptides and the restriction on the use of natural amino acids, there is great interest in developing synthetic procedures that rival genetically encoded synthesis to produce polypeptides with similar control of size and uniformity [56]. Although the genetic polymerization methods may offer greater control of sequence, synthetic methods do allow for the incorporation of artificial amino acids, stereochemically unique amino acids, and β-amino acids, all of which can prevent protease degradation. Conventional polymerization methods are also ideal for combining polypeptides with other synthetic polymers such as poly(ethylene glycol). Although progress has been made in the site specific incorporation of unnatural amino acids into proteins during recombinant synthesis [6, 7], these approaches are still difficult to use and do not, in general, lend themselves to the large scale production of polypeptides with unnatural amino acids.
2.3.2 Genetically encoded polymers
Conceptually, genetically encoded synthesis has three steps: creation of a recombinant gene segment that codes for the protein of interest, insertion of this segment into a DNA vector, which is typically a plasmid to create a recombinant DNA molecule, and transformation of this recombinant DNA molecule into a host cell (Figure 1). Cells that are successfully transformed or transfected with the recombinant DNA molecule are grown in culture, and the polypeptide produced by the cell is purified from the cell. The rate-limiting step in genetically encoded synthesis lies mostly in assembly of the gene to insert into the desired vector, especially if the desired polypeptide product (and consequently the gene that encodes it) is comprised of a large number of repeats. Short DNA sequences of up to 100 nucleotides can be directly synthesized chemically on an automated solid phase DNA synthesizer. These short single stranded, chemically synthesized DNA sequences must be assembled into larger pieces that encode for repetitive polypeptides of the desired MW (typically several tens of thousands or larger MW). These methods include concatenation of oligonucleotides, recursive directional ligation (RDL), and mutagenesis or amplification of existing gene segments using polymerase chain reaction (PCR). In this section, we focus on concatenation of oligonucleotides and RDL.
Figure 1.

Schematics of concatemerization (A) and RDL (B). During concatemerization, a statistical distribution of genes encoding a monomer is ligated together, the gene inserts are sorted based on size, and desired insert is ligated into a vector. RDL incorporates the insert produced during a given step as the insert for the next step to ensure a given size. Figure reproduced from [62] with permission from Elsevier.
The concatemerization method and RDL are both designed to generate repetitive DNA sequences [50]. Both ELP and silk genes are candidates for these methods of gene design, as they consist of pentamer repeat units [57, 58]. Even with variation of the guest residue within pentamers, the distribution of guest residues can typically be captured within one synthetic insert. In the concatemerization method, a library of these oligonucleotide inserts is built, and the vectors are cut at specific sites with different restriction enzymes. The inserts of various discrete sizes are then ligated into vectors at a specific site to recircularize the vector-insert combinations. The ligation products are sorted according to their size by gel electrophoresis and transformed into cells for polypeptide expression. The advantage of using this method is that different sizes of the repetitive gene are obtained in a single experiment. The disadvantage is that concatemerization is a statistical process, so it is difficult to, a priori, design a polypeptide of a certain size, and it is difficult to synthesize genes that encode for large polypeptides (> 100 kDa). This method is useful when a library of genes of different sizes is desired, but not for the creation of a gene of a specific size or for very large genes.
RDL is an alternative method that provides a greater level of control over the size of the insert [59]. In this method, a single double stranded oligonucleotide is inserted into a vector to create a circular, recombinant DNA molecule with two different restriction sites flanking the insert. After in vivo amplification of the recombinant DNA molecules within a host cell by replication to obtain sufficient quantities for the next step, one population of this recombinant DNA molecule is cut at both sites and the insert is extracted. A second population is cut at a single site (linearized) so that the resultant ends are compatible with the extracted insert. The single oligonucleotide insert is then ligated to the linearized parent molecule to create a recombinant DNA molecule that now contains a dimerized insert in a head-to-tail orientation; by choice of the two restriction sites, this process seamlessly joins the 2 monomeric inserts and also eliminates the restriction site at the junction between the two monomeric inserts, so that the two unique restriction sites are now on either end of the dimerized gene. This process is then repeated to double the size of the gene, though we note that at the second iteration of RDL, the monomer can be combined with the dimer to create a trimer. In the iterative ligation process, any two oligomers from previous rounds can be combined, so that a library of genes can be obtained wherein each recombinant molecule contains a unique gene, and the library can contain genes ranging from the monomer to n-mer oligomer.
This deterministic method of synthesis has several advantages over chemical synthesis. First, polypeptides synthesized by this method have a defined sequence, stereochemistry, and molecular weight based on the genetic template. Second, transformed cells can provide a perpetual supply of the polypeptide. Third, in the event that the polypeptide has secondary or tertiary structural elements, the in vivo folding machinery of the cell can assist in forming the correct secondary and tertiary structure.
Recombinant synthesis techniques have several disadvantages as well. Genetically encoded polymers have a significant lead-time, mainly due to the work involved in the molecular biology necessary to assemble the gene in the desired vector and to optimize expression levels in the host cell. Second, typically only the 20 proteinogenic L-amino acids can be incorporated using standard cellular components, thus limiting the monomers that can be incorporated through genetically encoded synthesis, though as we have pointed out, these restrictions are slowly being relaxed due to advances in the incorporation of unnatural amino acids in genetically encoded synthesis [6, 60, 61].
2.4 Characterization techniques
Many techniques, originally developed to characterize the solution properties opf peptides and proteins and the mechanical properties of hydrogels, can be used to determine the biophysical, structural, and rheological properties of peptide-based materials. The techniques highlighted below are a partial compendium of useful techniques, and are by no means comprehensive. Additional techniques may be necessary for individual applications in order to fully determine the functionality of the biomaterials; these will be discussed separately for each application.
2.4.1 Biophysical properties
Many characterization techniques originally developed for the characterization of proteins are useful for the characterization of repetitive peptides as well [63]. Protein concentrations of peptide-based biomaterials can be rapidly determined spectrophotometrically or colorimetrically or by using a Coomassie brilliant blue or bicinchonic acid assay. Molecular weights of these materials can be measured by electrophoresis-based methods such as SDS-PAGE, mass spectrometry (MS) methods such as matrix-assisted laser desorption ionisation (MALDI-MS) and electrospray MS, or analytical ultracentrifugation based on sediment coefficients.
For ELPs, the inverse temperature transition behavior is usually represented by the lower critical solution temperature (LCST) or transition temperature (Tt) (the two terms are used interchangeably), which is often determined by the turbidity (optical density at 350 nm) as a function of temperature (Figure 3) [64, 65].
Figure 3.

(A) Relative turbidity measurements for free ELP and ELP fusion proteins of various size. (B) The relationship between transition temperature and length of thioredoxin-ELP fusion proteins. (C) The relationship between transition temperature and NaCl concentration for the thioredoxin/60-mer ELP fusion protein (25 μM) in 50 mM phosphate buffer, pH 8.0. [69]
Surface plasmon resonance spectroscopy (SPR) provides a quick and convenient way to determine the binding behavior of peptide-based biomaterials. Peptides can be easily immobilized on a gold coated glass chip using a common conjugation chemistry such as 11-mercaptohexadecanoic acid (MHA). Binding constants such as ka, kd, and Kd are easily calculated based on the binding curves of the peptides [66]. Isothermal titration calorimetry (ITC) can also be used to study the binding thermodynamics of peptides with their binding partner. Thermodynamic parameters such as the binding enthalpy, entropy, free energy, and binding constant can be calculated from calorimetric data.
2.4.2 Structural properties
Structural properties such as particle size and proper folding play an important role in protein purification, tissue engineering, and drug delivery applications. Nuclear magnetic resonance (NMR) and X-ray crystallography are the methods of choice to obtain comprehensive structural information for proteins. In addition, protein folding can be studied using circular dichroism (CD) spectrometry, which identifies the unfolding and folding transitions of peptides. In particular, CD measures the absorption of left- and right-handed circularly polarized light to determine the fraction of alpha-helices and beta sheets in a polypeptide. Protein folding is an important determinant in the functionality of peptide-based biomaterials, especially those with enzymatic and biological functions. Differential scanning microcalorimetry (DSC) can be used to elucidate the folding and refolding properties of peptides during cooling and heating. In principle, DSC measures the amount of heat required to increase the temperature of the peptide during a physical transformation such as a phase transition, from which the state of protein folding can be determined [66].
Dynamic light scattering (DLS), also known as photon correlation spectroscopy, can be used to determine the hydrodynamic radius (Rh) of polypeptides in aqueous solution. In DLS, a beam of light is passed through the aqueous sample, and the particles scatter some of the light in all directions. Because the particles are small enough to undergo random thermal (Brownian) motion, the distance between them is constantly varying, and constructive and destructive interference of the light scattered by neighboring particles causes the measured scattered intensity to fluctuate with time. Analysis of the intensity time correlation function can yield the diffusion coefficient of the particles and, via the Stokes Einstein equation, Rh [67]. DLS is especially useful for examining the formation of self-assembled polypeptide micelles.
2.4.3 Rheological properties
Rheological properties of peptide-based biomaterials are particularly important in tissue engineering scaffolds, because their primary role is as a structural component to support cell growth. The load supporting ability of crosslinked or self-assembled polypeptides can be determined by oscillatory rheological characterization in a solution state (Figure 4). The rheological properties can be estimated by measuring G′ (elastic (storage) modulus), G″ (viscous (loss) modulus), G* (dynamic (complex) shear modulus), η* (dynamic (complex) viscosity), and tan δ (loss angle) as a function of strain, frequency, temperature, time, and other parameters in order to elucidate a relationship between the molecular structure of a polypeptide and its rheological properties. From basic rheological principles, the elastic modulus G′ represents the solid-like component of a viscoelastic hydrogel, and the viscous modulus G″ represents the liquid-like component. If G″ is much larger than G′, the liquid-like behavior of the solution predominates. On the other hand, if G′ is larger than G″, solid-like behavior prevails. The gelation point is defined as the crossover between G′ and G″. The loss angle, δ, is a measure of the dissipation of energy inherent in the material (δ = 90 ° for an elastic solid; δ = 90 ° for a Newtonian viscous fluid), and is a useful parameter for quantifying the viscoelasticity of a material.
Figure 4.
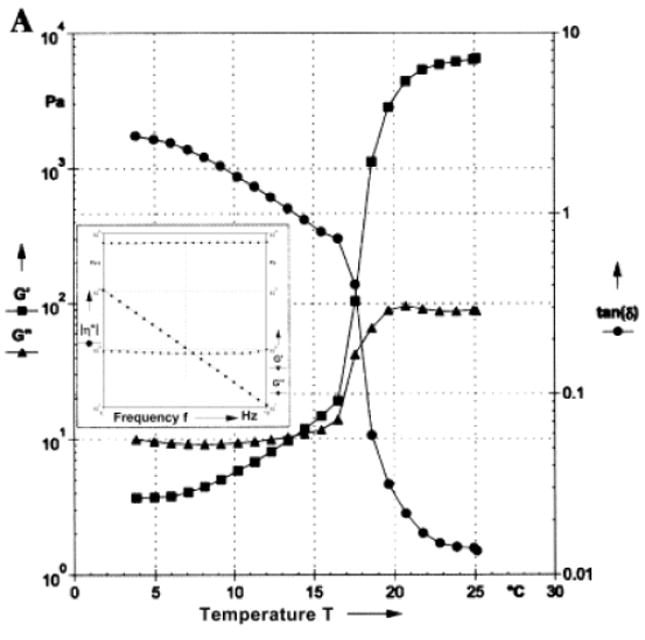
Dynamic rheological data describing the variation of the storage (G′) and loss (G″) moduli and loss angle (tan(δ)) as a function of temperature for a concentrated aqueous (25 wt.%) solution of an elastin-mimetic triblock polypeptide. The inset within the figure depicts the frequency sweep for the solution at 25 °C at a strain amplitude of 1 %. The gelation point is defined as the point where G′ and G″ cross. Below the gelation point, this polypeptide is a viscoelastic liquid (G′ < G″), and above the gelation point, it is a viscoelastic solid (G′ > G″). A loss angle approaching zero also indicates solid-like behavior. Reproduced from [70].
For elastin-like polypeptides, gel swelling is another important rheological parameter that can be quantitatively described by the swelling ratio. A swelling ratio of greater than one indicates that the gel undergoes swelling, and a ratio of less than one indicates that the gel undergoes contraction. By simply weighing the peptide-based gels before and after incubation with buffer, the swelling ratio can be easily calculated by normalizing the measured weight to the original weight [68].
3 Applications
In this section, we discuss a variety of applications of peptide-based polymers in protein purification, drug delivery, tissue engineering, and biointerface science.
3.1 Protein purification
3.1.1 Introduction
The ready availability of recombinant proteins is essential for their many biomedical applications as therapeutics and diagnostics, and in regenerative medicine and biosensing. Proteins are also important reagents for drug discovery, lead identification and validation, and high-throughput screening. Although many proteins can be produced in large quantities by recombinant expression, the cost of the final product, most of which (~70%) is due to purification, can sometimes be prohibitive for routine use. In addition, the production of many other proteins in quantities relevant for biopharmaceutical applications remains difficult, mainly due to problems in protein expression and purification. To a great extent, these constraints also apply to peptide-based polymers.
Many strategies have been developed to improve protein expression and have been thoroughly discussed in other reviews [71, 72]. Different expression hosts such as bacteria, mammalian cells, and insect cells are commonly used, and their selection is largely based on the properties of the proteins to be expressed. The choice is also dictated by the requirement of proper folding and post-translational modifications, which determine the bioactivity of the purified protein [71]. Once the expression host has been selected, the yield of the protein must be maximized by the optimization of expression conditions such as medium formulation, feeding schedule, and metabolic waste removal [72].
3.1.2 Protein purification methods
Most protein purification techniques are based on the intrinsic differences in the physico-chemical properties of proteins, such as solubility, size, charge, hydrophobicity, and shape. Commonly used methods that take advantage of these properties include precipitation, dialysis, electrophoresis, and chromatography.
3.1.2.1 Chromatography
Among all available methods, chromatography is by far the most widely used for purifying proteins. Chromatography encompasses a family of techniques that use columns to separate proteins based on size [73], charge [74], hydrophobicity [75], or shape [76]. A single chromatographic separation based on a single physical parameter of a protein can be applied to all proteins, but will usually only provide a modest gain in purity. Multiple chromatographic steps, each using an orthogonal separation property (e.g., separation based on charge followed by size) are needed to attain the desired purity. These methods, when used either individually or in combination, provide a means to isolate and enrich proteins of interest in a quantity relevant for biopharmaceutical and biomedical uses.
As an alternative to conventional chromatography methods that exploit differences in the physico-chemical properties of proteins, recombinant proteins can be purified through the combination of genetic engineering and affinity chromatography. An affinity tag, typically a short peptide, is tagged on a target protein at the N- or C-terminus using genetic engineering methods. The tag serves as a binding partner to capture molecules anchored on chromatography beads. Over the years, many affinity tags such as short peptide sequences and binding proteins have been developed and are now routinely used in protein purification (Table 2). Although affinity chromatography can produce proteins of high purity, the use of affinity tags is limited to recombinant proteins (with the notable exception of using protein A chromatography to purify antibodies). In addition, the presence of the affinity tag can change the structure and bioactivity of the isolated protein. In some cases, affinity tags can adversely influence crystallization (e.g. His-tag) [72] or make proteins hard to solubilize (e.g. polyarginine) [72]. The tags are also susceptible to proteolytic cleavage (e.g. CBD) [77]. Removal of the affinity tag from the purified protein by site-specific proteolysis requires an additional round of protein purification and may leave behind extraneous residues.
Table 2.
Affinity tags used in recombinant protein expression and purification [91].
| Tag | Size | Binding partner | Ref. |
|---|---|---|---|
| Short peptide sequences | |||
| FLAG™ peptide (DYKDDDDK) | 8 aa | Antibody with Ca2+ | [92] |
| HA tag (YPYDVPDYA, influenza virus hemagglutinin) | 9 aa | Antibody | |
| Myc/c-myc (EQKLISEED) | 10 aa | Antibody | |
| Poly-arginine | 5-15 aa | S-sepharose (cationic) | |
| Poly-aspartic acid | 5-16 aa | Anionic | |
| Poly-cysteine | 4 aa | Thiopropyl-sepharose | |
| Poly-histidine (His-tag) | 6/8/10 aa | Nickel | [93, 94] |
| Poly-phenyalanine | 11 aa | Phenyl-superose (hydrophobic) | |
| S-tag | 15 aa | S-protein (modified ribonuclease A) | |
| Strep-tag (AWRHPQFGG) | 9 aa | Streptavidin | [95, 96] |
| Strep-tag II (WSHPQFEK) | 8 aa | Strep•Tactin (modified streptavidin) | [97] |
| T7-tag | 11/16 aa | Antibody | |
| V5 tag (GKPIPNPLLGLDST, SV5 virus) | 14 aa | Antibody | |
| VSV tag (YTDIEMNRLGK, vesicular stomatitis virus) | 11 aa | Antibody | |
| Binding proteins | |||
| Calmodulin binding protein (CBP) | 4 kDa | Calmodulin/Ca2+ | |
| Cellulose binding domains (CBD) | 156/114/107 aa | Cellulose | [98-100] |
| Dihydrofolate reductase (DHFR) | 25 kDa | Methotrexate | |
| Galactose-binding protein (GBP) | 33.5 kDa | Galactose-sepharose | |
| Glutathione-S-transferase (GST) | 26 kDa | Glutathione/ GST antibody | |
| Maltose binding protein (MBP) | 40 kDa | Amylose | |
| Thioredoxin | 11.7 kDa | Phenylarsine oxide (ThioBond) | [101] |
| Streptavidin/avidin | 60/ 68 kDa | Biotin | |
| Staphylococcal protein A | 14/ 31 kDa | IgG antibody | [102] |
| Streptococcal protein G | 28 kDa | Albumin | |
Overall, although chromatography-based protein purification methods have been successfully applied to the production of many recombinant proteins and peptide-based polymers, challenges remain in several important areas: 1) Chromatography packing materials can be quite costly, especially those for affinity chromatography when biological ligands are used. Although column packing materials can be reused after regeneration, the chromatography column may suffer from a deterioration in performance and require regular replacement to ensure high recovery. 2) The sample throughput of chromatography is volume-limited, because the loading capability is governed by the amount of packing materials used, which in turn is limited by the physical size of the column. 3) Products isolated by chromatography are usually diluted by elution buffer. A concentration step is frequently necessary after chromatography and may cause a loss in yield. Because of these issues, especially cost, affinity chromatography is largely a bench top laboratory tool, and its use in industry is confined to the use of protein A chromatography for the purification of antibodies.
3.1.2.2 ELP-based protein purification
Research and development efforts have been made to alleviate purification problems through incremental improvements in the design of equipment and chromatography column packing material. In our view, new approaches are needed to overcome all of these difficulties. Protein purification using environmentally responsive polypeptides such as thermally sensitive elastin-like polypeptides (ELPs) circumvents many of the problems associated with chromatography based methods. Similar to an affinity tag, an ELP tag can be genetically engineered into recombinant proteins, and it imparts its phase transition behavior to the fusion protein. Because ELP fusion proteins are soluble at temperatures lower than their transition temperature Tt and insoluble at temperatures above the Tt, ELP fusion proteins can be purified in solution without chromatography using a technique termed inverse thermal cycling (ITC). In ITC, the soluble proteins are suspended in buffer, and the solution is centrifuged at a temperature below Tt to remove insoluble cellular proteins. The supernatant is then heated and centrifuged at a temperature above Tt in order to capture the insoluble ELP fusion protein. The pellet can then be resuspended in buffer and subjected to several more centrifugation cycles to obtain recombinant fusion protein at a high yield and purity. To recover the target protein, the ELP tag can be cleaved by an enzyme or by a change in acidity, depending on the cleavage site used [78] (Figure 5).
Figure 5.
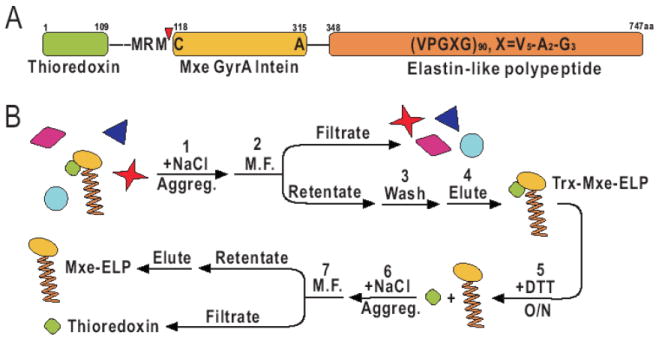
Purification of self-cleaving ELP. (A) The target protein, thioredoxin, is fused to an intein-ELP construct. (B) Purification scheme utilizing microfiltration (M.F.) The intein-ELP tag is removed from the target protein at the self-cleavage site (red arrow) by the addition of dithiothreitol (DTT). Figure reprinted from [103].
Protein purification using an ELP tag has several advantages over conventional chromatography: 1) Although the choice of ELP tag depends on the characteristics of the fusion partners and the desired transition temperature, purification of proteins with ELP tags by ITC appears to be universal for soluble recombinant proteins. 2) The ELP tag that is co-expressed as a fusion partner with the target protein acts as a capture mechanism, so no chromatography beads are needed. This reduces the expenses associated with protein purification. 3) The ELP-based purification method does not require a concentration step to recover the final product. Because the recombinant protein is precipitated and concentrated during each step of the purification process, loss of the final product is lower than in chromatography.
3.1.3 Applications of ELP-based protein purification
As the principle of ITC purification is quite simple, many proteins have been successfully expressed as ELP fusions and isolated using ITC for a variety of biotechnology and biomedical applications. Most applications of ELP-based protein purification can be categorized into two major groups: direct ELP tagging and ELP-mediated affinity capture (Table 3). In direct ELP tagging, an ELP is fused to the protein of interest and expressed in host cells. This method offers simplicity of purification without multiple binding and desorption steps, high yield and high recovery, and superior purity. Fusion proteins with ELP tags of different sizes and compositions have also been synthesized and used for applications other than protein purification, such as tissue engineering and drug delivery [69]. ELP-tagged green fluorescence protein (GFP), blue fluorescence protein (BFP), thioredoxin, chloramphenicol acetyl transferase (CAT), and calmodulin (CaM) are a few examples of proteins that have been purified by ITC [44, 79, 80].
Table 3.
Applications of ELP-based purification.
| Examples | Ref. | |
|---|---|---|
| Direct ELP tagging | ||
|
|
[44] |
|
[69] | |
|
|
[82] |
|
|
[78, 103] |
| ELP-mediated affinity capture | ||
|
HIV antigen | [104] |
|
Antibodies | [83] |
|
His-tagged protein | [84] |
|
DNA | [85] |
|
Sugar group | [86] |
|
Mercury, arsenic, and cadmium | [87-90] |
Direct ELP tagging suffers from several drawbacks: 1) The ELP must be cleaved from the fusion protein if the ELP is not necessary in subsequent applications. 2) Enzymatic cleavage of the ELP tag can be expensive in large-scale production and the enzyme used must be removed from the final product. 3) The design of ELP must be tailored to the protein of interest and the transition temperature must be fine-tuned for each protein. 4) The recovery of the target protein is not satisfactory when its concentration is very low. However, new techniques and methodologies in direct ELP tagging have been developed to overcome some of these hurdles. For example, the requirement of an enzyme such as thrombin to remove the ELP tag can be eliminated with the use of a pH-dependent self-cleavage sequence (intein) [78, 81] (Figure 5). As the efficiency of ELP-based protein purification largely depends on the concentration of the fusion protein itself, those expressed at very low concentrations may present difficulties in protein purification. To circumvent this problem, it has been demonstrated that the addition of excess ELP to the cell lysate is sufficient to isolate target proteins of concentrations as low as 1 – 10 nM, which is equivalent to a few fusion protein molecules per E. coli cell [82]. Some of these techniques can also be performed together with microfiltration to increase the recovery of fusion proteins [81] (Figure 6).
Figure 6.

Different configurations for microfiltration purification of ELP fusion proteins by ITC. A) Syringe with a microfilter. B) AKTA system (Amersham Biosciences, Piscataway, NJ) with a microfilter. C) AKTA system with a stirred cell module. [81]
In ELP-mediated affinity capture, ELP is either recombinantly expressed or chemically attached to a capture partner that binds specifically to the target protein in solution. After binding, ITC may be used to purify the protein. This strategy circumvents the need to individually produce the target protein together with an ELP tag, thus shortening the development and optimization cycle. Furthermore, ELP-mediated affinity capture also opens the possibility of recycling the capture molecule, which further reduces the cost of producing the target protein. Also, ELP-mediated affinity capture does not require enzymatic or chemical cleavage of the ELP tag.
Representative examples of capture ligands that could used for purification by ITC are listed in Table 3. ELP-tagged proteins A, G, and L provide a general platform to capture antibody molecules of different subclasses [83] and allow complex multiplexing of protein purification and detection schemes. ELP-nickel conjugates can be used successfully to isolate proteins tagged with a short histidine sequence [84]. To purify unmodified proteins, ELP-tagged single-chain antibodies can be selectively bound to their protein partners. The underlying principle of this method is very similar to immuno-precipitation; however, it is more efficient due to the ELP-tag that facilitates the isolation and enrichment of the target protein. Recently, Kostal et al reported the successful purification of plasmid DNA using an ELP-tagged bacterial metalloregulatory protein MerR [85] (Figure 7). Biomolecules other than protein and DNA can also be isolated by this technique. It is possible to isolate glyco-polypeptides using the interaction between the capture molecule and the sugar group [86] (Figure 8). It has also been shown that ELPs fused to an appropriate fusion partner can bind to heavy metals such as mercury [87], arsenic [88] or cadmium [89, 90], and facilitate the rapid isolation of these toxic metal contaminants.
Figure 7.

Affinity purification of plasmid DNA using thermally triggered precipitation of ELP-tagged bacterial metalloregulatory protein MerR [85].
Figure 8.

Protein purification by glyco-affinity precipitation [86].
3.2 Controlled drug delivery
3.2.1 Introduction
Many experimental therapeutics have shown activity in vitro, but do not have significant in vivo efficacy. This discrepancy in performance can usually be attributed to problems in transport; many therapeutics have limited efficacy due to sub-optimal pharmacokinetics or systemic toxicity. Advances in drug delivery are hence needed to improve the pharmacokinetics of promising drugs for many diseases, and peptide-based biomaterials have great potential for in vivo delivery of pharmaceuticals [105]. In this section, we focus on methods for the delivery of cancer therapeutics to solid tumors.
In 1906, Paul Ehrlich proposed the use of a carrier capable of delivering “therapeutically active groups to the organ in question,” thus effectively starting the field of targeted drug delivery. In this approach, drug carriers are designed to enhance availability of the drug at a specific location, while minimizing systemic non-specific exposure of a drug. This method offers improvement over delivery of free drug by targeting the drug to a specific site. The efficacy of the drug is increased, and the accumulation at off-target sites is lowered, which lowers the toxicity of the compound. [106].
Targeted drug delivery, however, must overcome the transport barriers in the body, which are quite complex. Systemic delivery of compounds can be divided into three transport phases: from injection to tissue site, from vasculature to interstitium, and from interstitium to molecular site of action [107] (Figure 9). Furthermore, those drugs that have subcellular targets (i.e. nucleus or lysosome) will require a fourth transport phase depending on the site of action [108, 109]. When a drug is administered systemically, the compound will accumulate at the site of action, but will also accumulate in healthy tissue, and may concurrently be cleared through the reticuloendothelial system (RES). Low MW drugs will readily diffuse from the bloodstream into healthy tissues, as suggested by studies that reveal uniform distribution throughout the body [110]. This homogeneous localization of the drug leads to low concentrations of the drug at the tumor site and causes most of the negative side effects associated with systemic therapy. Furthermore, small molecules are rapidly cleared from the bloodstream, requiring multiple administrations to achieve therapeutic effects [106].
Figure 9.
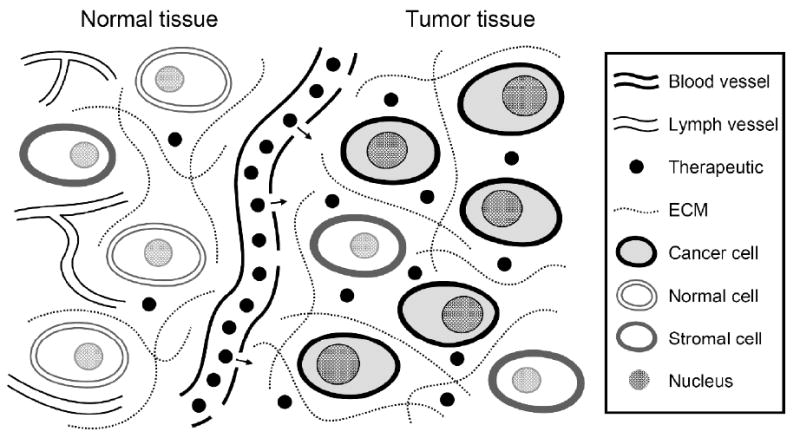
Diagram illustrating systemic drug delivery for therapy of solid tumors using the enhanced permeability and retention (EPR) effect. The drug will extravasate from the permeable tumor vasculature and diffuse across the interstitium to reach the target cells. A small portion of the drug is lost due to clearance in the bloodstream and the interstitium and due to uptake by normal tissue, but retention is enhanced within the tumor due to the lack of functional lymphatics. The goal of drug delivery is to maximize the drug concentration in the target cells while minimizing uptake by healthy cells.
One property of tumors that can be exploited in drug delivery is that the physiology of tumor vasculature is different from normal vasculature. While normal blood vessels have low permeability, the irregularity of tumor vasculature leads to leaky sections, which increases the overall permeability. There is also a lack of functional lymphatics within the tumor interstitium. These two properties of tumors combine to create the enhanced permeability and retention (EPR) effect, which causes an increase in the uptake of blood-borne molecules into the tumor [111, 112].
Despite the EPR effect, there are many factors opposing the delivery of drugs to tumors. The pressure in the interstitium is high relative to the vasculature, creating a barrier for diffusion into the tumor. Also, blood vessels within the tumor are heterogeneously distributed [113]. While the evenly-spaced vessels of healthy tissue allow for perfusion to all tissues, not all regions of a large tumor can be reached via simple diffusion [114]. The lack of lymphatics does reduce transport of the drug away from the tumor, but it also reduces any convection-based transport within the tumor. Yet another barrier is the multi-drug resistance effect that allows cells to actively pump a wide variety of hydrophobic drugs out of the cytoplasm via membrane-bound efflux pump proteins [115]. This leads to low intracellular concentrations as well as the prevention of subcellular trafficking. All of these barriers combine to create a challenge for successful administration of anticancer drugs.
Two distinct methods have been used to meet this challenge. First, soluble macromolecular carriers can be used for systemic delivery of antitumor agents. Soluble polymer carriers have a long plasma half-life, which allows them more time to accumulate in tumors by diffusing through their permeable vasculature. Knowledge of the exact location of the tumor is not necessary for administration; ideally the carriers can reach any vascularized tumors, provided that the compound is not quickly cleared from circulation. The limit of this approach is that the carriers must overcome the multiple transport barriers to reach the target cells.
The second method, local delivery, sidesteps transport issues by sustaining delivery of low MW compounds from a controlled release depot at the site of action. By releasing the drug directly at the tumor, delivery does not need to rely on the limited vasculature. Also, the composition of the matrices used for sustained release can be tailored for a wide variety of compounds. The limitation of local delivery is that the exact tumor site must be known and accessible via direct injection for minimally invasive administration. In this section we will discuss the use of peptide-based polymers as both soluble carriers and local depots.
3.2.2 Soluble carriers
Polymers have recently gained attention as macromolecular carriers that are capable of overcoming transport barriers that limit drug delivery to tumors [106, 110, 116, 117]. Polymer conjugates for drug delivery are typically large hydrophilic molecules linked to a therapeutic agent. These conjugates can target tumors either “passively” through the EPR effect or “actively” through a triggered stimulus or affinity towards the site of therapy [116, 118, 119]. These macromolecular carriers have a longer plasma half-life, show reduced systemic toxicity, retain activity against multiple drug resistant cell lines, and increase the solubility of poorly-soluble drugs. All of these attributes have led to higher anticancer efficiency for passively-targeted polymer conjugates compared to free drug [116, 120-127].
Recent advances in peptide-based polymer design have led to the use of polypeptides as polymeric carriers for anticancer therapeutics. There are several inherent advantages to using polypeptides as opposed to synthetic polymers. First, polypeptides comprised of natural amino acids are likely to maintain biocompatibility throughout the degradation process, breaking down into metabolites excreted through normal metabolic pathways [128]. Second, genetically encoded peptides exhibit molecular weight and sequence uniformity, which are properties that will control pharmacokinetics, transport, biodistribution, and degradation [120, 129]. Third, targeting moieties such as short peptide segments can be incorporated at the genetic level at predetermined locations on the protein. Furthermore, amphiphilic polypeptides can be used to encapsulate drugs in self-assembling structures such as micelles and vesicles similar to those made from synthetic polymers.
3.2.2.1 Elastin-like polypeptides
The thermosensitive properties of ELPs make them an interesting class of polypeptides for use in drug delivery. Because ELPs are macromolecules, they are useful as carriers for passive targeting. More importantly, certain ELPs have been shown to undergo the inverse solubility transition in vivo at temperatures between 37 °C, body temperature, and 42°C [64, 130, 131], the clinically permitted temperature used in hyperthermia treatment [132].
Over the past decade, much work has been done to develop ELPs as soluble carriers that can be thermally targeted to tumors. In an effort to identify ELPs with desired transition characteristics in vivo, a series of ELPs of different MWs and guest residues were synthesized based on the LCSTs of ELPs containing a single guest residue [36]. ELP genes designed for thermally targeted drug delivery were cloned using the RDL method followed by expression and characterization of the resulting proteins [133]. Two ELPs were chosen for these studies: ELP1, which has a guest residue distribution of Val, Ala, and Gly in a 5:2:3 ratio, and ELP2, which has a distribution of Val, Ala, and Gly in a 1:8:7 ratio. ELP1 with a MW of 50-60 kDa was shown to have an LCST slightly above body temperature at concentrations between 5-50 μM. ELP2, a more hydrophilic protein, has an LCST outside the hyperthermia range at the same conditions.
ELP1 and ELP2 were monitored in vivo using intravital microscopy, a technique that allows for monitoring the kinetics of the accumulation of flouorophore labels within the tumor and determination of the 3D spatial distribution with micron resolution [134]. The ELPs were conjugated to different fluorophores and injected into nude mice implanted with human tumor xenografts. ELP1 formed micron-sized aggregates in the vasculature while ELP2 did not, showing for the first time that a thermosensitive polymer could transition at a specific temperature inside tumor vasculature. The ELP was also compared to pNIPAAM, a synthetic polymer that also exhibits an LCST [64]. Both ELP and pNIPAAM showed a two-fold concentration increase within the tumor at temperatures above the Tt. However, the biocompatibility, monodispersity, and sequence control of the ELP make it more useful than pNIPAAM as a thermally targeted carrier.
More recently, studies have been performed to further develp ELPs as a drug carriers. One example is the development of an acid-labile ELP1-doxorubicin conjugate for lysosomal release [130]. Doxorubicin, a hydrophobic anticancer drug, acts at the nucleus by inhibiting topoisomerase, thus killing the tumor cell. Its intranuclear mode of action requires that it be released from the carrier upon cellular uptake to allow entry into the nucleus. ELP1-150 (~60 kDa) was conjugated via a pH-sensitive hydrazone bond to a modified doxorubicin that contains a maleimide linker. In vivo studies showed similar levels of cytotoxicity during administration of free doxorubicin and ELP-doxorubicin despite varied distribution within the cell, suggesting that the ELP-doxorubicin conjugate likely has a different mode of action. Further optimization studies of the ELP-Dox maleimide linker showed that the spacer length affects the LCST and aggregate size without significantly changing pH-sensitive drug release, leading to the selection of B-Dox, which has an 8.1 Å spacer [135]. This linker minimally affected the LCST and will be used for future investigation of ELP-doxorubicin conjugates.
Other studies were performed to evaluate the pharmacokinetics and biodistribution of ELP within the tumor. Dreher et al. used confocal fluorescence microscopy to quantify the effects of the ELP phase transition in vivo. ELP1 (green) and ELP2 (red) were co-injected into nude mice, creating a yellow color within the lumen of the vasculature (Figure 10) [134, 136]. Upon introduction of hyperthermia, bright green ELP1 aggregates formed along the epithelium of the vessel as well as along branch points in the vasculature. These aggregates disappeared after cooling, demonstrating the reversibility of the process over several cycles.
Figure 10.
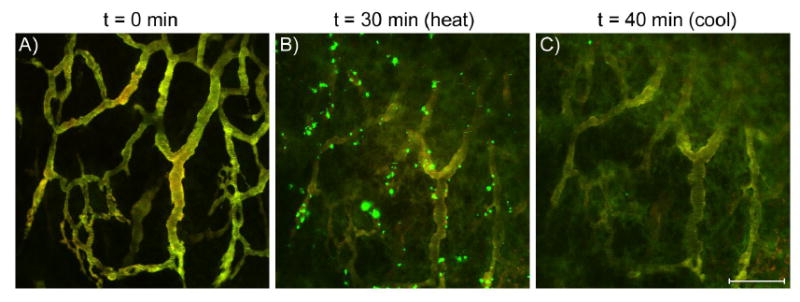
Images of ELP1 (green) and ELP2 (red) in a solid tumor before, during, and after hyperthermia treatment. These images are maximum projections along the z-axis of about 50 μm of tumor tissue, and imaging parameters were selected such that the vascular intensities of ELP1 and ELP2 were balanced to produce a yellow color. A) 0 min, no heat; B) tumor heated to 41.5 °C. The green particles indicate accumulation of ELP1; C) tumor cooled to 37 °C. The bar corresponds to 100 μm for all images. [136]
Biodistribution studies were performed on nude mice implanted with FaDu flank tumors using an ELP1/carbon-14 conjugate. This conjugate, formed at the expression level, was used to detect the amount of tumor uptake as well as the spatial distribution of the ELP upon hyperthermia [137]. The conjugate proved especially useful, as it allows for facile monitoring of the localization and degradation of ELP1 in vivo. Since the protein is covalently labeled with the radioactive nuclide, it also limits the potential for artifacts resulting from free radiolabel. Autoradiography of ELP1 shows that the product is homogenous and highly pure [138] (Figure 11). In two complementary, separate sets of experiments, 14C-ELP was used to monitor the plasma half-life of ELP1 in vivo as well as image the distribution of ELP within the tumor. The plasma half-life of ELP1-150 was 8.4 hr as determined in nude mice (Figure 11), and autoradiographs of tumor cross-sections showed that heated ELP1 accumulated 2-fold more and penetrated further into the tumor than non-heated and thermally-insensitive controls (Figure 12). Both of these recent studies suggest the potential of targeted drug delivery using thermosensitive ELP as a drug carrier.
Figure 11.
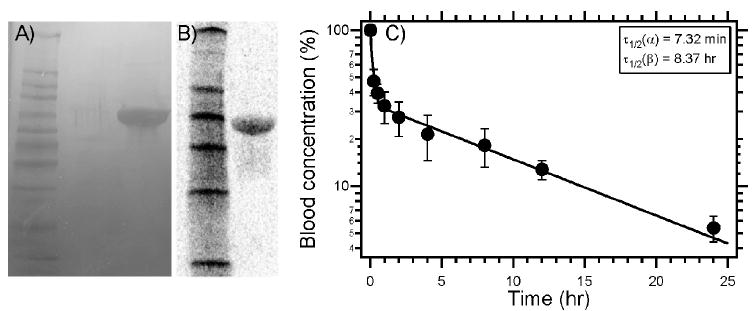
SDS-PAGE analysis of (A) C-ELP1 visualized by copper staining. (B) Radiolabeled ELP autoradiography after SDS-PAGE. (C) Pharmacokinetic analysis of radiolabeled ELP in mice (Balb/c nu/nu) exhibits a characteristic distribution and elimination response with a terminal half-life of 8.4 hr. Reprinted from [138] with permission from Elsevier.
Figure 12.

(A) Autoradiography images of 14C-ELP1 (ELPactive) with and without heat and 14C-ELP2 (ELPpassive)with heat from 20 μm tumor sections after 1 h of hyperthermia. (B) Scintillation analysis of tumor accumulation of the 14C-ELPs. ELP1 accumulates to a greater extent in heated tumors than unheated ELP1 or heated ELP2 controls. Data are shown as mean±SEM, n=5. *Significant difference compared with both controls (Fischer’s PLSD). Reprinted from [137] with permission from Elsevier.
ELP derivatives have also been used as soluble protein carriers by other investigators. Silk elastin-like polypeptides (SELPs) are polypeptides that contain repeating motifs from both silk and elastin proteins. SELPs with a minimal silk component do not form crosslinked networks and display the stimuli-sensitive behavior seen in ELPs, making them good candidates for use as soluble carriers. Nagresekar et al. observed a decrease in Tt when hydrophobic residues were added to an otherwise hydrophilic SELP. When glutamic acid residues were added, SELPs also exhibited pronounced pH and ionic transition temperature sensitivity [139]. The ability to adjust the sensitivity to ionic effects makes non-crosslinking SELPs good vehicles for systemic gene delivery.
Genetic templating allows for the incorporation of other peptide units with great precision at the gene level. This has led to the development of ELP-fusion peptides not only for protein purification, but also for therapeutic applications. These fusion proteins, unlike those designed for protein purification, do not necessarily require the eventual detachment of the bound peptide. For example, peptides that will enhance accumulation at the tumor site can be attached to the carrier. Raucher et al. attached the cell penetrating peptides (CPP) penetratin (AntP), MTS, and Tat to the 5′ end of an ELP [140]. The addition of a CPP to the ELP increased uptake in both SKOV-3 ovarian cancer cells and HeLa cervical cancer cells. The CPP AntP showed the greatest in vitro increase in uptake in the unimer phase. An AntP-ELP fusion protein was also conjugated to the H1 peptide that is used to disrupt transcriptional activation of cells by inhibiting the c-Myc oncogene [141]. AntP-ELP-H1 caused a 2-fold decrease in cell proliferation when administered in hyperthermic versus normothermic conditions.
3.2.2.2 Polymeric micelles
Polymeric micelles have recently received great attention as potential carriers for hydrophobic drugs or genes [142-146]. These self-assembling nanostructures are made from amphiphilic block copolymers (BCPs) that consist of two blocks with differing polarity. This difference drives the formation of micellar structures in aqueous solution with the hydrophobic block of each unimer partitioning into the core and the hydrophilic block forming the corona. Other physical differences between the blocks, most notably charge, can influence the formation or destruction of nanoparticles (Figure 13). The hydrophobic core can be used to encapsulate hydrophobic compounds and increase their solubility in an aqueous environment. Polymeric micelles typically have a critical micelle concentration that is lower than for surfactant micelles, allowing for self-assembly at concentrations relevant for drug delivery [147]. Micelles also retain the positive aspects of polymeric delivery, such as prolonged plasma half-life, because the size of a typical micelle allows it to evade the reticuloendothelial system (Rh< 100 nm). Furthermore, the predictable self-assembly behavior allows for the incorporation of functional groups in the corona that range from passive components such as PEG to active groups such as targeting ligands or CPPs.
Figure 13.
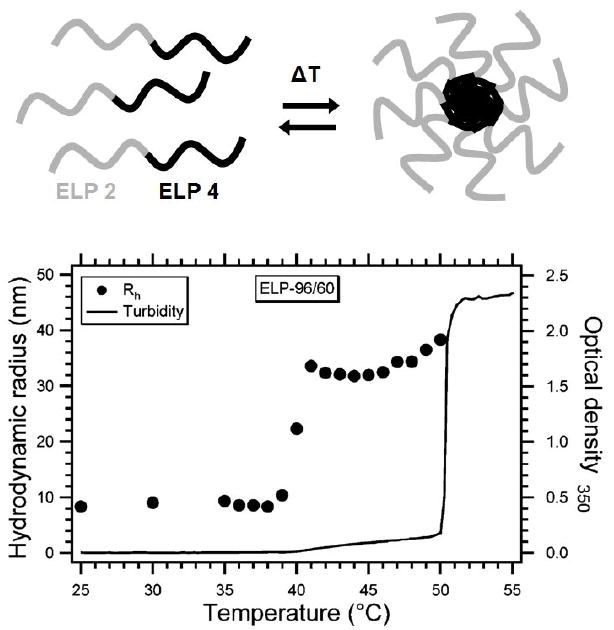
Formation of thermosensitive ELP block copolymer micelles. At temperatures below the transition temperature of the ELP4 block (< 37 °C), the copolymer remains in unimer form. At temperatures between the transition temperatures of the ELP2 and ELP4 blocks, the block copolymers self assemble into spherical nanoparticles, possibly micelles. At temperatures above the transition temperature of the ELP2 block (> 37 °C), the corona of the micelle undergoes its phase transition, and micron-sized aggregates form. The turbidity (line) and DLS (points) data show the progression of the ELP from monomer to aggregate. [182]
The use of biopolymers has created a new class of “smart” micelles capable of triggered response to outside stimuli. For example, pH-sensitive micelles have been created using synthetic methods to create poly(histidine)-PEG and poly(histidine co-phenylalanine) block copolymers [133, 148, 149]. These micelles have a PEG corona and a hydrophobic core formed by the poly(histidine) block (pKa ~ 6.0). At low pH, the histidine becomes protonated, making the core more hydrophilic and thus disrupting the micelle. The addition of hydrophobic amino acids such as phenylalanine can be used to tune the pH sensitivity. Early studies have shown that these micelles, when coated with folate, can reduce the multi drug resistance effect by destabilizing endosomes within MCF-7 tumor cells[150, 151].
Both bio- and synthetic polymers have been used to produce thermosensitive self-assembling block copolymer micelles [133, 152-154]. The use of biopolymers provides the ability to design block copolymers that have specific LCSTs and precise MWs. Thermosensitive ELPs can be used to make block copolymers by seamlessly fusing multiple ELP genes at the genetic level. If the two blocks have different transition temperatures, each block will transition independently, and amphiphilic structures will form in the temperature range lying between the two transitions. Multiple groups have successfully created ELP BCPs using standard cloning techniques. Lee et al. concatemerized ELP oligonucleotides for hydrophilic and hydrophobic blocks and sorted them via gel electrophoresis to obtain a variety of block sizes [152]. Thermally induced formation of micelles was verified by dynamic light scattering (DLS) and differential scanning calorimetry (DSC). TEM images showed the formation of spherical and cylindrical micelles with core diameters of 54±9 nm and 50±10 nm, respectively. Chilkoti et al. expanded this idea by applying RDL to form blocks of precise size and composition [133] (Figure 13). ELP2 (guest residues: V1A8G7) and ELP4 (guest residue: V) blocks of varying molecular weight were characterized by turbidity and DLS measurements. The DLS data suggest that ELP2-ELP4 block copolymers exist in four distinct phases: unimer at T < Tt1, micelle at Tt1 < T < Tt2, larger nanoparticle at T ~ Tt2, and aggregate at T > Tt2. ELP BCP micelles with transitions Tt1 < 37 °C and Tt2 > 37 °C show potential as carriers for triggered and targeted delivery.
Checot and coworkers designed and characterized a series of polypeptide-polymer hybrids for drug delivery using poly(butadiene-b) (hydrophobic block) and poly(l-glutamic acid) (hydrophilic block) (Figure 14). These diblock copolymers self-assemble into micelles at hydrophilic to hydrophobic ratios of 3:1 [155]. The hydrodynamic radius of the micelles depends on both pH and salt concentration, because the poly(l-glutamic acid) in the corona transitions from a helix to an extended coil structure. These block copolymers contain a synthetic polymer but show greater stimulus sensitivity than other supramolecular assemblies due to their protein component.
Figure 14.
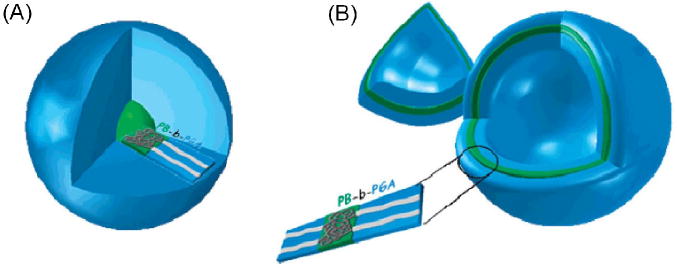
Diagram of pH-sensitive (A) micelles and (B) vesicles formed from poly(butadiene-b)-poly(glutamic acid) diblock copolymers. The hydrophilic to hydrophobic ratio of the blocks determines whether a micelle or vesicle will form. Deprotonation of glutamic acid residues in the hydrophilic corona imparts pH sensitivity to these structures. Reproduced from [155].
3.2.2.3 Vesicles
Polymer vesicles are another type of self-assembled nanoscale carrier for hydrophobic drugs. Vesicles maintain the geometry and flexibility of naturally-occurring lipid bilayer cellular membranes, but can include a wider variety of amphiphiles [156]. They are typically larger than micelles, and vesicles can fully encapsulate the compound of interest, protecting it from the outer environment. For example, “stealth” lipid-based vesicles, or liposomes, have been successfully used for systemic doxorubicin delivery [157].
Bellomo et al. produced synthetic diblock copolymers consisting of one lysine block and one leucine/lysine block using an existing synthetic peptide formation process [158]. Fluorescence emission measurements indicated no release of particles for several days at pH 10.6, but the measurements show near instantaneous degradation of the vesicles at pH 3.0. Checot et al. developed a series of pH-responsive poly(butadiene)-poly(glutamic acid) block copolymer vesicles intended for nano-encapsulation (Figure 14) [159]. They found that the copolymers form vesicles at near 1:1 hydrophilic to hydrophobic ratios and micelles at a 3:1 ratio. The vesicles are stimuli-responsive, and their size depends on both the pH and ionic strength of the solution [160]. Vesicles designed from these copolymers that degrade at pH 4 are physiologically relevant for lysosomal-triggered drug release [155].
3.2.3 Local Delivery
The second major class of controlled drug delivery methods is local delivery. In local delivery, the local drug concentration is increased by implanting a drug reservoir near the site of interest. This enhances diffusion into the desired site, mitigating the negative effects seen in systemic delivery. However, this method has some limitations. For example, not all tumor sites can easily be identified or are accessible within the body. Also, many implantable polymer structures require invasive implantation. Self-assembling injectable hydrogels are an alternative to rigid polymer structures and do not require an invasive surgical implantation. These hydrogels spontaneously crosslink and quickly form a gel in vivo following injection at the site. Self-assembling hydrogels can also incorporate a number of stimuli such as temperature or pH to control the matrix assembly [139, 161].
Injectable hydrogels must posess a unique set of properties. Gelation kinetics, geometry, swelling, density, stability, charge, and release rates are all important characteristics to consider when designing a matrix that will appropriately release therapeutic compounds [162]. Fortunately, these are all properties that can be controlled through the rational design of peptide-based biopolymers. In addition, most peptide-based hydrogels are biocompatible and resorbable upon degradation. Furthermore, control over polypeptide folding and organization can be exploited to create unique crosslinking systems that do not require chemical modification. It has been demonstrated that self-assembling protein hydrogels can be used to deliver both hydrophobic drugs as well as plasmid DNA for gene therapy applications.
3.2.3.1 Peptide-ELP fusion proteins
Many protein drugs have been developed to treat the degenerative joint disease osteoarthritis. While these drugs have the potential to modify progression of the disease upon intra-articular injection, their administration is limited by very short half lives [163, 164]. The Setton group has proposed a solution where the protein drug interleukin-1 receptor antagonist (IL1Ra) is fused to an elastin-like polypeptide (ELP). The ELPs aggregate and form a ‘drug depot’ upon injection at physiological temperatures, which prolongs the release time of the drug. In a study by Betre et al., an aggregating ELP was shown to have a 25-fold longer half-life in the injected joint than an equivalent molecular weight protein that remains soluble and does not aggregate [165]. In an accompanying study by Shamji et al., an ELP-IL1Ra fusion displayed a complete recovery of IL1Ra activity, matching that of the commercial antagonist [166]. They also demonstrated the facile collagenase degradation of the ELP domain, which may further promote the therapeutic efficacy of this drug delivery system. The results of these two studies show that an ELP-IL1Ra fusion injected within the intra-articular joint space will prolong the delivery of the active IL1Ra drug and improve the quality of treatment for osteoarthritis.
3.2.3.2 Silk-ELP hybrids
Silk is a useful biomaterial due to its high mechanical strength and biocompatibility. However, silk undergoes strong intramolecular hydrogen bonding that leads to rapid crystallization and can complicate its use for injection-based applications [53, 167]. One route to increasing its solubility is the incorporation of peptide sequences from other proteins, such as elastin, that disrupt β-sheet formation [50]. Silk elastin-like polypeptides (SELPs) are chimeric proteins designed to retain the mechanical properties of silk, while incorporating the high solubility and environmental sensitivity of ELPs [139, 161, 168]. Some SELPs can be injected through hypodermic needles, and they then form insoluble hydrogels at the injection site without chemical assistance [162, 169-171].
The main parameters of interest for SELP hydrogels are the rate of gelation, release rate of encapsulated compounds, swelling capability, stability, and biocompatibility of the hydrogel. Initial SELPs capable of self-gelation were identified by Cappello et al. They have the general composition (SmEn)o, where S is the silk-like block, GAGAGS, and E is the elastin-like block, GVGVP. The parameter m varies from 2 to 8, n from 1 to 16, and o from 2 to 100, so that the MW is between 60 and 85 kDa [171].
A series of SELP characterization studies support the use of these hydrogels for drug delivery. DSC was initially used to determine the gelation kinetics, and it showed that decreasing the number of silk blocks leads to a delay in the gelation rate at room temperature. Conversely, hydration studies showed the addition of silk blocks also reduces the swelling ratio [172, 173]. Encapsulation studies have shown that the presence of either charged or uncharged solutes disrupts the formation of the gel. Release studies were performed using fluorescent dextran, various proteins, and DNA [171, 174]. While an increase in polymer concentration greatly reduced the release rate, the MW of the released compound had no effect on the release rate. For charged molecules such as DNA, the ionic strength of the hydrogel affects the release rate, but encapsulation does not reduce the bioactivity of the encapsulated compounds [174]. Furthermore, degradation of SELPs does not appear to produce cytotoxic intermediates, as shown by histological data [175].
3.2.3.3 Coiled coil domains
The coiled coil is one of the basic folding patterns of native proteins, consisting of two or more right-handed amphiphilic α-helices wound together non-covalently to form a slightly left-handed superhelix (Figure 15) [176-178]. The coiled coil domain is based on a heptad repeat unit abcdefg, where a and d are typically hydrophobic amino acids (leucine), and e and g are charged (glutamic acid) [179]. Helical stability is maintained by electrostatic interactions between e and g [180]. Coiled coil motifs intended for drug delivery utilize the reversible hydrophobic association of α-helices within the core as well as the electrostatic interaction from the outer charged residues. These interactions determine the stability of the bundles upon changes in pH and temperature. The peptide sequence in the heptad unit of the coiled coil can be tailored so that the electrostatic interactions are disrupted and protein collapse is induced under the influence of stimuli such as pH, temperature, light, and binding of ligands. This behavior has been exploited to create pH-sensitive and temperature-sensitive hydrogels [176, 181]. These genetically encoded hydrogels demonstrated tunable diffusion, which is controlled by the residues composing the heptad sequence as well as the state of the hydrogel (soluble versus collapsed) [178, 181].
Figure 15.
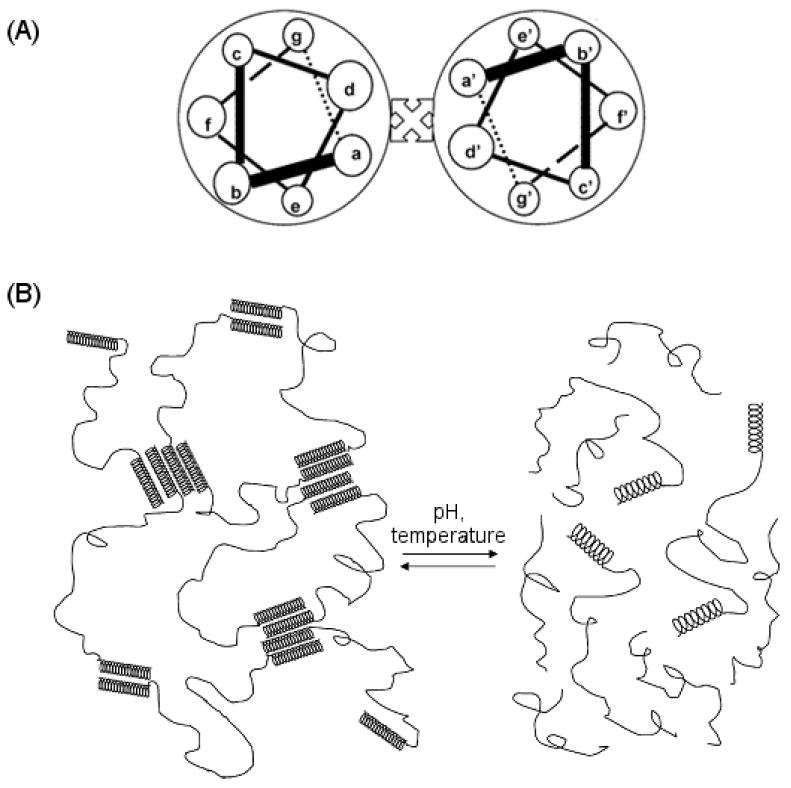
Schematic showing the design of coiled-coil polypeptide hydrogels. (A) The α-helices are formed from heptamer repeats. The helix bundles are formed by interhelical hydrophobic interaction between residues a and d and stabilized by interhelical electrostatic interactions between residues g and e. (B) Variations in pH and temperature can disrupt these interactions and cause reversible dissolution of the hydrogel. Reprinted with permission from [176]. Copyright 2005 American Chemical Society.
3.3 Tissue engineering
Peptide-based biomaterials are widely used for regenerative and reparative medicine. Biological materials such as elastin-based polypeptides, collagens derived from extracellular matrices, fibrins, and spider silk proteins [59, 62, 153, 183-187] are useful for tissue engineering because they have good chemical compatibility in aqueous solutions, good in vivo biocompatibility, a controllable degradation rate in vivo, the ability to break down into natural amino acids that can be metabolized by the body, and minimal cytotoxicity, immune response, and inflammation [174, 188-194]. In addition, biopolymers can be easily functionalized to enhance their interactions with cells and provide an optimal platform for cellular activities and tissue functions.
The main application of peptide-based biopolymers for tissue engineering is as injectable scaffolds that form gels in vivo via physical or chemical means. These biomaterials provide a minimally invasive route to deliver nano- to micron-scale tissue scaffolds. Prior to use, the liquid-like precursors can be easily manipulated and then injected directly at tissue defect sites. Upon injection, the polymers quickly form a hydrogel that can serve as a three-dimensional artificial extracellular matrix to provide embedded cells with structural integrity and functionality for tissue repair, a defect filler for tissue reconstitution, or a controlled drug release reservoir. Following injection, three different gelation mechanisms can be used: (1) self-assembly triggered by environmental stimuli such as temperature, pH, or ionic strength, (2) chemical crosslinking methods using enzymes, radiation, radical polymerization, or photo-polymerization, or (3) physical crosslinking methods. In this section, we discuss two main classes of peptide-based biopolymers in tissue engineering: self-assembling polypeptides that form gels by environmental stimuli and polypeptides that form gels via chemical crosslinking.
3.3.1 Self-assembling polypeptides
Protein folding, or the transitions between primary, secondary, tertiary, and quaternary structures, serves as the basic rationale for designing self-assembling polypeptides. By understanding how proteins undergo conformational changes in response to different conditions, it is possible to design biomaterials that are responsive to environmental stimuli. The complexity of protein folding provides many interesting possibilities in materials science, and a new class of peptide-based biomaterials for biomedical applications has been derived by the principles that govern protein folding. In this section, we summarize examples of self-assembling polypeptide hydrogels that are based on both genetically engineered and chemically synthesized biopolymers.
3.3.1.1 Genetically engineered polypeptides
3.3.1.1.1 Elastin-like polypeptides
Elastin-like polypeptides are useful for thermally sensitive injectable hydrogels because they undergo an inverse temperature phase transition and can be designed at the genetic level. By carefully selecting the guest residues, an ELP can be designed that is liquid at room temperature and forms a gel-like material in situ when the temperature is raised upon injection. Multiple physical or chemical crosslinking sites can be genetically encoded so that the ELPs form networks, and reactive sites can be incorporated for controlled degradation. Specific ligands can also be added to impart functionality for cell adhesion and tissue growth [168, 180, 189, 195-197], and initial work by Urry et al. demonstrated that ELPs cause minimal cytotoxicity and immune response when implanted [189, 190].
Studies using ELPs as substrates for tissue growth have focused on cartilage repair. Betre et al. showed that chondrocytes can be encapsulated in the gel-like material formed by aggregated ELPs above Tt (coacervate). The encapsulated chondrocytes maintained their unique morphology and phenotype in vitro for 15 days (Figure 16-1), demonstrating that ELP coacervates support the growth of chondrocytes. The cells were able to produce and accumulate both sulfated glycoaminoglycan (GAG) and collagen type II, the characteristic proteins of cartilage ECM [198]. In addition to supporting cell growth, the ELP coacervates also promoted the differentiation of human adipose-derived adult stem cells into chondrocytes without exogeneous chondrogenic supplements [199] (Figure 16-2). After encapsulation in ELP coacervates, the stem cells upregulated SOX9 and type II collagen, indicating that genes that control the cartilaginous phenotype were upregulated, while the type I collagen gene, which is characteristic of the fibroblast phenotype, was downregulated.
Figure 16.
(1) Histological section of ELP-chondrocyte constructs after 15 days of culture. Sections stained for: (A) cell morphology, H&E, (B), S-GAG using toluidine blue, and (C) collagen and extracellular matrix using Masson’s trichrome. The scale bar is 50 μm [198]. (2) Immunohistochemical staining of the human adipose-derived adult stem cells (hADAS). ELP constructs after 14 days of culture in standard (A, C, E and G) or chondrogenic (B, D, F and H) media at 5% O2. Sections stained for anti-type I collagen (A and B), anti-type II collagen (C and D) and antichondroitin-4-sulfate (E and F). Negative control (no primary antibody) is shown in G and H. (bar, 50 mm) [199].
While the ELP coacervates were able to support chondrocyte growth, their mechanical properties did not closely match those of normal cartilage. When the ELP solution undergoes its thermal transition above the LCST, the solution phase separates into a solid-like ELP coacervate and a separate aqueous phase. The phase separated ELP coacervate has a complex shear modulus (G*) of about 100 Pa at 10 rad/s, which is nearly 4 orders of magnitude lower than that of articular cartilage (100-500 kPa) [200]. In an effort to prevent the ELP solution from phase separating upon injection in vivo and to expand the range of accessible rheological and mechanical properties, Lim et al. designed a series of ELP triblock copolymers. The ABA triblock copolymers are composed of a hydrophobic A end block (aliphatic- or charged ELP block) with a low LCST (25 °C < Tt < 37 °C), a hydrophilic B middle block with a high LCST (> 90 °C), and the hydrophobic A end block. The triblock copolymers were genetically synthesized using RDL, so the block lengths and ratios were precisely controlled. At temperatures above the lower LCST, the hydrophobic A blocks aggregate and form physically crosslinked micellar domains under physiological conditions. The hydrophilic B blocks remain soluble in aqueous solution and function as bridges connecting the interspersed hydrophobic, aggregated domains. Lim et al. found that instead of phase separating into coacervate and solution, the ELP triblocks formed hydrogel networks in aqueous solution with optimized salt concentration.. Their studies showed that the rheological properties, mechanical properties, and thermal transition temperatures of the triblock copolymers were largely controlled by (1) the amino acid sequences and charged state of the hydrophobic A blocks, (2) the ratio of the hydrophilic B middle block length as compared to a monoblock ELP, and (3) the salt concentration under physiological conditions. This rheological study of ELP triblock copolymers is important for future applications of the copolymers as in situ gel formers for tissue repair (unpublished work).
3.3.1.1.2 Elastin-mimetic polypeptides
Wright et al. and Nagapudi et al. have reported systematic rheological studies of genetically synthesized self-assembling elastin-mimetic triblock polypeptides [153, 154, 201, 202]. The copolymers composed of a plastic domain (its consensus repetitive sequence: VPAVG) as the end blocks and an elastomeric domain (its consensus repetitive sequence: VPGVG) as the middle block. The single substitution of an alanine residue for a glysine residue in the third amino acid position of the repeating sequence converts the block’s mechanical behavior from elastic to plastic. This dramatic change is caused by the structural change from the Pro-Gly type II β-turn structure to the Pro-Ala type I β-turn structure [153]. Rheological measurements of an aqueous triblock copolymer solution as a function of temperature showed that the copolymers would be well-suited for biomedical applications. The loss modulus (G″) is higher than the storage modulus (G′) below the LCST, and G′ is higher than G″ above the LCST, indicating that above the LCST the solution converts from liquid-like to solid-like viscoelastic behavior [70, 153, 201, 202]. They found that changing the length or hydrophilicity of the middle largely affected the viscoelastic and mechanical behavior of the copolymers when the same plastic domain was used [202]. They also used different solvent environments, such as mixtures of trifluoroethanol (TFE) and water, to change the secondary structure of the proteins. When the protein solution was electrospun to form nanofiber scaffolds, using solvent mixtures resulted in varied microstructures and mechanical properties [70, 201, 202]. As florinated alcohols are known to form strong solid-state complexes with polyamides, TFE is a good solvent for all three blocks of the copolymer. Water only solvates the hydrophilic block, hence using solvent mixtures of TFE and water allows the fabrication of various nanofiber scaffolds with different mechanical properties [201].
3.3.1.1.3 Human tropoelastin-based polypeptides
Insoluble elastin is a major component of the elastic fibers found in the extracellular matrix. While the biosynthesis of these fibers is a complex process, the soluble precursor tropoelastin is known to play a key role in fiber formation. Tropoelastin forms coacervates at temperatures above its LCST, and it is thought that this behavior is partially responsible for elastin’s ability to form fibrils [31-33, 203]. In addition, Tamburro et al. have conducted many studies on the structures of tropoelastin, and their results provide further insight into the role secondary structure has on to the self-assembly of elastin fibers [204-207].
The gene of human tropoelastin is composed of 36 exons that encode alternating hydrophobic and hydrophilic peptide domains. The hydrophilic sequences are rich in lysine, which allows them to form covalent crosslinks with similar domains in neighboring molecules. These crosslinks facilitate the formation of the fibrillar polymeric structures that give elastin its extensibility and elastic recoil properties. While most elastin-like polypeptides utilize only the VPGXG repeating sequence that is found in the hydrophobic exons, Keeley et al. have developed various recombinant tropoelastin-based multi-block copolymers that also incorporate sequences from the hydrophilic exons [34, 35, 208]. The copolymers were built from exons 20 and 24, which are hydrophobic domains, and exons 21 and 23, which are hydrophilic crosslinking domains. The tri-, penta-, and hepta-block copolymers with molecular weights ranging from 10 to 31 kDa exhibited inverse phase transition behavior due to the presence of the hydrophobic blocks with repeating VPGXG sequences. Above the transition temperature, the hydrophobic interactions caused self-assembly of the copolymers into fibrillar structures, aligning the lysine-rich domains so that covalent crosslinks could form. These strong interactions result in the formation of polymeric matrices with solubility and mechanical properties similar to those of native elastin.
3.3.1.1.4 Leucine zipper based triblock proteins
DNA-binding leucine zipper proteins can be incorporated into polypeptides to create self-assembling copolymers. Tirrell et al. have demonstrated that genetically synthesized triblock copolymers consisting of leucine zipper helix endblocks and a water-soluble polyelectrolyte midblock will self-assemble into pH- and temperature-sensitive hydrogels upon dimerization of the leucine zipper coils [181, 209-211]. The leucine zipper domains are composed of a repeating heptad motif designated abcdefg, where a and d are hydrophobic amino acids (leucine is preferred at position d), and e and g are charged amino acids (glutamic acid is common). The repeating domain has an α-helix structure and easily forms inter- and intra-chain coiled-coil dimers due to the hydrophobic interaction between the a and d residues, which are positioned on a single face of the helix. The charged e and g residues positioned on the opposite face of the helix impart pH-sensitivity to the coiled-coil dimers. Upon elevation of the pH or temperature, the leucine zipper domains reversibly dissociate and create a viscous polymeric solution.
Leucine zipper based hydrogels have been explored as depots for the controlled release of therapeutics. In order to control the erosion rate of the gels and thus the rate of drug release, Tirrell et al. investigated two approaches: (1) introduction of cysteine residues into each coil end block of the triblock proteins in an asymmetric way, resulting in favorable intermolecular disulfide bond formation in addition to the coiled-coil dimers, and (2) introduction of dissimilar coil domains in each triblock protein, resulting in the preferential formation of intermolecular coiled-coil dimers while discouraging the formation of intramolecular loops (Figure 17) [209-211]. They observed that the erosion rate of mixed-endblock hydrogels is reduced by two to three orders of magnitude relative to the symmetric-endblock hydrogels. Adjusting the ratio of mixed-endblock to symmetric-endblock proteins can provide control over the hydrogel network topology and greatly increase the range of accessible materials properties.
Figure 17.

(A) Schematic representations of triblock proteins and the amino acid sequences of major domains. The major domains of each triblock protein are joined by short sequences of amino acids introduced in the construction of the cloning and expression vectors. (B) Structural and dynamic properties underlying the fast erosion of symmetric-endblock hydrogels. (a) Disengaged clusters form readily in the system because of the strong tendency towards intramolecular association. The clusters are lost from the surface through diffusion before reconnecting to the network. (b) Three possible states of tetrameric aggregates designated by the number of loops i. Reproduced from [211].
3.3.1.2 Chemically synthesized polypeptides
3.3.1.2.1 Peptide amphiphile nanofibers
Stupp et al. have synthesized peptide amphiphiles (PAs) that self-assemble into nanostructured materials and have a wide range of uses [212-219]. The PAs are obtained chemically using an automated peptide synthesizer and consist of an alkyl tail connected to a short peptide sequence. The peptide sequence always ends in a hydrophilic head group, giving the PA its amphiphilic character. Because of their amphiphilic character and tapered shape, PAs self-assemble into nanofibers that are nanometers in diameter but microns in length. The nanofibers have been shown to form networks that behave like hydrogels and can be used as scaffolds for cell growth.
The PA’s peptide sequence can be encoded to provide a variety of functions. Some of the commonly used sequences include: (1) four consecutive cysteine residues that form intermolecular disulfide bonds and stabilize the nanofiber structure, (2) three glycine residues that provide hydrophilicity and fliexibility for bonding, (3) a short sequence of alanine and glycine resudes that creates a gradual transition to the hydrophobic region, (4) a single phosphorylated serine residue that strongly interacts with calcium ions and induces the mineralization of hydroxyapatite, (5) acidic amino acids that produce a negative charge in the head group at neutral pH and allow for assembly at acidic pH, (6) basic amino acids that produce a net positive charge at neutral pH and allow for assembly at basic pH, and (7) biological ligands such as RGD for cell adhesion or IKVAV for promoting and directing neurite growth. Niece et al. have also shown that mixtures of acidic and basic PAs will self-assemble at neutral pH, which is essential for applications where the PA hydrogels will be used in vivo [217]. Hartgerink et al. showed that cysteine residues can be used to reversibly cross-link the nanofibers. The cross-linked nanofibers can then direct the mineralization of hydroxyapatite to form a composite material [219]. Silva et al. used PAs with the biological ligand IKVAV in the head group to encapsulate neural progenitor cells within the nanofiber network. The neurite-promoting laminin epitope IKVAV induced differentiation of the neural progenitor cells into neurons. The PA solution was used as an injectable scaffold and tested in vivo, as shown in Figure 18 [212]. The versatile nature of PAs and the ease of incorporating biological epitopes into their hydrogels makes them highly useful as biomaterials for tissue engineering.
Figure 18.
(A) Molecular graphics illustration of an IKVAV-containing peptide amphiphile molecule and its self-assembly into nano-fibers. (B) Scanning electron micrograph of an IKVAV nanofiber network formed by adding cell media (DMEM) to a peptide amphiphile aqueous solution. The sample in the image was obtained by network dehydration and critical point drying of samples caged in a metal grid to prevent network collapse. Micrographs of the gel formed by adding IKVAV peptide amphiphile solutions to (C) cell culture media and (D) cerebral spinal fluid. (E) Micrograph of an IKVAV nanofiber gel surgically extracted from an enucleated rat eye after intraocular injection of the peptide amphiphile solution [212].
3.3.1.2.2 β-sheet forming ionic oligopeptides
Zhang et al. have synthesized short oligopeptides having 12 to 16 amino acids that spontaneously form stable β-sheet structures under physiological solution conditions [220-235]. As shown in Figure 19a, these ionic self-complementary oligopeptides have amphiphilic character; one face of the molecule consists of nonpolar, hydrophobic amino acids (such as Ala, Phe, or Leu), and the other face consists of alternating oppositely charged amino acids (such as positively charged Lys or Arg and negatively charged Asp or Glu). The molecules self-assemble into β-sheets with the the charged residues on one side of the sheet and the nonpolar residues on the other. The alternate positive and negative charges assemble in a checkerboard-like pattern on polar side of the sheet. Then, two β-sheets come together with their nonpolar sides facing each other, away from the aqueous solution, and form nanofibers. The nanofibers assemble into interwoven matrices that form hydrogels with very high water content. These structures are very stable because they are formed by strong intermolecular interactions: ionic interactions between repetitive positive and negative charges on one side of the sheet as well as van der Waals interactions between the hydrophobic residues on the other side.
Figure 19.
(A) Molecular model of a single β-sheet forming ionic oligopeptide. The alternating hydrophobic and hydrophilic residues on the backbone promote β-sheet formation. The positively charged lysines (K) and negatively charged aspartic acids (D) are on the lower side of the β-sheet, and the hydrophobic leucines (L) are on the upper side. This molecular structure facilitates self-assembly into nanofibers and hydrogel matrices through intermolecular interactions. (B) A 12-mm chondrocyte-seeded peptide hydrogel plug, punched from a 1.6-mm-thick slab. (C) Light microscope image of chondrocytes encapsulated in a peptide hydrogel [225].
The hydrogels are sensitive to temperature, salt concentration, and pH, so these variables can be used modulate the properties of the self-assembled oligopeptide hydrogels for optimum performace as biomaterials for tissue engineering. It was shown that these self-assembled hydrogels can support cell attachment of a variety of mammalian cells, including endothelial cells [233], osteoblasts [234], neural cells [227, 235], hepatocytes [232], and chondrocytes [225]. Kisiday et al. used oligopeptides containing leucine, lysine, and aspartic acid (Figure 19a) to form hydrogel matrices that supported the growth and accumulation of chondrocytes (Figure 19b). The chondrocytes retained their morphology and produced a cartilage-like ECM, indicating that these hydrogels have potential as scaffolds for cartilage tissue repair [225]. Ellis-Behnke et al. used a similar peptide nanofiber scaffold to demonstrate axon regeneration and the functional return of vision in hamsters with severed optic tracts [235].
3.3.1.2.3 De novo designed β-hairpin polypeptides
Pochan and Schneider have demonstrated that short amphiphilic peptides that fold into β-hairpin structures will self-assemble into injectable hydrogels that can be used for tissue engineering [236-245]. The 20-amino acid peptide is composed of two sequences of alternating valine and lysine residues, which have a high propensity for β-sheet formation, adjoining a strong β-turn forming tetrapeptide (-VDPPT-). The molecule is induced into an intramolecularly folded β-hairpin structure by various external stimuli including pH [236], temperature [237], salt [238], and light [240, 244]. The alternating valine and lysine residues are oriented such that the molecule is amphiphilic and has one polar, lysine-rich face and one nonpolar, valine-rich face. Due to their amphiliphilic nature, the β-hairpin molecules can form intermolecular associations through both lateral hydrogen bond formation and facial hydrophobic interactions. The molecules form fibrils with well-defined cross-sections and self-assemble into supermolecular network hydrogels that are three-dimensional and rich in β-sheets.
The folding of the peptides into b-hairpin structures occurs upon elimination of charge repulsion between neighboring lysine residues. This can be accomplished via deprotonation at basic pH (9.0) or charge screening by ionic salts at physiological pH (7.4). Kretsinger et al. showed that the addition of cell culture media provides sufficient salt to induce β-hairpin folding while at the same time provides the nutrients that are necessary for cell growth in tissue engineering applications [241]. It was also shown that the β-hairpin hydrogels are non-cytotoxic and support fibroblast adhesion and proliferation. Rheological studies showed that the hydrogels possess storage moduli (G′) of 1-10 kPa under various solution conditions, which is in a suitable range for use as tissue engineering scaffolds.
Haines-Butterick et al. used β-hairpin molecules with a slightly lower net positive charge to homogeneously encapsulate C3H10t1/2 mesenchymal stem cells within the hydrogel network [245]. The resulting gel/cell constructs were shear-thin delivered via syringe directly to target sites, where the cells remained viable and the re-solidified construct remained in tact. An outline of the self-assembly and shear-thinning delivery process is shown in Figure 20.
Figure 20.
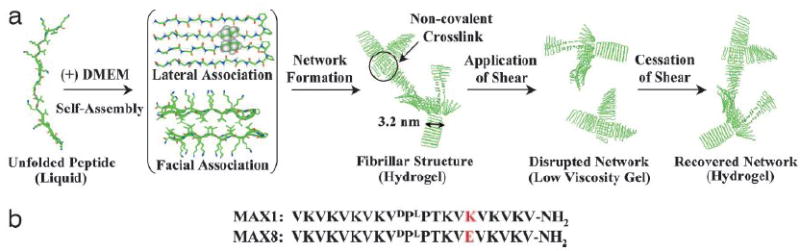
Self-assembly, shear-thinning, and self-healing mechanism allowing rapid formation of hydrogels that can be subsequently syringe-delivered. (a) Addition of cell culture buffer DMEM (pH 7.4, 37°C) to a buffered solution (25 mM Hepes, pH 7.4) of unfolded peptide induces formation of a β-hairpin structure that undergoes lateral and facial self-assembly affording a rigid hydrogel with a fibrillar supramolecular structure. Subsequent application of shear stress disrupts the noncovalently stabilized network, leading to the conversion of the hydrogel to a low-viscosity gel. Upon cessation of shear stress, the network structure recovers, converting the liquid back to a rigid hydrogel. (b) Peptide sequences of two β-hairpin molecules: MAX1 and MAX8. MAX8 has a net positive charge that is two less than that of MAX1 due to the substitution of one negatively charged glutamic acid residue for one positively charged lysine residue. The lower net positive charge allows MAX8 to undergo rapid gelation. Reproduced from [245].
3.3.2 Chemically crosslinked polypeptides
3.3.2.1 Chemically crosslinked elastin-based hydrogels
Elastin-like polypeptides are good candidates for chemical crosslinking, because it is easy to incorporate chemically active amino acids at the guest residue position in the elastin-based repeat unit, Val-Pro-Gly-Xaa-Gly. Also, because ELPs can be designed at the molecular level and genetically synthesized, unique properties can be introduced by incorporating other biologically active peptide sequences. Examples can be found of ELP hydrogels that are formed by irradiation[246-248], photoinitiation [249], amine-reactive chemical crosslinking [37, 68, 250-260], and enzymatic crosslinking by tissue transglutaminase [261]. The hydrogels have been successfully used for cartilage and intervertebral disc tissue repair, small-diameter vascular grafts, urinary bladders, stem cell matrices, neural guides, stem cell sheets, and post-surgical wound treatment [250-256, 262-264]. McMillan et al. used the electrophilic crosslinker bis(sulfosuccinimidyl) suberate to join lysine residues in a lysine-rich ELP to form hydrogels in either phosphate buffer at pH 8.5 or anhydrous dimethylsulfoxide. They found that choice of solvent affected the crosslinking density and the resulting microstructure of the gel [258, 259]. Trabbic-Carlson et al. also used lysine-based ELPs to form hydrogels by chemically crosslinking with tri-succinimidyl aminotriacetate in an organic solvent mixture of dimethyl sulfoxide and dimethylformamide. The physical properties of the gel were found to be tunable according to three parameters: ELP molecular weight, concentration, and lysine content. At low molecular weight, low concentration, or low lysine content, the formation of intramolecular, non-functional crosslinks is promoted, which generally leads to weaker gels. The ability to synthesize an array of hydrogels with well-defined and varied properties allows these ELPs to be used in a wide range of applications [68]. McHale et al. showed that ELPs containing glutamine and lysine residues can be crosslinked by the enzymatic activity of tissue transglutaminase in a biocompatible process. The resulting ELP hydrogels were used to encapsulate chondrocytes, which were then able to synthesize a cartilage matrix rich in sulfated glycosaminoglycans and type II collagen. They also recorded an increase in mechanical integrity after the incubation with chondrocytes, suggesting that the ELP matrix had been restructured by the deposition of cartilage ECM components [261].
Although there are many examples of chemically crosslinked ELP hydrogels, the application of ELPs for in situ gelation by chemical crosslinking has been limited by poor solubility in water, concerns about toxicity, lack of biocompatible crosslinking reagents and byproducts, or slow gelation kinetics. However, it was reported by Lim et al. that an aqueous, biocompatible Mannich type condensation reaction [265-267] can be used to crosslink lysine-containing ELPs over a wide range in pH (2-13), and it produces only water as a byproduct [260, 268]. In this reaction, an organophosphorous crosslinker, β-[tris(hydroxymethyl)phosphino]propionic acid (THPP), reacts with the amines of the lysine residues in the ELP to create trifunctional intra- or intermolecular crosslinks, as shown in Figure 21. The ELPs undergo gelation within minutes under physiological conditions, indicating that they are suitable for use as injectable biomaterials. The shear modulus of the crosslinked hydrogels is comparable to or higher than that of some connective tissues, such as nucleus pulposus or meniscus (see Table 4), and the modulus can be modulated by adjusting the lysine density or arrangement. The hydrogels were also tested for biocompatibility, and it was found that they are non-cytotoxic and can maintain cell survival. These results showed that THPP-crosslinked ELPs provide a biocompatible and injectable biomaterial that can support tissue regeneration in a load-bearing environment.
Figure 21.

Schematic of the inter- or intramolecular crosslinking mechanism using THPP to connect lysine residues in ELPs. This biocompatible reaction can be carried out in aqueous solution, has only water as a byproduct, and is non-cytotoxic. The gelation occurs rapidly, allowing for the use of these gels as injectable biomaterials, and the mehanical properties of fully stabilized gels approach those of cartilaginous tissues. In addition, the THPP crosslinking sites present reactive carboxylic acids that may be used for the further introduction of biological moieties into the hydrogels. [260]
Table 4.
Mechanical properties of elastin-based polymer networks and natural tissues
| Polymer Networks | Concentration (mg/ml) | |G*| (kPa)a or E (kPa)b | Ref.c |
|---|---|---|---|
| ELP coacervate, 37 °C | 324 | 0.08 a | [198] |
| tTG-crosslinked ELP | 100 | 0.26 a | [261] |
| TSAT-crosslinked ELP | 180 | 8 - 10 a | [68] |
| THPP-crosslinked ELP | 200 | 5.8 - 45.8 a | [260] |
| γ-irradiated ELP | 500 | 10 - 200 a | [246] |
| BS3-crosslinked ELP fusions | 200-400 | 80 - 700 b | [252] |
| HMDI-crosslinked ELP fusions | 100 | 400 - 930 b | [255] |
| Nucleus pulposus | NA | 11.0 a | [286] |
| Articular cartilage | NA | 440.0 a | [200] |
| Elastin | NA | 300 - 600 b | [252] |
Abbreviations: ELP, elastin-like polypeptide; tTG, tissue transglutaminase; TSAT, tris-succinimidyl aminotriacetate; THPP, β-[tris(hydroxymethyl)phosphino]-propionic acid; BS3, bis(sulfosuccinimidyl) suberate; HDMI, hexamethylene diisocyanate; NA, not available.
|G*|, Dynamic shear modulus (angular frequency of 10 rad/s).
E, Tensile modulus.
Some data estimated from graphical plots.
Urry has suggested that biomaterials containing ELPs with cell adhesion signals may be useful as vascular grafts for the reconstruction of blood vessels [269, 270]. The Tirrell group has followed through with this idea, and they have demonstrated that chemically crosslinked hydrogels formed from ELPs containing fibronectin cell-binding domains are successful at supporting the growth and spreading of endothelial cells [250-257]. The artificial ECM (aECM) proteins have a repeating structure composed of alternating lysine-containing ELP domains and integrin-binding fibronectin domains. The cell-binding domain is usually the CS5 region of fibronectin, which contains the REDV sequence that has been shown to promote the attachment and spreading of endothelial cells [271, 272]. The RGD integrin-binding domain from fibronectin has also been used, and it has been shown that this sequence is preferable over the CS5 domain when rapid cell spreading is required [254]. The aECM fusion proteins were crosslinked with glutaraldehyde [251], bis(sulfosuccinimidyl) suberate (BS3) [252], and hexamethylene diisocyanate [255], and it was found that the elastic moduli of the resulting hydrogels are very close to the modulus of native elastin, 0.3 to 0.6 MPa (see Table 4). In addition, the modulus can be tuned by altering the protein molecular weight, distance between lysine crosslinking sites, positioning of lysine crosslinking sites, molar ratio of crosslinker to primary amine, the type of crosslinker used, and the solvent conditions. Endothelial cells cultured on films of the aECM proteins successfully grew and formed monolayers by attachment to the integrin-binding domains. Their growth and spreading was shown to be largely dependent on the frequency of RGD or CS5 cell-binding domains as well as the location of the crosslinkable lysine residues [253, 254, 256]. Richman et al. also showed that endothelial cells adhered to ELP-RGD aECM proteins stimulated the intracellular focal adhesion kinase (FAK) signaling pathway, which demonstrates successful tethering to the α5β1 integrin [257]. The tunable, elastin-like mechanical properties of the aECM hydrogels and their ability to mediate cellular adhesion and proliferation make them suitable candidates for use in small diameter vascular grafts.
3.3.2.2 Enzymatically cleavable peptide-based hydrogels
The Hubbell group has turned their attention to designing and synthesizing hydrogels that mimic the bi-directional biomolecular interactions that naturally occur between cells and the extracellular matrix (ECM) surrounding them [184, 273-285]. They have found that signals such as cell adhesion sites, bound growth factors, and cleavage sites for proteolytic activity are essential for cells to be able to remodel the ECM and sustain tissue regeneration. To impart these additional functionalities to artifical ECMs, peptides are covalently incorporated into one of two types of chemically crosslinked hydrogels. The first class of hydrogels is based on naturally occurring fibrin, which is spontaneously formed by the polymerization of fibrinogen in the presence of thrombin and further cross-linked by the transglutaminase activity of factor XIIIa [273-278]. Ehrbar et al. engineered a vascular endothelial growth factor (VEGF) fusion protein that includes a substrate for binding to the fibrin matrix by transglutaminase during polymerization as well as a plasmin cleavage site for invading cells to release the bound growth factor [276]. The injectable hydrogels containing bound VEGF were able to induce endothelial cell proliferation better than hydrogels containing only native soluble VEGF. Fibrin matrices were also successfully augmented with bone morphogenetic protein-2 (BMP-2) to promote bone growth and healing [275] and with heparin-binding proteins to promote neurite extension [277].
In the second class of hydrogels, chemically crosslinked three-dimensional networks are formed by Michael-type addition reactions between thiol-bearing bioactive peptides and conjugated unsaturations on single- or multi-armed poly(ethylene glycol (PEG) chains endfunctionalized with vinyl sulfone [273, 279-285]. In studies by Rizzi et al., peptide sequences containing multiple cysteine residues, integrin-binding RGD domains, and substrates for degradation by the cell-secreted proteases matrix metalloproteinase (MMP) and plasmin were synthesized recombinantly [284, 285]. The cysteine residues provide reactive thiols for conjugation to synthetically polymerized vinyl sulfone-terminated PEG. Human fibroblasts cultured in the protein-PEG matrixes were found to be adhesive to the integrin-binding moieties, and the hydrogels were susceptible to degradation by secreted proteases. The cells exhibited three-dimensional migration patterns, and by comparison to non-degradable networks, it was found that proteolytic sensitivity was the most important factor in the cells’ ability to migrate within the hydrogel and promote degradation. The hydrogels were tested in vivo as carriers of bone morphogenetic protein (rhBMP-2) to heal bone defects, and they were successful at healing the defects by the remodeling and replacement of the artificial ECM (Figure 22). Similar protein-PEG composite hydrogels containing VEGF as well as cell adhesion and protease degradation sites were shown to promote angiogenesis and the remodeling and healing of vascular tissue [281, 283]. These designs for cell-responsive, smart hydrogel systems are an important advancement for functional tissue engineering and controlled drug delivery.
Figure 22.

(A) Schematic of protein-PEG network formation. The recombinant protein polymer contains RGD integrin-binding cell adhesion sites, MMP and plasmin degradation sites, and several cysteine residues for crosslinking. (B) Radiographic (left) and three-dimensional microcomputed tomography images (right) of the bone formation in rat calvarial defects after treatment with protein-PEG hydrogels. Group I: Degradable matrix with BMP-2; Group II: Non-degradable matrix with BMP-2; Group III: Degradable matrix without BMP-2; Group IV: Non-degradable matrix without BMP-2. In Group I, bone was prevalently observed to have replaced the matrix. A similar bone volume was measured in Group II, but the bone was confined at matrix surface. Reproduced from [285].
3.4 Surface engineering
Surface modifications play an important role in biomedical devices, biosensors, biomaterials, and implants [287-292]. In most biomedical applications, surfaces are the primary “agents” that come into contacts with biomolecules, body fluids, cells, and tissues. They also act as both passive and active “modulators” of detection, biocompatibility, and cell adhesion. Therefore, the success of many biomedical applications hinges on the ability to control and manipulate the interface between biotic and abiotic components. This biomedical research area remains an active area due to the complex and poorly understood dynamics between biological components and surfaces.
Many chemical techniques have been developed for the surface modification of ceramics [293-295], metals [295-298], and synthetic polymers [299-301]. These modifications typically only modify the physico-chemical properties of the material and act as “passive” components such as a physical barrier between the body and bulk material or a protective coating. Recently, there has been a shift in research focus to develop “active” biomaterials that are responsive to cellular behaviors and changes in the microenvironment. To this end, biomaterials need to be functionalized with biological ligands such as peptides or proteins, and their presentation needs to be both spatially and temporally controlled. This task is not trivial, due to the complex nature of biomolecules and their fragility when in contact with surfaces.
Although the successful use of biomolecules in surface modification requires extra care during the process, using peptide-based biopolymers offers several advantages over the conventional surface modification techniques because of 1) better biocompatibility with cells and tissues than their synthetic counterparts, 2) the ease of incorporating biologically useful ligands, 3) the precise control of the structures and functions of biopolymers, and 4) biodegradability in most of the cases. In this section, we provide an overview of the use of peptide-based biomaterials in biointerface science and present several examples of the current development in this emerging research area.
3.4.1 Biosensing
Detection of biomolecules is very important for the diagnosis, prevention, and treatment of many diseases. However, improvements are required in many areas, such as prevention of non-specific binding [302-306], sensitivity [307-315], site-specific conjugation [316-318], bioactivity [319-322], multiplexing [309, 323], and reusability [324-326]. Some of these problems can be addressed by using different chemical surface modification approaches. For example, polymeric brushes created with surface-initiated polymerization provide a superior non-fouling surface that dramatically reduces background signals and further improves the signal-to-noise ratios of an assay [303]. Surface enhanced laser desorption and ionization time-of-flight mass spectrometry (SELDI ToF MS) utilizes surfaces that are hydrophobic, cationic, anionic, or ligand-specific to fractionate biological samples for mass characterization of target proteins, thus eliminating pre-processing of complex samples [327-330]. The continuous advancement of patterning techniques for proteins and DNA has progressively reduced the spot size of biomolecules and allowed the simultaneous detection of numerous targeted molecules in a massively parallel manner with a reduced consumption of reagents [331-333]. Sensing chips with immobilized with nickel-nitrilotriacetic acid (NTA) have an excellent surface for repetitive binding of histidine-tagged proteins and provide a platform for continuous replacement of the sensing element [325]. There are many other examples, but these suffice to illustrate the broad range of this field.
The use of novel environmentally responsive biomaterials such as elastin-like polypeptides in biosensing opens up several new possibilities. For example, a fusion protein of ELP with protein A, G, or L can bind to an antibody and act as a concentration mechanism to enhance signals [334] (Figure 23). A similar strategy can be used to immobilize recombinant proteins through the interaction between the leucine zipper pairs [335]. In addition, this makes it possible to individually address biomolecules on surfaces by immobilizing the biomolecules in a spatial controlled manner using local environmental changes [336]. Biosensing surfaces can also be quickly regenerated by exploiting the reversible phase transition behavior of ELPs to create an actively controlled surface in which the surface properties such as hydrophobicity and functionality can be rapidly modulated by a simple temperature switch [337].
Figure 23.

Fabrication of multiplex antibody microarrays. (a) Antibodies are captured by ELP-Protein A, G, or L fusions and collected by centrifugation. Separation of bound and unbound antibodies was followed by the resolubilization of the pellet to yield purified proteins, which are then spotted on microarrays. (b) Image of microarrays with immobilized antibodies of different species (rabbit, goat, and mouse) captured by ELP-Protein A, G or L fusion proteins, respectively. (c) Comparison of the relative fluorescence intensities detected from the immobilized complexes with no NaCl added, with 2 M NaCl added, and after washing with 4 °C PBS. Reproduced from [83].
3.4.2 Micro- and nano-structures
The combination of advanced fabrication technologies in the semi-conductor industry with new methods in genetic and protein engineering has led to the development of a research area known as bionanofabrication. Using conventional lithography and patterning techniques, peptides and proteins can be directly deposited in a spatially specific manner. This provides us with the capability to manipulate molecules at the nanometer or atomic length scale and create advanced materials for biosensing, biology, and materials science. Detection time, sensitivity, sample size, and cost can be substantially reduced when the length scale of a biosensing system shrinks dramatically [338-342]. In addition, bionanofabrication potentially allows direct interrogation of numerous biomolecules in a parallel manner. In materials science, molecular recognition of biomolecules such as DNA and antibodies can direct the assembly of nanoscale components such as gold nanoparticles, quantum dots, and proteins [23, 343-346]. Generation of more complex biomolecular ensembles is also possible when using self-assembling biomolecules.
Several researchers have successfully shown the feasibility of bionanofabrication using biopolymers. Hyun and coworkers generated an array of elastin-like polypeptides (ELPs) using dip-pen nanolithography, in which an AFM cantilever was used to generate a nanopatterned template to conjugate ELPs on a surface substrate [347] (Figure 24). This provides a foundation to spatially control the deposition of biopolymers on a surface. While most bionanofabrication techniques produce two-dimensional patterns, Hill and Shear have created three-dimensional protein scaffolds that can be functionalized for catalytic activity [348]. They used a direct-write process in which multiphoton excitation promotes photochemical crosslinking of protein molecules from aqueous solution within specified volume elements. Using this method, topographically complex sensors and dosing sources can be produced with potential applications in microfluidics, sensor array fabrication, and real-time chemical modification of cell culture environments.
Figure 24.

ELP nanoarray generated by dip pen nanolithography (DPN). (a) Atomic force microscopy tapping mode image of ELP dot array in PBS buffer at room temperature. (b) Close-up image of the area highlighted in (a) and a cross section showing a feature height of 5-6 nm and a lateral feature size of about 200 nm. Reproduced from [347].
3.4.3 Affinity tags for surface modification
In the previous sections, we highlighted the importance of peptide-based biopolymers in surface modifications for biosensing, fluidic systems, and materials science, but the use of these materials can be limited by their size and secondary structure. On the other hand, short peptide sequences, also known as affinity tags, are structurally stable and easy to generate, and provide an excellent alternative to bulky biopolymers for surface engineering purposes. Their benefits also include longer shelf lives for biomedical products and the ease of functionalization of many materials such as metal, ceramics, inorganics, synthetic polymers, and biopolymers. Affinity tags are used in many biomedical and clinical applications, including functionalization of surfaces with biological moieties, better integration of implants with tissues, promotion of cell adhesion and cell growth, and specific binding of biomolecules of interest. Many peptide sequences have been identified as affinity tags, so there are now many choices of affinity tags available for specific surface functionalization (Table 5).
Table 5.
Commonly used affinity tags for surface modifications
| Peptide | Surface modifications |
|---|---|
| RGD-containing peptide |
|
| RGD-linked polymers |
|
| QQKFQFQFEQQ | |
| GHK |
|
| KNEED |
|
| IKVAV |
|
| YIGSR |
Among all affinity tags, Arg-Gly-Asp (RGD) is one of the most commonly used peptide sequences in surface modifications of biomaterials. RGD sequences present on peptide amphiphiles [349], hyaluronan hydrogels [350], dextran [351], collagen [352], poly-l-lysine-graft-(polyethylene glycol) copolymers [353], poly(lactic acid-co-lysine) [354], poly(ethylene glycol)-poly(lactic acid) diblock copolymers [355], acrylic terpolymers [356], and polyurethanes [357] have been reported to promote cell adhesion to a variety of cell types, including neurons [353, 358], osteoblasts [359, 360], endothelial cells [361], and fibroblasts [350]. In some cases, the RGD sequences can also promote other cellular functions such as matrix mineralization of osteogenic cells [359, 360] and neurite outgrowth [358]. Other peptide sequences representing important domains of laminin and fibronectin such as IKVAV, YIGSR, and KNEED also promote the attachment of neuronal and endothelial cells [8-11]. At the tissue level, the peptide sequence GHK was found to promote dermal wound healing and tissue repair [12]. Peptide sequences containing a glutamine residue can be used to generate self-assembling tissue engineering scaffolds after in situ crosslinking by tissue transglutaminase [261, 362]. This mechanism is also useful for incorporating cell adhesion peptides into tissue engineering scaffolds [363].
Affinity tags have also been used in the biotechnology and pharmaceutical industries, mainly for protein purification and recovery. Detailed descriptions of these affinity tags can be found elsewhere in comprehensive reviews [364-368]. Recently, affinity tags that can specifically and strongly bind to non-biological molecules, including bulk materials, have been developed by phage display and other combinatorial techniques. The advances in these areas further extend the impact of affinity tags into semi-conducting materials and conducting polymers [369], thus providing a much-needed biointerface for applications involving both biological and non-biological elements (Figure 25). The selectivity and affinity of many of these tags remains unclear for their putative targets, but these newly identified tags can also potentially be incorporated into peptide-based biopolymers to improve their functionalities.
Figure 25.

Specific recognition of semiconductor materials by bacterial phages. (a) Fluorescent image showing minimal background fluorescent signal due to the presence of primary antibody and streptavidin-tetramethyl rhodamine. (b) Fluorescent image showing binding of fluorescently labeled phage clone G12-3 to GaAs. One mm GaAs lines spaced with 4 mm SiO2 are patterned on the surface. (c) Schematic of bacterial phages binding specifically to the GaAs semi-conductor instead of the silicon substrate. Reproduced from [369].
4 Conclusion
Peptide-based biomaterials have emerged as a new class of biomaterials that possesses unique and often superior properties when compared to conventional materials. A basic understanding of their design principles and synthesis methodologies is crucial to bringing these biomaterials to biomedical and biotechnological applications. We have summarized the synthesis methods and characterization techniques that are used to study the biophysical, structural, and rheological properties of peptide-based biomaterials. The prevalence of these materials is exemplified by the wide range of biomedical and biotechnological applications of these materials in protein purification, drug delivery, tissue engineering, and surface engineering. Although peptide-based biomaterials have become increasingly important, they also have limitations, such as short shelf life and thermal instability. Many of these limitations can be addressed by emerging technologies, thus further expanding the uses of peptide-based biomaterials into applications for which they are currently impractical.
Acknowledgments
The authors would like to acknowledge financial support from NIH Grants GM61232, EB00188, and EB001630.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Burkhard P, Stetefeld J, Strelkov SV. Trends In Cell Biology. 2001;11:82. doi: 10.1016/s0962-8924(00)01898-5. [DOI] [PubMed] [Google Scholar]
- 2.DeGrado WF, Summa CM, Pavone V, Nastri F, Lombardi A. Annual Review Of Biochemistry. 1999;68:779. doi: 10.1146/annurev.biochem.68.1.779. [DOI] [PubMed] [Google Scholar]
- 3.Debelle L, Tamburro AM. International Journal Of Biochemistry & Cell Biology. 1999;31:261. doi: 10.1016/s1357-2725(98)00098-3. [DOI] [PubMed] [Google Scholar]
- 4.Iozzo RV. Annual Review Of Biochemistry. 1998;67:609. doi: 10.1146/annurev.biochem.67.1.609. [DOI] [PubMed] [Google Scholar]
- 5.Bryson JW, Betz SF, Lu HS, Suich DJ, Zhou HXX, Oneil KT, Degrado WF. Science. 1995;270:935. doi: 10.1126/science.270.5238.935. [DOI] [PubMed] [Google Scholar]
- 6.Connor RE, Tirrell DA. Polymer Reviews. 2007;47:9. [Google Scholar]
- 7.Chen S, Schultz PG, Brock A. Journal of Molecular Biology. 2007;371:112. doi: 10.1016/j.jmb.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 8.Jun HW, West JL. Journal Of Biomedical Materials Research Part B-Applied Biomaterials. 2005;72B:131. doi: 10.1002/jbm.b.30135. [DOI] [PubMed] [Google Scholar]
- 9.Wong JY, Weng ZP, Moll S, Kim S, Brown CT. Biomaterials. 2002;23:3865. doi: 10.1016/s0142-9612(02)00127-8. [DOI] [PubMed] [Google Scholar]
- 10.Ranieri JP, Bellamkonda R, Bekos EJ, Vargo TG, Gardella JA, Aebischer P. Journal Of Biomedical Materials Research. 1995;29:779. doi: 10.1002/jbm.820290614. [DOI] [PubMed] [Google Scholar]
- 11.Matsuda A, Kobayashi H, Itoh S, Kataoka K, Tanaka J. Biomaterials. 2005;26:2273. doi: 10.1016/j.biomaterials.2004.07.032. [DOI] [PubMed] [Google Scholar]
- 12.Arul V, Gopinath D, Gomathi K, Jayakumar R. Journal Of Biomedical Materials Research Part B-Applied Biomaterials. 2005;73B:383. doi: 10.1002/jbm.b.30246. [DOI] [PubMed] [Google Scholar]
- 13.Zheng YJ, Huo Q, Kele P, Andreopoulos FM, Pham SM, Leblanc RM. Organic Letters. 2001;3:3277. doi: 10.1021/ol0101638. [DOI] [PubMed] [Google Scholar]
- 14.Valenti LE, De Pauli CP, Giacomelli CE. Journal of Inorganic Biochemistry. 2006;100:192. doi: 10.1016/j.jinorgbio.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 15.Stuart DI, Acharya KR, Walker NPC, Smith SG, Lewis M, Phillips DC. Nature. 1986;324:84. doi: 10.1038/324084a0. [DOI] [PubMed] [Google Scholar]
- 16.Slocik JM, Stone MO, Naik RR. Small. 2005;1:1048. doi: 10.1002/smll.200500172. [DOI] [PubMed] [Google Scholar]
- 17.Slocik JM, Naik RR. Advanced Materials. 2006;18:1988. [Google Scholar]
- 18.Rousselot-Pailley P, Seneque O, Lebrun C, Crouzy S, Boturyn D, Dumy P, Ferrand M, Delangle P. Inorganic Chemistry. 2006;45:5510. doi: 10.1021/ic060430b. [DOI] [PubMed] [Google Scholar]
- 19.Rainer MJA, Rode BM. Inorganica Chimica Acta-Bioinorganic Chemistry. 1984;92:1. [Google Scholar]
- 20.Perkins CM, Rose NJ, Weinstein B, Stenkamp RE, Jensen LH, Pickart L. Inorganica Chimica Acta-Articles and Letters. 1984;82:93. [Google Scholar]
- 21.Pazirandeh M, Wells BM, Ryan RL. Applied and Environmental Microbiology. 1998;64:4068. doi: 10.1128/aem.64.10.4068-4072.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lane TF, Iruelaarispe ML, Johnson RS, Sage EH. Journal of Cell Biology. 1994;125:929. doi: 10.1083/jcb.125.4.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim SB, Lim JW. Asian-Australasian Journal of Animal Sciences. 2004;17:1459. [Google Scholar]
- 24.Ferrari R, Bernes S, de Barbarin CR, Mendoza-Diaz G, Gasque L. Inorganica Chimica Acta. 2002;339:193. [Google Scholar]
- 25.Dong J, Liu C, Zhang J, Xin ZT, Yang G, Gao B, Mao CQ, Liu NL, Wang F, Shao NS, Fan M, Xue YN. Chemical Biology & Drug Design. 2006;68:107. doi: 10.1111/j.1747-0285.2006.00421.x. [DOI] [PubMed] [Google Scholar]
- 26.Deo S, Godwin HA. Journal of the American Chemical Society. 2000;122:174. [Google Scholar]
- 27.Bayer E. Angewandte Chemie-International Edition in English. 1991;30:113. [Google Scholar]
- 28.Aimoto S. Current Organic Chemistry. 2001;5:45. [Google Scholar]
- 29.Deming TJ. Polypeptide and polypeptide hybrid copolymer synthesis via NCA polymerization. Peptide Hybrid Polymers. 2006;202:1–18. [Google Scholar]
- 30.Tsutsumiuchi K, Aoi K, Okada M. Macromolecules. 1997;30:4013. [Google Scholar]
- 31.Vrhovski B, Weiss AS. European Journal Of Biochemistry. 1998;258:1. doi: 10.1046/j.1432-1327.1998.2580001.x. [DOI] [PubMed] [Google Scholar]
- 32.Vrhovski B, Jensen S, Weiss AS. European Journal Of Biochemistry. 1997;250:92. doi: 10.1111/j.1432-1033.1997.00092.x. [DOI] [PubMed] [Google Scholar]
- 33.Wise SG, Mithieux SM, Raftery MJ, Weiss AS. Journal Of Structural Biology. 2005;149:273. doi: 10.1016/j.jsb.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 34.Bellingham CM, Lillie MA, Gosline JM, Wright GM, Starcher BC, Bailey AJ, Woodhouse KA, Keeley FW. Biopolymers. 2003;70:445. doi: 10.1002/bip.10512. [DOI] [PubMed] [Google Scholar]
- 35.Keeley FW, Bellingham CM, Woodhouse KA. Philosophical Transactions Of The Royal Society Of London Series B-Biological Sciences. 2002;357:185. doi: 10.1098/rstb.2001.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Urry DW. Journal Of Physical Chemistry B. 1997;101:11007. [Google Scholar]
- 37.Martino M, Tamburro AM. Biopolymers. 2001;59:29. doi: 10.1002/1097-0282(200107)59:1<29::AID-BIP1003>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 38.Martino M, Coviello A, Tamburro AM. International Journal Of Biological Macromolecules. 2000;27:59. doi: 10.1016/s0141-8130(99)00118-x. [DOI] [PubMed] [Google Scholar]
- 39.Spezzacatena C, Perri T, Guantieri V, Sandberg LB, Mitts TF, Tamburro AM. European Journal Of Organic Chemistry. 2002:95. [Google Scholar]
- 40.Urry DA. Journal of Physical Chemistry B. 1997;101:11007. [Google Scholar]
- 41.Luan CH, Urry DW. Journal of Physical Chemistry. 1991;95:7896. [Google Scholar]
- 42.Urry DW, Luan CH, Parker TM, Gowda DC, Prasad KU, Reid MC, Safavy A. Journal Of The American Chemical Society. 1991;113:4346. [Google Scholar]
- 43.Word JM, Lovell SC, LaBean TH, Taylor HC, Zalis ME, Presley BK, Richardson JS, Richardson DC. Journal of Molecular Biology. 1999;285:1711. doi: 10.1006/jmbi.1998.2400. [DOI] [PubMed] [Google Scholar]
- 44.Trabbic-Carlson K, Meyer DE, Liu L, Piervincenzi R, Nath N, LaBean T, Chilkoti A. Protein Engineering, Design & Selection. 2004;17:57–66. doi: 10.1093/protein/gzh006. [DOI] [PubMed] [Google Scholar]
- 45.Vepari C, Kaplan DL. Progress in Polymer Science. 2007;32:991. doi: 10.1016/j.progpolymsci.2007.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang J, Foo CWP, Kaplan DL. Polymer Reviews. 2007;47:29. [Google Scholar]
- 47.Tsujimoto Y, Suzuki Y. Cell. 1979;16:425. doi: 10.1016/0092-8674(79)90018-7. [DOI] [PubMed] [Google Scholar]
- 48.Xu M, Lewis RV. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:7120. doi: 10.1073/pnas.87.18.7120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mello CM, Senecal K, Yeung B, Vouros P, Kaplan D. Initial Characterization of Nephila-Clavipes Dragline Protein. Silk Polymers. 1994;544:67–79. [Google Scholar]
- 50.Cappello J, Crissman J, Dorman M, Mikolajczak M, Textor G, Marquet M, Ferrari F. Biotechnology Progress. 1990;6:198. doi: 10.1021/bp00003a006. [DOI] [PubMed] [Google Scholar]
- 51.McGrath KP, Fournier MJ, Mason TL, Tirrell DA. Journal of the American Chemical Society. 1992;114:727. [Google Scholar]
- 52.Krejchi MT, Atkins EDT, Waddon AJ, Fournier MJ, Mason TL, Tirrell DA. Science. 1994;265:1427. doi: 10.1126/science.8073284. [DOI] [PubMed] [Google Scholar]
- 53.Winkler S, Szela S, Avtges P, Valluzzi R, Kirschner DA, Kaplan D. International Journal Of Biological Macromolecules. 1999;24:265. doi: 10.1016/s0141-8130(98)00088-9. [DOI] [PubMed] [Google Scholar]
- 54.Hood EE, Jilka JM. Current Opinion in Biotechnology. 1999;10:382. doi: 10.1016/S0958-1669(99)80069-X. [DOI] [PubMed] [Google Scholar]
- 55.Lazaris A, Arcidiacono S, Huang Y, Zhou JF, Duguay F, Chretien N, Welsh EA, Soares JW, Karatzas CN. Science. 2002;295:472. doi: 10.1126/science.1065780. [DOI] [PubMed] [Google Scholar]
- 56.Deming TJ. Advanced Drug Delivery Reviews. 2002;54:1145. doi: 10.1016/s0169-409x(02)00062-5. [DOI] [PubMed] [Google Scholar]
- 57.Haider M, Megeed Z, Ghandehari H. Journal Of Controlled Release. 2004;95:1. doi: 10.1016/j.jconrel.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 58.Prince JT, McGrath KP, Digirolamo CM, Kaplan DL. Biochemistry. 1995;34:10879. doi: 10.1021/bi00034a022. [DOI] [PubMed] [Google Scholar]
- 59.Chilkoti A, Dreher MR, Meyer DE. Advanced Drug Delivery Reviews. 2002;54:1093. doi: 10.1016/s0169-409x(02)00060-1. [DOI] [PubMed] [Google Scholar]
- 60.Kim WY, George A, Evans M, Conticello VP. Chembiochem. 2004;5:928. doi: 10.1002/cbic.200400052. [DOI] [PubMed] [Google Scholar]
- 61.Xie JM, Schultz PG. Nature Reviews Molecular Cell Biology. 2006;7:775. doi: 10.1038/nrm2005. [DOI] [PubMed] [Google Scholar]
- 62.Haider M, Megeed Z, Ghandehari H. Journal Of Controlled Release. 2004;95:1. doi: 10.1016/j.jconrel.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 63.Crans DC, Rithner CD, Baruah B, Gourley BL, Levinger NE. Journal Of The American Chemical Society. 2006;128:4437. doi: 10.1021/ja0583721. [DOI] [PubMed] [Google Scholar]
- 64.Meyer DE, Shin BC, Kong GA, Dewhirst MW, Chilkoti A. J Control Release. 2001;74:213. doi: 10.1016/s0168-3659(01)00319-4. [DOI] [PubMed] [Google Scholar]
- 65.Meyer DE, Chilkoti A. Biomacromolecules. 2004;5:846. doi: 10.1021/bm034215n. [DOI] [PubMed] [Google Scholar]
- 66.Piervincenzi RT, Chilkoti A. Biomolecular Engineering. 2004;21:33. doi: 10.1016/s1389-0344(03)00080-7. [DOI] [PubMed] [Google Scholar]
- 67.Raucher D, Chilkoti A. Cancer Research. 2001;61:7163. [PubMed] [Google Scholar]
- 68.Trabbic-Carlson K, Setton LA, Chilkoti A. Biomacromolecules. 2003;4:572. doi: 10.1021/bm025671z. [DOI] [PubMed] [Google Scholar]
- 69.Meyer DE, Chilkoti A. Nature Biotechnology. 1999;17:1112. doi: 10.1038/15100. [DOI] [PubMed] [Google Scholar]
- 70.Wright ER, McMillan RA, Cooper A, Apkarian RP, Conticello VP. Advanced Functional Materials. 2002;12:149. [Google Scholar]
- 71.Baneyx F. Current Opinion In Biotechnology. 1999;10:411. doi: 10.1016/s0958-1669(99)00003-8. [DOI] [PubMed] [Google Scholar]
- 72.Makrides SC. Microbiological Reviews. 1996;60:512. doi: 10.1128/mr.60.3.512-538.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lathe GH, Ruthven CRJ. Biochem J. 1956;62:665. doi: 10.1042/bj0620665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Weiss J, Weiss T. Handbook of Ion Chromatography. Wiley-VCH; Weinheim: 2004. [Google Scholar]
- 75.Regnier FE. Science. 1983;222:245. doi: 10.1126/science.6353575. [DOI] [PubMed] [Google Scholar]
- 76.Bailon P, Berthold W, Fung WJ, Ehrlich GK. Affinity Chromatography: Methods and Protocols. Springer-Verlag; New York: 2000. [Google Scholar]
- 77.Greenwood JM, Ong E, Gilkes NR, Warren RAJ, Miller RC, Jr, Kilburn DG. Protein Eng. 1992;5:361. doi: 10.1093/protein/5.4.361. [DOI] [PubMed] [Google Scholar]
- 78.Banki MR, Feng LA, Wood DW. Nature Methods. 2005;2:659. doi: 10.1038/nmeth787. [DOI] [PubMed] [Google Scholar]
- 79.Lim DW, Trabbic-Carlson K, MacKay JA, Chilkoti A. Biomacromolecules. 2007;8:1417. doi: 10.1021/bm060849t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chow DC, Dreher MR, Trabbic-Carlson K, Chilkoti A. Biotechnology Progress. 2006;22:638. doi: 10.1021/bp0503742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ge X, Trabbic-Carlson K, Chilkoti A, Filipe CDM. Biotechnology And Bioengineering. 2006;95:424. doi: 10.1002/bit.21046. [DOI] [PubMed] [Google Scholar]
- 82.Christensen T, Trabbic-Carlson K, Liu WG, Chilkoti A. Analytical Biochemistry. 2007;360:166. doi: 10.1016/j.ab.2006.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim JY, O’Malley S, Mulchandani A, Chen W. Analytical Chemistry. 2005;77:2318. doi: 10.1021/ac0484326. [DOI] [PubMed] [Google Scholar]
- 84.Stiborova H, Kostal J, Mulchandani A, Chen W. Biotechnology and Bioengineering. 2003;82:605. doi: 10.1002/bit.10609. [DOI] [PubMed] [Google Scholar]
- 85.Kostal J, Mulchandani A, Chen W. Biotechnology And Bioengineering. 2004;85:293. doi: 10.1002/bit.10890. [DOI] [PubMed] [Google Scholar]
- 86.Sun XL, Chaikof EL. Glycobiology. 2004;14:1203. [Google Scholar]
- 87.Kostal J, Mulchandani A, Gropp KE, Chen W. Environmental Science & Technology. 2003;37:4457. doi: 10.1021/es034210y. [DOI] [PubMed] [Google Scholar]
- 88.Kostal J, Yang R, Wu CH, Mulchandani A, Chen W. Applied And Environmental Microbiology. 2004;70:4582. doi: 10.1128/AEM.70.8.4582-4587.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Prabhukumar G, Matsumoto M, Mulchandani A, Chen W. Environmental Science & Technology. 2004;38:3148. doi: 10.1021/es035150z. [DOI] [PubMed] [Google Scholar]
- 90.Kostal J, Mulchandani A, Chen W. Macromolecules. 2001;34:2257. [Google Scholar]
- 91.Stevens RC. Structure. 2000;8:R177. doi: 10.1016/s0969-2126(00)00193-3. [DOI] [PubMed] [Google Scholar]
- 92.Hopp TP, Conlon PJ. Bio/Technology. 1988;6:1204. [Google Scholar]
- 93.Crowe J, Döbeli H, Gentz R, Hochuli E, Stüber D, Henco K. 6xHis-Ni-NTA chromatography as a superior technique in recombinant protein expression/purification. In: Harwood AJ, editor. Methods in Molecular Biology. Vol. 31. Humana Press, Inc.; Totawa: 1994. [DOI] [PubMed] [Google Scholar]
- 94.Sherwood R. Trends Biotechnol. 1991;9:1. doi: 10.1016/0167-7799(91)90002-Y. [DOI] [PubMed] [Google Scholar]
- 95.Schmidt TGM, Skerra A. Protein Eng. 1993;6:109. doi: 10.1093/protein/6.1.109. [DOI] [PubMed] [Google Scholar]
- 96.Skerra A, Schmidt TGM. Methods in Enzymology. 1993;326:271. doi: 10.1016/s0076-6879(00)26060-6. [DOI] [PubMed] [Google Scholar]
- 97.Maier T, Drapal N, Thanbichler M, Böck A. Anal Biochem. 1998;259:68. doi: 10.1006/abio.1998.2649. [DOI] [PubMed] [Google Scholar]
- 98.Greenwood JM, Gilkes NR, Kilburn DG, Miller RCJ, Warren RAJ. FEBS Lett. 1989;244:127. doi: 10.1016/0014-5793(89)81177-9. [DOI] [PubMed] [Google Scholar]
- 99.Ong E, Gilkes NR, Warren RAJ, Miller RC, Jr, Kilburn DG. BioTechnol. 1989;7:604. [Google Scholar]
- 100.Greenwood JM, Ong E, Gilkes NR, Warren RAJ, Miller RC, Jr, Kilburn DG. Protein Eng. 1992;5:361. doi: 10.1093/protein/5.4.361. [DOI] [PubMed] [Google Scholar]
- 101.Lu ZJ, DiBlasioSmith EA, Grant KL, Warne NW, LaVallie ER, CollinsRacie LA, Follettie MT, Williams MJ, McCoy JM. Journal of Biological Chemistry. 1996;271:5059. [PubMed] [Google Scholar]
- 102.Nilsson B, Abrahmsen L, Uhlen M. EMBO J. 1985;4:1075. doi: 10.1002/j.1460-2075.1985.tb03741.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ge X, Yang DSC, Trabbic-Carlson K, Kim B, Chilkoti A, Filipe CDM. Journal Of The American Chemical Society. 2005;127:11228. doi: 10.1021/ja0531125. [DOI] [PubMed] [Google Scholar]
- 104.Scheller J, Leps M, Conrad U. Plant Biotechnology Journal. 2006;4:243. doi: 10.1111/j.1467-7652.2005.00176.x. [DOI] [PubMed] [Google Scholar]
- 105.Hart DS, Gehrke SH. Journal of Pharmaceutical Sciences. 2007;96:484. doi: 10.1002/jps.20755. [DOI] [PubMed] [Google Scholar]
- 106.Langer R. Nature. 1998;392:5. [PubMed] [Google Scholar]
- 107.Jain RK. Annual Review of Biomedical Engineering. 1999;1:241. doi: 10.1146/annurev.bioeng.1.1.241. [DOI] [PubMed] [Google Scholar]
- 108.Murphy MP, Smith RAJ. Advanced Drug Delivery Reviews. 2000;41:235. doi: 10.1016/s0169-409x(99)00069-1. [DOI] [PubMed] [Google Scholar]
- 109.Merdan T, Kopecek J, Kissel T. Advanced Drug Delivery Reviews. 2002;54:715. doi: 10.1016/s0169-409x(02)00046-7. [DOI] [PubMed] [Google Scholar]
- 110.Putnam D, Kopecek J. Polymer conjugates with anticancer activity. Biopolymers Ii. 1995;122:55–123. [Google Scholar]
- 111.Matsumura Y, Maeda H. Cancer Research. 1986;46:6387. [PubMed] [Google Scholar]
- 112.Maeda H, Seymour LW, Miyamoto Y. Bioconjugate Chemistry. 1992;3:351. doi: 10.1021/bc00017a001. [DOI] [PubMed] [Google Scholar]
- 113.Lewis WH. Bulletin of the Johns Hopkins Hospital. 1927;41:156. [Google Scholar]
- 114.Jain RK. Scientific American. 1994;271:58. doi: 10.1038/scientificamerican0794-58. [DOI] [PubMed] [Google Scholar]
- 115.Litman T, Druley TE, Stein WD, Bates SE. Cellular and Molecular Life Sciences. 2001;58:931. doi: 10.1007/PL00000912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Duncan R. Anti-Cancer Drugs. 1992;3:175. doi: 10.1097/00001813-199206000-00001. [DOI] [PubMed] [Google Scholar]
- 117.Jain RK. Advanced Drug Delivery Reviews. 2001;46:149. doi: 10.1016/s0169-409x(00)00131-9. [DOI] [PubMed] [Google Scholar]
- 118.Ringsdorf H. Journal of Polymer science. 1975;51:135. [Google Scholar]
- 119.Tomlinson E. Journal of Controlled Release. 1985;2:385. [Google Scholar]
- 120.Ghandehari H, Cappello J. Pharmaceutical Research. 1998;15:813. doi: 10.1023/a:1011999810298. [DOI] [PubMed] [Google Scholar]
- 121.Kopecek J, Kopeckova P, Minko T, Lu ZR. European Journal of Pharmaceutics and Biopharmaceutics. 2000;50:61. doi: 10.1016/s0939-6411(00)00075-8. [DOI] [PubMed] [Google Scholar]
- 122.Lukyanov AN, Torchilin VP. Advanced Drug Delivery Reviews. 2004;56:1273. doi: 10.1016/j.addr.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 123.Torchilin VP, Lukyanov AN, Gao ZG, Papahadjopoulos-Sternberg B. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:6039. doi: 10.1073/pnas.0931428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nan A, Ghandehari H, Hebert C, Siavash H, Nikitakis N, Reynolds M, Sauk JJ. Journal of Drug Targeting. 2005;13:189. doi: 10.1080/10611860500065187. [DOI] [PubMed] [Google Scholar]
- 125.Weissig V, Whiteman KR, Torchilin VP. Pharmaceutical Research. 1998;15:1552. doi: 10.1023/a:1011951016118. [DOI] [PubMed] [Google Scholar]
- 126.Kopecek J, Kopeckova P, Minko T, Lu ZR, Peterson CM. Journal Of Controlled Release. 2001;74:147. doi: 10.1016/s0168-3659(01)00330-3. [DOI] [PubMed] [Google Scholar]
- 127.Kabanov AV, Batrakova EV, Sriadlbhatla S, Yang ZH, Kelly DL, Alakov VY. Journal of Controlled Release. 2005;101:259. doi: 10.1016/j.jconrel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 128.Cappello J. Trends In Biotechnology. 1990;8:309. doi: 10.1016/0167-7799(90)90207-e. [DOI] [PubMed] [Google Scholar]
- 129.Nagarsekar A, Ghandehari H. Journal Of Drug Targeting. 1999;7:11. doi: 10.3109/10611869909085489. [DOI] [PubMed] [Google Scholar]
- 130.Dreher MR, Raucher D, Balu N, Colvin OM, Ludeman SM, Chilkoti A. Journal of Controlled Release. 2003;91:31. doi: 10.1016/s0168-3659(03)00216-5. [DOI] [PubMed] [Google Scholar]
- 131.Meyer DE, Kong GA, Dewhirst MW, Zalutsky MR, Chilkoti A. Cancer Research. 2001;61:1548. [PubMed] [Google Scholar]
- 132.Feyerabend T, Steeves R, Jager B, Wiedemann GJ, Sommer K, Richter E, Katschinski DM, Robins HI. International Journal of Oncology. 1997;10:591. doi: 10.3892/ijo.10.3.591. [DOI] [PubMed] [Google Scholar]
- 133.Meyer DE, Chilkoti A. Biomacromolecules. 2002;3:357. doi: 10.1021/bm015630n. [DOI] [PubMed] [Google Scholar]
- 134.Dreher MR, Liu WG, Michelich CR, Dewhirst MW, Yuan F, Chilkoti A. Journal of the National Cancer Institute. 2006;98:335. doi: 10.1093/jnci/djj070. [DOI] [PubMed] [Google Scholar]
- 135.Furgeson DY, Dreher MR, Chilkoti A. Journal of Controlled Release. 2006;110:362. doi: 10.1016/j.jconrel.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 136.Simnick AJ, Lim DW, Chow D, Chilkoti A. Polymer Reviews. 2007;47:121. [Google Scholar]
- 137.Liu WE, Dreher MR, Furgeson DY, Peixoto KV, Yuan H, Zalutsky MR, Chilkoti A. Journal of Controlled Release. 2006;116:170. doi: 10.1016/j.jconrel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 138.Liu W, Dreher MR, Chow DC, Zalutsky MR, Chilkoti A. Journal of Controlled Release. 2006;114:184. doi: 10.1016/j.jconrel.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 139.Nagarsekar A, Crissman J, Crissman M, Ferrari F, Cappello J, Ghandehari H. Journal Of Biomedical Materials Research. 2002;62:195. doi: 10.1002/jbm.10272. [DOI] [PubMed] [Google Scholar]
- 140.Massodi I, Bidwell GL, Raucher D. Journal of Controlled Release. 2005;108:396. doi: 10.1016/j.jconrel.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 141.Bidwell GL, Raucher D. Molecular Cancer Therapeutics. 2005;4:1076. doi: 10.1158/1535-7163.MCT-04-0253. [DOI] [PubMed] [Google Scholar]
- 142.Torchilin V. Expert Opinion on Therapeutic Patents. 2005;15:63. [Google Scholar]
- 143.Kataoka K, Harada A, Nagasaki Y. Advanced Drug Delivery Reviews. 2001;47:113. doi: 10.1016/s0169-409x(00)00124-1. [DOI] [PubMed] [Google Scholar]
- 144.Kwon GS. Critical Reviews in Therapeutic Drug Carrier Systems. 2003;20:357. doi: 10.1615/critrevtherdrugcarriersyst.v20.i5.20. [DOI] [PubMed] [Google Scholar]
- 145.Adams ML, Lavasanifar A, Kwon GS. Journal of Pharmaceutical Sciences. 2003;92:1343. doi: 10.1002/jps.10397. [DOI] [PubMed] [Google Scholar]
- 146.Kabanov AV, Batrakova EV, Alakhov VY. Advanced Drug Delivery Reviews. 2002;54:759. doi: 10.1016/s0169-409x(02)00047-9. [DOI] [PubMed] [Google Scholar]
- 147.Kataoka K, Kwon GS, Yokoyama M, Okano T, Sakurai Y. Journal of Controlled Release. 1993;24:119. doi: 10.1016/s0168-3659(99)00133-9. [DOI] [PubMed] [Google Scholar]
- 148.Kim GM, Bae YH, Jo WH. Macromolecular Bioscience. 2005;5:1118. doi: 10.1002/mabi.200500121. [DOI] [PubMed] [Google Scholar]
- 149.Lee ES, Shin HJ, Na K, Bae YH. Journal of Controlled Release. 2003;90:363. doi: 10.1016/s0168-3659(03)00205-0. [DOI] [PubMed] [Google Scholar]
- 150.Lee ES, Na K, Bae YH. Journal of Controlled Release. 2003;91:103. doi: 10.1016/s0168-3659(03)00239-6. [DOI] [PubMed] [Google Scholar]
- 151.Lee ES, Na K, Bae YH. Journal of Controlled Release. 2005;103:405. doi: 10.1016/j.jconrel.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 152.Lee TAT, Cooper A, Apkarian RP, Conticello VP. Advanced Materials. 2000;12:1105. [Google Scholar]
- 153.Wright ER, Conticello VP. Advanced Drug Delivery Reviews. 2002;54:1057. doi: 10.1016/s0169-409x(02)00059-5. [DOI] [PubMed] [Google Scholar]
- 154.Wright ER, Conticello VP, Apkarian RP. Microscopy And Microanalysis. 2003;9:171. doi: 10.1017/S1431927603030150. [DOI] [PubMed] [Google Scholar]
- 155.Checot F, Brulet A, Oberdisse J, Gnanou Y, Mondain-Monval O, Lecommandoux S. Langmuir. 2005;21:4308. doi: 10.1021/la0468500. [DOI] [PubMed] [Google Scholar]
- 156.Torchilin VP. Nature Reviews Drug Discovery. 2005;4:145. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 157.Gabizon A, Catane R, Uziely B, Kaufman B, Safra T, Cohen R, Martin F, Huang A, Barenholz Y. Cancer Research. 1994;54:987. [PubMed] [Google Scholar]
- 158.Bellomo EG, Wyrsta MD, Pakstis L, Pochan DJ, Deming TJ. Nature Materials. 2004;3:244. doi: 10.1038/nmat1093. [DOI] [PubMed] [Google Scholar]
- 159.Checot F, Lecommandoux S, Gnanou Y, Klok HA. Angewandte Chemie-International Edition. 2002;41:1339. doi: 10.1002/1521-3773(20020415)41:8<1339::aid-anie1339>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 160.Checot F, Lecommandoux S, Klok HA, Gnanou Y. European Physical Journal E. 2003;10:25. doi: 10.1140/epje/e2003-00006-1. [DOI] [PubMed] [Google Scholar]
- 161.Nagarsekar A, Crissman J, Crissman M, Ferrari F, Cappello J, Ghandehari H. Biomacromolecules. 2003;4:602. doi: 10.1021/bm0201082. [DOI] [PubMed] [Google Scholar]
- 162.Megeed Z, Cappello J, Ghandehari H. Pharmaceutical Research. 2002;19:954. doi: 10.1023/a:1016406120288. [DOI] [PubMed] [Google Scholar]
- 163.Arend WP, Dayer JM. Arthritis and Rheumatism. 1995;38:151. doi: 10.1002/art.1780380202. [DOI] [PubMed] [Google Scholar]
- 164.Gouze E, Pawliuk R, Gouze JN, Pilapil C, Fleet C, Palmer GD, Evans CH, Leboulch P, Ghivizzani SC. Molecular Therapy. 2003;7:460. doi: 10.1016/s1525-0016(03)00024-8. [DOI] [PubMed] [Google Scholar]
- 165.Betre H, Liu W, Zalutsky MR, Chilkoti A, Kraus VB, Setton LA. Journal of Controlled Release. 2006;115:175. doi: 10.1016/j.jconrel.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 166.Shamji MF, Betre H, Kraus VB, Chen J, Chilkoti A, Pichika R, Masuda K, Setton LA. Arthritis & Rheumatism. 2007;56:3650. doi: 10.1002/art.22952. [DOI] [PubMed] [Google Scholar]
- 167.Jin HJ, Kaplan DL. Nature. 2003;424:1057. doi: 10.1038/nature01809. [DOI] [PubMed] [Google Scholar]
- 168.Megeed Z, Cappello J, Ghandehari H. Advanced Drug Delivery Reviews. 2002;54:1075. doi: 10.1016/s0169-409x(02)00063-7. [DOI] [PubMed] [Google Scholar]
- 169.Hatefi A, Cappello J, Ghandehari H. Pharmaceutical Research. 2007;24:773. doi: 10.1007/s11095-006-9200-5. [DOI] [PubMed] [Google Scholar]
- 170.Boyd LM, Carter AJ. European Spine Journal. 2006;15:S414. doi: 10.1007/s00586-006-0172-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Cappello J, Crissman JW, Crissman M, Ferrari FA, Textor G, Wallis O, Whitledge JR, Zhou X, Burman D, Aukerman L, Stedronsky ER. Journal Of Controlled Release. 1998;53:105. doi: 10.1016/s0168-3659(97)00243-5. [DOI] [PubMed] [Google Scholar]
- 172.Dinerman AA, Cappello J, Ghandehari H, Hoag SW. Biomaterials. 2002;23:4203. doi: 10.1016/s0142-9612(02)00164-3. [DOI] [PubMed] [Google Scholar]
- 173.Megeed Z, Cappello J, Ghandehari H. Biomacromolecules. 2004;5:793. doi: 10.1021/bm0343491. [DOI] [PubMed] [Google Scholar]
- 174.Megeed Z, Haider M, Li DQ, O’Malley BW, Cappello J, Ghandehari H. Journal Of Controlled Release. 2004;94:433. doi: 10.1016/j.jconrel.2003.10.027. [DOI] [PubMed] [Google Scholar]
- 175.Cappello J. Genetically engineered protein polymers. In: Domb AJ, Kost J, Wiseman DM, editors. Handbook of Biodegradable Polymers. Harwood Academic Publishers; Amsterdam: 1997. pp. 387–416. [Google Scholar]
- 176.Xu CY, Breedveld V, Kopecek J. Biomacromolecules. 2005;6:1739. doi: 10.1021/bm050017f. [DOI] [PubMed] [Google Scholar]
- 177.Xu CY, Joss L, Wang C, Pechar M, Kopecek J. Macromolecular Bioscience. 2002;2:395. [Google Scholar]
- 178.Tang A, Wang C, Stewart RJ, Kopecek J. Journal of Controlled Release. 2001;72:57. doi: 10.1016/s0168-3659(01)00262-0. [DOI] [PubMed] [Google Scholar]
- 179.Lupas AA, Van Dyke MM, Stock JJ. Science. 1991;252:1162. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- 180.Van Hest J, Tirrell DA. Chemical Communications. 2001;19:1897. doi: 10.1039/b105185g. [DOI] [PubMed] [Google Scholar]
- 181.Petka WA, Harden JL, McGrath KP, Wirtz D, Tirrell DA. Science. 1998;281:389. doi: 10.1126/science.281.5375.389. [DOI] [PubMed] [Google Scholar]
- 182.Dreher MR, Simnick AJ, Fischer K, Smith RJ, Patel A, Schmidt M, Chilkoti A. J Am Chem Soc. 2007 [Google Scholar]
- 183.Langer R, Tirrell DA. Nature. 2004;428:487. doi: 10.1038/nature02388. [DOI] [PubMed] [Google Scholar]
- 184.Lutolf MP, Hubbell JA. Nature Biotechnology. 2005;23:47. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- 185.Cappello J, Ghandehari H. Advanced Drug Delivery Reviews. 2002;54:1053. doi: 10.1016/s0169-409x(02)00063-7. [DOI] [PubMed] [Google Scholar]
- 186.Foo CWP, Kaplan DL. Advanced Drug Delivery Reviews. 2002;54:1131. doi: 10.1016/s0169-409x(02)00061-3. [DOI] [PubMed] [Google Scholar]
- 187.van Hest JCM, Tirrell DA. Chemical Communications. 2001:1897. doi: 10.1039/b105185g. [DOI] [PubMed] [Google Scholar]
- 188.Karle IL, Urry DW. Biopolymers. 2005;77:198. doi: 10.1002/bip.20214. [DOI] [PubMed] [Google Scholar]
- 189.Urry DW. Trends In Biotechnology. 1999;17:249. doi: 10.1016/s0167-7799(99)01306-2. [DOI] [PubMed] [Google Scholar]
- 190.Urry DW, Pattanaik A, Xu J, Woods TC, McPherson DT, Parker TM. Journal Of Biomaterials Science-Polymer Edition. 1998;9:1015. doi: 10.1163/156856298x00316. [DOI] [PubMed] [Google Scholar]
- 191.Nicol A, Gowda DC, Urry DW. Journal Of Biomedical Materials Research. 1992;26:393. doi: 10.1002/jbm.820260309. [DOI] [PubMed] [Google Scholar]
- 192.Nicol A, Gowda DC, Parker TM, Urry DW. Journal Of Biomedical Materials Research. 1993;27:801. doi: 10.1002/jbm.820270613. [DOI] [PubMed] [Google Scholar]
- 193.Altman GH, Diaz F, Jakuba C, Calabro T, Horan RL, Chen JS, Lu H, Richmond J, Kaplan DL. Biomaterials. 2003;24:401. doi: 10.1016/s0142-9612(02)00353-8. [DOI] [PubMed] [Google Scholar]
- 194.Horan RL, Antle K, Collette AL, Huang YZ, Huang J, Moreau JE, Volloch V, Kaplan DL, Altman GH. Biomaterials. 2005;26:3385. doi: 10.1016/j.biomaterials.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 195.Maskarinec SA, Tirrell DA. Current Opinion In Biotechnology. 2005;16:422. doi: 10.1016/j.copbio.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 196.Langer R. Nature. 2004;428:487. doi: 10.1038/nature02388. [DOI] [PubMed] [Google Scholar]
- 197.Lutolf MMP, Hubbell JJA. Nature biotechnology. 2005;23:47. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- 198.Betre H, Setton LA, Meyer DE, Chilkoti A. Biomacromolecules. 2002;3:910. doi: 10.1021/bm0255037. [DOI] [PubMed] [Google Scholar]
- 199.Betre H, Ong SR, Guilak F, Chilkoti A, Fermor B, Setton LA. Biomaterials. 2006;27:91. doi: 10.1016/j.biomaterials.2005.05.071. [DOI] [PubMed] [Google Scholar]
- 200.Setton LA, Mow VC, Howell DS. Journal Of Orthopaedic Research. 1995;13:473. doi: 10.1002/jor.1100130402. [DOI] [PubMed] [Google Scholar]
- 201.Nagapudi K, Brinkman WT, Leisen J, Thomas BS, Wright ER, Haller C, Wu XY, Apkarian RP, Conticello VP, Chaikof EL. Macromolecules. 2005;38:345. [Google Scholar]
- 202.Nagapudi K, Brinkman WT, Thomas BS, Park JO, Srinivasarao M, Wright E, Conticello VP, Chaikof EL. Biomaterials. 2005;26:4695. doi: 10.1016/j.biomaterials.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 203.Petke S, Kozel BA, Wachi H. Molecular Biology of the Cell. 2000;11:265A. [Google Scholar]
- 204.Tamburro AM. Matrix Biology. 2004;23:413. [Google Scholar]
- 205.Tamburro AM, Bochicchio B, Pepe A. Biochemistry. 2003;42:13347. doi: 10.1021/bi034837t. [DOI] [PubMed] [Google Scholar]
- 206.Tamburro AM, Bochicchio B, Pepe A. Pathologie Biologie. 2005;53:383. doi: 10.1016/j.patbio.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 207.Tamburro AM, Pepe A, Bochicchio B, Quaglino D, Ronchetti IP. Journal Of Biological Chemistry. 2005;280:2682. doi: 10.1074/jbc.M411617200. [DOI] [PubMed] [Google Scholar]
- 208.Miao M, Cirulis JT, Lee S, Keeley FW. Biochemistry. 2005;44:14367. doi: 10.1021/bi0510173. [DOI] [PubMed] [Google Scholar]
- 209.Shen W, Lammertink RGH, Sakata JK, Kornfield JA, Tirrell DA. Macromolecules. 2005;38:3909. [Google Scholar]
- 210.Stevens MM, Allen S, Davies MC, Roberts CJ, Sakata JK, Tendler SJB, Tirrell DA, Williams PM. Biomacromolecules. 2005;6:1266. doi: 10.1021/bm049369x. [DOI] [PubMed] [Google Scholar]
- 211.Shen W, Zhang KC, Kornfield JA, Tirrell DA. Nature Materials. 2006;5:153. doi: 10.1038/nmat1573. [DOI] [PubMed] [Google Scholar]
- 212.Silva GA, Czeisler C, Niece KL, Beniash E, Harrington DA, Kessler JA, Stupp SI. Science. 2004;303:1352. doi: 10.1126/science.1093783. [DOI] [PubMed] [Google Scholar]
- 213.Guler MO, Hsu L, Soukasene S, Harrington DA, Hulvat JF, Stupp SI. Biomacromolecules. 2006;7:1855. doi: 10.1021/bm060161g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Stendahl JC, Rao MS, Guler MO, Stupp SI. Advanced Functional Materials. 2006;16:499. [Google Scholar]
- 215.Beniash E, Hartgerink JD, Storrie H, Stendahl JC, Stupp SI. Acta Biomaterialia. 2005;1:387. doi: 10.1016/j.actbio.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 216.Behanna HA, Donners J, Gordon AC, Stupp SI. Journal Of The American Chemical Society. 2005;127:1193. doi: 10.1021/ja044863u. [DOI] [PubMed] [Google Scholar]
- 217.Niece KL, Hartgerink JD, Donners J, Stupp SI. Journal Of The American Chemical Society. 2003;125:7146. doi: 10.1021/ja028215r. [DOI] [PubMed] [Google Scholar]
- 218.Hartgerink JD, Beniash E, Stupp SI. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:5133. doi: 10.1073/pnas.072699999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Hartgerink JD, Beniash E, Stupp SI. Science. 2001;294:1684. doi: 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]
- 220.Altman M, Lee P, Rich A, Zhang SG. Protein Science. 2000;9:1095. doi: 10.1110/ps.9.6.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Caplan MR, Moore PN, Zhang SG, Kamm RD, Lauffenburger DA. Biomacromolecules. 2000;1:627. doi: 10.1021/bm005586w. [DOI] [PubMed] [Google Scholar]
- 222.Caplan MR, Schwartzfarb EM, Zhang SG, Kamm RD, Lauffenburger DA. Biomaterials. 2002;23:219. doi: 10.1016/s0142-9612(01)00099-0. [DOI] [PubMed] [Google Scholar]
- 223.Holmes TC, de Lacalle S, Su X, Liu GS, Rich A, Zhang SG. Proceedings Of The National Academy Of Sciences Of The United States Of America. 2000;97:6728. doi: 10.1073/pnas.97.12.6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Hong YS, Legge RL, Zhang S, Chen P. Biomacromolecules. 2003;4:1433. doi: 10.1021/bm0341374. [DOI] [PubMed] [Google Scholar]
- 225.Kisiday J, Jin M, Kurz B, Hung H, Semino C, Zhang S, Grodzinsky AJ. Proceedings Of The National Academy Of Sciences Of The United States Of America. 2002;99:9996. doi: 10.1073/pnas.142309999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Narmoneva DA, Oni O, Sieminski AL, Zhang SG, Gertler JP, Kamm RD, Lee RT. Biomaterials. 2005;26:4837. doi: 10.1016/j.biomaterials.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 227.Semino CE, Kasahara J, Hayashi Y, Zhang SG. Tissue Engineering. 2004;10:643. doi: 10.1089/107632704323061997. [DOI] [PubMed] [Google Scholar]
- 228.Vauthey S, Santoso S, Gong HY, Watson N, Zhang SG. Proceedings Of The National Academy Of Sciences Of The United States Of America. 2002;99:5355. doi: 10.1073/pnas.072089599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Yokoi H, Kinoshita T, Zhang SG. Proceedings Of The National Academy Of Sciences Of The United States Of America. 2005;102:8414. doi: 10.1073/pnas.0407843102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Zhang SG. Nature Biotechnology. 2003;21:1171. doi: 10.1038/nbt874. [DOI] [PubMed] [Google Scholar]
- 231.Zhao XJ, Zhang SG. Trends In Biotechnology. 2004;22:470. doi: 10.1016/j.tibtech.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 232.Semino CE, Merok JR, Crane GG, Panagiotakos G, Zhang SG. Differentiation. 2003;71:262. doi: 10.1046/j.1432-0436.2003.7104503.x. [DOI] [PubMed] [Google Scholar]
- 233.Davis ME, Motion JPM, Narmoneva DA, Takahashi T, Hakuno D, Kamm RD, Zhang SG, Lee RT. Circulation. 2005;111:442. doi: 10.1161/01.CIR.0000153847.47301.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Bokhari MA, Akay G, Zhang SG, Birch MA. Biomaterials. 2005;26:5198. doi: 10.1016/j.biomaterials.2005.01.040. [DOI] [PubMed] [Google Scholar]
- 235.Ellis-Behnke RG, Liang YX, You SW, Tay DKC, Zhang SG, So KF, Schneider GE. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:5054. doi: 10.1073/pnas.0600559103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Schneider JP, Pochan DJ, Ozbas B, Rajagopal K, Pakstis L, Kretsinger J. Journal Of The American Chemical Society. 2002;124:15030. doi: 10.1021/ja027993g. [DOI] [PubMed] [Google Scholar]
- 237.Pochan DJ, Schneider JP, Kretsinger J, Ozbas B, Rajagopal K, Haines L. Journal Of The American Chemical Society. 2003;125:11802. doi: 10.1021/ja0353154. [DOI] [PubMed] [Google Scholar]
- 238.Ozbas B, Kretsinger J, Rajagopal K, Schneider JP, Pochan DJ. Macromolecules. 2004;37:7331. [Google Scholar]
- 239.Ozbas B, Rajagopal K, Schneider JP, Pochan DJ. Physical Review Letters. 2004;93:268106. doi: 10.1103/PhysRevLett.93.268106. [DOI] [PubMed] [Google Scholar]
- 240.Haines LA, Rajagopal K, Ozbas B, Salick DA, Pochan DJ, Schneider JP. Journal Of The American Chemical Society. 2005;127:17025. doi: 10.1021/ja054719o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Kretsinger JK, Haines LA, Ozbas B, Pochan DJ, Schneider JP. Biomaterials. 2005;26:5177. doi: 10.1016/j.biomaterials.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 242.Rajagopal K, Ozbas B, Pochan DJ, Schneider JP. European Biophysics Journal With Biophysics Letters. 2006;35:162. doi: 10.1007/s00249-005-0017-7. [DOI] [PubMed] [Google Scholar]
- 243.Veerman C, Rajagopal K, Palla CS, Pochan DJ, Schneider JP, Furst EM. Macromolecules. 2006;39:6608. [Google Scholar]
- 244.Rughani RV, Lamm MS, Pochan DJ, Schneider JP. Biopolymers. 2007;88:629. [Google Scholar]
- 245.Haines-Butterick L, Rajagopal K, Branco M, Salick D, Rughani R, Pilarz M, Lamm MS, Pochan DJ, Schneider JP. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:7791. doi: 10.1073/pnas.0701980104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Lee J, Macosko CW, Urry DW. Macromolecules. 2001;34:5968. [Google Scholar]
- 247.Lee J, Macosko CW, Urry DW. Journal Of Biomaterials Science-Polymer Edition. 2001;12:229. doi: 10.1163/156856201750180942. [DOI] [PubMed] [Google Scholar]
- 248.Lee J, Macosko CW, Urry DW. Macromolecules. 2001;34:4114. [Google Scholar]
- 249.Nagapudi K, Brinkman WT, Leisen JE, Huang L, McMillan RA, Apkarian RP, Conticello VP, Chaikof EL. Macromolecules. 2002;35:1730. [Google Scholar]
- 250.Panitch A, Yamaoka T, Fournier MJ, Mason TL, Tirrell DA. Macromolecules. 1999;32:1701. [Google Scholar]
- 251.Welsh ER, Tirrell DA. Biomacromolecules. 2000;1:23. doi: 10.1021/bm0002914. [DOI] [PubMed] [Google Scholar]
- 252.Di Zio K, Tirrell DA. Macromolecules. 2003;36:1553. [Google Scholar]
- 253.Heilshorn SC, DiZio KA, Welsh ER, Tirrell DA. Biomaterials. 2003;24:4245. doi: 10.1016/s0142-9612(03)00294-1. [DOI] [PubMed] [Google Scholar]
- 254.Liu JC, Heilshorn SC, Tirrell DA. Biomacromolecules. 2004;5:497. doi: 10.1021/bm034340z. [DOI] [PubMed] [Google Scholar]
- 255.Nowatzki PJ, Tirrell DA. Biomaterials. 2004;25:1261. doi: 10.1016/s0142-9612(03)00635-5. [DOI] [PubMed] [Google Scholar]
- 256.Heilshorn SC, Liu JC, Tirrell DA. Biomacromolecules. 2005;6:318. doi: 10.1021/bm049627q. [DOI] [PubMed] [Google Scholar]
- 257.Richman GP, Tirrell DA, Asthagiri AR. Journal Of Controlled Release. 2005;101:3. doi: 10.1016/j.jconrel.2004.07.034. [DOI] [PubMed] [Google Scholar]
- 258.McMillan RA, Caran KL, Apkarian RP, Conticello VP. Macromolecules. 1999;32:9067. [Google Scholar]
- 259.McMillan RA, Conticello VP. Macromolecules. 2000;33:4809. [Google Scholar]
- 260.Lim DW, Nettles DL, Setton LA, Chilkoti A. Biomacromolecules. 2007;8:1463. doi: 10.1021/bm061059m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.McHale MK, Setton LA, Chilkoti A. Tissue Engineering. 2005;11:1768. doi: 10.1089/ten.2005.11.1768. [DOI] [PubMed] [Google Scholar]
- 262.Arias FJ, Reboto V, Martin S, Lopez I, Rodriguez-Cabello JC. Biotechnology Letters. 2006;28:687. doi: 10.1007/s10529-006-9045-3. [DOI] [PubMed] [Google Scholar]
- 263.Girotti A, Reguera J, Rodriguez-Cabello JC, Arias FJ, Alonso M, Testera AM. Journal Of Materials Science-Materials In Medicine. 2004;15:479. doi: 10.1023/b:jmsm.0000021124.58688.7a. [DOI] [PubMed] [Google Scholar]
- 264.Zhang HL, Iwama M, Akaike T, Urry DW, Pattanaik A, Parker TM, Konishi I, Nikaido T. Tissue Engineering. 2006;12:391. doi: 10.1089/ten.2006.12.391. [DOI] [PubMed] [Google Scholar]
- 265.Berning DE, Katti KV, Barnes CL, Volkert WA. Journal Of The American Chemical Society. 1999;121:1658. [Google Scholar]
- 266.Katti KV, Pillarsetty N, Raghuraman K. New vistas in chemistry and applications of primary phosphines. New Aspects In Phosphorus Chemistry III. 2003;229:121–141. [Google Scholar]
- 267.Tramontini M, Angiolini L. Tetrahedron. 1990;46:1791. [Google Scholar]
- 268.Ong SR, Trabbic-Carlson KA, Nettles DL, Lim DW, Chilkoti A, Setton LA. Biomaterials. 2006;27:1930. doi: 10.1016/j.biomaterials.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 269.Nicol A, Gowda C, Urry DW. Journal Of Vascular Surgery. 1991;13:746. doi: 10.1016/0741-5214(91)90373-3. [DOI] [PubMed] [Google Scholar]
- 270.Urry DW. Research & Development. 1988;30:56. [Google Scholar]
- 271.Hubbell JA, Massia SP, Desai NP, Drumheller PD. Bio-Technology. 1991;9:568. doi: 10.1038/nbt0691-568. [DOI] [PubMed] [Google Scholar]
- 272.Massia SP, Hubbell JA. Journal of Biological Chemistry. 1992;267:14019. [PubMed] [Google Scholar]
- 273.Hubbell JA. Current Opinion in Biotechnology. 2003;14:551. doi: 10.1016/j.copbio.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 274.Zisch AH, Zeisberger SM, Ehrbar M, Djonov V, Weber CC, Ziemiecki A, Pasquale EB, Hubbell JA. Biomaterials. 2004;25:3245. doi: 10.1016/j.biomaterials.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 275.Schmoekel HG, Weber FE, Schense JC, Gratz KW, Schawalder P, Hubbell JA. Biotechnology And Bioengineering. 2005;89:253. doi: 10.1002/bit.20168. [DOI] [PubMed] [Google Scholar]
- 276.Ehrbar M, Metters A, Zammaretti P, Hubbell JA, Zisch AH. Journal Of Controlled Release. 2005;101:93. doi: 10.1016/j.jconrel.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 277.Pittier R, Sauthier F, Hubbell JA, Hall H. Journal Of Neurobiology. 2005;63:1. doi: 10.1002/neu.20116. [DOI] [PubMed] [Google Scholar]
- 278.Urech L, Bittermann AG, Hubbell JA, Hall H. Biomaterials. 2005;26:1369. doi: 10.1016/j.biomaterials.2004.04.045. [DOI] [PubMed] [Google Scholar]
- 279.Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, Hubbell JA. Proceedings Of The National Academy Of Sciences Of The United States Of America. 2003;100:5413. doi: 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Lutolf MP, Raeber GP, Zisch AH, Tirelli N, Hubbell JA. Advanced Materials. 2003;15:888. [Google Scholar]
- 281.Zisch AH, Lutolf MP, Ehrbar M, Raeber GP, Rizzi SC, Davies N, Schmokel H, Bezuidenhout D, Djonov V, Zilla P, Hubbell JA. Faseb Journal. 2003;17:2260. doi: 10.1096/fj.02-1041fje. [DOI] [PubMed] [Google Scholar]
- 282.Lutolf MR, Weber FE, Schmoekel HG, Schense JC, Kohler T, Muller R, Hubbell JA. Nature Biotechnology. 2003;21:513. doi: 10.1038/nbt818. [DOI] [PubMed] [Google Scholar]
- 283.Seliktar D, Zisch AH, Lutolf MP, Wrana JL, Hubbell JA. Journal of Biomedical Materials Research Part A. 2004;68A:704. doi: 10.1002/jbm.a.20091. [DOI] [PubMed] [Google Scholar]
- 284.Rizzi SC, Hubbell JA. Biomacromolecules. 2005;6:1226. doi: 10.1021/bm049614c. [DOI] [PubMed] [Google Scholar]
- 285.Rizzi SC, Ehrbar M, Halstenberg S, Raeber GP, Schmoekel HG, Hagenmuller H, Muller R, Weber FE, Hubbell JA. Biomacromolecules. 2006;7:3019. doi: 10.1021/bm060504a. [DOI] [PubMed] [Google Scholar]
- 286.Iatridis JC, Setton LA, Weidenbaum M, Mow VC. Journal Of Orthopaedic Research. 1997;15:318. doi: 10.1002/jor.1100150224. [DOI] [PubMed] [Google Scholar]
- 287.Castner DG, Ratner BD. Surface Science. 2002;500:28. [Google Scholar]
- 288.Tirrell M, Kokkoli E, Biesalski M. Surface Science. 2002;500:61. [Google Scholar]
- 289.Chu PK, Chen JY, Wang LP, Huang N. Materials Science & Engineering R-Reports. 2002;36:143. [Google Scholar]
- 290.Klee D, Hocker H. Polymers for biomedical applications: Improvement of the interface compatibility. Biomedical Applications: Polymer Blends. 1999;149:1–57. [Google Scholar]
- 291.Elbert DL, Hubbell JA. Annual Review Of Materials Science. 1996;26:365. [Google Scholar]
- 292.Ikada Y. Interfacial Biocompatibility. Polymers Of Biological And Biomedical Significance. 1994;540:35–48. [Google Scholar]
- 293.Ducheyne P, Qiu Q. Biomaterials. 1999;20:2287. doi: 10.1016/s0142-9612(99)00181-7. [DOI] [PubMed] [Google Scholar]
- 294.Ohgushi H, Caplan AI. Journal Of Biomedical Materials Research. 1999;48:913. doi: 10.1002/(sici)1097-4636(1999)48:6<913::aid-jbm22>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 295.Brown IG. Annual Review Of Materials Science. 1998;28:243. [Google Scholar]
- 296.Gray JE, Luan B. Journal Of Alloys And Compounds. 2002;336:88. [Google Scholar]
- 297.Wittstock G. Fresenius Journal Of Analytical Chemistry. 2001;370:303. doi: 10.1007/s002160100795. [DOI] [PubMed] [Google Scholar]
- 298.Freund HJ, Kuhlenbeck H, Staemmler V. Reports On Progress In Physics. 1996;59:283. [Google Scholar]
- 299.Uyama Y, Kato K, Ikada Y. Surface modification of polymers by grafting. Grafting/Characterization Techniques/Kinetic Modeling. 1998;137:1–39. [Google Scholar]
- 300.Chan CM, Ko TM, Hiraoka H. Surface Science Reports. 1996;24:3. [Google Scholar]
- 301.Liston EM, Martinu L, Wertheimer MR. Journal Of Adhesion Science And Technology. 1993;7:1091. [Google Scholar]
- 302.Nath N, Hyun J, Ma H, Chilkoti A. Surface Science. 2004;570:98. [Google Scholar]
- 303.Ma HW, Hyun JH, Stiller P, Chilkoti A. Advanced Materials. 2004;16:338. [Google Scholar]
- 304.Ishihara K, Ishikawa E, Iwasaki Y, Nakabayashi N. Journal Of Biomaterials Science-Polymer Edition. 1999;10:1047. doi: 10.1163/156856299x00676. [DOI] [PubMed] [Google Scholar]
- 305.Zhang MQ, Desai T, Ferrari M. Biomaterials. 1998;19:953. doi: 10.1016/s0142-9612(98)00026-x. [DOI] [PubMed] [Google Scholar]
- 306.Kyrolainen M, Rigsby P, Eddy S, Vadgama P. Acta Anaesthesiologica Scandinavica. 1995;39:55. doi: 10.1111/j.1399-6576.1995.tb04255.x. [DOI] [PubMed] [Google Scholar]
- 307.Drummond TG, Hill MG, Barton JK. Nature Biotechnology. 2003;21:1192. doi: 10.1038/nbt873. [DOI] [PubMed] [Google Scholar]
- 308.Bustin SA. Journal Of Molecular Endocrinology. 2000;25:169. doi: 10.1677/jme.0.0250169. [DOI] [PubMed] [Google Scholar]
- 309.Nikitin PI, Grigorenko AN, Beloglazov AA, Valeiko MV, Savchuk AI, Savchuk OA, Steiner G, Kuhne C, Huebner A, Salzer R. Sensors And Actuators A-Physical. 2000;85:189. [Google Scholar]
- 310.Johansen K, Arwin H, Lundstrom I, Liedberg B. Review Of Scientific Instruments. 2000;71:3530. [Google Scholar]
- 311.Patton WF. Electrophoresis. 2000;21:1123. doi: 10.1002/(SICI)1522-2683(20000401)21:6<1123::AID-ELPS1123>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 312.Lyon LA, Musick MD, Natan MJ. Analytical Chemistry. 1998;70:5177. doi: 10.1021/ac9809940. [DOI] [PubMed] [Google Scholar]
- 313.Figeys D, Aebersold R. Electrophoresis. 1998;19:885. doi: 10.1002/elps.1150190603. [DOI] [PubMed] [Google Scholar]
- 314.Jungblut P, Thiede B. Mass Spectrometry Reviews. 1997;16:145. doi: 10.1002/(SICI)1098-2787(1997)16:3<145::AID-MAS2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 315.Hayashi K, Yandell DW. Human Mutation. 1993;2:338. doi: 10.1002/humu.1380020503. [DOI] [PubMed] [Google Scholar]
- 316.Hoffman AS, Stayton PS, Bulmus V, Chen GH, Chen JP, Cheung C, Chilkoti A, Ding ZL, Dong LC, Fong R, Lackey CA, Long CJ, Miura M, Morris JE, Murthy N, Nabeshima Y, Park TG, Press OW, Shimoboji T, Shoemaker S, Yang HJ, Monji N, Nowinski RC, Cole CA, Priest JH, Harris JM, Nakamae K, Nishino T, Miyata T. Journal Of Biomedical Materials Research. 2000;52:577. doi: 10.1002/1097-4636(20001215)52:4<577::aid-jbm1>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 317.Hoffman AS. Clinical Chemistry. 2000;46:1478. [PubMed] [Google Scholar]
- 318.Stayton PS, Shimoboji T, Long C, Chilkoti A, Chen GH, Harris JM, Hoffman AS. Nature. 1995;378:472. doi: 10.1038/378472a0. [DOI] [PubMed] [Google Scholar]
- 319.Veiseh M, Zareie MH, Zhang MQ. Langmuir. 2002;18:6671. [Google Scholar]
- 320.Yu JH, Ju HX. Analytical Chemistry. 2002;74:3579. doi: 10.1021/ac011290k. [DOI] [PubMed] [Google Scholar]
- 321.Wadu-Mesthrige K, Amro NA, Liu GY. Scanning. 2000;22:380. doi: 10.1002/sca.4950220607. [DOI] [PubMed] [Google Scholar]
- 322.Li J, Chia LS, Goh NK, Tan SN. Analytica Chimica Acta. 1998;362:203. [Google Scholar]
- 323.Blohm DH, Guiseppi-Elie A. Current Opinion In Biotechnology. 2001;12:41. doi: 10.1016/s0958-1669(00)00175-0. [DOI] [PubMed] [Google Scholar]
- 324.Asanov AN, Wilson WW, Odham PB. Analytical Chemistry. 1998;70:1156. doi: 10.1021/ac970805y. [DOI] [PubMed] [Google Scholar]
- 325.Nieba L, NiebaAxmann SE, Persson A, Hamalainen M, Edebratt F, Hansson A, Lidholm J, Magnusson K, Karlsson AF, Pluckthun A. Analytical Biochemistry. 1997;252:217. doi: 10.1006/abio.1997.2326. [DOI] [PubMed] [Google Scholar]
- 326.Pollema CH, Ruzicka J. Analytical Chemistry. 1994;66:1825. doi: 10.1021/ac00083a008. [DOI] [PubMed] [Google Scholar]
- 327.Tolson J, Bogumil R, Brunst E, Beck H, Elsner R, Humeny A, Kratzin H, Deeg M, Kuczyk M, Mueller GA, Mueller CA, Flad T. Laboratory Investigation. 2004;84:845. doi: 10.1038/labinvest.3700097. [DOI] [PubMed] [Google Scholar]
- 328.Li JN, Zhang Z, Rosenzweig J, Wang YY, Chan DW. Clinical Chemistry. 2002;48:1296. [PubMed] [Google Scholar]
- 329.Issaq HJ, Veenstra TD, Conrads TP, Felschow D. Biochemical And Biophysical Research Communications. 2002;292:587. doi: 10.1006/bbrc.2002.6678. [DOI] [PubMed] [Google Scholar]
- 330.Wright GL, Cazares LH, Leung SM, Nasim S, Adam BL, Yip TT, Schellhammer PF, Gong L, Vlahou A. Prostate Cancer And Prostatic Diseases. 1999;2:264. doi: 10.1038/sj.pcan.4500384. [DOI] [PubMed] [Google Scholar]
- 331.Lynch M, Mosher C, Huff J, Nettikadan S, Johnson J, Henderson E. Proteomics. 2004;4:1695. doi: 10.1002/pmic.200300701. [DOI] [PubMed] [Google Scholar]
- 332.Bruckbauer A, Zhou DJ, Kang DJ, Korchev YE, Abell C, Klenerman D. Journal Of The American Chemical Society. 2004;126:6508. doi: 10.1021/ja0317426. [DOI] [PubMed] [Google Scholar]
- 333.Demers LM, Ginger DS, Park SJ, Li Z, Chung SW, Mirkin CA. Science. 2002;296:1836. doi: 10.1126/science.1071480. [DOI] [PubMed] [Google Scholar]
- 334.Gao D, McBean N, Schultz JS, Yan YS, Mulchandani A, Chen WF. Journal Of The American Chemical Society. 2006;128:676. doi: 10.1021/ja056364e. [DOI] [PubMed] [Google Scholar]
- 335.Zhang KC, Diehl MR, Tirrell DA. Journal Of The American Chemical Society. 2005;127:10136. doi: 10.1021/ja051457h. [DOI] [PubMed] [Google Scholar]
- 336.Frey W, Meyer DE, Chilkoti A. Advanced Materials. 2003;15:248. [Google Scholar]
- 337.Frey W, Meyer DE, Chilkoti A. Langmuir. 2003;19:1641. [Google Scholar]
- 338.Wu RZ, Bailey SN, Sabatini DM. Trends In Cell Biology. 2002;12:485. doi: 10.1016/s0962-8924(02)02354-1. [DOI] [PubMed] [Google Scholar]
- 339.Wolcke J, Ullmann D. Drug Discovery Today. 2001;6:637. doi: 10.1016/s1359-6446(01)01807-4. [DOI] [PubMed] [Google Scholar]
- 340.Jakeway SC, de Mello AJ, Russell EL. Fresenius Journal Of Analytical Chemistry. 2000;366:525. doi: 10.1007/s002160051548. [DOI] [PubMed] [Google Scholar]
- 341.Silverman L, Campbell R, Broach JR. Current Opinion In Chemical Biology. 1998;2:397. doi: 10.1016/s1367-5931(98)80015-x. [DOI] [PubMed] [Google Scholar]
- 342.Jones VW, Kenseth JR, Porter MD, Mosher CL, Henderson E. Analytical Chemistry. 1998;70:1233. doi: 10.1021/ac971125y. [DOI] [PubMed] [Google Scholar]
- 343.Yan H, Park SH, Finkelstein G, Reif JH, LaBean TH. Science. 2003;301:1882. doi: 10.1126/science.1089389. [DOI] [PubMed] [Google Scholar]
- 344.Hyun J, Ahn SJ, Lee WK, Chilkoti A, Zauscher S. Nano Letters. 2002;2:1203. [Google Scholar]
- 345.Mirkin CA, Letsinger RL, Mucic RC, Storhoff JJ. Nature. 1996;382:607. doi: 10.1038/382607a0. [DOI] [PubMed] [Google Scholar]
- 346.Niemeyer CM, Sano T, Smith CL, Cantor CR. Nucleic Acids Research. 1994;22:5530. doi: 10.1093/nar/22.25.5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Hyun J, Lee WK, Nath N, Chilkoti A, Zauscher S. Journal of the American Chemical Society. 2004;126:7330. doi: 10.1021/ja049721e. Amer Chemical Soc. [DOI] [PubMed] [Google Scholar]
- 348.Hill RT, Shear JB. Analytical Chemistry. 2006;78:7022. doi: 10.1021/ac061102w. [DOI] [PubMed] [Google Scholar]
- 349.Dillow AK, Ochsenhirt SE, McCarthy JB, Fields GB, Tirrell M. Biomaterials. 2001;22:1493. doi: 10.1016/s0142-9612(00)00304-5. [DOI] [PubMed] [Google Scholar]
- 350.Shu XZ, Ghosh K, Liu YC, Palumbo FS, Luo Y, Clark RA, Prestwich GD. Journal Of Biomedical Materials Research Part A. 2004;68A:365. doi: 10.1002/jbm.a.20002. [DOI] [PubMed] [Google Scholar]
- 351.Massia SP, Stark J. Journal Of Biomedical Materials Research. 2001;56:390. doi: 10.1002/1097-4636(20010905)56:3<390::aid-jbm1108>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 352.Myles JL, Burgess BT, Dickinson RB. Journal Of Biomaterials Science-Polymer Edition. 2000;11:69. doi: 10.1163/156856200743508. [DOI] [PubMed] [Google Scholar]
- 353.VandeVondele S, Voros J, Hubbell JA. Biotechnology And Bioengineering. 2003;82:784. doi: 10.1002/bit.10625. [DOI] [PubMed] [Google Scholar]
- 354.Cook AD, Hrkach JS, Gao NN, Johnson IM, Pajvani UB, Cannizzaro SM, Langer R. Journal Of Biomedical Materials Research. 1997;35:513. doi: 10.1002/(sici)1097-4636(19970615)35:4<513::aid-jbm11>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 355.Lieb E, Hacker M, Tessmar J, Kunz-Schughart LA, Fiedler J, Dahmen C, Hersel U, Kessler H, Schulz MB, Gopferich A. Biomaterials. 2005;26:2333. doi: 10.1016/j.biomaterials.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 356.Fussell GW, Cooper SL. Biomaterials. 2004;25:2971. doi: 10.1016/j.biomaterials.2003.09.062. [DOI] [PubMed] [Google Scholar]
- 357.Lin HB, Garciaecheverria C, Asakura S, Sun W, Mosher DF, Cooper SL. Biomaterials. 1992;13:905. doi: 10.1016/0142-9612(92)90113-3. [DOI] [PubMed] [Google Scholar]
- 358.Zhang ZP, Yoo R, Wells M, Beebe TP, Biran R, Tresco P. Biomaterials. 2005;26:47. doi: 10.1016/j.biomaterials.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 359.Rezania A, Healy KE. Biotechnology Progress. 1999;15:19. doi: 10.1021/bp980083b. [DOI] [PubMed] [Google Scholar]
- 360.Rezania A, Healy KE. Journal Of Biomedical Materials Research. 2000;52:595. doi: 10.1002/1097-4636(20001215)52:4<595::aid-jbm3>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 361.Chung TW, Liu DZ, Wang SY, Wang SS. Biomaterials. 2003;24:4655. doi: 10.1016/s0142-9612(03)00361-2. [DOI] [PubMed] [Google Scholar]
- 362.Collier JH, Messersmith PB. Bioconjugate Chemistry. 2003;14:748. doi: 10.1021/bc034017t. [DOI] [PubMed] [Google Scholar]
- 363.Ito A, Mase A, Takizawa Y, Shinkai M, Honda H, Hata K, Ueda M, Kobayashi T. Journal Of Bioscience And Bioengineering. 2003;95:196. [PubMed] [Google Scholar]
- 364.Lee WC, Lee KH. Analytical Biochemistry. 2004;324:1. doi: 10.1016/j.ab.2003.08.031. [DOI] [PubMed] [Google Scholar]
- 365.Terpe K. Applied Microbiology And Biotechnology. 2003;60:523. doi: 10.1007/s00253-002-1158-6. [DOI] [PubMed] [Google Scholar]
- 366.Hearn MTW, Acosta D. Journal Of Molecular Recognition. 2001;14:323. doi: 10.1002/jmr.555. [DOI] [PubMed] [Google Scholar]
- 367.Gaberc-Porekar V, Menart V. Journal Of Biochemical And Biophysical Methods. 2001;49:335. doi: 10.1016/s0165-022x(01)00207-x. [DOI] [PubMed] [Google Scholar]
- 368.Arnold FH. Bio-Technology. 1991;9:151. [Google Scholar]
- 369.Sanghvi AB, Miller KPH, Belcher AM, Schmidt CE. Nature Materials. 2005;4:496. doi: 10.1038/nmat1397. [DOI] [PubMed] [Google Scholar]
- 370.Alsberg E, Anderson KW, Albeiruti A, Rowley JA, Mooney DJ. Proceedings Of The National Academy Of Sciences Of The United States Of America. 2002;99:12025. doi: 10.1073/pnas.192291499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Barber TA, Harbers GM, Park S, Gilbert M, Healy KE. Biomaterials. 2005;26:6897. doi: 10.1016/j.biomaterials.2005.04.043. [DOI] [PubMed] [Google Scholar]
- 372.Gilbert M, Giachelli CM, Stayton PS. Journal Of Biomedical Materials Research Part A. 2003;67A:69. doi: 10.1002/jbm.a.10053. [DOI] [PubMed] [Google Scholar]