

Abstract
Background
The brain is increasingly being recognized as a sanctuary site for metastatic tumor cells in women with HER2-overexpressing breast cancer who receive trastuzumab therapy. There are no approved or widely accepted treatments for brain metastases other than steroids, cranial radiotherapy, and surgical resection. We examined the efficacy of lapatinib, an inhibitor of the epidermal growth factor receptor (EGFR) and HER2 kinases, for preventing the outgrowth of breast cancer cells in the brain in a mouse xenograft model of brain metastasis.
Methods
EGFR-overexpressing MDA-MB-231-BR (231-BR) brain-seeking breast cancer cells were transfected with an expression vector that contained or lacked the HER2 cDNA and used to examine the effect of lapatinib on the activation (ie, phosphorylation) of cell signaling proteins by immunoblotting, on cell growth by the tetrazolium salt 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide assay, and on cell migration using a Boyden chamber assay. The outgrowth of large (ie, >50 μm2) and micrometastases was counted in brain sections from nude mice that had been injected into the left cardiac ventricle with 231-BR cells and, beginning 5 days later, treated by oral gavage with lapatinib or vehicle (n = 22–26 mice per treatment group). All statistical tests were two-sided.
Results
In vitro, lapatinib inhibited the phosphorylation of EGFR, HER2, and downstream signaling proteins; cell proliferation; and migration in 231-BR cells (both with and without HER2). Among mice injected with 231-BR-vector cells, those treated with 100 mg lapatinib/kg body weight had 54% fewer large metastases 24 days after starting treatment than those treated with vehicle (mean number of large metastases per brain section: 1.56 vs 3.36, difference = 1.80, 95% confidence interval [CI] = 0.92 to 2.68, P < .001), whereas treatment with 30 mg lapatinib/kg body weight had no effect. Among mice injected with 231-BR-HER2 cells, those treated with either dose of lapatinib had 50%–53% fewer large metastases than those treated with vehicle (mean number of large metastases per brain section, 30 mg/kg vs vehicle: 3.21 vs 6.83, difference = 3.62, 95% CI = 2.30 to 4.94, P < .001; 100 mg/kg vs vehicle: 3.44 vs 6.83, difference = 3.39, 95% CI = 2.08 to 4.70, P < .001). Immunohistochemical analysis revealed reduced phosphorylation of HER2 in 231-BR-HER2 cell–derived brain metastases from mice treated with the higher dose of lapatinib compared with 231-BR-HER2 cell–derived brain metastases from vehicle-treated mice (P < .001).
Conclusions
Lapatinib is the first HER2-directed drug to be validated in a preclinical model for activity against brain metastases of breast cancer.
CONTEXT AND CAVEATS
Prior knowledge
The brain is increasingly being recognized as a sanctuary site for metastatic tumor cells in women with HER2-overexpressing breast cancer who receive trastuzumab therapy. Treatment options for brain metastases are currently limited to steroids, cranial radiotherapy, and surgical resection.
Study design
A preclinical mouse model was used to test the efficacy of lapatinib, an inhibitor of the epidermal growth factor receptor (EGFR) and HER2 kinases, for preventing the outgrowth of breast cancer cells in the brain. EGFR-overexpressing brain-seeking breast cancer cells with or without HER2 overexpression were used to examine the effect of lapatinib on the phosphorylation of cell signaling proteins, on cell growth, and on cell migration.
Contribution
In vitro, lapatinib inhibited the phosphorylation of EGFR, HER2, and downstream signaling proteins; cell proliferation; and migration in brain-seeking breast cancer cells (both with and without HER2 overexpression). In vivo, lapatinib inhibited the formation of large brain metastases by HER2-overexpressing brain-seeking breast cancer cells by 50%–53% and reduced phosphorylation of HER2 in the brain metastases.
Implications
Lapatinib is the first HER2-directed drug to be validated in a preclinical prevention model for activity against brain metastases of breast cancer.
Limitations
Lapatinib did not completely inhibit the formation of large brain metastases, which suggests that some breast cancer cells may be resistant to this drug.
From the Editors
Historically, symptomatic brain metastases develop in 10%–20% of patients with metastatic breast cancer, most often following disease progression at other sites. Patients whose breast cancer has metastasized to the brain have an estimated 1-year survival rate of 20% and considerable associated morbidity. Treatment options for patients with brain metastases include combinations of cranial stereotactic radiosurgery, neurosurgery, whole-brain radiotherapy, and steroids [reviewed in (1,2)]. Recently, prophylactic whole-brain radiation was tested in a randomized trial of small-cell lung cancer patients with brain metastases and found to increase disease-free and overall survival by only 2.7 weeks and 1.3 months, respectively (3). Other studies have reported adverse effects of whole-brain irradiation on quality of life, particularly with respect to cognitive function (4–6). Other treatment options for cancer patients who have, or are at risk of developing, brain metastases are needed.
Specific subpopulations of breast cancer patients appear to be at an increased risk of developing brain metastases. Retrospective studies of breast cancer patients with brain metastases found that younger age at diagnosis; primary tumors that are hormone receptor negative, overexpress HER2, and/or are epidermal growth factor receptor (EGFR) positive; and the presence of lymph node or distant metastases were associated with the development of brain metastatic disease (7–15). In particular, patients with HER2-overexpressing tumors have been the subject of several investigations that have documented an increased incidence of brain metastases. For example, Bendell et al. (16) reported that 34% of patients with HER2-overexpressing breast tumors developed brain metastases within 16 months after being diagnosed with metastatic disease and within 6 months after the initiation of trastuzumab treatment; at the time of central nervous system (CNS) relapse, 50% of patients were responding to treatment or had stable disease throughout the rest of the body (non-CNS sites). Similar results have been reported by other groups (13,17–20). In addition, we previously reported that among resected brain metastases from 123 breast cancer patients, 36% overexpressed HER2, indicating an enrichment in the frequency of HER2 overexpression in breast cancer cells in the brain compared with those in primary tumors (21).
Previously, we tested the hypothesis that HER2 overexpression could functionally affect the colonization of tumor cells in the brain. Overexpression of HER2 in a brain-seeking derivative of the MDA-MB-231 breast carcinoma cell line (231-BR cells) increased the number of large metastases (>50 μm2) that formed by 2.5- to 3-fold (21), identifying one reason for the preponderance of brain metastases in this subpopulation of breast cancer patients.
HER2 is also a prominent target for the development of therapeutic compounds for the treatment of breast cancer. Trastuzumab, a recombinant humanized monoclonal antibody against the extracellular domain of HER2, has been approved in combination with cytotoxic chemotherapy for the treatment of HER2-overexpressing breast cancer in the metastatic and adjuvant settings (22–24). However, trastuzumab levels in cerebrospinal fluid are 300-fold lower than those in plasma (25,26), indicating that trastuzumab cannot cross the blood–brain barrier. This inability of trastuzumab to cross the blood–brain barrier may also contribute to the increased incidence of brain metastases in patients with HER2-overexpressing breast cancer. Thus, new drugs are needed that can disrupt the HER2 signaling pathway in breast cancer patients who are at risk for the development of brain metastatic disease.
One such drug is lapatinib, a small-molecule competitive tyrosine kinase inhibitor that binds reversibly to the cytoplasmic ATP-binding site in the kinase domains of EGFR and HER2, thereby inhibiting receptor phosphorylation (ie, activation) (27). In a randomized phase 3 study, lapatinib added to capecitabine improved the response rate and statistically significantly (P = .002) prolonged the time to disease progression compared with capecitabine alone in patients with metastatic HER2-overexpressing breast cancer whose disease had progressed following trastuzumab-based therapy (28). However, lapatinib has not yet demonstrated clinical efficacy against breast tumors that express EGFR (29). Modest single-agent lapatinib activity has been observed in patients with recurrent brain metastases from HER2-overexpressing breast cancer following trastuzumab and cranial radiotherapy, with 2.6%–6% partial responses and stable disease for a period of 16 weeks in 13%–14.7% of patients [(30); N.U. Lin, S.D. Rubin, V. Die’ras, D. Paul, D. Lossignol, C. Christodoulou, et al., unpublished observations]. In this study, we used a preclinical mouse model to test the hypothesis that lapatinib can prevent the metastatic colonization of brain by a brain-seeking derivative of the MDA-MB-231 human breast carcinoma cell line (ie, 231-BR cells). We also report results of in vitro and in vivo analyses of the effects of lapatinib on brain-seeking breast cancer cells that express EGFR or both HER2 and EGFR.
Materials and Methods
Drugs and Cell Lines
Lapatinib was provided by GlaxoSmithKline (Philadelphia, PA) and trastuzumab was provided by Genentech (San Francisco, CA) through a Materials Transfer Agreement.
The human MDA-MB-231-BR “brain-seeking” breast cancer cell line (hereafter referred to as 231-BR cells; kindly provided by Dr Toshyaki Yoneda, University of Texas at San Antonio, San Antonio, TX) was previously described (31). The 231-BR cells were transduced to express enhanced green fluorescent protein (EGFP) and transfected to overexpress HER2 as described in Palmieri et al. (21). Briefly, the retroviral vector pLEGFP-C1 (BD Biosciences, San Jose, CA) was transfected into the murine fibroblast PT67 packaging cell line. After 24 hours, EGFP-expressing cells were selected in the presence of 1 mg/mL G418 (Invitrogen, Carlsbad, CA) and colonies were expanded. EGFP virus was harvested from the PT67 cells and used to infect 231-BR cells. The following day, 231-BR cells were selected in the presence of 0.8 mg/mL G418. EGFP-expressing cells were then co-transfected with pCMV4.HER2 full-length human cDNA (kindly provided by Dr Dihua Yu, M. D. Anderson Cancer Center, Houston, TX) and pSVzeo to confer antibiotic resistance. The sequence of the HER2 insert in pCMV4.HER2 was confirmed by sequencing. Stable colonies were selected in the presence of 0.750 mg/mL zeocin. A vector control cell line was simultaneously established by transfecting both pCMV4 that lacked inserted cDNA and pSVzeo into the 231-BR-EGFP cells and selecting stable colonies in the presence of 0.750 mg/mL zeocin. The 231-BR cells that were transfected with vectors that contained or lacked the HER2 cDNA were maintained in Dulbecco's modified Eagle Medium (DMEM, Invitrogen) supplemented with 10% fetal bovine serum (FBS, Invitrogen) and 1% penicillin–streptomycin solution (Invitrogen). The human breast cancer SKBr3 cell line was purchased from the American Type Culture Collection (Manassas, VA) and maintained in DMEM with 10% FBS.
Anchorage-Independent Cell Proliferation
231-BR-HER2 and SKBr3 cells (10 000 per well) were plated in 1 mL of culture medium containing 0.3% (wt/vol) top agar in 24-well plates as described previously (21). After 14 days in culture, colonies (>50 cells) were counted. Results are representative of three independent experiments, each performed in triplicate.
EGFR Gene Silencing by Transfection With Small Interfering RNA
231-BR-vector and 231-BR-HER2 cells (3 × 105) were seeded in 10-cm plates and incubated overnight. The cells were transiently transfected with a small interfering RNA (siRNA) targeted against the EGFR gene or a control siRNA containing a sequence not homologous to any human, mouse, or rat gene (siGENOME SMARTpool Human ERBB1 and siCONTROL Non-Targeting siRNA Pool, respectively; Dharmacon, Chicago, IL). siRNAs were transfected into cells using HiPerFect reagent (Qiagen, Valencia, CA) according to the manufacturer's instructions. Twenty-four hours after transfection, the cells were seeded into 6-well plates (for immunoblot analysis) or 96-well plates (for cell proliferation assays).
Immunoblot Analysis of HER Family Members and Downstream Signaling Proteins
231-BR-vector, 231-BR-HER2, and SKBr3 cells (3 × 106) were seeded in 10-cm plates and serum starved overnight. The cells were then treated with 0.5 or 1.0 μM lapatinib (in dimethyl sulfoxide [DMSO]) or an equal volume of diluent (DMSO) for 24 hours, followed by stimulation with with 100 ng/mL epidermal growth factor (EGF; Peprotech Inc., Rocky Hill, NJ) for 10 minutes before lysing. The cells were lysed in RIPA buffer (20 mM Tris–HCl [pH 8], 137 mM NaCl, 10% glycerol, 1% Nonidet P-40, 2 mM EDTA, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate [SDS]) containing Complete Mini EDTA-free Protease Inhibitor Cocktail (Roche Molecular Biochemicals, Bassel, Switzerland) and Phosphatase Inhibitor Cocktails 1 and 2 (Sigma, St Louis, MO). Total lysates (50 μg per lane) were resolved by SDS–polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. Immunoblot analysis was performed using the following rabbit polyclonal antibodies (Cell Signaling Technology, Danvers, MA), all at a 1:1000 dilution: anti-HER2, anti-phospho HER2 (Tyr877), anti-phospho HER2 (Tyr1221/1222), anti-EGFR, anti-phospho EGFR (Tyr845, 992, 1045, and 1068), anti-p42/44 mitogen activated pathway kinase (MAPK), anti-phospho p42/44 MAPK (Thr202/Tyr204), anti-AKT, anti-phospho AKT (Ser473), anti-p38 MAPK, anti-PLCγ1, and anti-phospho PLCγ1 (Tyr771). In addition, the following rabbit monoclonal antibodies (Cell Signaling Technology) were used at a 1:1000 dilution: anti-HER3, anti-phospho HER3 (Tyr1289), and anti-phospho p38 MAPK (Thr180/Tyr182). Mouse monoclonal antibodies against p21 and tubulin were purchased from Calbiochem (Gibbstown, NJ) and used at a 1:1000 and 1:2000 dilution, respectively.
Horseradish peroxidase–conjugated secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) were used at dilutions of 1:5000. Antibody binding was detected using enhanced chemiluminescence (Cell Signaling Technology) and autoradiography.
Cell Proliferation Assay
231-BR-vector and 231-BR-HER2 cells were plated at a density of 5 × 103 cells per well in 96-well plates in DMEM plus 10% FBS and incubated overnight to allow cells to adhere to the substratum. The cells were treated with various concentrations (3–10 μM) of lapatinib or with DMSO (ie, the diluent for lapatinib) as a control. We determined the number of viable cells at 72, 96, and 120 hours after lapatinib addition by adding 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT; Sigma) at a final concentration of 0.5 mg/mL to each well. After a 2-hour incubation at 37°C, DMSO was added to the wells to dissolve the cells and solubilize the MTT, and absorbance was measured at 570 nm. Data are shown as a percentage of the vehicle-treated control cells at each time point tested. Three separate experiments were performed, with six replicate wells for each data point.
Cell Migration Assay
Cell migration was examined with the use of 48-well Boyden chemotaxis chambers, as previously described (21). Briefly, the top and bottom compartments of the chambers were separated by polycarbonate (polyvinylprrolidone-free) nucleopore filters (8-μm pore size, Neuro Probe, Gaithersburg, MD) coated with 0.01% collagen (BD Biosciences). FBS (1%) in DMEM was used as the chemoattractant in the bottom chamber. 231-BR-vector and 231-BR-HER2 cells were pretreated for 24 hours with lapatinib (1 or 3 μM) or diluent (DMSO). The pretreated cells (3 × 105 cells/mL) were added to the top chamber in DMEM supplemented with 1 or 3 μM lapatinib or diluent (DMSO). The chambers were incubated for 4 hours in a 37°C incubator with 5% CO2. The chambers were disassembled and the filters were fixed and stained with the use of a Diff-Quik kit (Fischer Scientific, Pittsburgh, PA). Cells that had migrated to the undersurface of the membrane were counted with the use of a light microscope. Three separate experiments were performed with four replicate wells for each data point in experiment 1 and three replicate wells in experiments 2 and 3.
Mice and Imaging
Animal experiments were conducted under a National Cancer Institute–approved Animal Use Agreement. In two experiments, a total of 140 female BALB/c nude mice (5–7 weeks old; Charles River Laboratories, Frederick, MD) were anesthetized with isoflurane/O2 and injected in the left cardiac ventricle with 231-BR-vector or 231-BR-HER2 cells (1.75 × 105 cells in 0.1 mL serum-free medium; n = 35 mice per cell line per experiment). Lapatinib treatment began 5 days after cell injection. Mice were randomly assigned to receive vehicle (0.5% hydroxypropylmethylcellulose with 0.1% Tween 80 in water) or lapatinib (30 or 100 mg/kg body weight) twice daily by oral gavage for 24 days (n = 22–26 mice per treatment group). Mice were euthanized by CO2 asphyxiation at the end of treatment or when they showed signs of neurological impairment. The whole brain was removed from the skull and subjected to fluorescence imaging to detect the presence of the injected 231-BR cells. EGFP fluorescence was detected in whole brains with the use of a Maestro 420 In Vivo Spectral Imaging System (Cambridge Research and Instrumentation, Woburn, MA) and data acquisition and processing software that could distinguish or unmix images of fluorescence from multiple sources (Nuance Technology, Burlington, MA).
After fluorescence imaging, each brain was bisected along the sagittal plane and the left hemisphere was immediately frozen in Tissue-Tek OCT compound (Sakura Finetek USA, Torrance, CA); these samples were used for histology. The right hemisphere was fixed in 4% paraformaldehyde for 24 hours at 4°C, transferred to 20% sucrose and incubated overnight at 4°C, and then frozen; these samples were used for immunohistochemistry (see below). Brain sections (10 μm thick) were serially cut from the left hemisphere and stained with hematoxylin and eosin (H & E) according to standard procedures.
Ten H & E–stained serial sections every 300 μm through the left hemisphere of the brain were analyzed for the presence of metastatic lesions with the use of a Zeiss microscope outfitted with a 5× objective that contained an ocular grid with 0.8-mm2 squares. We counted micrometastases (ie, those ≤50 μm2) to a maximum of 300 per section and every large metastasis (ie, those >50 μm2) in each section. The >50-μm2 metric for large metastases represents the mouse equivalent of the proportion of a magnetic resonance imaging–detectable brain metastasis (5 mm) to the length of a human brain. All analyses were carried out by two investigators who were blinded to experimental group assignment. Two separate experiments were performed, and the data were pooled for statistical analysis.
Immunohistochemistry
Sections (5 μm thick) of frozen OCT-embedded mouse brains were fixed and permeabilized in ice-cold methanol. Sections from five mice per treatment group were then incubated overnight at 4°C with a primary antibody specific for a form of HER2 that is phosphorylated at tyrosine residues 1221 and 1222 (p-HER2, rabbit monoclonal, 1:25 dilution, Cell Signaling Technology) or a primary antibody specific for EGFR that is phosphorylated at tyrosine residue 1068 (p-EGFR, rabbit polyclonal, 1:25 dilution, Cell Signaling Technology). Binding of the primary antibodies was detected with the use of an EnVision+ HRP system (Dako, Carpinteria, CA) according to the manufacturer's instructions followed by hematoxylin counterstaining. The staining assay was confirmed by a pathologist (M.J. Merino), who compared the staining results in the mouse samples with those in human breast tumors that were known to overexpress HER2 or EGFR.
One stained section per mouse (n = 5 mice per treatment group) was evaluated at ×100 magnification. Each large metastasis and 25 randomly chosen micrometastases per section were scored. No more than three micrometastases were scored from any group of multiple micrometastatic lesions in a given region so that a representative sampling of the entire brain section could be obtained. Staining for p-HER2 and p-EGFR was scored on an intensity scale of 0–3+ (defined a priori): 0 corresponded to background staining intensity; 1+ lesions contained tumor cells with visible cytoplasmic and membrane staining above background but often contained some tumor cells that had background levels of staining (ie, unstained); 2+ staining appeared homogeneous and darker than 1+ staining; and 3+ staining was the darkest.
Statistical Analysis
Generally, for both the in vitro and in vivo experiments, an analysis of variance (ANOVA) was performed on the data with experiment specified as the random effect. When appropriate, residuals were examined for normality with the Shapiro–Wilk test and were found to be normally distributed. For all ANOVAs, residuals were examined for homogeneity both graphically and numerically (the model with no variance partitioning was compared with models with partitioned variance with likelihood ratio test). Residuals were partitioned into homogeneous groups if heterogeneous variance was detected. P values for a priori comparisons to a control group were adjusted using Dunnett's method. Statistical significance was defined as P < .05. All statistical tests were two-sided. All analyses were performed with SAS statistical software (version 9.1; SAS Institute, Cary, NC).
For cell proliferation assays, a one-way ANOVA was performed, by time (72, 96, and 120 hours), with cell line (231-BR-vector, 231-BR-HER2) specified as the fixed effect. For analysis of cell proliferation in response to transfection with siRNA, a three-factor factorial ANOVA was performed, with lapatinib treatment (7, 8, 9, and 10 μM), cell line (231-BR-vector, 231-BR-HER2), and siRNA (EGFR, control) specified as the factors. Cell line by siRNA simple effect comparisons were specified a priori. For migration assay analysis, a one-way ANOVA was performed, by cell line (231-BR-vector, 231-BR-HER2), with lapatinib level (0, 1, and 3 μM) specified as the fixed effect. For the in vivo mouse experiments, data were pooled from two experiments and a two-factor factorial ANOVA was performed for each outcome, with cell line and lapatinib dose specified as the factors. A priori hypotheses tested were as follows: for each cell line 1) mean outcome was equal between lapatinib doses 0 and 30 mg/kg body weight and 2) mean outcome was equal between lapatinib doses 0 and 100 mg/kg body weight; and between the two cell lines 3) for each level of lapatinib, the mean outcome was equal between cell lines. For ANOVA of the immunohistochemisty data, we used a binomial distribution for the outcome variable and a logit link function. Higher-order effects (eg, three-way interactions) were dropped from the model if P was greater than .05.
Results
Effect of Lapatinib on Expression and Activation of Proteins Involved in HER2 and EGFR Signaling Pathways in 231-BR Cells
The goal of this study was to examine the efficacy of lapatinib, a small-molecule inhibitor of EGFR and HER2 tyrosine kinases, in a preclinical model of breast cancer brain metastasis. The model used was a brain-seeking derivative of human MDA-MB-231 cells (ie, 231-BR cells), which display increased EGFR expression and a propensity to metastasize to brain when injected into mice. We previously showed that 231-BR cells that were transfected with an expression vector containing the HER2 cDNA (231-BR-HER2) express 20-fold more HER2 protein than 231-BR cells transfected with empty vector (231-BR-vector) (21). In an experimental metastasis xenograft experiment, 231-BR-HER2 cells produced 2.5- to 3-fold more large brain metastases than 231-BR-vector cells (21).
Because lapatinib is approved for patients with HER2-overexpressing breast tumors with disease progression after receiving trastuzumab, we first examined the sensitivity of the 231-BR-HER2 cells to trastuzumab. In an anchorage-dependent growth assay, treatment of the cells with 1 mg/mL trastuzumab for 6 days had no effect on their growth (data not shown). By contrast, the same dose of trastuzumab inhibited the anchorage-dependent growth of SKBr3 cells, which display high endogenous expression of HER2, by 40%–50% (data not shown). In an anchorage-independent growth assay, SKBr3 colony formation was inhibited by 49% in the presence of 0.2 mg/mL trastuzumab, whereas no effect was observed on 231-BR-HER2 cells. Therefore, in vitro, the 231-BR-HER2 cells were intrinsically resistant to growth inhibition by trastuzumab.
Next, we examined the effects of lapatinib on HER2 and EGFR signaling pathways. After serum starvation overnight, 231-BR-vector and 231-BR-HER2 cells were cultured in the presence or absence of lapatinib for 24 hours, stimulated with 100 ng/mL EGF for 10 minutes to trigger activation (ie, phosphorylation) of EGF family receptor tyrosine kinases, and then lysed for immunoblot analysis. Figure 1 shows the overexpression of total HER2 protein in the 231-BR-HER2 cells (lanes 4–6) compared with the 231-BR-vector cell lines (lanes 1–3); total EGFR protein levels were the same in both cell lines. In this model system, expression of the HER3 receptor family member is below the limit of detection in both cell lines. In the absence of lapatinib, HER2 overexpression led to complex patterns of activation of downstream signaling proteins compared with 231-BR-vector cells (Figure 1, lanes 1 and 4). We observed increased levels of phosphorylated AKT, total p21, and phosphorylated PLCγ1, and decreased levels of phosphorylated p38.
Figure 1.
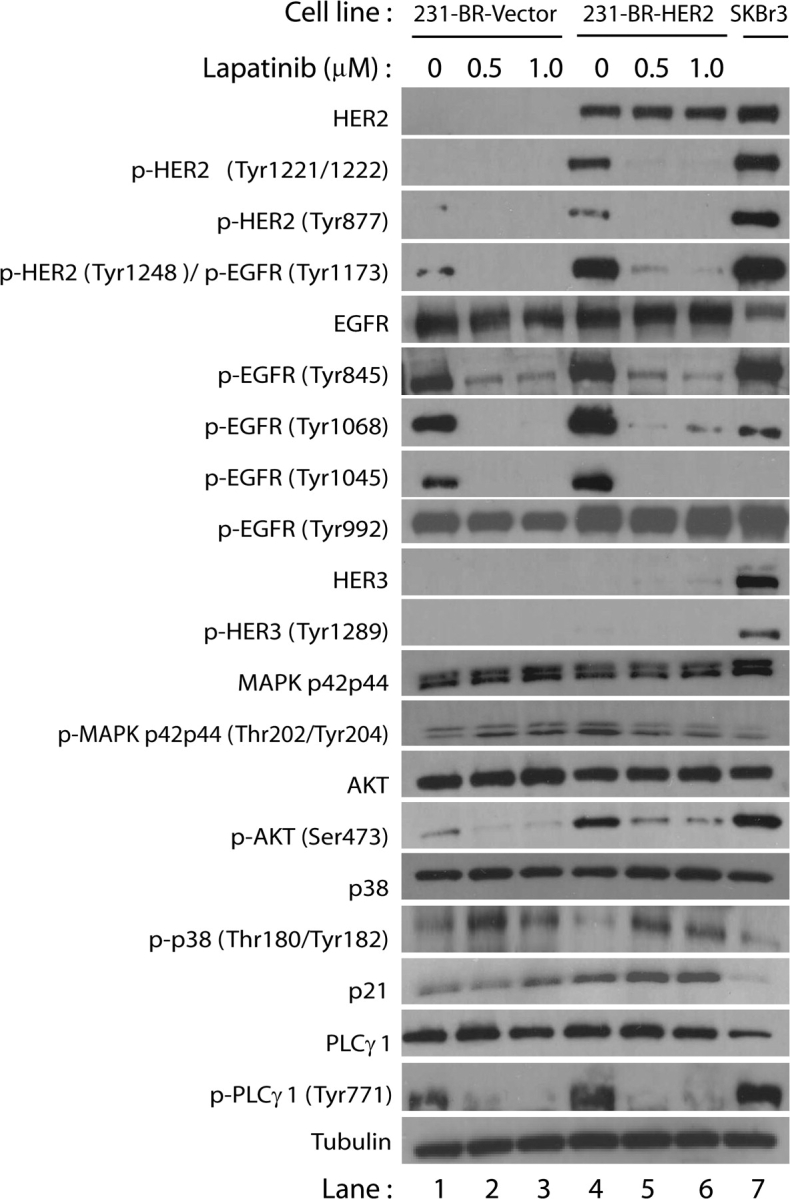
Effect of lapatinib on expression of proteins involved in HER2 and epidermal growth factor receptor (EGFR) signaling pathways in 231-BR cells. 231-BR-vector control (lanes 1–3) or 231-BR-HER2 (lanes 4–6) cells were serum starved overnight and subsequently treated with either 0.5 or 1.0 μM lapatinib for 24 h. Lapatinib was diluted in dimethyl sulfoxide, which served as the vehicle control (lanes 1 and 4). After lapatinib or vehicle treatment, cells were stimulated with 100 ng/mL epidermal growth factor for 10 min and total cell lysates were then prepared for immunoblot analysis. SKBr3 cells (lane 7), which endogenously overexpress HER2, were used as a positive control for antibody detection of HER family members. Proteins analyzed were HER2, EGFR, HER3, mitogen activated protein kinase (MAPK p42/p44), AKT, p38, p21, PLCγ1, tubulin. The prefix “p-” refers to phosphorylation of the residues in parentheses. The data shown are representative of results obtained in at least three separate experiments.
We confirmed the inhibition of HER2 and EGFR receptor autophosphorylation by lapatinib (Figure 1). For the 231-BR-HER2 cells, lapatinib inhibited phosphorylation of tyrosines 1221/1222 of HER2. In both the 231-BR-HER2 and 231-BR-vector cell lines, EGFR autophosphorylation was inhibited at tyrosines 1068 and 1045, but not at tyrosine 992. In addition, lapatinib inhibited autophosphorylation of tyrosine 1248 of HER2 and tyrosine 1173 of EGFR as detected by an antibody that recognizes both sites on the respective receptors. Lapatinib also inhibited phosphorylation of the Src phosphorylation sites on HER2 (Y877) and EGFR (Y845). Lapatinib is therefore an effective inhibitor of the activation of both the EGFR and HER2 tyrosine kinase receptors in vitro, except for EGFR tyrosine 992.
We also examined the effect of lapatinib on the expression and activation of proteins involved in signaling downstream of EGFR and HER2. Phosphorylated tyrosines 1221/1222 and 1248 of HER2 are binding sites for the adaptor proteins Shc and Grb2, which can induce the Ras p42/44 MAPK pathway (32,33). Phosphorylated tyrosine 1068 of EGFR is a Grb2-binding site that leads to activation of the Ras p42/44 MAPK pathway (32,33). Inhibition of phosphorylation of these sites by lapatinib should prevent the binding of Grb2, thus inhibiting activation of the Ras p42/44 MAPK pathway. We observed that 231-BR-HER2 cells treated with lapatinib had slightly less p42/44 MAPK phosphorylation than those treated with diluent (lanes 5–6 vs lane 4, Figure 1). Although lapatinib would be expected to inhibit the phosphorylation of p42/44 MAPK, this pathway is constitutively activated in the MDA-MB-231 cells because they express mutant Ras (k-ras codon 13) which is upstream of p42/44 MAPK (34). Phosphorylation of EGFR at tyrosine 1068 can also activate the PI3-K/AKT pathway by interacting directly with the p85 subunit of PI3-K or by interacting with the Gab-1 adaptor protein (32,33). Lapatinib, at both concentrations tested, inhibited AKT phosphorylation (serine 473) in both 231-BR-vector and 231-BR-HER2 cells (lane 1 vs lanes 2–3 and lane 4 vs lanes 5–6, respectively).
We also examined the effect of lapatinib on the activation of another member of the MAPK signaling pathway in both cell lines. Lapatinib increased phosphorylation of tyrosine residues 180 and 182 of p38, a stress-induced member of the MAPK pathway that is involved in apoptosis. Lapatinib treatment was also associated with a slight increase in the protein level of the cyclin-dependent kinase inhibitor p21. In the 231-BR-vector cells, this increase was only apparent at the highest lapatinib concentration tested.
Recently, Zhan et al. (35) reported that activation of PLCγ1 plays a role in the invasiveness of breast cancer cells that overexpress HER2 and EGFR. We observed that lapatinib inhibited phosphorylation of tyrosine 771 of PLCγ1 in 231-BR-vector and 231-BR-HER2 cells (lane 1 vs lanes 2–3 and lane 4 vs lanes 5–6, respectively). In summary, lapatinib inhibited the activation of the three main EGFR and HER2 downstream signaling pathways, decreasing phosphorylation of MAPK, AKT, and PLCγ1 in vitro.
Effect of Lapatinib on 231-BR-HER2 Cell Proliferation and Migration In Vitro
We next examined the effects of lapatinib on the proliferation and migration of 231-BR cells in vitro. The mean lapatinib concentration that caused 50% inhibition of growth at 96 hours of culture was 7.5 μM (95% confidence interval [CI] = 7.2 to 7.6 μM) for 231-BR-HER2 cells and 8.5 μM (95% CI = 8.3 to 8.8 μM) for 231-BR-vector cells (data not shown). Treatment of 231-BR-HER2 and 231-BR-vector cells with 8 μM lapatinib for various times resulted in differential growth inhibition; 231-BR-HER2 cells were 20%–59% more sensitive to 8 μM lapatinib than 231-BR-vector cells (mean percentage of control cell proliferation at 72 hours: 63.7% vs 79.2%, P = .003; at 96 hours: 37.2% vs 67.7%, P = .003; at 120 hours: 16.4% vs 40.4%, P = .001) (Figure 2, A). In vitro, the growth inhibitory effects of lapatinib were greatest when both cell lines were treated with fresh drug on a daily basis compared with experiments where cells were treated with lapatinib only at the beginning of the experiment (data not shown).
Figure 2.

Effects of lapatinib on 231-BR cell proliferation and migration. A) Time course of lapatinib (8 μM) inhibition of the proliferation of 231-BR-vector (V)– and 231-BR-HER2 (H)–overexpressing cell lines. The mean percentage proliferation versus diluent (dimethyl sulfoxide, DMSO) control treatment for each cell line and time point is shown, and error bars represent 95% confidence intervals. B) and C) Dependence of the antiproliferative activity of lapatinib on the number of target receptors expressed. 231-BR-vector cells or 231-BR-HER2 cells were incubated with small interfering RNA (siRNA) targeting EGFR or a nontargeting control (a sequence not homologous to any human, mouse, or rat gene) siRNA. After 24 h of siRNA treatment, cells were cultured in the presence of various doses of lapatinib or diluent (DMSO) for 96 h. Half of the diluent-treated cells were lysed for immunoblot analysis of epidermal growth factor receptor expression (B), and the remaining cells were assayed for cell viability using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide assay (C). The P values represent data averaged over the 7- to 10-μM doses of lapatinib. D) In vitro cell migration assay. 231-BR-vector (V) or 231-BR-HER2 cells (H) were pretreated with 1 or 3 μM lapatinib or diluent (DMSO). Cell migration in response to 1.0% fetal bovine serum was examined using a Boyden chamber assay. The graph represents percentage of migration compared with DMSO control treatment. For the 231-BR-vector cell line, no statistically significant difference was observed between DMSO and 1 μM lapatinib treatment (P = .27). All P values are two-sided (analysis of variance).
To examine the basis for the increased sensitivity of 231-BR-HER2 cells to lapatinib, we used siRNAs to knock down expression of EGFR in 231-BR-vector and 231-BR-HER2 cells. Each cell line was transiently transfected with an siRNA against EGFR or a nontargeting control siRNA to create cell populations that expressed EGFR alone (231-BR-vector cells transfected with control siRNA), HER2 alone (231-BR-HER2 cells transfected with EGFR siRNA), both EGFR and HER2 (231-BR-HER2 cells transfected with control siRNA), or neither receptor (231-BR-vector cells transfected with EGFR siRNA). The cells were cultured for 24 hours after siRNA transfection, treated with different doses of lapatinib for 96 hours, and subjected to the MTT assay to assess cell proliferation. In parallel, immunoblot analysis of the cells 120 hours after siRNA transfection showed that EGFR protein levels remained low in cells that were transfected with EGFR siRNA (Figure 2, B). EGFR siRNA–transfected 231-BR-vector cells displayed some growth inhibition in response to lapatinib (14%–42% inhibition at 7–9 μM lapatinib), possibly because they expressed a very low endogenous level of HER2 that was below the limit of detection for the immunoblots (Figure 2, C). The EGFR-only–expressing (231-BR-vector cells transfected with nontargeting siRNA) and the HER2-only–expressing cells (231-BR-HER2 cells transfected with EGFR siRNA) were equally sensitive to lapatinib. However, cells that expressed high levels of both EGFR and HER2 were approximately 30% more sensitive to lapatinib than cells that expressed high levels of either receptor alone (mean percent inhibition of cell growth compared with diluent (DMSO)-treated control at 8 μM lapatinib: for EGFR-only expression, 45.6%, 95% CI = 40.9% to 50.3%; for HER2-only expression = 48.8%, 95% CI = 42.0% to 55.6%; both EGFR and HER2 expression, 72.5%, 95% CI = 59.9% to 85.1%) (Figure 2, C). Therefore, in vitro, sensitivity to growth inhibition by lapatinib appears to reflect the number of target receptor tyrosine kinases expressed by the cell line.
We next examined the effect of lapatinib on the migration of 231-BR cells by using a Boyden chamber assay (Figure 2, D). 231-BR-vector and 231-BR-HER2 cells were pretreated for 24 hours with 1 or 3 μM lapatinib, and migration in the presence of the same concentration of lapatinib was quantified using 1% FBS as the chemoattractant. Treatment of 231-BR-vector cells with 3 μM lapatinib inhibited cell migration by 42.6% (95% CI = 24.6% to 60.6%; P = .013) compared with DMSO treatment. By contrast, migration of the 231-BR-HER2 cell line was statistically significantly inhibited at both 1 and 3 μM lapatinib treatments (mean percent inhibition at 1 μM = 23.7%, 95% CI = 20.1% to 27.3%, P = .030; at 3 μM = 31.2%, 95% CI = 24.7% to 37.7%, P = .009). Thus, 231-BR-HER2 cells were more sensitive to lapatinib inhibition of cell proliferation and migration than 231-BR-vector cells.
Effect of Lapatinib on Outgrowth of Metastatic Breast Cancer Cells in the Brain
We next used the 231-BR-HER2 and 231-BR-vector cells in an in vivo metastasis assay in mice to examine whether lapatinib could prevent metastastic outgrowth of breast cancer cells in the brain. Five days after intracardiac injection of 231-BR cells, mice were randomly assigned to receive lapatinib (30 or 100 mg/kg body weight) or vehicle solution twice daily by oral gavage (n = 22–26 mice per group). Mice received 24 days of lapatinib treatment; all mice were then killed and their brains were harvested for ex vivo whole-brain fluorescence imaging of EGFP-positive brain metastases. Brains of control mice that were not injected with breast cancer cells showed a minor amount of nonspecific diffuse autofluorescence at both the anterior and posterior ends of the brain (data not shown), whereas brains of mice injected with 231-BR cells showed discrete foci of fluorescence throughout the brain. In general, mice treated with lapatinib had fewer metastatic foci than mice treated with vehicle as determined by whole-brain imaging (Figure 3).
Figure 3.

Lapatinib inhibition of metastatic colonization of mouse brain by 231-BR breast carcinoma cells. 231-BR-HER2 cells or 231-BR-vector cells, both of which were transduced with a retrovirus that expressed enhanced green fluorescent protein (EGFP), were injected into the left cardiac ventricle of BALB/c nude mice. Five days after injection, lapatinib (30 or 100 mg/kg body weight) or vehicle (0.5% hydroxypropylmethylcellulose with 0.1% Tween 80 in water) was administered by twice-daily oral gavage for 24 days (n = 22–26 mice per treatment group). Brains dissected at necropsy were imaged using a Maestro 420 Spectral Imaging System to detect the presence of EGFP expressing metastases derived from the injected 231-BR cells (metastatic foci on a green to white [greater intensity] fluorescent intensity spectrum). Representative dorsal whole-brain images from two mice in each treatment group are shown.
To quantify the effect of lapatinib on tumor cell colonization of brain, we counted the number of large metastatic lesions (ie, >50 μm2) and micrometastases (ie, ≤50 μm2) in H & E–stained brain sections with the aid of an ocular micrometer (Table 1). The 50-μm2 size cutoff was selected based on proportionality to a magnetic resonance imaging–detectable lesion in the brain of a patient with metastatic breast cancer. The vehicle-treated mice confirmed our previously published (21) finding that overexpression of HER2 in the injected breast cancer cells increased the number of large metastases in brain by twofold (6.83 vs 3.36; difference = 3.47; 95% CI = 2.33 to 4.61; P < .001). In addition, among vehicle-treated mice, those injected with 231-BR-HER2 cells had 37% more micrometastases than those injected with 231-BR-vector cells (138 vs 101, difference = 37, 95% CI = 8 to 65, P = .012). Among mice injected with 231-BR-HER2 cells, those treated with 100 mg/kg lapatinib had 50% fewer large metastases than those treated with vehicle (3.44 vs 6.83, difference = 3.39, 95% CI = 2.08 to 4.70, P < .001); those treated with 30 mg/kg lapatinib had 53% fewer large metastases (3.21 vs 6.83, difference = 3.62, 95% CI = 2.30 to 4.94, P < .001). These data indicate that lapatinib suppressed the outgrowth of large brain metastases at both doses tested.
Table 1.
In vivo analysis of the effect of lapatinib on the growth of breast cancer brain metastases*
| Cell line | Lapatinib dose (mg/kg body weight) | No. of mice | Mean No. of large metastases† (95% CI) | P‡ | Mean No. of micrometastases (95% CI) | P‡ |
| 213-BR-Her2 | 0 (vehicle) | 22 | 6.83 (5.86 to 7.79) | (referent) | 138 (101 to 175) | (referent) |
| 30 | 25 | 3.21 (2.31 to 4.11) | <.001 | 109 (72 to 146) | .090 | |
| 100 | 26 | 3.44 (2.55 to 4.32) | <.001 | 127 (90 to 164) | .51 | |
| 231-BR-vector | 0 (vehicle) | 22 | 3.36 (2.73 to 3.98) | (referent) | 101 (52 to 151) | (referent) |
| 30 | 22 | 2.80 (2.18 to 3.43) | .22 | 96 (59 to 133) | .70 | |
| 100 | 23 | 1.56 (0.94 to 2.17) | .001 | 65 (14 to 116) | <.001 |
Data shown were pooled from two independent experiments. Mice were injected with 1.75 x 105 cells on day 0, and treatment with lapatinib or vehicle started 5 days later. Mice were killed 24 days after treatment began. Mean number of large metastases and micrometastases was determined by counting the number of metastases in 10 step-sections from one hemisphere of the brain. The size of each metastasis was determined with the use of a 16-mm2 ocular grid. CI = confidence interval.
>50 μm2.
A two-factor factorial analysis of variance was performed for each outcome with cell line and lapatinib dose specified as the factors. A priori hypotheses tested were as follows: for each cell line 1) mean outcome was equal between lapatinib doses 0 and 30 and 2) mean outcome was equal between lapatinib doses 0 and 100; and between each cell line 3) for each level of lapatinib, the mean outcome was equal between cell lines. All P values are two-sided.
We also observed a distinct trend in the metastatic colonization of brain by 231-BR-vector cells, which endogenously express high levels of EGFR but not HER2. Mice injected with these cells and treated with 100 mg/kg lapatinib showed statistically significantly fewer micrometastases (65 vs 101, difference = 36, 95% CI = 16 to 57, P < .001) and large metastases (1.56 vs 3.36, difference = 1.80, 95% CI = 0.92 to 2.68, P < .001) than vehicle-treated mice; the 30 mg/kg dose of lapatinib had no statistically significant effect on the numbers of 231-BR–derived micrometastases or large metastases.
Effect of Lapatinib Treatment on Phosphorylation of HER2 and EGFR In Vivo
We next subjected brain sections from the treated mice to immunohistochemistry to examine the relative activation levels of HER2 and EGFR in vivo. A single brain section from five randomly chosen mice per treatment arm was stained with either an antibody specific for pHER2 (tyrosines 1221 and 1222) or an antibody specific for pEGFR (tyrosine 1068); 25 micrometastases and all large metastases per section were scored for staining by each antibody on a 0–3+ intensity scale (Figure 4 and Table 2).
Figure 4.
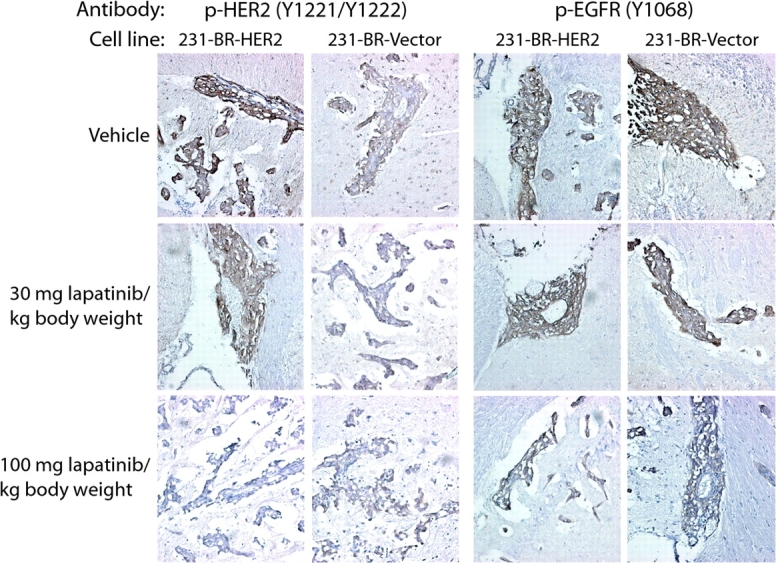
Immunohistochemical evaluation of HER2 and epidermal growth factor receptor (EGFR) activation in vivo in response to lapatinib treatment. Frozen sections (5 µm thick) of brains from mice injected with 231-BR-vector or 231-BR-HER2 cells and treated with lapatinib (30 or 100 mg/kg body weight) or vehicle (n = 5 mice per group) were stained with antibodies specific for phosphorylated HER2 (p-HER2; tyrosines 1221 and 1222) or phosphorylated EGFR (p-EGFR; tyrosine 1068). The staining of all large metastases (ie, >50 μm2) and 125 micrometastases per treatment group was scored on a 0–3+ intensity scale by two investigators who were blinded to the treatment group assignment. Representative images of large metastases for each group are shown (×200 magnification). The presence of p-HER2 or p-EGFR antigen is indicated by brown staining; nuclei were counterstained purple with hematoxylin.
Table 2.
Effect of lapatinib treatment on the level of HER2 phosphorylation (p-HER2) and the level of epidermal growth factor receptor phosphorylation (p-EGFR) in breast cancer brain metastases by immunohistochemical staining*
| Lapatinib dose (mg/kg body weight) | Fraction (%) of large metastases with staining intensity† |
Fraction (%) of micrometastases with staining intensity‡ |
||||||||
| Antibody | Cell line | 0 | 1+ | 2+ | 3+ | 0 | 1+ | 2+ | 3+ | |
| p-HER2 (Y1221/ Y1222) | 231-BR-HER2 | 0 (vehicle) | 0 | 13/54 (24) | 32/54 (59) | 9/54 (17) | 0 | 13/125 (10) | 51/125 (41) | 61/125 (49) |
| 30 | 4/35 (11) | 11/35 (31) | 12/35 (34) | 8/35 (23) | 22/125 (18) | 35/125 (28) | 45/125 (36) | 23/125 (18) | ||
| P = .10 | P < .001 | |||||||||
| 100 | 16/42 (38) | 19/42 (45) | 7/42 (17) | 0 | 34/125 (27) | 73/125 (58) | 18/125 (14) | 0 | ||
| P < .001 | P < .001 | |||||||||
| 231-BR-vector | 0 (vehicle) | 4/45 (9) | 28/45 (62) | 13/45 (29) | 0 | 25/175 (20) | 71/125 (57) | 28/125 (22) | 1/125 (1) | |
| 30 | 2/35 (6) | 22/35 (63) | 11/35 (31) | 0 | 17/125 (14) | 63/125 (50) | 44/125 (35) | 1/125 (1) | ||
| P = .70 | P = .037 | |||||||||
| 100 | 10/23 (44) | 12/23 (52) | 1/23 (4) | 0 | 48/125 (38) | 66/125 (53) | 11/125 (9) | 0 | ||
| P = .081 | P = .012 | |||||||||
| p-EGFR (Y1068) | 231-BR-HER2 | 0 (vehicle) | 2/30 (7) | 17/30 (57) | 6/30 (20) | 5/30 (17) | 8/125 (6) | 46/125 (37) | 43/125 (34) | 28/125 (22) |
| 30 | 2/71 (3) | 20/71 (28) | 36/71 (51) | 13/71 (18) | 8/125 (6) | 50/125 (40) | 47/125 (38) | 20/125 (16) | ||
| P = .36 | P = .81 | |||||||||
| 100 | 7/57 (12) | 36/57 (63) | 14/57 (25) | 0 | 10/125 (13) | 76/125 (61) | 30/125 (24) | 9/125 (7) | ||
| P = .36 | P = .041 | |||||||||
| 231-BR-vector | 0 (vehicle) | 0 | 12/32 (38) | 11/32 (34) | 9/32 (28) | 3/125 (2) | 33/125 (25) | 56/125 (45) | 33/125 (26) | |
| 30 | 1/55 (2) | 21/55 (38) | 22/55 (40) | 11/55 (20) | 9/125 (7) | 30/125 (24) | 56/125 (45) | 30/125 (24) | ||
| P = .84 | P = .84 | |||||||||
| 100 | 3/22 (14) | 13/22 (59) | 5/22 (23) | 1/22 (5) | 14/125 (11) | 50/125 (40) | 48/125 (38) | 13/125 (10) | ||
| P = .13 | P = .30 | |||||||||
All P values listed represent the difference between the fraction with 0–1+ staining for the indicated treatment group compared with the vehicle control for that group. For each antibody (p-HER2 and p-EGFR) and for both sizes of metastases (micro and large), the fraction of low level staining (0, 1+) was calculated (ie, a binomial outcome). For each of the four analyses, we performed a two-factor factorial analysis of variance to test the nine simple effects created by crossing cell line with lapatinib.
All large metastases per section were scored.
In total, 125 micrometastatic lesions per treatment arm were scored.
The majority of lesions in the brains of mice injected with 231-BR-HER2 cells and treated with vehicle—76% of the large metastases and 90% of the micrometastases—had a staining intensity of 2+ or 3+ for p-HER2. By contrast, there were fewer lesions with a staining intensity of 2+ or 3+ for p-HER2 in the brains of mice injected with 231-BR-HER2 cells and treated with either dose of lapatinib (57% of large metastases and 54% of micrometastases at 30 mg lapatinib/kg body weight; 17% of large metastases and 14% of micrometastases at 100 mg lapatinib/kg body weight); in particular, we observed no lesions with a 3+ staining intensity in mice treated with the higher dose of lapatinib (Table 2). Both doses of lapatinib increased the percentage of large lesions with staining intensities of 0 or 1+ over that seen in vehicle-treated mice (100 mg lapatinib/kg body weight vs vehicle: 83% vs 24%, P < .001; 30 mg lapatinib/kg body weight vs vehicle: 43% vs 24%, P = .10). Similar results were observed for micrometastases derived from the injection of the 231-BR-HER2 cell line (P < .001 for vehicle vs either dose of lapatinib). Lapatinib therefore effectively decreased the phosphorylation of HER2 in vivo.
We observed some p-HER2 staining in 231-BR-vector cell–derived brain metastases (Figure 4), possibly as a result of transphosphorylation of low endogenous levels of HER2 by heterodimerization with abundant EGFR. Virtually none (ie, 1 of 170 [0.6%]) of the brain metastases produced by the 231-BR-vector cells had a p-HER2 staining intensity of 3+ (Table 2). Both doses of lapatinib increased the frequency of micrometastases with p-HER2 staining intensities of 0 or 1+ over that seen in vehicle-treated mice (100 mg lapatinib/kg body weight vs vehicle: 91% vs 10%, P < .001; 30 mg lapatinib/kg body weight vs vehicle: 91% vs 46%, P < .001). However, neither dose of lapatinib had a statistically significant effect on the frequency of low-staining large metastases compared with vehicle.
Staining for p-EGFR was distributed throughout the 0–3+ intensity scale in the brain metastases from mice injected with both cell lines (Table 2 and Figure 4). Few lesions (6%–7%) were negative for p-EGFR. Neither dose of lapatinib had a statistically significant effect on frequency of metastases with p-EGFR staining intensity of 0 or 1+ in mice injected with either cell line. These data indicate that EGFR activation was unaffected by lapatinib therapy and therefore could contribute to lapatinib resistance.
Discussion
Brain metastases of breast cancer represent an unmet medical need that affects approximately 35% of patients with metastatic HER2-overexpressing breast cancer. The increasing incidence of brain metastases in patients with metastatic breast cancer whose tumors overexpress HER2 reflects a “perfect storm” of 1) HER2 overexpression, which increases the colonization of breast cancer cells in the brain (21), and 2) the poor pharmacokinetics of the large monoclonal antibody trastuzumab in the brain. Brain metastastic relapses were reported in an adjuvant clinical trial of trastuzumab (22), indicating that earlier trastuzumab treatment does not eliminate this complication. Therefore, new strategies for the prevention and treatment of brain metastasis from HER2-overexpressing breast tumors must be devised. We propose a scenario in which standard treatments such as neurosurgery and stereotactic radiosurgery are used to treat clinical metastases and currently unavailable molecular therapeutics are then used to hold the remaining micrometastases in check. One possible molecular therapeutic is lapatinib, a dual inhibitor of EGFR and HER2 kinases. We tested the effect of lapatinib on the outgrowth of metastatic tumor cells in the brain in a preclinical mouse model. Lapatinib inhibited the formation of large metastases by breast cancer cells that overexpressed HER2 by 50%–53%, suggesting that this agent may demonstrate clinical efficacy in the adjuvant and preventive clinical settings. The inhibition of brain metastatic colonization of the HER2-overexpressing cell line by lapatinib was paralleled by reduced pHER2 staining in the remaining lesions in vivo, confirming that the drug affected its intended target.
The 231-BR cell model that we used to test the efficacy of lapatinib in the prevention of brain metastatic colonization by breast cancer cells has four strengths. First, 231-BR-HER2 cells were resistant to trastuzumab in vitro, which suggests that they are a good model of an aggressive breast cancer in vivo. Second, the quantification system was designed to account for clinical metastasis formation. Metastases were scored using an ocular micrometer and quantified using a cutoff of >50 μm2, which is proportional to a magnetic resonance imaging-detectable lesion in a human brain. Third, in independent studies, the 231-BR model system faithfully replicated histological and pharmacokinetic aspects of human brain metastases of breast cancer. For example, approximately 53% of cells in 231-BR cell–derived tumors are positive for the cell proliferation marker Ki67 in the mouse model, which is comparable to the percentage of Ki67-positive tumor cells in human brain metastases of breast cancer as assessed in a cohort of 16 surgically excised brain metastases (ie, 47%). Apoptosis was minimal in both the mouse model and the resected human metastases (D. Fitzgerald, D. Palmieri, E. Hua, E. Hargrave, J. Herring, Y. Qian, et al., unpublished observations). Thus, the mouse model mimics the proliferative and apoptotic rates of human brain metastases. In addition, an analysis of the distribution of 14C-labeled lapatinib and paclitaxel in the brains of mice injected with 231-BR-HER2 cells revealed an approximately fourfold increase in the amount of 14C-labeled paclitaxel in the large metastases compared with normal brain tissue from the same mouse, whereas 14C-lapatinib levels in the brain metastases were at least 20-fold higher than those in normal brain (Q. Smith, P. Lockman, Texas Tech University, personal communication). These data indicate selective drug uptake by the 231-BR-HER2-overexpressing model.
Finally, the 231-BR model system takes into account EGFR expression, which has been reported to be an important molecular characteristic of both brain metastases and HER2 signaling. The potential importance of EGFR expression was suggested by Hicks et al. (14), who reported that breast cancer patients whose primary tumors were positive for EGFR expression had an increased risk of CNS metastasis. Gaedcke et al. (36) reported that EGFR was detectable by immunostaining in only 16% of primary breast cancers from patients with known CNS involvement vs 35% of unmatched CNS metastases; this trend was confirmed in a smaller cohort of matched primary tumors and CNS metastases, with EGFR expression in 13% and 38%, respectively. Thus, EGFR expression appears concentrated in brain metastases. The relationship between EGFR and HER2 expression was reported by DiGiovanna et al. (37), who found that 87% of EGFR-positive primary breast tumors also overexpressed HER2 and that 97% of tumors with p-HER2 expression co-overexpressed also overexpressed EGFR. Similarly, Gschwantler-Kaulich et al. (38) reported a statistically significant correlation between expression of p-tyrosine 845 EGFR and expression of either total HER2 or p-tyrosine 1248 HER2 in HER2-overexressing metastatic breast tumors. Based on much smaller numbers, similar trends have been reported in brain metastases. For example, we found that two of five HER2-amplified resected human brain metastases of breast cancer coordinately expressed high levels of EGFR mRNA (21); similar trends at the protein level have also been reported (39).
The data validate, in a preclinical model, the efficacy of lapatinib for the inhibition of brain metastatic colonization by HER2-overexpressing human breast cancer cells. The lapatinib doses used herein are thought to be relevant because they produce serum drug levels in mice that are comparable to those achieved in clinical trials. Human serum concentrations at 6 hours after administration of oral daily doses of 1600 and 1200 mg lapatinib were approximately 2500 and 1000 ng/mL, respectively (40). The serum concentrations of lapatinib in mice 6 hours after both single and repeated oral administration of 100 and 60 mg/kg were 2160 and 1591 ng/mL, respectively (T. Gilmer, GlaxoSmithKline, unpublished data). Using a lapatinib dose of 100 mg/kg body weight and a lower dose of 30 mg/kg body weight inhibited the formation of large brain metastases by 231-BR-HER2 cells by 50%–53% (Table 1).
Only the highest dose of lapatinib tested was effective in preventing the formation of large metastases by the 231-BR cell line expressing only EGFR. This was in contrast to the data for the 231-BR-HER2 cell line. It is unclear why lapatinib was more effective in preventing metastasis formation by cells that expressed both HER2 and EGFR than by cells that expressed EGFR alone, given that the drug is equally effective in inhibiting the phosphorylation of EGFR and HER2 in vitro (Figure 1). There are conflicting data in the literature on the efficacy of lapatinib in breast cancer cells that overexpress HER2 and/or EGFR. Several reports have suggested a preferential antiproliferative activity of lapatinib in HER2-overexpressing cells in vitro (27,41). However, a recent report suggested that expression levels of both HER2 and EGFR and tissue type were associated with the IC50 of lapatinib in a panel of 61 tumor cell lines (42). Data from our in vitro studies using EGFR siRNA agree with the latter report: we found that the antiproliferative activity of lapatinib in 231-BR cells that expressed EGFR only was similar to that in 231-BR cells that expressed HER2 only, and that cells that expressed both targets were more sensitive to lapatinib than those that expressed either one alone. This trend was also observed in vivo: lapatinib was more effective in inhibiting the development of large metastases by cells that expressed two targets (EGFR and HER2) than by cells that expressed only one target (EGFR). We are currently attempting to derive a brain-metastatic breast cancer cell line that expresses HER2 without overexpressing EGFR to further investigate this finding.
A limitation of our study is that lapatinib at either dose did not completely inhibit the formation of large brain metastases, which suggests that some breast cancer cells were resistant, or not as sensitive to this drug as other cells. Three potential sources of resistance may contribute to this finding. First, in vitro, lapatinib failed to inhibit the phosphorylation of tyrosine 992 of EGFR. This residue is located outside the catalytic domain and is thought to be a secondary site for association with PLCγ1 as well as a binding site for Src and Ras GTPase-activating protein (43–45). We could not ascertain the phosphorylation status of EGFR tyrosine 992 in sections of brain tissue from lapatinib-treated mice because the phosphorylation-specific antibody did not work on frozen sections. Confirmation with another antibody specific for phosphorylation at this site would strengthen this observation. Second, the MDA-MB-231 cell line harbors an oncogenic Ras mutation, which may interfere with the ability of lapatinib to completely inhibit this signaling axis. Third, our data suggest that phosphorylation of EGFR is not statistically significantly inhibited by lapatinib in vivo. We analyzed the phosphorylation of both HER2 (Y1221–1222) and EGFR (Y1068) in the brain metastases using immunohistochemistry. A reduced intensity of p-HER2 staining, in terms of a loss of 3+ staining and a gain of 0–1+ staining, was observed in the 231-BR-HER2 cells. In contrast to in vitro signaling data, p-EGFR staining intensities were spread over the 1–3+ range in all cell lines and treatment groups in vivo. This inability to shut down EGFR phosphorylation may also contribute to lapatinib resistance.
Funding
P. S. Steeg receives research support from the Intramural program of the National Cancer Institute (NCI) and by grant W81XWH-062-0033 from the Department of Defense Breast Cancer Research Program. S. D. Rubin owns stock in GlaxoSmithKline (the maker of lapatinib).
References
- 1.Weil R, Palmieri D, Bronder J, Stark A, Steeg P. Breast cancer metastasis to the central nervous system. Am J Pathol. 2005;167(4):913–920. doi: 10.1016/S0002-9440(10)61180-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lin N, Bellon J, Winer E. CNS metastases in breast cancer. J Clin Oncol. 2004;22(17):3608–3617. doi: 10.1200/JCO.2004.01.175. [DOI] [PubMed] [Google Scholar]
- 3.Slotman B, Faive-Finn C, Kramer G, et al. Prophylactic cranial irradiation in extensive small-cell lung cancer. N Engl J Med. 2007;357(7):664–672. doi: 10.1056/NEJMoa071780. [DOI] [PubMed] [Google Scholar]
- 4.Sneed PK, Lamborn KR, Forstner JM, et al. Radiosurgery for brain metastases: is whole brain radiotherapy necessary? Int J Radiat Oncol Biol Phys. 1999;43(3):549–558. doi: 10.1016/s0360-3016(98)00447-7. [DOI] [PubMed] [Google Scholar]
- 5.Patchell RA, Regine WF. The rationale for adjuvant whole brain radiation therapy with radiosurgery in the treatment of single brain metastases. Technol Cancer Res Treat. 2003;2(2):111–115. doi: 10.1177/153303460300200206. [DOI] [PubMed] [Google Scholar]
- 6.Lo SS, Chang EL, Suh JH. Stereotactic radiosurgery with and without whole-brain radiotherapy for newly diagnosed brain metastases. Expert Rev Neurother. 2005;5(4):487–495. doi: 10.1586/14737175.5.4.487. [DOI] [PubMed] [Google Scholar]
- 7.Ryberg M, Nielsen D, Osterlind K, Andersen PK, Skovsgaard T, Dombernowsky P. Predictors of central nervous system metastasis in patients with metastatic breast cancer. A competing risk analysis of 579 patients treated with epirubicin-based chemotherapy. Breast Cancer Res Treat. 2005;91(3):217–225. doi: 10.1007/s10549-005-0323-x. [DOI] [PubMed] [Google Scholar]
- 8.Carey LA, Ewend MG, Metzger R, et al. Central nervous system metastases in women after multimodality therapy for high risk breast cancer. Breast Cancer Res Treat. 2004;88(3):273–280. doi: 10.1007/s10549-004-0999-3. [DOI] [PubMed] [Google Scholar]
- 9.Slimane K, Andre F, Delaloge S, et al. Risk factors for brain relapse in patients with metastatic breast cancer. Ann Oncol. 2004;15(11):1640–1644. doi: 10.1093/annonc/mdh432. [DOI] [PubMed] [Google Scholar]
- 10.Evans A, James J, Cornford E, et al. Brain metastases from breast cancer: identification of a high-risk group. Clin Oncol. 2004;16(5):345–349. doi: 10.1016/j.clon.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 11.Clark G, Sledge GW, Jr, Osborne C, McGuire W. Survival from first recurrence: relative importance of prognostic factors in 1,015 breast cancer patients. J Clin Oncol. 1987;5(1):55–61. doi: 10.1200/JCO.1987.5.1.55. [DOI] [PubMed] [Google Scholar]
- 12.Tham YL, Sexton K, Kramer R, Hilsenbeck S, Elledge R. Primary breast cancer phenotypes associated with propensity for central nervous system metastases. Cancer. 2006;107(4):696–704. doi: 10.1002/cncr.22041. [DOI] [PubMed] [Google Scholar]
- 13.Pestalozzi BC, Zahrieh D, Price KN, et al. Identifying breast cancer patients at risk for Central Nervous System (CNS) metastases in trials of the International Breast Cancer Study Group (IBCSG) Ann Oncol. 2006;17(6):935–944. doi: 10.1093/annonc/mdl064. [DOI] [PubMed] [Google Scholar]
- 14.Hicks D, Short S, Prescott N, et al. Breast cancers with brain metastases are more likely to be estrogen receptor negative, express teh basal cytokeratin CK5/6 and over-express Her-2 or EGFR. Am J Surg Pathol. 2006;30(9):1097–1104. doi: 10.1097/01.pas.0000213306.05811.b9. [DOI] [PubMed] [Google Scholar]
- 15.Miller K, Weathers T, Hanley L, et al. Occult central nervous system involvement in patients with metastatic breast cancer: prevalence, predictive factors and impact on overall survival. Ann Oncol. 2003;14(7):1072–1077. doi: 10.1093/annonc/mdg300. [DOI] [PubMed] [Google Scholar]
- 16.Bendell J, Domchek S, Burstein H, et al. Central nervous system metastases in women who receive trastuzumab-based therapy for metastatic breast carcinoma. Cancer. 2003;97(12):2972–2977. doi: 10.1002/cncr.11436. [DOI] [PubMed] [Google Scholar]
- 17.Gabos Z, Sinha R, Hanson J, et al. Prognostic significance of human epidermal growth factor receptor positivity for the development of brain metastasis after newly diagnosed breast cancer. J Clin Oncol. 2006;24(36):5658–5663. doi: 10.1200/JCO.2006.07.0250. [DOI] [PubMed] [Google Scholar]
- 18.Shmueli E, Wigler N, Inbar M. Central nervous system progression among patients with metastatic breast cancer responding to trastuzumab treatment. Eur J Cancer. 2004;40(3):379–382. doi: 10.1016/j.ejca.2003.09.018. [DOI] [PubMed] [Google Scholar]
- 19.Clayton A, Danson S, Jolly S, et al. Incidence of cerebral metastases in patients treated with trastuzumab for metastatic breast cancer. Br J Cancer. 2004;91(4):639–643. doi: 10.1038/sj.bjc.6601970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lower E, Blau R, Bismayer J, et al. Increased brain metastasis detected in metastatic breast cancer patients receiving herceptin. Breast Cancer Res Treat. 2001;69(3):271. Abstract 351. [Google Scholar]
- 21.Palmieri D, Bronder JL, Herring JM, et al. Her-2 overexpression increases the metastatic outgrowth of breast cancer cells in the brain. Cancer Res. 2007;67(9):4190–4198. doi: 10.1158/0008-5472.CAN-06-3316. [DOI] [PubMed] [Google Scholar]
- 22.Romond EH, Perez EA, Bryant J, et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med. 2005;353(16):1673–1684. doi: 10.1056/NEJMoa052122. [DOI] [PubMed] [Google Scholar]
- 23.Piccart-Gebhart MJ, Procter M, Leyland-Jones B, et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med. 2005;353(16):1659–1672. doi: 10.1056/NEJMoa052306. [DOI] [PubMed] [Google Scholar]
- 24.Slamon D, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against Her2 for metastatic breast cancer that overexpresses Her2. N Engl J Med. 2001;344(11):783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 25.Pestalozzi B, Brignoli S. Traztuzumab in CSF. J Clin Oncol. 2000;18(11):2350–2351. doi: 10.1200/JCO.2000.18.11.2349. [DOI] [PubMed] [Google Scholar]
- 26.Pestalozzi BC. Correction: meningeal carcinomatosis from breast carcinoma responsive to trastuzumab. J Clin Oncol. 2001;19(20):4091. [PubMed] [Google Scholar]
- 27.Rusnak DW, Lackey K, Affleck K, et al. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol Cancer Ther. 2001;1(2):85–94. [PubMed] [Google Scholar]
- 28.Geyer C, Forster J, Lindquist D, et al. Lapatinib plus capecitabine for HER-2-positive advanced breast cancer. N Engl J Med. 2006;355(26):2733–2743. doi: 10.1056/NEJMoa064320. [DOI] [PubMed] [Google Scholar]
- 29.Spector N, Blackwell K, Hurley J, et al. EGF103009, a phase II trial of lapatinib monotherapy in patients with relapsed/refractory inflammatory breast cancer (IBC): clinical activity and biologic predictors of response. J Clin Oncol. 2006;24(18S):502. [Google Scholar]
- 30.Lin N, Carey L, Liu M, et al. Phase II trial of lapatinib for brain metastases in patients with HER2+ breast cancer. J Clin Oncol. 2006;24(18S):503. doi: 10.1200/JCO.2007.12.3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoneda T, Williams P, Hiraga T, Niewolna M, Nishimura R. A bone seeking clone exhibits different biological properties from the MDA-MB-231 parental human breast cancer cells and a brain-seeking clone in vivo and in vitro. J Bone Miner Res. 2001;16(8):1486–1495. doi: 10.1359/jbmr.2001.16.8.1486. [DOI] [PubMed] [Google Scholar]
- 32.Schulze W, Deng L, Mann M. Phosphotyrosine interactome of the ErbB-receptor kinase family. Mol Syst Biol. 2005;1(May):42–54. doi: 10.1038/msb4100012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5(5):341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 34.Warnberg F, White D, Anderson E, et al. Effect of farnesyl transfer inhibitor (R115777) on ductal carcinoma in situ of the breast in a human xenograft model and on breast and ovarian cancer cell growth in vitro and in vivo. Br Cancer Res. 2006;8(2):R21–R31. doi: 10.1186/bcr1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhan L, Xiang B, Muthuswamy SK. Controlled activation of ErbB1/ErbB2 heterodimers promote invasion of three-dimensional organized epithelia in an ErbB1-dependent manner: implications for progression of ErbB2-overexpressing tumors. Cancer Res. 2006;66(10):5201–5208. doi: 10.1158/0008-5472.CAN-05-4081. [DOI] [PubMed] [Google Scholar]
- 36.Gaedcke J, Traub F, Milde S, et al. Predominance of the basal type and HER-2/neu type in brain metastasis from breast cancer. Mod Pathol. 2007;20(8):864–870. doi: 10.1038/modpathol.3800830. [DOI] [PubMed] [Google Scholar]
- 37.DiGiovanna MP, Stern DF, Edgerton SM, Whalen SG, Moore D, II, Thor AD. Relationship of epidermal growth factor receptor expression to ErbB-2 signaling activity and prognosis in breast cancer patients. J Clin Oncol. 2005;23(6):1152–1160. doi: 10.1200/JCO.2005.09.055. [DOI] [PubMed] [Google Scholar]
- 38.Gschwantler-Kaulich D, Hudelist G, Koestler WJ, et al. EGFR activity in HER-2 over-expressing metastatic breast cancer: evidence for simultaneous phosphorylation of Her-2/neu and EGFR. Oncol Rep. 2005;14(2):305–311. [PubMed] [Google Scholar]
- 39.Grupka N, Lear-Kaul K, Kleinschmidt-DeMasters B, Singh M. Epidermal growth factor receptor status in breast cancer metastases to the central nervous system. Arch Pathol Lab Med. 2004;128(9):974–979. doi: 10.5858/2004-128-974-EGFRSI. [DOI] [PubMed] [Google Scholar]
- 40.Burris HA, III, Hurwitz HI, Dees EC, et al. Phase I safety, pharmacokinetics, and clinical activity study of lapatinib (GW572016), a reversible dual inhibitor of epidermal growth factor receptor tyrosine kinases, in heavily pretreated patients with metastatic carcinomas. J Clin Oncol. 2005;23(23):5305–5313. doi: 10.1200/JCO.2005.16.584. [DOI] [PubMed] [Google Scholar]
- 41.Konecny GE, Pegram MD, Venkatesan N, et al. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. 2006;66(3):1630–1639. doi: 10.1158/0008-5472.CAN-05-1182. [DOI] [PubMed] [Google Scholar]
- 42.Rusnak DW, Alligood KJ, Mullin RJ, et al. Assessment of epidermal growth factor receptor (EGFR, ErbB1) and HER2 (ErbB2) protein expression levels and response to lapatinib (Tykerb, GW572016) in an expanded panel of human normal and tumour cell lines. Cell Prolif. 2007;40(4):580–594. doi: 10.1111/j.1365-2184.2007.00455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chattopadhyay A, Vecchi M, Ji Q, Mernaugh R, Carpenter G. The role of individual SH2 domains in mediating association of phospholipase C-gamma1 with the activated EGF receptor. J Biol Chem. 1999;274(37):26091–26097. doi: 10.1074/jbc.274.37.26091. [DOI] [PubMed] [Google Scholar]
- 44.Luttrell DK, Lee A, Lansing TJ, et al. Involvement of pp60c-src with two major signaling pathways in human breast cancer. Proc Natl Acad Sci U S A. 1994;91(1):83–87. doi: 10.1073/pnas.91.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Agazie YM, Hayman MJ. Molecular mechanism for a role of SHP2 in epidermal growth factor receptor signaling. Mol Cell Biol. 2003;23(21):7875–7886. doi: 10.1128/MCB.23.21.7875-7886.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]