

Abstract
Molecular imaging is an important new direction in medical diagnosis; however, its success is dependent upon molecular probes that demonstrate selective tissue targeting. We report the design and chemical synthesis of a derivative of human amyloid-β (Aβ) peptide that is capable of selectively targeting individual amyloid plaques in the brain of Alzheimer’s disease transgenic mice after being intravenously injected. This derivative is based on the sequence of the first 30 amino acid residues of Aβ with asparagyl/glutamyl-4-aminobutane residues (N-4ab/Q-4ab) substituted at unique Asp and Glu positions and with Gd-DTPA-aminohexanoic acid covalently attached at the N-terminal Asp. The Gd[N-4ab/Q-4ab]Aβ30 peptide was homogeneous as shown by high-resolution analytical techniques with a mass of ±4385 Da determined by electrospray ionization mass spectrometry. This diamine- and gadolinium-substituted derivative of Aβ is shown to have enhanced in vitro binding to Alzheimer’s disease (AD) amyloid plaques and increased in vivo permeability at the blood–brain barrier because of the unique Asp/Glu substitutions. In addition, specific in vivo targeting to AD amyloid plaques is demonstrated throughout the brain of an APP, PS1 transgenic mouse after intravenous injection. Because of the magnetic resonance (MR) imaging contrast enhancement provided by gadolinium, this derivative should enable the in vivo MR imaging of individual amyloid plaques in the brains of AD animals or patients to allow for early diagnosis and also provide a direct measure of the efficacy of anti-amyloid therapies currently being developed.
Molecular imaging by magnetic resonance requires a molecular probe containing a contrast agent that is capable of selective tissue targeting in labeling a specific molecular entity within the tissue of interest which can be detected by magnetic resonance imaging (MRI). One of the pathological hallmarks of Alzheimer’s disease (AD) is the extracellular accumulation of amyloid-β (Aβ) peptide into plaques. These plaques are essential for the definitive diagnosis of AD, which is usually confirmed only post-mortem. At present, there is no method for direct imaging of individual β-amyloid plaques in humans that would provide a definitive pre-mortem diagnosis of this disease or a method of assessing disease progression. MRI has a spatial resolution of 30–50 μm, which at least theoretically, has the capacity to resolve individual plaques (the neuritic plaque size in an AD patient varies from 2 to 200 μm).
Radioiodinated human Aβ40 has been used as a molecular probe which binds to β-amyloid plaques both in vitro and in vivo (1). This in vivo binding of plaques was demonstrated with radioiodinated, polyamine-modified, human Aβ40 following intravenous injection into a transgenic mouse model of AD. Furthermore, by covalently attaching gadolinium-DTPA to polyamine-modified Aβ, we have been able to selectively enhance individual plaques by MRI performed on the ex vivo AD mouse brain at 7 T with a spatial resolution approximating plaque size (62.5 μm3) (2).
The ability to quantify the permeability of peptides and proteins at the blood–brain barrier (BBB) (3) has allowed the evaluation of different protein modifications that might be used to enhance this permeability (4–6). In particular, covalent modification with the naturally occurring polyamines, such as putrescine, spermidine, or spermine, has resulted in dramatic increases in the BBB permeability of a number of proteins (6–11). Indeed, polyamine modification of human Aβ40 as described above not only resulted in a significant increase in the BBB permeability but also resulted in enhanced binding to amyloid plaques in AD brain sections (1, 2).
The chemical modification reaction that we have been using to modify polyamine proteins has been to target carboxyl groups of aspartic and glutamic acid residues utilizing water-soluble carbodiimide. This two-step reaction sequence involves the condensation between carboxyl groups of proteins with a nucleophile, such as the polyamine putrescine. In the initial reaction, the carbodiimide adds to ionized carboxyl groups to form an O-acylisourea intermediate (12). Subsequent reaction of the intermediate with the amine yields the corresponding amide. Because this reaction depends on the ionization of the individual carboxyl groups, the extent of modification can be controlled by maintaining a desired pH to limit the ionization of the carboxyl groups and, hence, preserve the bioactivity of the protein. One of the difficulties with this reaction is that the reactive intermediate also undergoes hydrolysis slowly, which may in turn react with other nucleophiles to form different carboxylated derivatives. In particular, reaction with an amino group from a second protein or the same protein may lead to a cross-link between the two proteins. This is less problematic with high-molecular mass proteins; however, it can be more problematic with lower-molecular mass peptides or synthetic peptides lacking post-translational modifications, particularly those with unblocked N- and C-termini. This is particularly evident for a peptide such as Aβ40, which readily forms aggregates and fibrils leading to increased insolubility.
One way to avoid the problems associated with carbodiimide-mediated modification is to create unique protein isoforms by directly synthesizing the amine-modified carboxyl groups of glutamic and aspartic acid to create a glutamyl-4-aminobutane or asparagyl-4-aminobutane (Figure 1) which is then incorporated into the synthesis of the protein. By substitution of glutamic acid at positions 3, 11, and 22 with glutamyl-4-aminobutane and substitution of aspartic acid at positions 7 and 23 with asparagyl-4-aminobutane, the same derivative can be synthesized without the inherent problems of peptide cross-linking and decreased solubility. Of course, the number and position of this diamine substitution within the peptide will determine its BBB permeability and its ability to efficiently target amyloid plaques. In the study presented here, we characterize this diamine- and Gd-DTPA-substituted Aβ1–30 by mass spectrometry, protein electrophoresis, its blood–brain barrier (BBB) permeability, its ability to bind plaques in AD tissue sections, and also its ability to target amyloid plaques in the AD mouse after intravenous injection. Our focus in this investigation is on Aβ1–30, because the cell surface binding domain (Aβ31–34) is excluded and the neurotoxic domain (Aβ25–35) is truncated (13). It is particularly important to develop a derivative of Aβ that is nontoxic as this contrast agent might have clinical application for the definitive pre-mortem diagnosis of AD in human patients.
Figure 1.

Molecular structure of diamine-substituted Aβ1–30. Amino acid sequence of Aβ peptides and chemical structure of modified glutamine and asparagine residues.
EXPERIMENTAL PROCEDURES
Subjects
These evaluations were performed using transgenic mice that express two mutant human proteins associated with familial AD and have been described in detail elsewhere (14). Hemizygous transgenic mice (Tg2576) expressing mutant human amyloid precursor protein (APP695) (15) were mated with a second strain of hemizygous transgenic mice (M146L6.2) expressing mutant human presenilin 1 (PS1) (14). The animals were genotyped for the expression of both transgenes by a PCR method using a sample of mouse tail DNA. The mice were housed in a virus-free barrier facility under a 12 h light-dark cycle, with ad libidum access to food and water. All procedures were performed in accordance with the NIH Guidelines for the Care and Use of Laboratory Animals.
Synthesis Reactions
Unless otherwise stated, all reactions were carried out under an argon or nitrogen atmosphere. Commercially available materials were used without purification. The dichloromethane was distilled over CaH2. The diethyl ether and tetrahydrofuran were dried by distillation over sodium benzophenone ketyl. NMR spectra were recorded with a Brüker Avance-300 instrument at 300 MHz for 1H NMR. Chemical shifts are given using tetramethyl-silane as an internal standard. Kieselgel 60 (230–400 mesh) silica gel was used in the flash chromatography.
Preparation of N-α-Fmoc-L-aspartyl-γ-N-(4-aminobutyl)-carbamic Acid tert-Butyl Ester (3a)
The N-α-Fmoc-L-aspartyl α-allyl ester (1a) (3.67 g, 9.276 mmol) was dissolved in 100 mL of acetonitrile (ACN). To this mixture, under an inert atmosphere, were sequentially added the BOP reagent (4.30 g, 9.74 mmol) and diisopropylethylamine (DIPEA, 3.31 mL, 18.55 mmol). After being stirred at room temperature (RT) for 5 min, the mixture was cooled to 0 °C. While the mixture was being stirred, N-(4-aminobutyl)carbamic acid tert-butyl ester (1.75 g, 9.28 mmol) was added. The reaction mixture was stirred for 1 h. The ACN was removed under vacuum, and the solid residue was dissolved in 100 mL of water. After acidification of the solution to pH 3 with 1 N HCl, the aqueous phase was extracted with dichloromethane (DCM) three times, and the combined DCM layers were washed first with aqueous NaHCO3 and then with brine and dried (Na2SO4). Addition of 200 mL of hexane to the DCM solution and cooling it at −14 °C for 30 min resulted in the formation of a precipitate which was filtered and lyophilized. The product N-α-Fmoc-L-aspartyl-β-N-(4-aminobutyl)carbamic acid tert-butyl ester α-allyl ester (2a) (5.1 g, 97.2%) was obtained as a white solid: 1H NMR (CDCl3) δ 1.43 (9H, s), 1.57 (4H, m), 2.74 (1H, dd, J = 15.8, 4.2 Hz), 2.98 (1H, dd, J = 16.1, 4.4 Hz), 3.09 (2H, d, J = 4.9 Hz), 3.41 (2H, t, J = 5.1 Hz), 4.6 (2H, m), 4.67 (2H, d, J = 5.47 Hz), 5.24 (2H, dd, J = 25.1, 17.2 Hz), 5.9 (2H, m), 6.17 (1H, d, J = 8.39 Hz), 7.31 (2H, t, J = 7.24 Hz), 7.41 (2H, t, J = 7.24 Hz), 7.61 (2H, d, J = 7.29 Hz), 7.76 (2H, d, J = 7.4 Hz).
The allyl ester (2a) (5.00 g, 8.60 mmol) and Pd(Ph3)4 (400 mg, 0.39 mmol) were suspended in 40 mL of THF, and the suspension was stirred at RT for 5 min. p-Toluenesulfinic acid (as sodium salt, 1.70 g, 9.46 mmol) dissolved in 20 mL of water was added, and the heterogeneous mixture was stirred for 2 h at RT. Removal of the THF in vacuo and washing the aqueous layer with ether (three times) left a colored aqueous layer which was decolorized after being treated with hot charcoal for 10 min and filtered through Celite. Then, the clear aqueous solution was cooled to 5 °C and acidified to pH 3 with 1 N HCl. The precipitate was isolated by centrifugation and dried overnight under high vacuum. The N-α-Fmoc-L-aspartyl-β-N-(4-aminobutyl)carbamic acid tert-butyl ester (3a) (3.7 g, 79.46%) was obtained as an off-white solid: 1H NMR (CDCl3) δ 1.42 (9H, s), 1.53 (4H, m), 2.71 (1H, dd, J = 16.28, 8.6 Hz), 2.98 (1H, d, J = 15.1 Hz), 3.19 (2H, br s), 3.27 (1H, br s), 3.37 (1H, br s), 4.21 (1H, t, J = 6.64 Hz), 4.36 (2H, t, J = 8.77 Hz), 4.51 (1H, d, J = 6.11 Hz), 6.24 (1H, br s), 6.68 (1H, br s), 7.31 (2H, t, J = 7.24 Hz), 7.41 (2H, t, J = 7.24 Hz), 7.61 (2H, d, J = 7.29 Hz), 7.76 (2H, d, J = 7.4 Hz).
Preparation of N-α-Fmoc-L-glutamyl-δ-N-(4-aminobutyl)-carbamic Acid tert-Butyl Ester (3b)
Following the same procedure as described above for 3a, N-α-Fmoc-L-glutamyl acid γ-N-(4-aminobutyl)carbamic acid tert-butyl ester α-allyl ester (2b) was obtained as a white powder (6.9 g, 97.5%) from N-α-Fmoc-L-glutamyl α-allyl ester (5.00 g, 12.19 mmol): 1H NMR (CDCl3) δ 1.42 (9H, s), 1.52 (4H, m), 1.82 (1H, s), 2.24 (3H, s), 3.11 (2H, d, J = 5.8 Hz), 3.26 (2H, t, J = 5.9 Hz), 4.19 (1H, t, J = 6.85 Hz), 4.41 (3H, m), 4.64 (3H, d, J = 5.67 Hz), 5.27 (2H, dd, J = 17.2, 12.71 Hz), 5.81 (1H, d, J = 14.44 Hz), 5.89 (1H, m), 6.11 (1H, br s), 7.31 (2H, t, J = 7.24 Hz), 7.41 (2H, t, J = 7.24 Hz), 7.61 (2H, d, J = 7.29 Hz), 7.76 (2H, d, J = 7.4 Hz).
The allyl group in 2b was removed using the procedure outlined above to furnish N-α-Fmoc-L-glutamyl-γ-N-(4-aminobutyl)carbamic acid tert-butyl ester (3b) as a pale yellow powder (5.4 g, 87.7%) from the corresponding allyl ester (6.60 g, 11.07 mmol): 1H NMR (CDCl3) δ 1.40 (9H, s), 1.51 (4H, m), 1.85 (1H, d, J = 4.52 Hz), 2.06 (1H, d, J= 6.3 Hz), 2.42 (2H, d, J = 17.5 Hz), 3.09 (2H, d, J = 4.67 Hz), 3.27 (2H, br s), 4.19 (1H, t, J = 6.674 Hz), 4.35 (3H, d, J = 4.85 Hz), 4.81 (1H, br s), 6.08 (1H, br s), 6.74 (1H, br s), 7.31 (2H, t, J = 7.24 Hz), 7.41 (2H, t, J = 7.24 Hz), 7.61 (2H, d, J = 7.29 Hz), 7.76 (2H, d, J = 7.4 Hz).
Synthesis of Diamine- and Gd-Substituted Aβ Derivatives
Aβ1–30, with the sequence Ahx-DAEFRHDSGYEVH-HQKLVFFAEDVGSNKGA (Ahx) Fmoc-6-aminohexanoic acid), was synthesized on an ABI 433 (Foster City, CA) peptide sythesizer using HBTU activation and the manufacturer’s suggested synthesis protocols. The starting resin was Ala-NovaSyn TGA (Calbiochem-Novabiochem, San Diego, CA). Glutamic acid residues 3, 11, and 22 were synthesized with N-α-Fmoc-L-glutamyl-δ-N-(4-aminobutyl)carbamic acid tert-butyl ester (3b), and aspartic acid residues 7 and 23 were synthesized with N-α-Fmoc-L-aspartyl-γ-N-(4-aminobutyl)-carbamic acid tert-butyl ester described above. After completion of the synthesis and final Fmoc deprotection, diethyl-enetriaminepentaacetic acid anhydride (DTPA) was added to the N-terminal Ahx residue by dissolving 120 mg of the DTPA in 2 mL of DMSO and 8 mL of DMF and reacting the DTPA solution with the peptide resin, which had been washed previously with DIEA and DCM. The coupling of DTPA was allowed to proceed overnight at RT. Completion of the reaction was verified by a negative ninhydrin reaction. The Aβ1–30 peptide was then cleaved from the resin support using 5% crystalline phenol, 5% water, 2.5% triisopropyl-silane, and 87.5% TFA for 2 h at RT. The peptide was purified by reverse-phase HPLC on a C18 Jupiter column (250 mm × 21.2 mm, Phenomonex Corp.) using a gradient system of 0.1% aqueous TFA containing 80% acetonitrile. The calculated mass of 3390 amu for Aβ1–30 and 4231 amu for DTPA[N-4ab/Q-4ab]Aβ30 was confirmed by electrospray ionization mass spectrometry (Sciex API 165). The element gadolinium (Gd) was chelated at an equimolar concentration to the DTPA functional group of the Aβ1–30 peptide using Gd(III) chloride hexahydrate (Sigma) in water at RT for 1 h (now designated Gd[N-4ab/Q-4ab]Aβ30) (Figure 1). Gadolinium- and diamine-substituted Aβ1–30 mixtures were analyzed by electrospray ionization mass spectrometry on a Sciex 165B instrument (Perkin-Elmer). Approximately 2.5 μL of each Gd/peptide mixture was injected utilizing the following parameters: start m/z of 500, stop m/z of 2000, and a dwell time of 0.1 ms at 40 scans/min. The deconvolution of each spectrum into a single mass was performed using the BioToolBox program supplied by the manufacturer. All Aβ peptides were labeled with 125I or 131I using a modified chloramine T procedure as described previously (16).
Determination of PS and Vp in Mice
PS and Vp measurements were performed as described previously (6–9, 17). This involved the IV bolus injection technique in which a bolus of PBS containing 125I-labeled protein was rapidly injected into the catheterized femoral vein of anesthetized mice (isoflurane, 1.5%). Blood was sampled from the femoral artery at several intervals over the next 15 min. Whole blood was sampled directly, using heparinized micro-hematocrit capillary tubes, and TCA extracted. The supernatant was separated from the pellet, and both were counted in a gamma counter. The radioactivity in the pellet was expressed as a percentage of the total radioactivity found in both the pellet and supernatant (17). An aliquot of the peptide labeled with 131I was then injected into the femoral vein 15 s prior to the sacrifice of the animal to serve as a measure of residual plasma volume (Vp, in microliters per gram). After collection of the final blood sample, the anesthetized animal was sacrificed. Several brain regions were dissected and assayed for 125I and 131I radioactivity in a two-channel gamma counter (Cobra II, Packard) with the activity corrected for background and crossover of I activity into the 125I channel. The products of the permeability coefficient and the surface area (PS, in ×10−6 milliliters per gram per second) were then calculated using the Vp as a measure of residual plasma volume based on equations that have been discussed in detail elsewhere (6–9, 17). Statistical evaluations of PS and Vp were performed using analysis of variance (ANOVA) followed by Bonferroni multiple comparisons.
Labeling of Amyloid Plaques in Human AD and APP, PS1 Mouse Brain Sections in Vitro with Diamine- and Gadolinium-Substituted Aβ30 Peptides
HPLC-purified [125I]Aβ40, [125I]Aβ30, [125I][N-4ab/Q-4ab]Aβ30, [125I]Gd[N-4ab/Q-4ab]Aβ30, or buffer was incubated with sections of unfixed AD temporal lobe cortex using the same procedure we used previously (18). Briefly, the 15 μm sections were incubated for 3 h at RT with 100 pM radioiodinated peptide or alone in 250 μL of TBS [50 mM Tris-HCl and 138 mM sodium chloride (pH 7.0)] containing 0.1% BSA, 0.6 mg/mL magnesium chloride, 0.04 mg/mL bacitracin, 0.002 mg/mL chymostatin, and 0.004 mg/mL leupeptin. The sections then underwent immunohistochemistry (IH) for amyloid using an anti-Aβ monoclonal mouse antibody (4G8, 1:1000, Signet Laboratories, Dedham, MA). Next, the sections were dipped in an autoradiographic emulsion (Type NTB-3, Kodak, Rochester, NY) for direct comparison of 125I-labeled amyloid deposits to anti-Aβ IH. The slides were dipped in emulsion, exposed for various durations, and developed according to the instructions. The sections were dehydrated with successive changes of ethanol and xylene and then coverslipped.
[125I]Aβ40 or [125I]Gd[N-4ab/Q-4ab]Aβ30 was also incubated in vitro with brain sections from APP, PS1 mice to verify that the radioiodinated peptides label amyloid deposits in AD transgenic mice in the same manner that they label amyloid plaques in human AD brain sections. Briefly, unfixed, frozen 15 μm brain sections from APP, PS1 mice at 12 and 52 weeks of age, or a nontransgenic mouse, were incubated with [125I]Aβ40, [125I]Gd[N-4ab/Q-4ab]Aβ30, or buffer as described above. The sections then underwent anti-Aβ IH and emulsion autoradiography as described above.
Labeling of Amyloid Plaques in Vivo
APP, PS1 transgenic mice (21 months of age) were catheterized in the femoral vein under general anesthesia (isoflurane, 1.5%) and injected with 750 μg of HPLC-purified [125I]Aβ40, [125I]Aβ30, [125I]-[N-4ab/Q-4ab]Aβ30, or [125I]Gd[N-4ab/Q-4ab]Aβ30. The specific activities of the HPLC-purified radiolabeled peptides were equal (8 μCi/μg). After 4 h, each animal was given an overdose of sodium pentobarbital (200 mg/kg, IP) and perfused with PBS, followed by neutral-buffered, 10% formalin, and then 10% sucrose in 0.1 M sodium phosphate (pH 7.2). Frozen sections (15 μm) of each brain were cut with a cryostat and then processed with anti-Aβ IH and emulsion autoradiography for the presence of radiolabeled amyloid deposits using the same methods described above. Silver grains from sections exposed for 6 days and 2 weeks were quantitated using unbiased, stereological techniques. Sections exposed for 4 or 8 weeks were somewhat overexposed, and as a result, over many of the plaques the exposed silver grains had become confluent and could not be counted accurately. Silver grains were counted over plaques in three sections from each animal in the retrosplenial cortex and CA1 region of the hippocampus using a 10 μm × 10 μm dissector at 400×. The mean background level of exposed silver grains was also determined for each section. The background was sampled over the tissue adjacent to each plaque that was analyzed and was on average 1.36 and 1.60 silver grains/100 μm2 after 6 days of exposure in the cortex and hippocampus, respectively, and 1.99 and 2.54 silver grains/100 μm2, respectively, after 2 weeks. The results were expressed as the mean number of silver grains per 100 μm2 minus the background. Since the specific activities of the radiolabeled peptides were equal, the grain counts did not need further correction. Statistical analysis was then performed using ANOVA followed by Bonferroni multiple comparisons.
RESULTS
Autoradiography following SDS-PAGE of [125I]Aβ Derivatives
Aliquots of radioiodinated Aβ40, Aβ30, [N-4ab/Q-4ab]Aβ30, and Gd[N-4ab/Q-4ab]Aβ30 containing 1000 dpm were run on SDS-polyacrylamide gel electrophoresis at concentrations of 15% T and 1% C until the indicator dye was 1 cm from the bottom of the gel. As shown in Figure 2, single bands were observed for each of the Aβ derivatives. This concentration of acrylamide does not have the resolving capacity to distinguish the differences in the molecular masses of the different derivatives.
Figure 2.

Autoradiography after SDS-PAGE of [125I]Aβ derivatives. SDS-polyacrylamide gel electrophoresis of radioiodinated Aβ peptides and derivatives.
Gadolinium Chelation to DTPA[N-4ab/Q-4ab]Aβ30 EValuated by Electrospray Ionization Mass Spectrometry
The relative quantitation of chelation of Gd to the DTPA- and diamine-substituted Aβ30 was evaluated by electrospray ionization mass spectroscopy (Figure 3). Increasing the ratio of Gd to DTPA[N-4ab/Q-4ab]Aβ30 showed a shift in the atomic mass unit from 4231 for DTPA[N-4ab/Q-4ab]Aβ30 to 4385, the value expected for Gd-DTPA[N-4ab/Q-4ab]Aβ30. These data demonstrate mole concentrations of Gd equal to that of the DTPA functional group of the Aβ30 peptide were sufficient for complete chelation of the peptide.
Figure 3.

Relative quantitation of Gd chelation to DTPA[N-4ab/Q-4ab]Aβ30 by electrospray ionization mass spectrometry. Mixtures of increasing mole ratios of Gd to DTPA[N-4ab/Q-4ab]Aβ30 were analyzed: (A) 0/1, (B) 0.25/1, (C) 0.5/1, (D) 0.75/1, and (E) 1/1. See the text for details.
BBB Permeability of the Radioiodinated Aβ Derivatives in the Normal Adult Mouse
Aβ30 had PS values ranging from 102 to 136 × 10−6 mL g−1s−1 in the six different brain regions (Table 1). The diamine-substituted Aβ30 had PS values ranging from 121 to 175 × 10−6 mL g−1s−1, which were 1.1–1.3-fold greater than the PS values for Aβ30. Gadolinium-substituted Aβ30 exhibited a significant 50–60% decrease in permeability relative to that of Aβ30. This decrease in permeability was at least partially rescued by the diamine substitution with the diamine- and gadolinium-substituted derivative displaying PS values that ranged from 66 to 103 × 10−6 mL g−1s−1, or 20–40% lower than that of Aβ30. In contrast, the Vp values were not significantly different among these derivatives compared to that of Aβ30. Although the gadolinium- and diamine-substituted derivative exhibited a PS value decreased relative to that of Aβ30, this value was still 1.5–1.8 times significantly higher than that of Aβ40 (Table 2), whose BBB transport is receptor-mediated (19). The high PS value for this derivative, therefore, may facilitate its delivery across the blood-brain barrier and, ultimately, its ability to target amyloid plaques in the AD transgenic mouse brain.
Table 1.
PS and Vp Aβ30, [N-4ab/Q-4ab]Aβ30, Gd Aβ30, and Gd[N-4ab/Q-4ab]Aβ30 at the BBB in the Normal Adult Mouse (B6SJL)a
| brain region | Aβ30 (n = 11) | [N-4ab/Q-4ab]Aβ30 (n = 5) | RI | P | GdAβ30 (n = 9) | RI | P | Gd[N-4ab/Q-4ab]Aβ30 (n = 6) | RI | P |
|---|---|---|---|---|---|---|---|---|---|---|
| PS | ||||||||||
| cortex | 117.5 ± 4.7 | 139.6 ± 9.4 | 1.2 | nsb | 49.8 ± 5.8 | 0.4 | e | 79.2 ± 4.1 | 0.7 | e |
| caudate-putamen | 101.8 ± 4.1 | 121.2 ± 5.9 | 1.2 | c | 39.7 ± 3.7 | 0.4 | e | 65.8 ± 4.7 | 0.7 | e |
| hippocampus | 113.8 ± 5.2 | 139.6 ± 10.3 | 1.2 | nsb | 50.7 ± 6.5 | 0.4 | e | 89.4 ± 6.2 | 0.8 | nsb |
| thalamus | 136.0 ± 6.1 | 147.0 ± 9.2 | 1.1 | nsb | 61.0 ± 8.7 | 0.4 | e | 84.4 ± 3.8 | 0.6 | e |
| brain stem | 134.4 ± 6.1 | 149.0 ± 9.2 | 1.1 | nsb | 62.0 ± 8.0 | 0.5 | e | 92.2 ± 5.9 | 0.7 | d |
| cerebellum | 131.9 ± 5.0 | 174.6 ± 14.1 | 1.3 | d | 60.2 ± 6.8 | 0.5 | e | 102.8 ± 7.4 | 0.8 | nsb |
| Vp | ||||||||||
| cortex | 24.0 ± 1.4 | 20.4 ± 1.2 | 0.9 | nsb | 23.3 ± 2.3 | 1.1 | nsb | 22.6 ± 1.1 | 0.9 | nsb |
| caudate-putamen | 12.3 ± 0.7 | 11.2 ± 0.8 | 0.9 | nsb | 14.6 ± 4.2 | 1.2 | nsb | 14.5 ± 1.2 | 1.2 | nsb |
| hippocampus | 25.6 ± 1.6 | 24.2 ± 2.1 | 1.0 | nsb | 26.7 ± 2.2 | 1.0 | nsb | 29.1 ± 1.4 | 1.1 | nsb |
| thalamus | 25.8 ± 1.4 | 19.3 ± 0.9 | 0.8 | nsb | 26.8 ± 2.3 | 1.0 | nsb | 22.6 ± 1.0 | 0.9 | nsb |
| brain stem | 32.7 ± 2.2 | 23.8 ± 1.8 | 0.7 | nsb | 37.8 ± 3.7 | 1.2 | nsb | 26.7 ± 1.8 | 0.8 | nsb |
| cerebellum | 31.0 ± 2.2 | 31.7 ± 2.3 | 1.0 | nsb | 36.5 ± 3.4 | 1.2 | nsb | 34.9 ± 1.0 | 1.1 | nsb |
PS is the product of the permeability coefficient and the surface area (×10−6 milliliters per gram per second, x̄ ± (SEM) determined with [125I]Aβ30 derivatives over the course of 15 min and corrected for Vp. Vp is the residual brain region blood volume (microliters per gram, x̄ ± SEM) determined with [131I]Aβ30 derivatives given 15–30 s prior to the end of the experiment. RI is the relative increase vs Aβ30P. P is the ANOVA with Bonferroni multiple comparisons vs Aβ30.
Not significant (P > 0.05).
P < 0.05.
P < 0.01.
P < 0.001.
Table 2.
PS of Aβ40, Aβ30, and Gd[N-4ab/Q-4ab]Aβ30 at the BBB in the Normal Adult Mouse (B6SJL)a
| brain region | Aβ40 (n = 14) | Aβ30 (n = 11) | RI | P | Gd[N-4ab/Q-4ab]Aβ30 (n = 6) | RI | P |
|---|---|---|---|---|---|---|---|
| cortex | 52.3 ± 3.4 | 117.5 ± 4.7 | 2.2 | b | 79.2 ± 4.1 | 1.5 | b |
| caudate-putamen | 41.0 ± 3.2 | 101.8 ± 4.1 | 2.5 | b | 65.8 ± 4.7 | 1.6 | c |
| hippocampus | 48.7 ± 3.2 | 113.8 ± 5.2 | 2.3 | b | 89.4 ± 6.2 | 1.8 | b |
| thalamus | 50.8 ± 3.4 | 136.0 ± 6.1 | 2.7 | b | 84.4 ± 3.8 | 1.7 | b |
| brain stem | 61.6 ± 4.1 | 134.4 ± 6.1 | 2.2 | b | 92.2 ± 5.9 | 1.5 | c |
| cerebellum | 65.5 ± 4.5 | 131.9 ± 5.0 | 2.0 | b | 102.8 ± 7.4 | 1.6 | b |
PS is the product of the permeability coefficient and the surface area (×10−6 milliliters per gram per second, (x̄ ± SEM) determined over the course of 15 min. RI is the relative increase vs Aβ40. P is the ANOVA with Bonferroni multiple comparisons vs Aβ40.
P < 0.001.
P < 0.01.
Labeling of Amyloid Plaques in Human AD Brain Sections in Vitro
HPLC-purified [125I]Aβ40, [125I]Aβ30, [125I][N-4ab/Q-4ab]Aβ30, or [125I]Gd[N-4ab/Q-4ab]Aβ30 was incubated with sections of unfixed AD temporal lobe cortex (Figure 4). [125I]Aβ40 labeled most dense-core, neuritic-type plaques (Figure 4C,D). Since this has been reported previously by us (1) and others (20), [125I]Aβ40 served as a positive control for this study. [125I]Aβ30 labeled only a very few neuritic plaques in human AD sections (Figure 4E,F), even with exposure times of >1 month. [125I][N-4ab/Q-4ab]Aβ30, however, labeled as many neuritic plaques as [125I]Aβ40 (Figure 4G,H). [125I][N-4ab/Q-4ab]Aβ30 also labeled many small amyloid deposits, while [125I]Aβ40 labeled only a few. Furthermore, relatively more [125I][N-4ab/Q-4ab]Aβ30 bound to plaques than did [125I]Aβ40 in the presence of equal amounts of peptide. This is apparent because while it required 7 days to detect exposed silver grains with [125I]Aβ40, it required only 1 day to expose an equal or greater number of silver grains with [125I][N-4ab/Q-4ab]Aβ30. [125I]Gd[N-4ab/Q-4ab]Aβ30, which includes the MRI contrast enhancement agent, Gd, showed the same pattern of labeling as [125I][N-4ab/Q-4ab]Aβ30, with only a slight reduction in the relative amount of exposed silver grains (Figure 4I,J).
Figure 4.
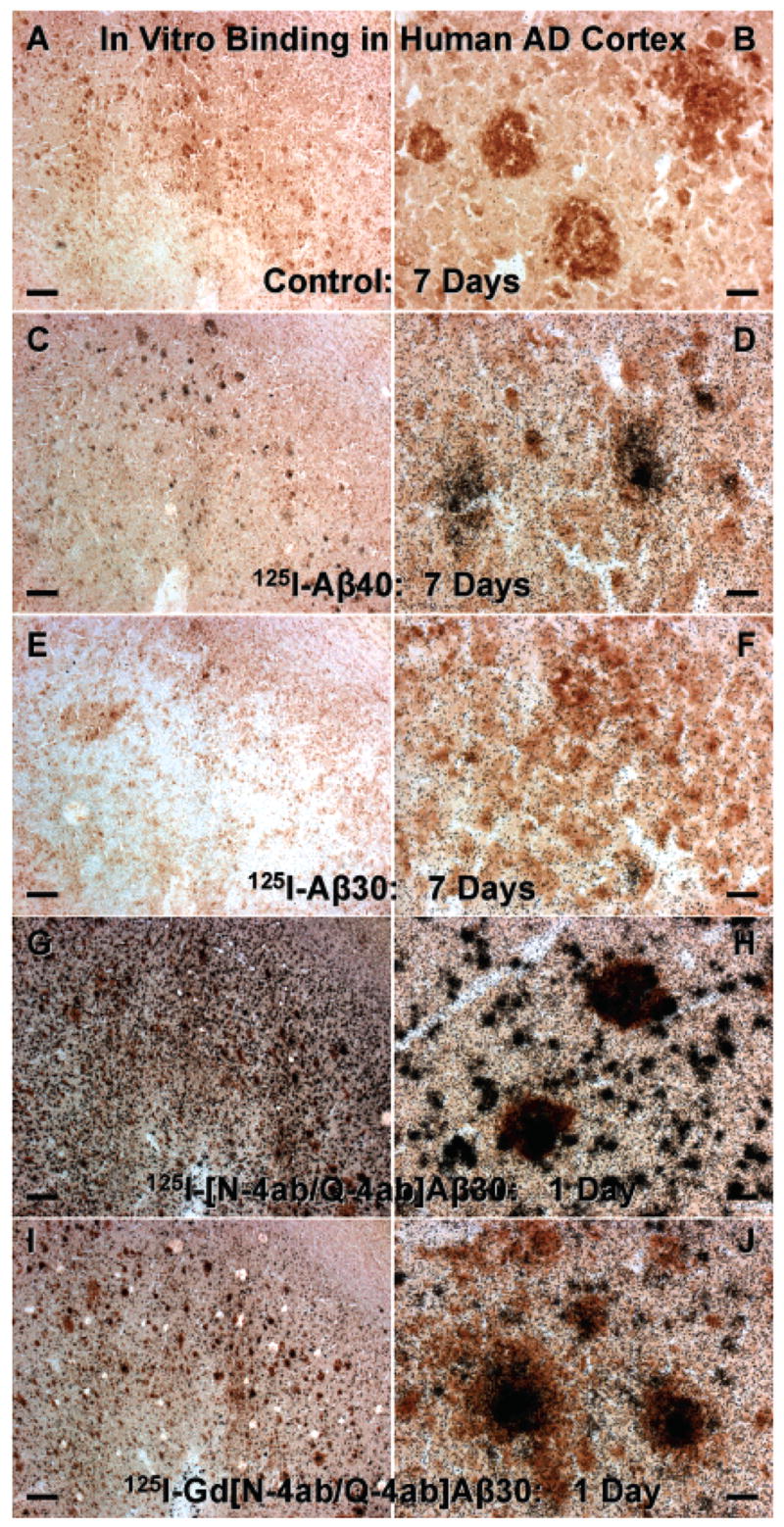
Labeling of amyloid plaques in human AD brain sections in vitro. (A and B) Unfixed temporal lobe cortex section incubated with buffer only and processed for anti-Aβ IH and emulsion autoradiography (exposed for 7 days). (C and D) Sections incubated with 100 pM [125I]Aβ40 (exposed for 7 days). (E and F) Section incubated with 100 pM [125I]Aβ30 (exposed for 7 days). (G and H) Section incubated with 100 pM [125I][N-4ab/Q-4ab]Aβ30 (exposed for 1 day). (I and J) Section incubated with 100 pM [125I]-Gd[N-4ab/Q-4ab]Aβ30 (exposed for 1 day). For panels A, C, E, G, and I, the scale bar is 250 μm. For panels B, D, F, H, and J, the scale bar is 25 μm.
Labeling of Amyloid Plaques in APP, PS1 Transgenic Mouse Brain in Vitro
[125I]Aβ40 or [125I]Gd[N-4ab/Q-4ab]Aβ30 was then incubated in vitro with brain sections from APP, PS1 mice to verify that the radioiodinated peptides label amyloid deposits in AD transgenic mice in the same manner that they label amyloid plaques in human AD brain sections (Figure 5). In contrast to the results with human AD sections, [125I]Aβ40 and [125I]Gd[N-4ab/Q-4ab]Aβ30 displayed very similar patterns of plaque labeling. With equal amounts of peptide, both labeled most of the neuritic plaques and required the same duration of exposure (1 day) to achieve a detectable amount of silver grains. The numerous small amyloid deposits present in the human AD sections were far less numerous in the APP, PS1 mouse sections, however. The relative increase in the level of labeling of [125I]Aβ40 in mouse sections compared to human sections could be ascribed to slight differences in the Aβ peptides that make up the plaques. It has been reported that the Aβ peptides contained in human plaques display a great deal of post-translational modifications such as N-terminal degradation and oxidation while those in mouse plaques do also, but to a far lesser extent (21). Therefore, there is a larger proportion of intact, unmodified Aβ40 for [125I]Aβ40 to bind to in mouse plaques than in human plaques. The reason [125I][N-4ab/Q-4ab]Aβ30 and [125I]Gd[N-4ab/Q-4ab]Aβ30 displayed increased levels of labeling in human sections in vitro compared to [125I]Aβ40 and [125I]Aβ30 is the addition of the diamine moieties. In previous studies, polyamine-modified [I]Aβ40 displayed an increased level of labeling of plaques compared to unmodified [125I]Aβ40, in both human AD sections in vitro and APP, PS1 mouse brain in vivo (1, 2). In competitive binding studies, we have shown that the polyamine itself does not bind to plaques (1). Alternatively, the addition of the diamine moieties to the peptide may slightly alter the conformation of the peptide, increasing the exposure of the hydrophobic domain, so that it can bind more readily to the highly hydrophobic β-amyloid in the plaques.
Figure 5.

Labeling of amyloid plaques in APP, PS1 transgenic mouse brain in vitro. (A and B) Unfixed APP, PS1 mouse brain section incubated with buffer only and processed for anti-Aβ IH and emulsion autoradiography. (C and D) Section incubated with 100 pM [125I]Aβ40 (exposed for 1 day). (E and F) Section incubated with 100 pM [125I]Gd[N-4ab/Q-4ab]Aβ30 (exposed for 1 day). For panels A, C, and E, the scale bar is 100 μm. For panels B, D, and F, the scale bar is 10 μm.
Labeling of Amyloid Plaques in APP, PS1 Transgenic Mouse Brain in Vivo
The main purpose of this study was to determine if [125I]Gd[N-4ab/Q-4ab]Aβ30 labels amyloid plaques in APP, PS1 transgenic mouse brain in vivo for potential use as an MRI contrast enhancement agent. One requirement of a putative MRI contrast enhancement agent for AD is that it must selectively label plaques. Since MRI studies are very expensive, a preliminary study using radioiodinated probes and autoradiographic detection could determine if the probe labels plaques, but at a fraction of the cost. The results of this study show that [125I]Gd[N-4ab/Q-4ab]Aβ30 does indeed label plaques in APP, PS1 mouse brain in vivo following IV injection (Figures 6–9). Most plaques throughout the cortex and hippocampus were labeled. A few labeled plaques could already be detected above background after only 1 day of exposure, and after 6 days of exposure, all labeled plaques were clearly apparent above background. After 4–8 weeks, the exposed silver grains reached confluence over many of the plaques and >90% of all plaques in the cortex and hippocampus had detectable labeling. Plaques near blood vessels and ventricles tended to be somewhat more intensely labeled. It is apparent from the results that [125I]Aβ40 and [125I][N-4ab/Q-4ab]Aβ30 also labeled plaques in vivo. As in the in vitro results, [125I]Aβ30 did not appear to label plaques at all, even after exposure for as much as 12 weeks. To determine if there was any difference in the plaque labeling efficiency of the peptides, exposed silver grains were then counted over plaques in the cortex and hippocampus.
Figure 6.
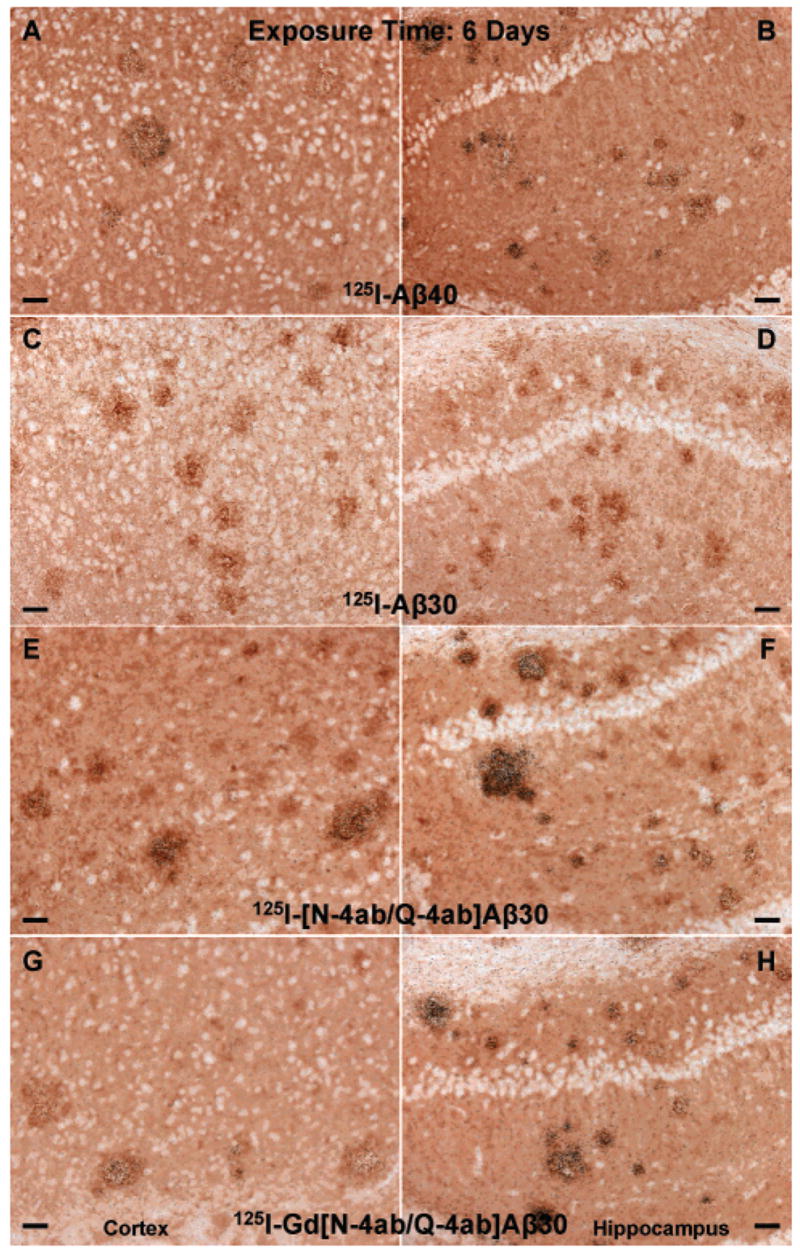
Labeling of amyloid plaques in APP, PS1 transgenic mouse brain in vivo after exposure for 6 days. Fixed, frozen brain sections from a 21-month-old APP, PS1 mouse after intravenous injection with 750 μg of [125I]Aβ40 (A and B), [125I]Aβ30 (C and D), [125I][N-4ab/Q-4ab]Aβ30 (E and F), or [125I]Gd[N-4ab/Q-4ab]Aβ30 (G and H) and after being processed for anti-Aβ IH and emulsion autoradiography. For panels A, C, E, and G (cortex), the scale bar is 50 μm. For panels B, D, F, and H (CA1 subfield of the hippocampus), the scale bar is 50 μm.
Figure 9.
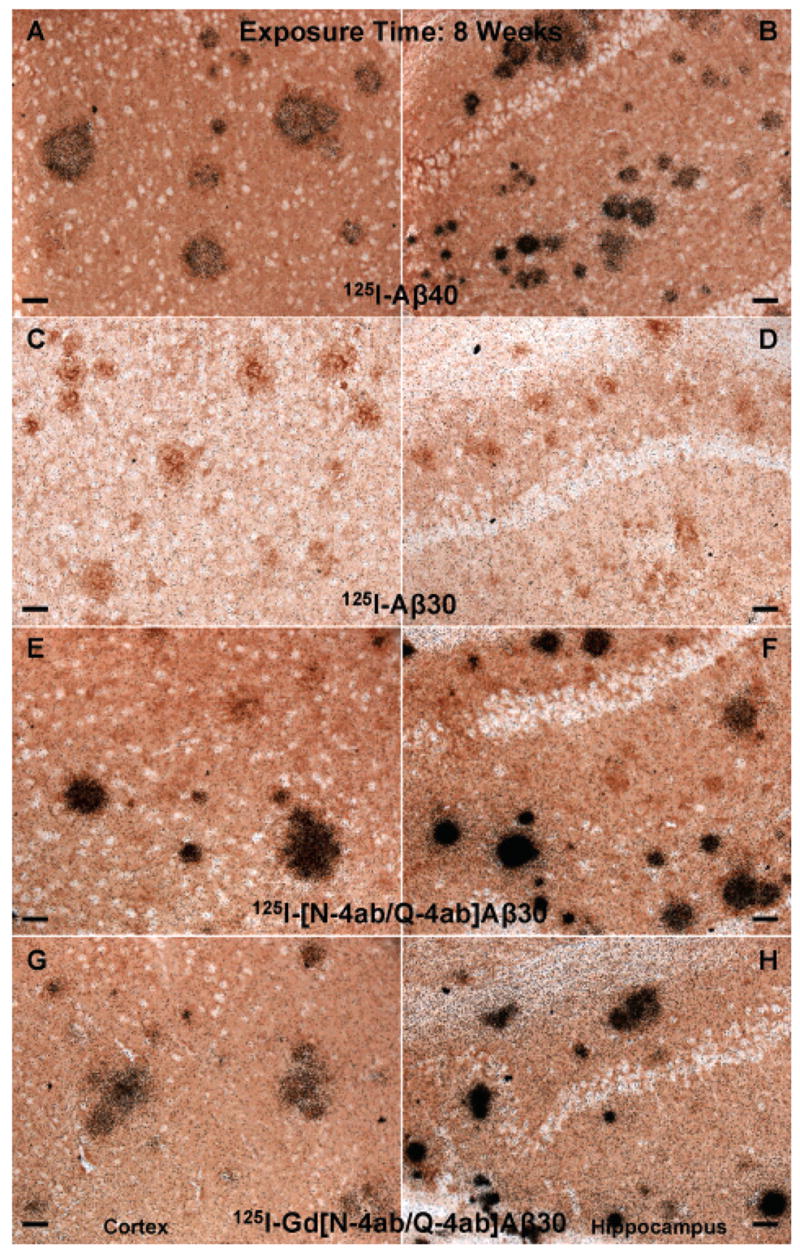
Labeling of amyloid plaques in APP, PS1 transgenic mouse brain in vivo after exposure for 8 weeks. Fixed, frozen brain sections from a 21-month-old APP, PS1 mouse after intravenous injection with 750 μg of [125I]Aβ40 (A and B), [125I]Aβ30 (C and D), [125I][N-4ab/Q-4ab]Aβ30 (E and F), or [125I]Gd[N-4ab/Q-4ab]Aβ30 (G and H) and after being processed for anti-Aβ IH and emulsion autoradiography. For panels A, C, E, and G (cortex), the scale bar is 50 μm. For panels B, D, F, and H (CA1 subfield of the hippocampus), the scale bar is 50 μm.
Silver grains from sections exposed for 6 days and 2 weeks were quantitated using unbiased, stereological techniques. The quantitative results revealed that the APP, PS1 mice treated IV with [125I]Gd[N-4ab/Q-4ab]Aβ30 and [125I][N-4ab/Q-4ab]Aβ30 generally had significantly more exposed silver grains over the plaques in the cortex and hippocampus than mice treated with [125I]Aβ40 (Figure 10). Two-way analysis of variance (four peptides with two exposure durations) of the number of exposed silver grains over plaques in the retrosplenial cortex indicated that there was a highly significant overall effect of peptides [F(3,208) = 36.49; P < 0.0001], as well as in the CA1 region of the hippocampus [F(3,208) = 41.52; P < 0.0001]. The mean number of exposed silver grains was greater for [125I][N-4ab/Q-4ab]Aβ30 and [125I]Gd[N-4ab/Q-4ab]Aβ30 than for [125I]Aβ40 in both brain regions and exposure durations. Half of those means reached significance according to Bonferroni multiple comparisons. The number of exposed silver grains for [125I]Aβ30 was similar to background and did not increase with exposure duration, indicating that no labeling occurred. The results of Bonferroni multiple comparisons of the individual means of [125I]Aβ40 versus [125I]Aβ30, [125I][N-4ab/Q-4ab]Aβ30, or [125I]Gd[N-4ab/Q-4ab]Aβ30 are given in Figure 10.
Figure 10.
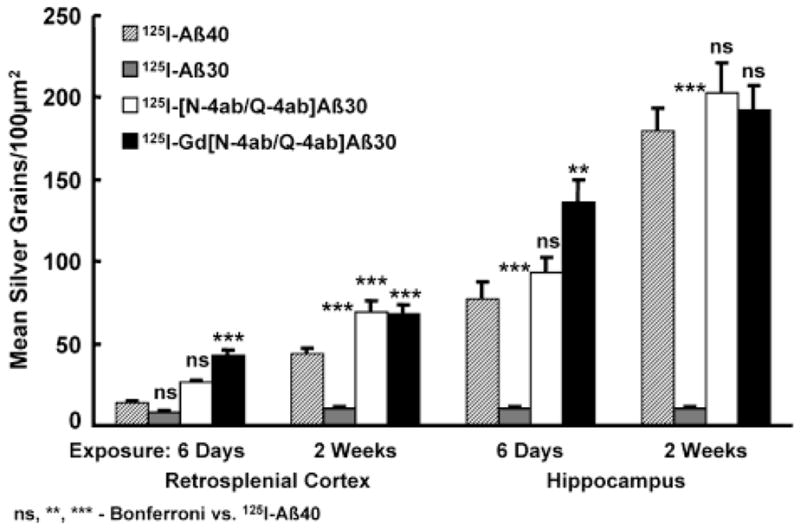
Quantitation of exposed silver grains on amyloid plaques in the retrosplenial cortex and hippocampus of 21-month-old APP, PS1 mice. On the ordinate is plotted the mean number of exposed silver grains per 100 μm2 quantitated by stereological methods. On the abscissa are plotted the brain region and exposure time. Analysis of variance was performed followed by Bonferroni tests of multiple comparisons. The results of Bonferroni comparisons of [125I]Aβ40 vs [125I]Aβ30 and its derivatives are included in the graph. See the text for details.
DISCUSSION
The purpose of this study was to develop a molecular probe capable of labeling amyloid plaques in vivo that is nontoxic and also provides contrast enhancement that can be detected by MRI for the potential use in clinical imaging for the pre-mortem diagnosis of AD in human patients. We have described such a probe, Gd[N-4ab/Q-4ab]Aβ30, that is a 30-amino acid derivative of Aβ40 which truncates the neurotoxic sequence of Aβ40, has select diamine substitutions to increase its BBB permeability and level of binding to plaques, and has a Gd-DTPA chelator arm to provide MRI contrast enhancement. In addition, the complete chemical synthesis of this probe eliminates formation of peptide cross-linking aggregates and insolubility that affected the carbodiimide-mediated modification of Aβ40 with putrescine. We have shown that although Gd-DTPA substitution decreases the BBB permeability of Aβ30, diamine substitution partially rescues the BBB permeability which is significantly higher than that of Aβ40. Furthermore, radioiodinated Gd[N-4ab/Q-4ab]Aβ30 labeled amyloid plaques in vitro in both human AD and APP, PS1 mouse brain sections. More importantly, Gd[N-4ab/Q-4ab]Aβ30 extensively labeled plaques throughout the brain of an APP, PS1 mouse in vivo following IV injection. These data provide promising results for the further development of Gd[N-4ab/Q-4ab]Aβ30 as a diagnostic probe for clinical MRI.
Diamine substitution enhances not only the permeability of [125I]Aβ30 at the BBB, as demonstrated by higher PS values, but also its ability to bind to amyloid plaques in human AD brain sections. While [125I]Aβ40 labeled amyloid plaques in human AD brain sections in vitro, as reported previously (1, 20), the study presented here showed that [125I]Aβ30 labeled only very few plaques. Diamine substitution, however, significantly enhanced the ability of [125I]Aβ30 to bind to amyloid plaques. Moreover, the enhanced level of labeling was greater than that of [125I]Aβ40, which required 7 days to expose a substantial amount of silver grains, compared to only 1 day for [125I][N-4ab/Q-4ab]Aβ30 or [125I]Gd[N-4ab/Q-4ab]Aβ30. This is not due to binding of the diamine itself to the amyloid, however. In a previous study, we demonstrated that when putrescine-modified [125I]Aβ40 was co-incubated on human AD brain sections with a 10-fold excess of cold putrescine, there was no decrease in the amount of exposed silver grains (1). It is possible that diamine substitution induces a conformational change in the peptide that enlarges its hydrophobic domain and thus its affinity for the highly hydrophobic amyloid plaques.
When [125I]Aβ40 or [125I]Gd[N-4ab/Q-4ab]Aβ30 was incubated with APP, PS1 mouse brain sections in vitro, however, a slightly different result was observed. Both peptides demonstrated relatively equal labeling of amyloid plaques, both requiring only 1 day of exposure. [125I]Aβ40, therefore, demonstrated enhanced binding to amyloid plaques of APP, PS1 mouse brain sections compared to human AD sections. As discussed briefly in the Results, the Aβ peptides contained within AD transgenic mouse plaques have a much lower extent of post-translational modifications such as N-terminal degradation and oxidation (21). Therefore, there is a larger proportion of intact, unmodified Aβ40 in mouse plaques, allowing more [125I]Aβ40 to bind than in human AD plaques, and thus requiring a shorter exposure duration to achieve the same relative density of exposed silver grains.
While it is apparent from the results that [125I]Aβ40, [125I]-[N-4ab/Q-4ab]Aβ30, and [125I]Gd[N-4ab/Q-4ab]Aβ30 all label plaques in vivo, only [125I]Gd[N-4ab/Q-4ab]Aβ30 includes a Gd-DTPA moiety which provides contrast enhancement that could be detected by MRI. In a previous study, we reported that [125I]Aβ40 did not label plaques in APP, PS1 mice in vivo, but did so only after polyamine modification (1). Even with polyamine modification, labeled plaques were observed primarily in the medial septum and hippocampus with few labeled plaques observed in the cortex. In that study, much smaller doses of peptide were used (200 μg vs 750 μg). Furthermore, APP, PS1 mice at 27 weeks of age were used in the earlier study, which would have far fewer and smaller neuritic plaques than the 21-month-old mice used in this study. While it might be hypothesized that the BBB of a 21-month-old APP, PS1 mouse may be more permeable than that of a 27-week-old mouse, accounting for in vivo labeling of plaques by [125I]Aβ40 in this study but not in the previous study, in another study, we found no alterations in BBB permeability of APP, PS1 mice up to 12 months of age (17). Even though in this study [125I]Aβ40 was shown to label amyloid plaques in vivo in APP, PS1 mice following IV injection, albeit to a slightly lesser extent based on the slightly smaller numbers of silver grains over the plaques labeled with [125I]Aβ40, the covalent attachment of Gd-DTPA would be necessary to provide the contrast enhancement for the labeled plaques to be able to be detected by MRI, which is the only diagnostic imaging technique with a resolution sufficiently high enough to be able to detect individual plaques. Gd-DTPA substitution alone, however, decreases the BBB permeability of Aβ40 and was shown not to provide contrast enhancement of plaques during MRI of APP, PS1 mouse brains ex vivo following IV injection of Gd-Aβ40 (2). Only after polyamine modification, which increases the BBB permeability of Gd-Aβ40, was (PUT-)Gd-Aβ40 shown to provide contrast enhancement of plaques following IV injection during MRI of APP, PS1 mouse brains ex vivo (2). Although we previously reported that polyamine-modified Gd-Aβ40 provided selective contrast enhancement of plaques in APP, PS1 mice following IV injection and ex vivo MRI (2), the study presented here supports the development of Gd[N-4ab/Q-4ab]Aβ30 as a putative MRI contrast enhancement agent over polyamine-modified Gd-Aβ40 because Aβ30 truncates the neurotoxic amino acid residues found in Aβ40 and is not amyloidogenic like Aβ40. Future toxicology studies might require further truncation (or possibly elongation) of Aβ30. The electrospray ionization mass spectrometry indicates that equimolar concentrations of Gd and the DTPA functional groups of the Aβ30 peptide were sufficient for complete chelation of the peptide. Because of the toxicity of free Gd, appropriate chromatographic procedures would be required to ensure that there is no free Gd in the preparation prior to administration to a patient. Our studies with Gd[N/Q-4ab]Aβ30 injected intravenously in mice at doses from 0.5 to 3.1 μg have not resulted in any obvious toxic effects. Also, it is not known if this contrast agent remains on the amyloid plaques over time and whether this might be detrimental to the patient. Rigorous toxicity studies would be required prior to use with patients.
The percent of injected dose of the radiolabeled peptides that entered the brain in the study presented here was similar to that observed for native and polyamine-modified Aβ40 in our previous report (1), ranging from 0.03 to 0.35% with Aβ30 and Aβ40 exhibiting the lowest level of uptake and the diamine-modified peptides the highest. These results are similar to the BBB permeability measurements in which polyamine- and diamine-modified peptides exhibit significantly increased BBB permeability. The percent of injected dose for radiolabeled Aβ40 in this study is similar to that reported by another laboratory (22). The amount of peptide that is retained on the plaques over time is unknown. Extensive time-course and pharmacokinetic studies are currently underway in our laboratory. In relation to MRI, most commonly used Gd-labeled contrast agents do not cross the BBB. The development of target and organ specific MRI contrast agents is a relatively new field of research, and few data are available. We have shown previously, however, that Gd-labeled, polyamine-modified Aβ40 labels amyloid plaques in quantities sufficient to be detected by MRI in ex vivo APP, PS1 transgenic mouse brain following IV injection (11).
The intent of this study was to design a contrast agent that would allow the detection by MRI of amyloid plaques in the brain of live AD mice and, ultimately, AD patients. Contrast-enhanced, clinical brain MRI examinations routinely use biologically stable gadolinium chelates, such as gadolinium diethylenetriaminepentaacetic acid (Gd-DTPA). Gd-DTPA causes a greater T1 acceleration effect than 2 acceleration; hence, it is primarily the T1 of Gd that is exploited in clinical imaging. The basis for the visualization of amyloid plaques via contrast enhancement in MRI, therefore, is the acceleration of the T1 rate of tissue water protons near amyloid plaques that have been specifically targeted by the Gd-tagged molecular probe. This has been particularly useful for detecting amyloid plaques in the living AD mouse in high-field strength MRI (manuscript in preparation).
While gadolinium complexes are routinely used as MRI contrast agents in clinical imaging, other lanthanide ion coordination complexes may allow for even greater enhanced relaxation at higher field strengths (23, 24). Paramagnetic CEST agents such as Eu3+, Tb3+, Dy3+, Er3+, Tm3+, or Yb3+ alter tissue contrast via chemical exchange saturation transfer of presaturated spins to bulk water (25, 26). The T1 acceleration and contrast enhancement of Gd and especially Fe have been shown to saturate at very high field strengths, however, while these other lanthanides do not (26), thus taking full advantage of the increased resolution of very high field strengths. In future studies, the diamine-modified Aβ derivative could readily be complexed to PARACEST agents for enhanced MRI contrast via water protein exchange of amyloid plaques at high field strengths. In the meantime, since these PARACEST agents have not been tested in human patients and Gd requires much higher field strength to saturate than Fe, Gd provides an adequate contrast enhancement agent for the development of a diagnostic probe.
The spatial resolution of MRI is approximately 30–50 μm, a resolution at which individual plaques that vary in size from 2 to 200 μm in human AD patients should be able to be resolved. This likely will require high magnetic field strength of ≥7 T. Using a high-field strength magnet at 9.4 T, we have been able to image individual plaques in the live AD transgenic mouse on T2-weighted relaxation with an imaging time of 67 min and a spatial resolution of 60 μm × 60 μm × 120 μm (C. R. Jack, Jr., et al., manuscript in preparation). Such imaging time can be used for patients, although this is not yet ideal. It is possible that with further optimization of the imaging parameters, we might be able to decrease this time. Since most clinical magnets are 1.5 or 3 T, our diamine- and gadolinium-substituted Aβ derivative would probably not be able to visualize enhancement of individual plaques at this lower field strength. Instead, the objective would be to visualize bulk tissue enhancement in cortical areas that contain plaques compared to brain areas devoid of plaques. This is analogous to the imaging by micro positron emission tomography (PET), which has a spatial resolution of ~2 mm using probes labeled with radioisotopes. For clinical imaging by PET, therefore, our diamine Aβ derivative could be labeled with radioisotopes such as I,F, In, Ga, and Tc, instead of Gd-DPTA. Again, visualization of bulk tissue enhancement of the diamine-substituted Aβ derivative labeled with a radioisotope by PET may provide an additional direction for this diagnostic probe.
In this study, we substituted with diamine all of the Asp and Glu residues of Aβ30, except for the N-terminal Asp, which was reserved for Gd addition. It has not been determined if fewer or more selective Asp and Glu substitutions will result in even more enhanced targeting to amyloid plaques. Selective substitution of the five Asp and Glu residues would require evaluation of 120 possible combinations. It is reasonable that at least one of these combinations will give improved targeting to the amyloid plaques. Indeed, selective substitutions of these residues may provide insight into the mechanism of enhanced binding to amyloid plaques.
Our previous studies have demonstrated 2–3-fold higher Vp values for the Dutch variant of Aβ than for normal human Aβ1–40 (16). The Dutch variant is caused by a missense mutation in the β-amyloid precursor protein that results in a Glu-to-Gln substitution at position 22 in the Aβ domain (27). This rare autosomal dominant disorder is characterized by extensive cerebral amyloid angiopathy with recurrent and often fatal intracerebral hemorrhages by 50 years of age (28–32). The finding of significantly increased p increased adherence to the vessel walls in different brain regions, which is consistent with the heavy Aβ deposition that has been described in intracerebral vessels with this variant. Because of the structural similarities of Gln at position 22 with glutamyl-4-aminobutane that was synthesized in this study, we thought that not modifying this position might significantly reduce the Vp for the possibility of even higher PS values and more efficient targeting to amyloid plaques. A comparison of [N-4ab/Q-4ab]Aβ30 with and without Q-4ab at position 22, however, revealed no significant differences in the PS or Vp (data not shown). This observation exemplifies the remarkable structural specificity of the missense mutations whereby elevated Vp values observed for the Dutch mutation (glutamine) are not duplicated with glutamyl-4-aminobutane at position 22.
In summary, Gd[N-4ab/Q-4ab]Aβ30 is a derivative of Aβ40 that demonstrates increased BBB permeability and the ability to label amyloid plaques in vitro in both human AD and APP, PS1 mouse brain sections, as well as in vivo in APP, PS1 mice following IV injection. The chemical synthesis of N-R-Fmoc-L-aspartyl-β-N-(4-aminobutyl)carbamic acid tert-butyl ester (3a) and N-R-Fmoc-L-glutamyl-δ-N-(4-aminobutyl)carbamic acid tert-butyl ester (3b) as described in this study and its substitution for Asp and Glu residues during peptide synthesis may be a general procedure for enhancing protein delivery across the blood–brain barrier. Of course, it is important that these substitutions not affect the bioactivity of the final product. This chemical synthesis approach might have wide applications for the delivery of diagnostic and therapeutic proteins across the blood–brain barrier for both the diagnosis and treatment of AD, as well as other neurodegenerative disorders.
Figure 7.

Labeling of amyloid plaques in APP, PS1 transgenic mouse brain in vivo after exposure for 2 weeks. Fixed, frozen brain sections from a 21-month-old APP, PS1 mouse after intravenous injection with 750 μg of [125I]Aβ40 (A and B), [125I]Aβ30 (C and D), [125I][N-4ab/Q-4ab]Aβ30 (E and F), or [125I]Gd[N-4ab/Q-4ab]Aβ30 (G and H) and after being processed for anti-Aβ IH and emulsion autoradiography. For panels A, C, E, and G (cortex), the scale bar is 50 μm. For panels B, D, F, and H (CA1 subfield of the hippocampus), the scale bar is 50 μm.
Figure 8.
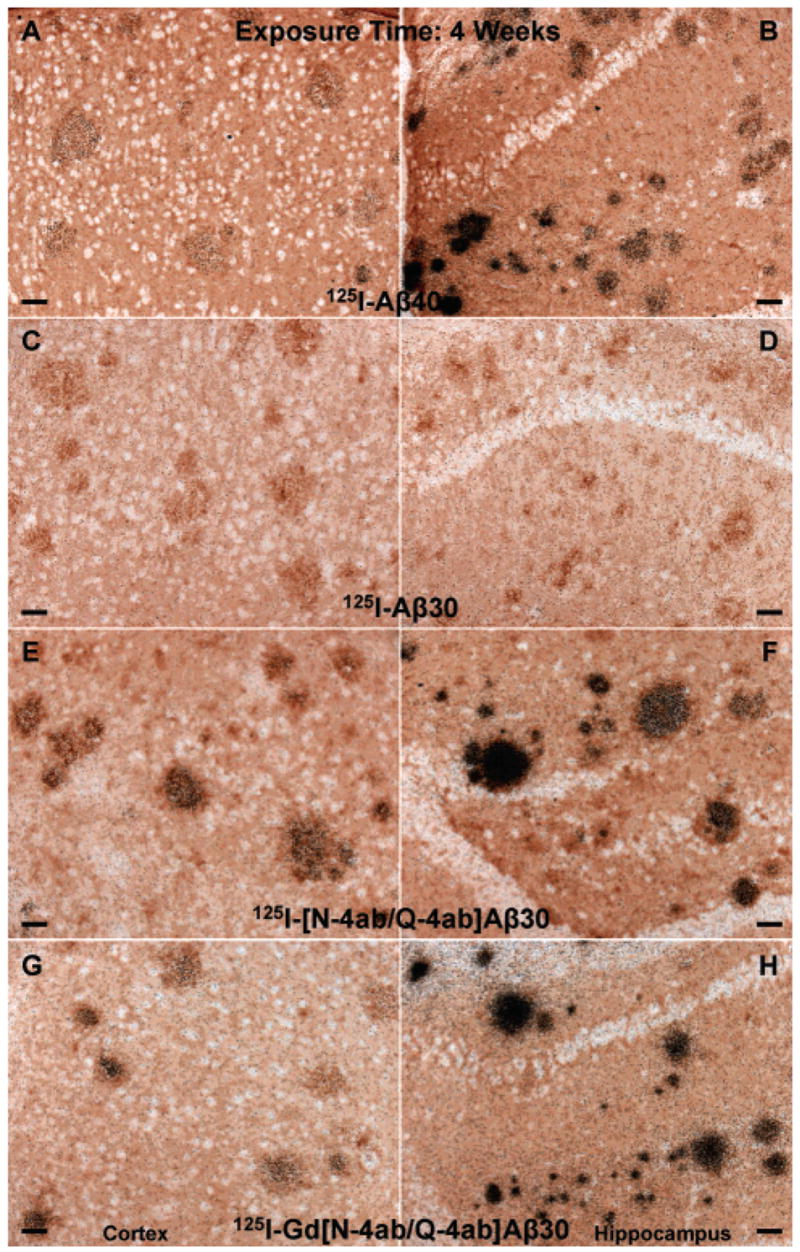
Labeling of amyloid plaques in APP, PS1 transgenic mouse brain in vivo after exposure for 4 weeks. Fixed, frozen brain sections from a 21-month-old APP, PS1 mouse after intravenous injection with 750 μg of [125I]Aβ40 (A and B), [125I]Aβ30 (C and D), [125I][N-4ab/Q-4ab]Aβ30 (E and F), or [125I]Gd[N-4ab/Q-4ab]Aβ30 (G and H) and after being processed for anti-Aβ IH and emulsion autoradiography. For panels A, C, E, and G (cortex), the scale bar is 50 μm. For panels B, D, F, and H (CA1 subfield of the hippocampus), the scale bar is 50 μm.
Acknowledgments
We thank Dr. Karen Duff for the PS1 transgenic mouse line and Dawn Gregor for her excellent technical assistance.
Footnotes
This work was supported in part by the Alzheimer’s Association (J.F.P.), NIA (AG22034) (J.F.P.), and the Mayo Foundation.
References
- 1.Wengenack TM, Curran GL, Poduslo JF. Targeting Alzheimer amyloid plaques in vivo. Nat Biotechnol. 2000;18:868–872. doi: 10.1038/78482. [DOI] [PubMed] [Google Scholar]
- 2.Poduslo JF, Wengenack TM, Curran GL, Wisniewski T, Sigurdsson EM, Macura SI, Borowski BJ, Jack CR., Jr Molecular targeting of Alzheimer’s amyloid plaques for contrast-enhanced magnetic resonance imaging. Neurobiol Dis. 2002;11:315–329. doi: 10.1006/nbdi.2002.0550. [DOI] [PubMed] [Google Scholar]
- 3.Poduslo JF, Curran GL, Berg CT. Macro-molecular permeability across the blood-nerve and blood-brain barriers. Proc Natl Acad Sci USA. 1994;91:5705–5709. doi: 10.1073/pnas.91.12.5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poduslo JF, Curran GL. Increased permeability across the blood-nerve barrier of albumin glycated in vitro and in vivo from patients with diabetic polyneuropathy. Proc Natl Acad Sci USA. 1992;89:2218–2222. doi: 10.1073/pnas.89.6.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poduslo JF, Curran GL. Glycation increases the permeability of proteins across the blood-nerve and blood-brain barriers. Mol Brain Res. 1994;23:157–162. doi: 10.1016/0169-328x(94)90222-4. [DOI] [PubMed] [Google Scholar]
- 6.Poduslo JF, Curran GL. A polyamine modification increases the permeability of proteins at the blood-nerve and blood-brain barriers. J Neurochem. 1996;66:1599–1609. doi: 10.1046/j.1471-4159.1996.66041599.x. [DOI] [PubMed] [Google Scholar]
- 7.Poduslo JF, Curran GL. Increased permeability of superoxide dismutase at the blood-nerve and blood-brain barriers with retained enzymatic activity after covalent modification with the naturally occurring polyamine, putrescine. JNeurochem. 1996;67:734–741. doi: 10.1046/j.1471-4159.1996.67020734.x. [DOI] [PubMed] [Google Scholar]
- 8.Wengenack TM, Curran GL, Olson EE, Poduslo JF. Putrescine-modified catalase with preserved enzymatic activity exhibits increased permeability at the blood-nerve and blood-brain barriers. Brain Res. 1997;767:128–135. doi: 10.1016/s0006-8993(97)00565-9. [DOI] [PubMed] [Google Scholar]
- 9.Poduslo JF, Curran GL, Gill JS. Putrescine-modified nerve growth factor: bioactivity, plasma pharmaco-kinetics, blood-brain/nerve barrier permeability, and nervous system biodistribution. J Neurochem. 1998;71:1651–1660. doi: 10.1046/j.1471-4159.1998.71041651.x. [DOI] [PubMed] [Google Scholar]
- 10.Poduslo JF, Curran GL, Kumar A, Frangione B, Soto C. β-Sheet breaker peptide inhibitor of Alzheimer’s amyloidogenesis with increased blood-brain barrier permeability and resistance to proteolytic degradation in plasma. J Neurobiol. 1999;39:371–382. [PubMed] [Google Scholar]
- 11.Poduslo JF, Whelan SL, Curran GL, Wengenack TM. Therapeutic benefit of polyamine-modified catalase as a scavenger of hydrogen peroxide and nitric oxide in familial amyotrophic lateral sclerosis transgenics. Ann Neurol. 2000;48:943–947. [PubMed] [Google Scholar]
- 12.Hoare DG, Koshland DE., Jr A method for the quantitative modification and estimation of carboxylic acid groups in proteins. J Biol Chem. 1967;242:2447–2453. [PubMed] [Google Scholar]
- 13.Yamada K, Nabeshima T. Animal models of Alzheimer’s disease and evaluation of anti-dementia drugs. Pharmacol Ther. 2000;88:93–113. doi: 10.1016/s0163-7258(00)00081-4. [DOI] [PubMed] [Google Scholar]
- 14.Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, Ocampo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- 15.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang FS, Cole G. Correlative memory deficits, A-β elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 16.Poduslo JF, Curran GL, Haggard JJ, Biere AL, Selkoe DJ. Permeability and residual plasma volume of human, Dutch variant, and rat amyloid β-protein 1–40 at the blood-brain barrier. Neurobiol Dis. 1997;4:27–34. doi: 10.1006/nbdi.1997.0132. [DOI] [PubMed] [Google Scholar]
- 17.Poduslo JF, Curran GL, Wengenack TM, Malester B, Duff K. Permeability of proteins at the blood-brain barrier in the normal adult mouse and double transgenic mouse model of Alzheimer’s disease. Neurobiol Dis. 2001;8:555–567. doi: 10.1006/nbdi.2001.0402. [DOI] [PubMed] [Google Scholar]
- 18.Wengenack TM, Whelan S, Curran GL, Duff KE, Poduslo JF. Quantitative histological analysis of amyloid deposition in Alzheimer’s double transgenic mouse brain. Neuroscience. 2000;101:939–944. doi: 10.1016/s0306-4522(00)00388-2. [DOI] [PubMed] [Google Scholar]
- 19.Poduslo JF, Curran GL, Sanyal B, Selkoe DJ. Receptor-mediated transport of human amyloid β-protein 1–40 and 1–42 at the blood-brain barrier. Neurobiol Dis. 1999;6:190–199. doi: 10.1006/nbdi.1999.0238. [DOI] [PubMed] [Google Scholar]
- 20.Maggio JE, Stimson ER, Ghilardi JR, Allen CJ, Dahl CE, Whitcomb DC, Vigna SR, Vinters HV, Labenski ME, Mantyh PW. Reversible in vitro growth of Alzheimer disease β-amyloid plaques by deposition of labeled amyloid peptide. Proc Natl Acad Sci USA. 1992;89:5462–5466. doi: 10.1073/pnas.89.12.5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuo YM, Kokjohn TA, Beach TG, Sue LI, Brune D, Lopez JC, Kalback WM, Abramowski D, Sturchlerpierrat C, Staufenbiel M, Roher AE. Comparative analysis of amyloid-β chemical structure and amyloid plaque morphology of transgenic mouse and Alzheimer’s disease brains. J Biol Chem. 2001;276:12991–12998. doi: 10.1074/jbc.M007859200. [DOI] [PubMed] [Google Scholar]
- 22.Saito Y, Buciak J, Yang J, Pardridge WM. Vector-mediated delivery of 125I-labeled β-amyloid peptide Aβ1–40through the blood-brain barrier and binding to Alzheimer disease amyloid of the Aβ1–40/vector complex. Proc Natl Acad Sci USA. 1995;92:10227–10231. doi: 10.1073/pnas.92.22.10227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aime S, Botta M, Fasano M, Terreno E. Lan-thanide(III) chelates for NMR biomedical applications. Chem Soc Rev. 1998;27:19–29. [Google Scholar]
- 24.Aime S, Cabella C, Colombatto S, Crich SG, Gianolio E, Maggiore F. Insights into the use of paramagnetic Gd(III) complexes in MR-molecular imaging investigations. J Magn Reson Imaging. 2002;16:394–406. doi: 10.1002/jmri.10180. [DOI] [PubMed] [Google Scholar]
- 25.Elst LV, Roch A, Gillis P, Laurent S, Botteman F, Bulte JWM, Muller RN. Dy-DTPA derivatives as relaxation agents for very high field MRI: The beneficial effects of slow water exchange on the transverse relaxivities. Magn Reson Med. 2002;47:1121–1130. doi: 10.1002/mrm.10163. [DOI] [PubMed] [Google Scholar]
- 26.Zhang S, Merritt M, Woessner DE, Lenkinski RE, Sherry AD. PARACEST Agents: Modulating MRI contrast via water proton exchange. Acc Chem Res. 2003;36:783–790. doi: 10.1021/ar020228m. [DOI] [PubMed] [Google Scholar]
- 27.Levy E, Carman MD, Fernandez-Madrid IJ, Power MD, Lieberburg I, van Duinen SG, Bots GT, Luyendijk W, Frangione B. Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science. 1990;248:1124–1126. doi: 10.1126/science.2111584. [DOI] [PubMed] [Google Scholar]
- 28.van Duinen SG, Castano EM, Prelli F, Bots GT, Luyendijk W, Frangione B. Hereditary cerebral hemorrhage with amyloidosis in patients of Dutch origin is related to Alzheimer disease. Proc Natl Acad Sci USA. 1987;84:5991–5994. doi: 10.1073/pnas.84.16.5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luyendijk W, Bots GT, Vegter-Van der Vlis M, Went LN, Frangione B. Hereditary cerebral haemorrhage caused by cortical amyloid angiopathy. J Neurol Sci. 1988;85:267–280. doi: 10.1016/0022-510x(88)90186-4. [DOI] [PubMed] [Google Scholar]
- 30.Van Broeckhoven C, Haan J, Bakker E, Hardy JA, Van Hul W, Wehnert A, Vegter-Van der Vlis M, Roos RA. Amyloid β protein precursor gene and hereditary cerebral hemorrhage with amyloidosis (Dutch) Science. 1990;248:1120–1122. doi: 10.1126/science.1971458. [DOI] [PubMed] [Google Scholar]
- 31.Haan J, Algra PR, Roos RA. Hereditary cerebral hemorrhage with amyloidosis-Dutch type. Clinical and computed tomographic analysis of 24 cases. Arch Neurol. 1990;47:649–653. doi: 10.1001/archneur.1990.00530060059018. [DOI] [PubMed] [Google Scholar]
- 32.Wattendorff AR, Frangione B, Luyendijk W, Bots GT. Hereditary cerebral haemorrhage with amyloidosis, Dutch type (HCHWA-D): clinicopathological studies. J Neurol Neurosurg Psychiatry. 1995;58:699–705. doi: 10.1136/jnnp.58.6.699. [DOI] [PMC free article] [PubMed] [Google Scholar]